
Indomethacin: Effect of Diffusionless Crystal Growth on Thermal Stability during Long-Term Storage

Abstract
:1. Introduction
2. Experimental
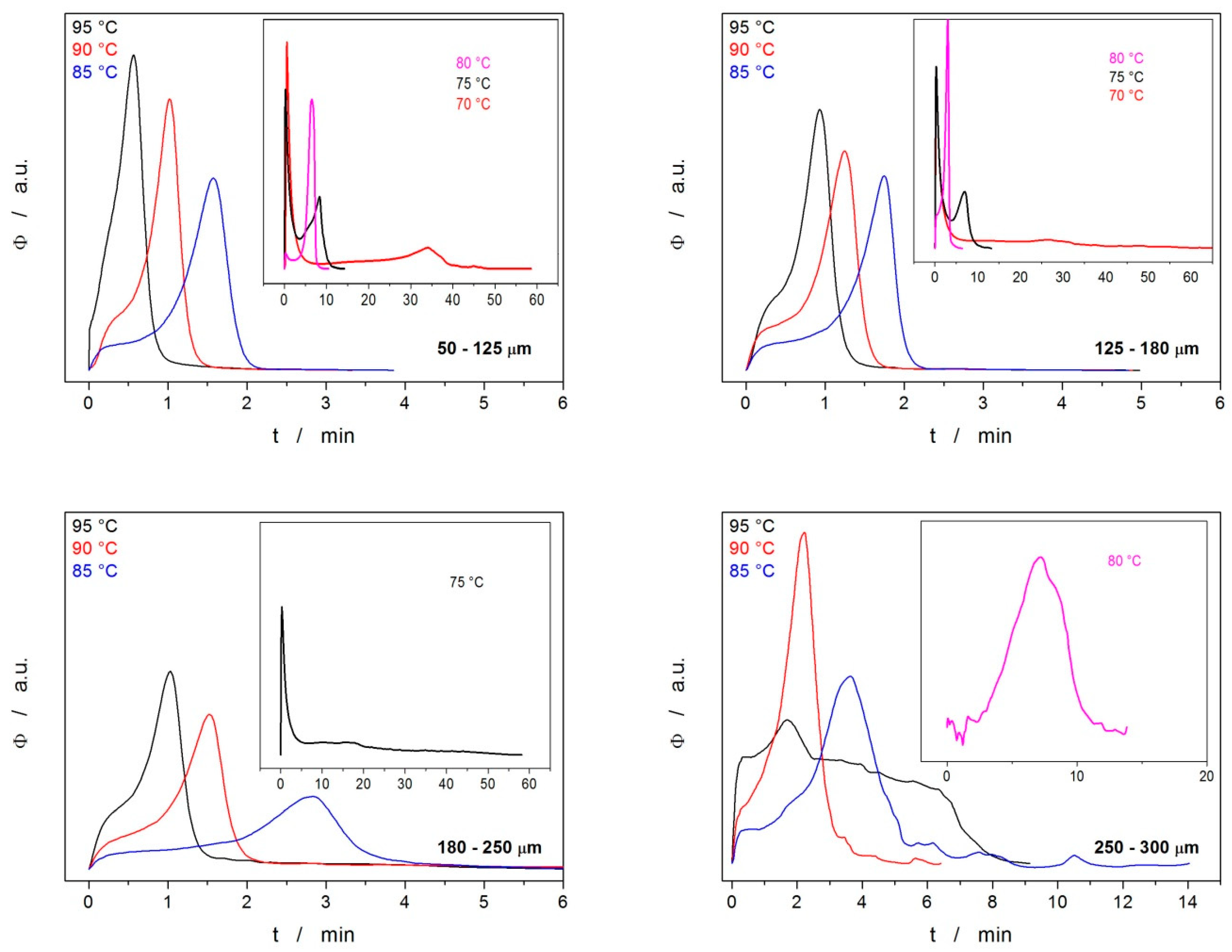
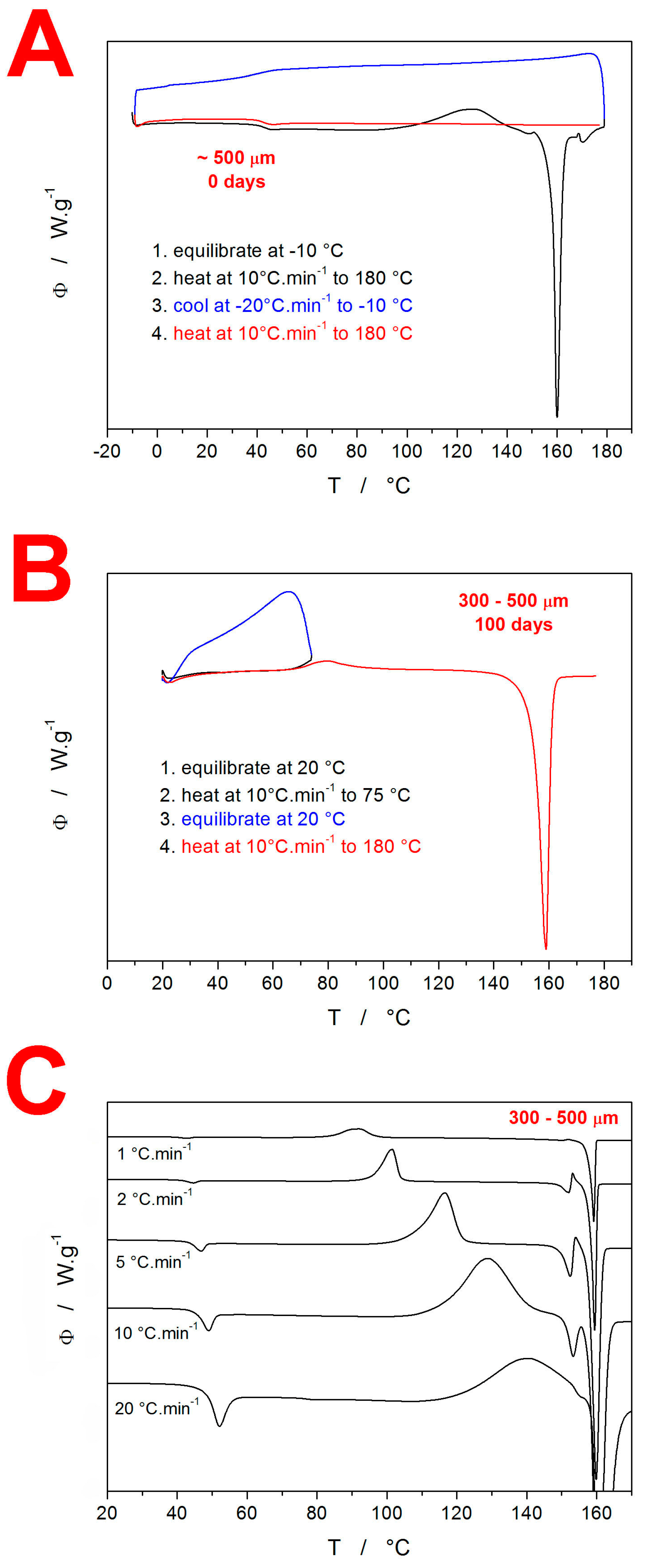
3. Results
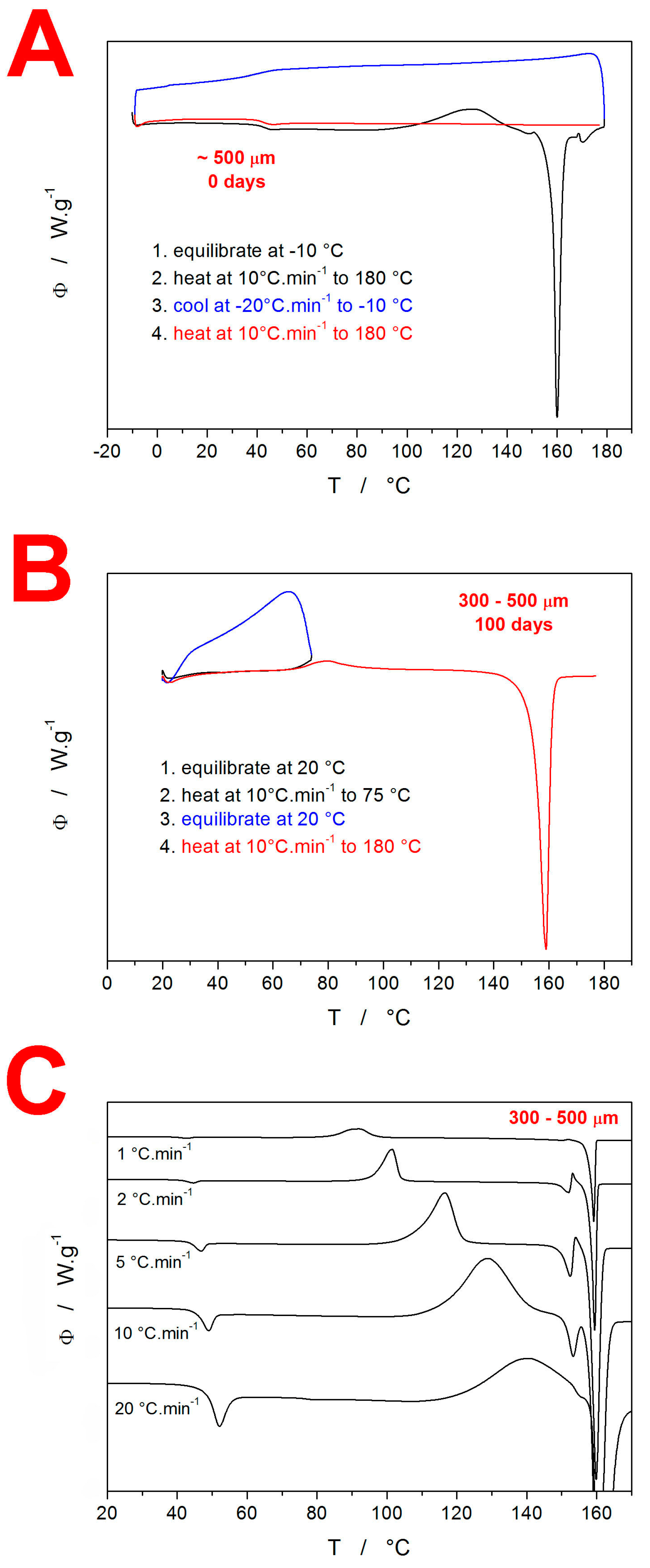
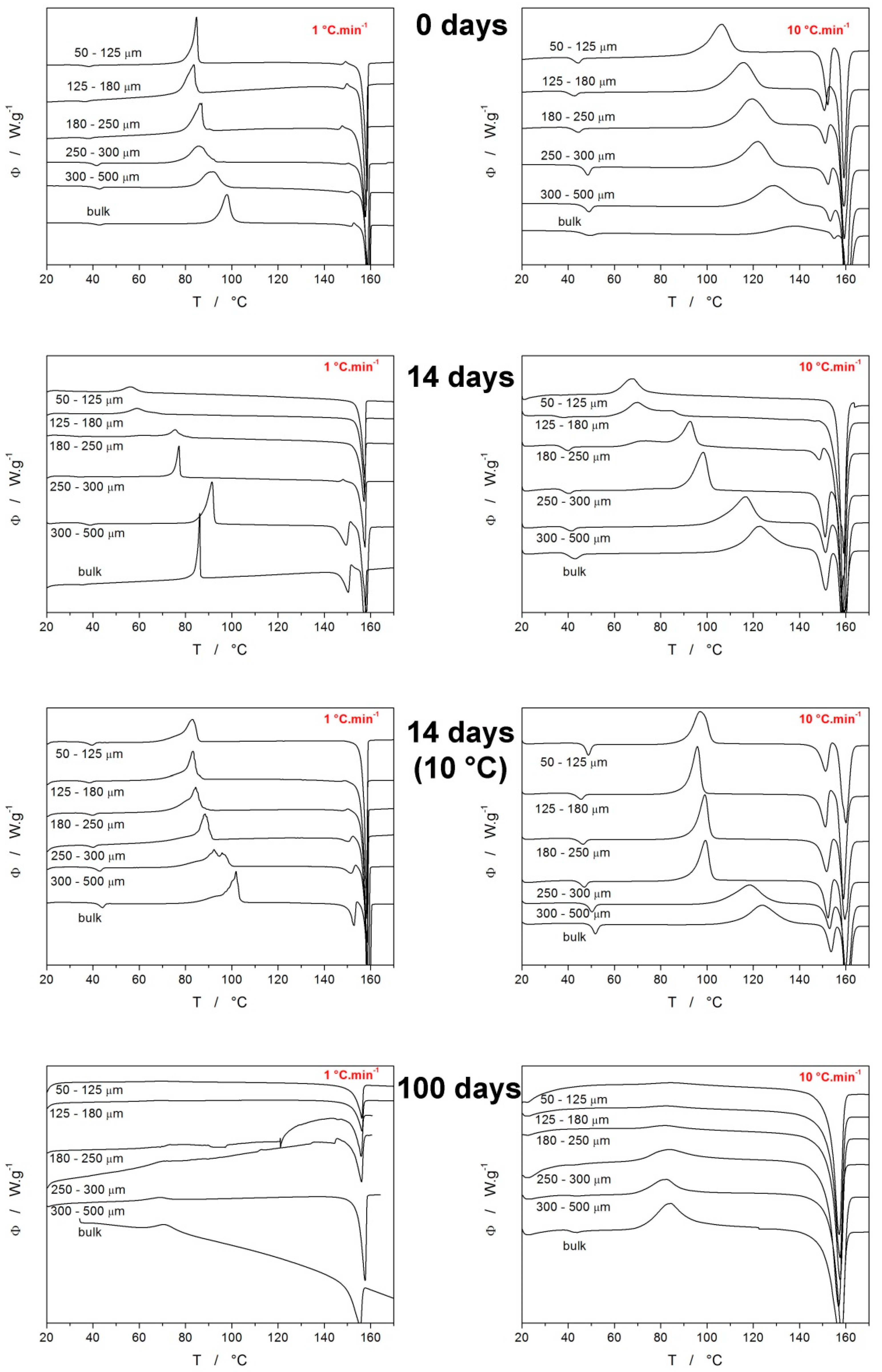
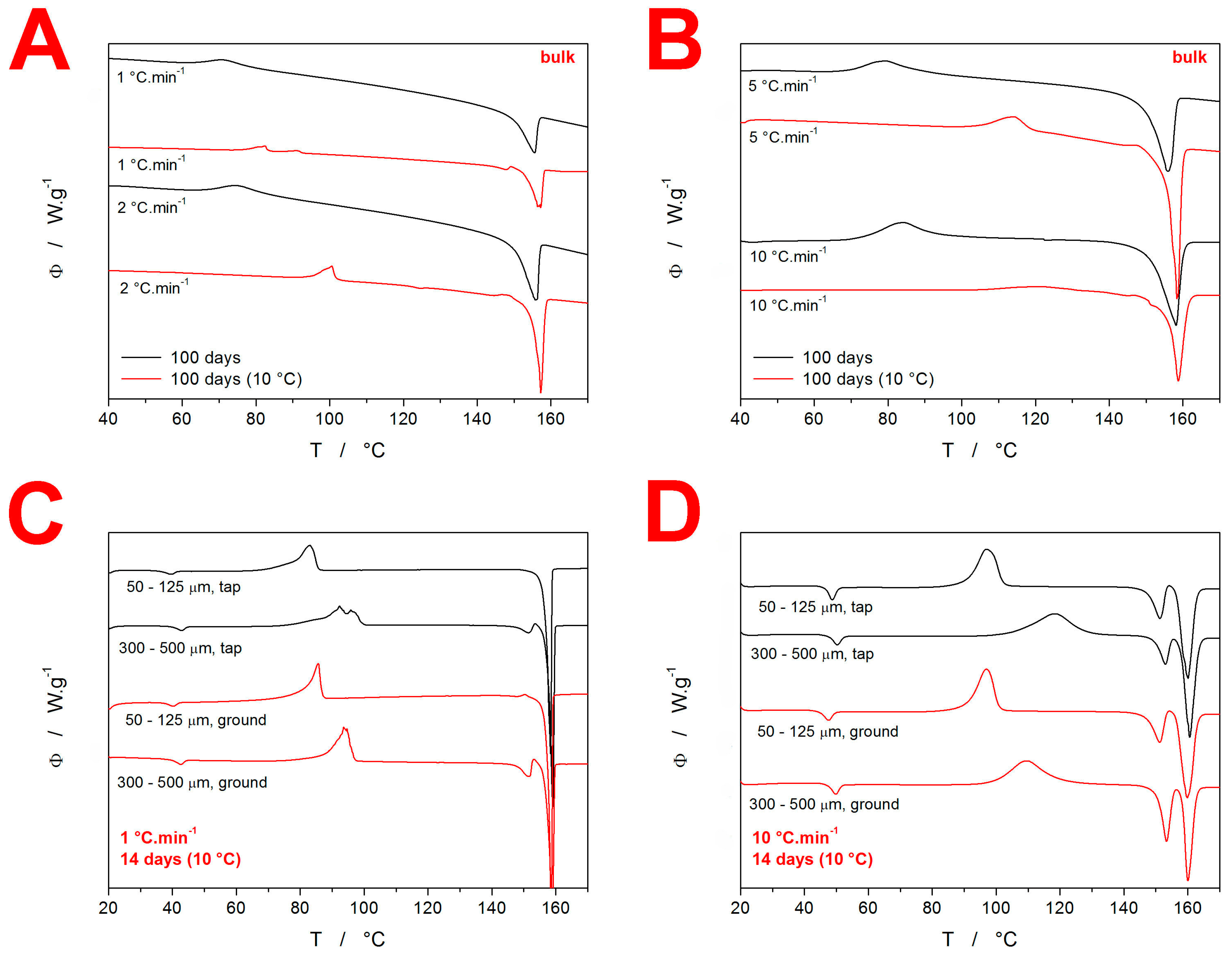
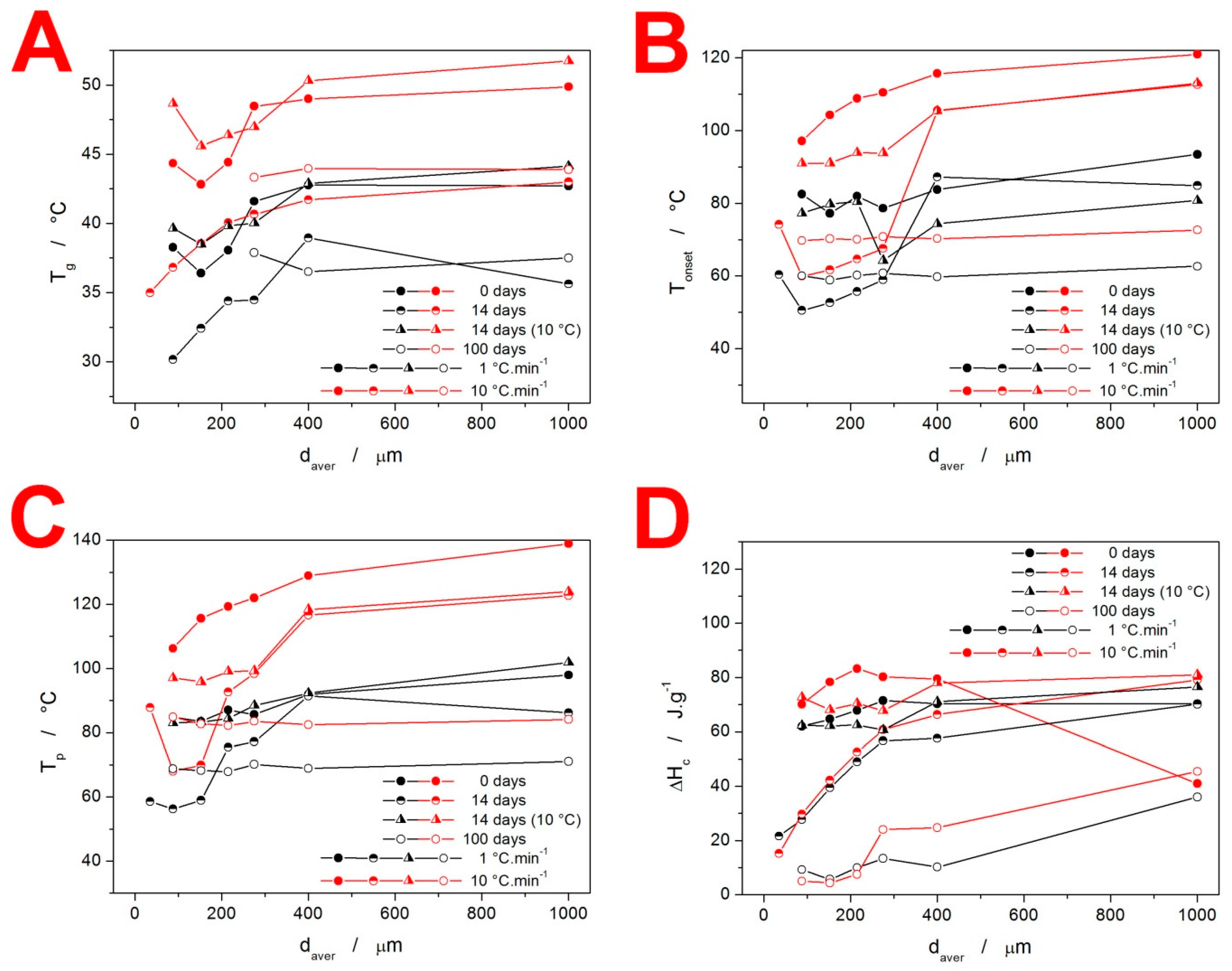
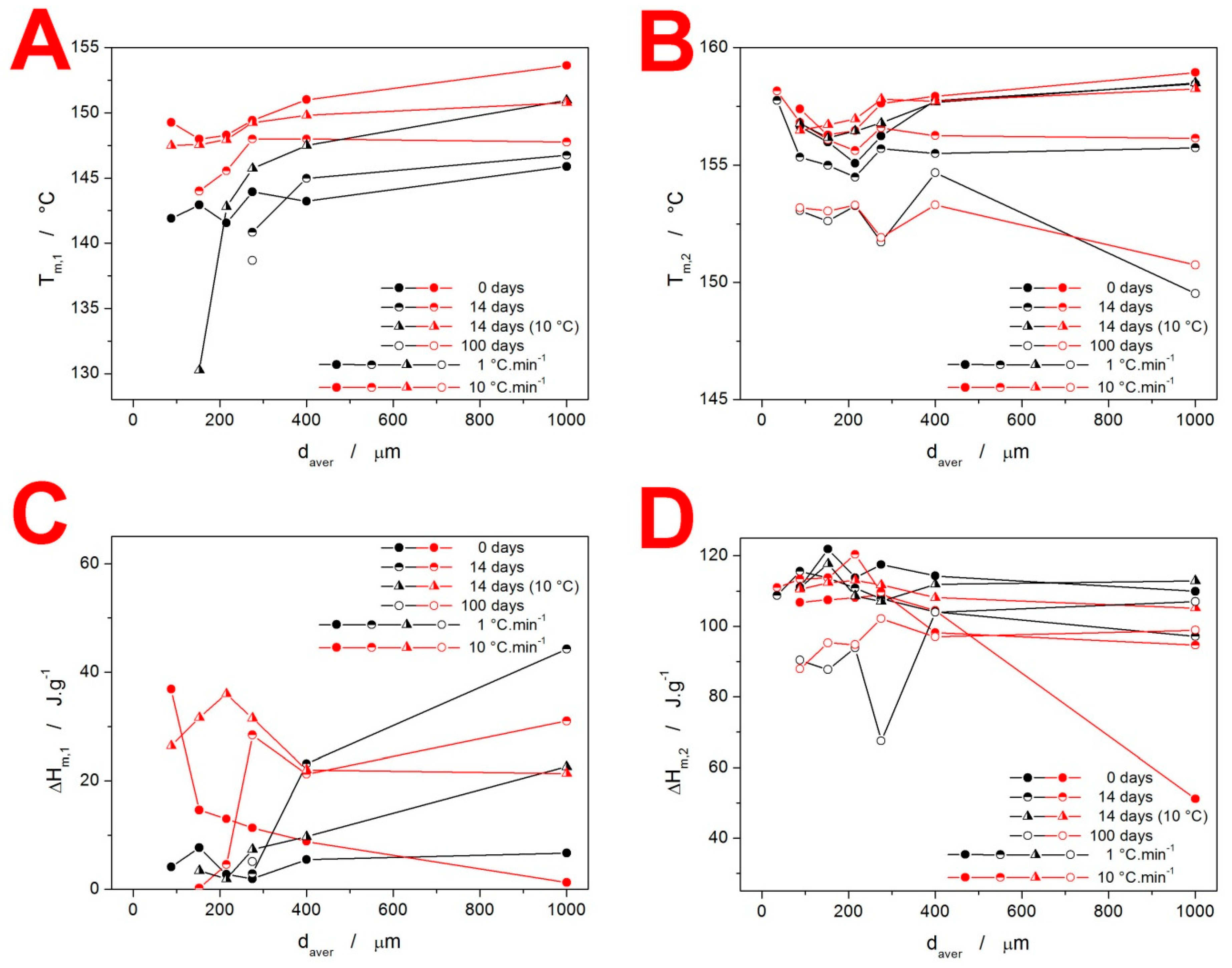
3.1. Differential Scanning Calorimetry
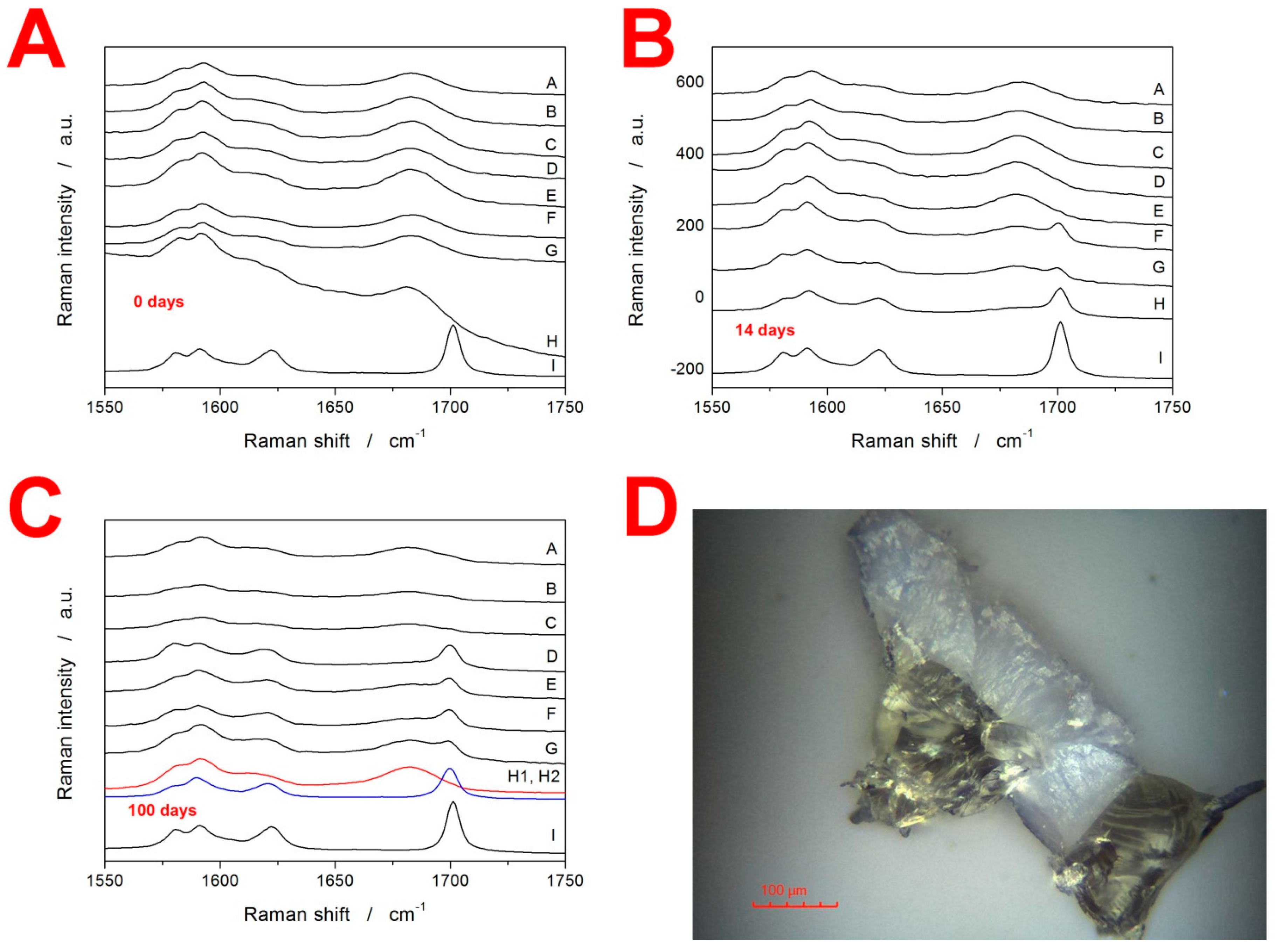
3.2. Raman Spectroscopy
3.3. X-ray Diffraction Analysis and Optical Microscopy
4. Discussion
4.1. Quantification of Thermal Behavior
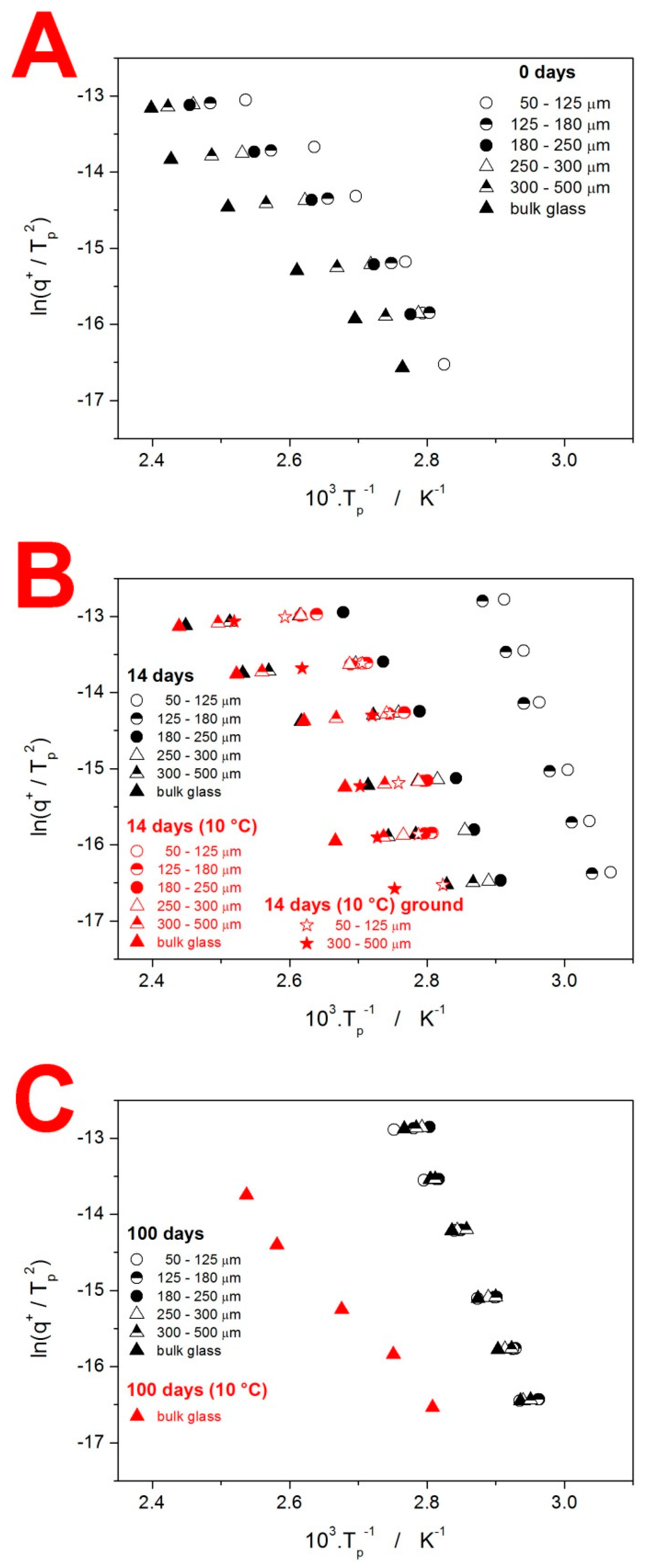
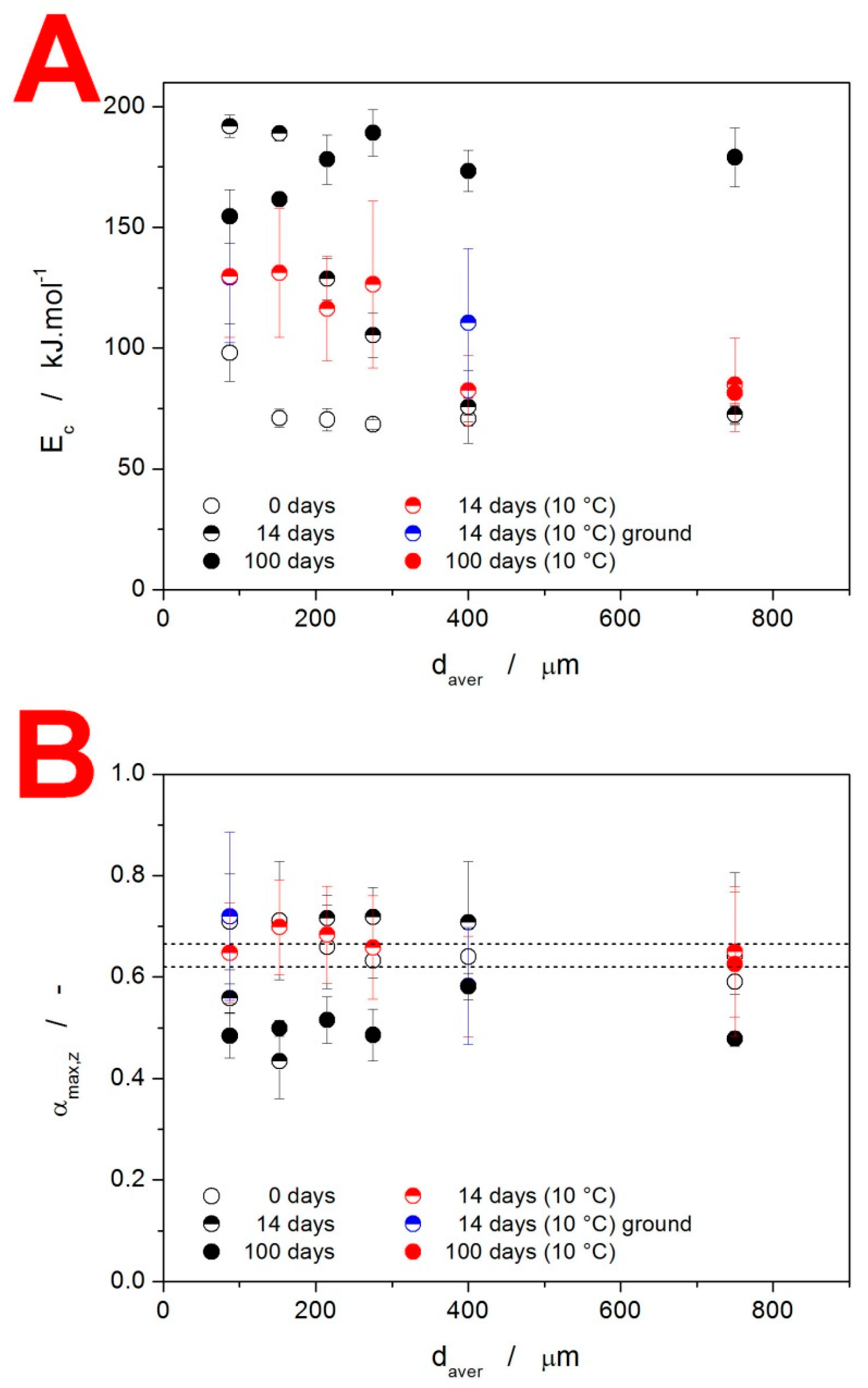
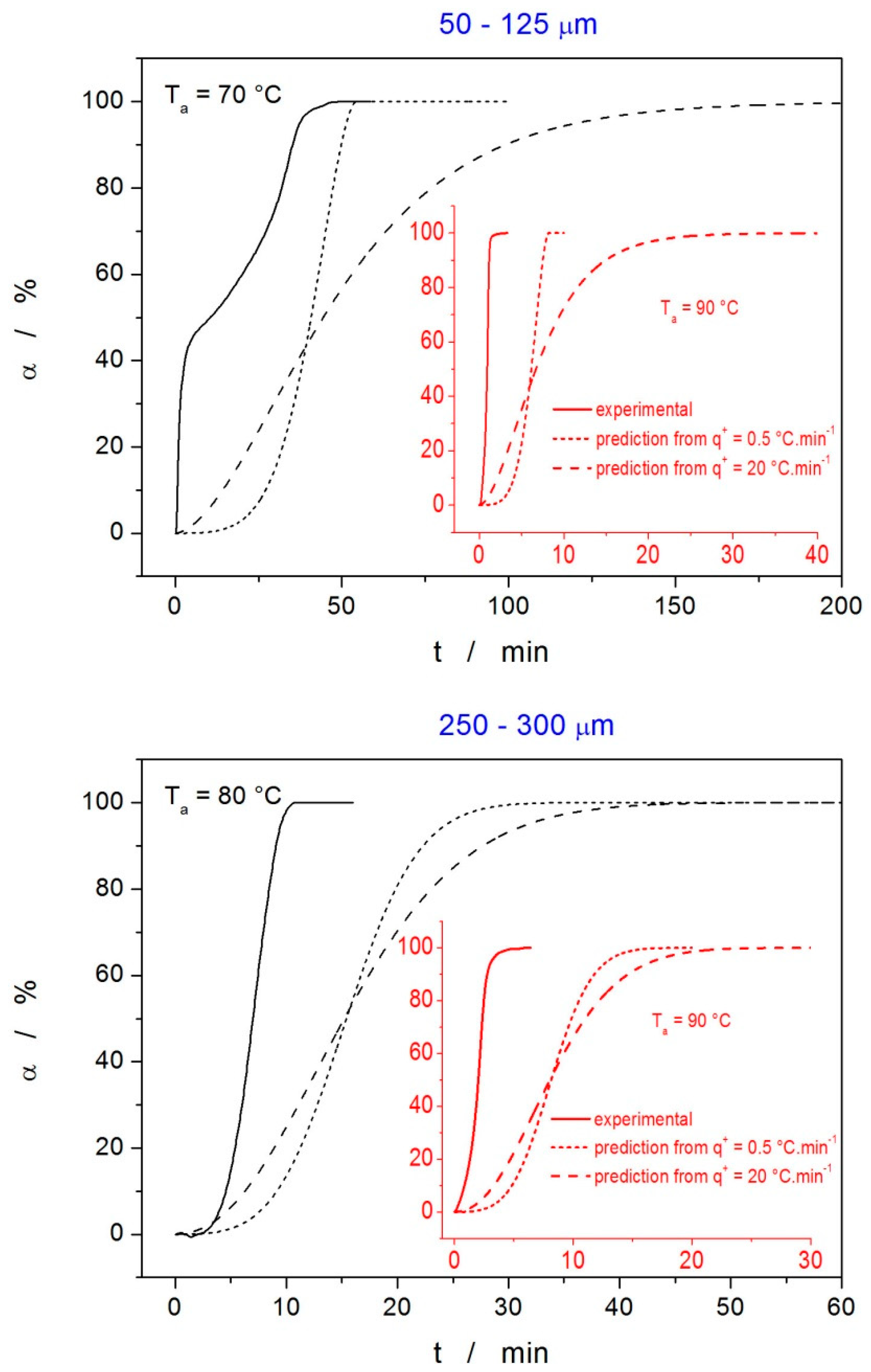
4.2. Crystallization Kinetics
4.3. Kinetic Predictions
5. Conclusions
- The preferential formation/dominance of particular polymorphs is consistent and well reproducible, with the increased formation of the α-IMC phase being associated with high T (and mobility in general), the presence of mechanical defects, and long-term nucleation at the free surface.
- Whereas the nonisothermal crystallization proceeds uniformly (in an apparent single process), isothermal DSC crystallization data show that below ~80 °C, the significantly faster crystal growth from the mechanical defects proceeds independently from the growth at the free surface of the IMC grains.
- At 10 °C and zero humidity, the GC growth proceeds during long-term storage in accordance with the literature reports on crystal growth rate (~100 µm of the γ-IMC phase in 100 days).
- At 25 °C (still well below Tg) and laboratory humidity, the freely stored fine powder samples (with daver ≤ 250–300 µm) exhibited practically no traces of the crystalline phase during the first 14 days of storage; after 100 days, the same was still true for powders with daver ≤ 125–180 µm. This indicates marked suppression of the GC growth by the presence of mechanical defects at these conditions. This finding may open a revolutionary route to the long-term storage of amorphous APIs.
- The freely stored bulk material with no mechanical damage and a smooth surface exhibited zero traces of GC growth (as confirmed by microscopy) after >150 days of storage.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schittny, A.; Huwyler, J.; Puchkov, M. Mechanisms of increased bioavailability through amorphous solid dispersions: A review. Drug Deliv. 2020, 27, 110–127. [Google Scholar] [CrossRef] [PubMed]
- Rumondor, A.C.F.; Dhareshwar, S.S.; Kesisoglou, F. Amorphous solid dispersions of prodrugs: Complementary strategies to increase drug absorption. J. Pharm. Sci. 2016, 105, 2498–2508. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, R.; Lobmann, K.; Strachan, C.J.; Grohganz, H.; Rades, T. Emerging trends in the stabilization of amorphous drugs. Int. J. Pharm. 2013, 453, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Okuda, T.; Lu, X.Y.; Chan, H.K. Amorphous powdrs for inhalation drug delivery. Adv. Drug Deliv. Rev. 2016, 100, 102–115. [Google Scholar] [CrossRef]
- Tomar, D.; Singh, P.K.; Hoque, S.; Modani, S.; Sriram, A.; Kumar, R.; Madan, J.; Khatri, D.; Dua, K. Amorphous systems for delivery of nutraceuticals: Challenges opportunities. Crit. Rev. Food Sci. Nutr. 2020, 62, 1204–1221. [Google Scholar] [CrossRef] [PubMed]
- Murdande, S.B.; Pikal, M.J.; Shanker, R.M.; Bogner, R.H. Aqueous solubility of crystalline and amorphous drugs: Challenges in measurement. Pharm. Dev. Technol. 2011, 16, 187–200. [Google Scholar] [CrossRef]
- Shah, N.; Sandhu, H.; Choi, D.S.; Chokshi, H.; Malick, A.W. Amorphous Solid Dispersions: Theory and Practice; Springer: New York, NY, USA, 2014. [Google Scholar]
- Rams-Baron, M.; Jachowicz, R.; Boldyreva, E.; Zhou, D.; Jamroz, W.; Paluch, M.; Rams-Baron, M.; Jachowicz, R.; Boldyreva, E.; Zhou, D.; et al. Amorphous Drugs—Benefits and Challenges; Springer: Berlin/Heidelberg, Germany, 2018; ISBN 978-3-319-72002-9. [Google Scholar]
- Yu, L. Amorphous pharmaceutical solids: Preparation, characterization and stabilization. Adv. Drug Deliv. Rev. 2001, 48, 27–42. [Google Scholar] [CrossRef]
- Kumar, N.S.K.; Suryanarayanan, R. Crystallization Propensity of Amorphous Pharmaceuticals: Kinetics and Thermodynamics. Mol. Pharm. 2022, 19, 472–483. [Google Scholar] [CrossRef]
- Bhugra, C.; Pikal, M.J. Role of thermodynamic, molecular, and kinetic factors in crystallization from the amorphous state. J. Pharm. Sci. 2008, 97, 1329–1349. [Google Scholar] [CrossRef]
- Lucas, S. The Pharmacology of Indomethacin. Headache 2016, 56, 436–446. [Google Scholar] [CrossRef]
- Draper, M.P.; Martell, R.L.; Levy, S.B. Indomethacin-mediated reversal of multidrug resistance and drug efflux in human and murine cell lines overexpressing MRP, but not P-glycoprotein. Br. J. Cancer. 1997, 75, 810–815. [Google Scholar] [CrossRef]
- Amici, C.; La Frazia, S.; Brunelli, C.; Balsamo, M.; Angelini, M.; Santoro, M.G. Inhibition of viral protein translation by indomethacin in vesicular stomatitis virus infection: Role of eIF2α kinase PKR. Cell Microbiol. 2015, 17, 1391–1404. [Google Scholar] [CrossRef] [PubMed]
- Yalkowsky, S.H.; Dannenfelser, R.M. Aquasol Database of Aqueous Solubility; Version 5; College of Pharmacy, University of Arizona: Tucson, AZ, USA, 1992. [Google Scholar]
- Wu, T.; Yu, L. Surface crystallization of indomethacin below Tg. Pharm. Res. 2006, 23, 2350–2355. [Google Scholar] [CrossRef] [PubMed]
- Rautaniemi, K.; Vuorimma-Laukkannen, E.; Strachan, C.J.; Laaksonen, T. Crystallization Kinetics of an Amorphous Pharmaceutical Compound Using Fluorescence-Lifetime-Imaging Microscopy. Mol. Pharm. 2018, 15, 1964–1971. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhu, L.; Kearns, K.L.; Ediger, M.D.; Yu, L. Glasses crystallize rapidly at free surfaces by growing crystals upward. Proc. Natl. Acad. Sci. USA 2011, 108, 5990–5995. [Google Scholar] [CrossRef]
- Einfalt, T.; Planinsek, O.; Hrovat, K. Methods of amorphization and investigation of the amorphous state. Acta Pharm. 2013, 63, 305–334. [Google Scholar] [CrossRef]
- Wu, T.; Sun, Y.; Li, N.; de Villiers, M.M.; Yu, L. Inhibiting Surface Crystallization of Amorphous Indomethacin by Nanocoating. Langmuir 2007, 23, 5148–5153. [Google Scholar] [CrossRef]
- Van Duong, T.; Van Humbeeck, J.; Van den Mooter, G. Crystallization kinetics of indomethacin/polyethylene glycol dispersions containing high drug loadings. Mol. Pharmaceutics 2015, 12, 2493–2504. [Google Scholar] [CrossRef]
- Planinsek, O.; Zadnik, J.; Kunaver, M.; Srcic, S.; Godec, A. Structural Evolution of Indomethacin Particles upon Milling: Time-Resolved Quantification and Localization of Disordered Structure Studied by IGC and DSC. J. Pharm. Sci. 2010, 99, 1968–1981. [Google Scholar] [CrossRef]
- Romanová, J.; Svoboda, R.; Obadalová, I.; Beneš, L.; Pekárek, T.; Krejčík, L.; Komersová, A. Amorphous Enzalutamide—non-isothermal recrystallization kinetics and thermal stability. Thermochim. Acta 2018, 655, 134–141. [Google Scholar] [CrossRef]
- Svoboda, R.; Romanová, J.; Šlang, S.; Obadalová, I.; Komersová, A. Influence of particle size and manufacturing conditions on the recrystallization of amorphous Enzalutamide. Eur. J. Pharm. Sci. 2020, 153, 105468. [Google Scholar] [CrossRef]
- Svoboda, R.; Košťálová, D.; Krbal, M.; Komersová, A. Indomethacin: The Interplay between Structural Relaxation, Viscous Flow and Crystal Growth. Molecules 2022, 27, 5668. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K. Crystallization Tendency of Pharmaceutical Glasses: Relevance to Compound Properties, Impact of Formulation Process, and Implications for Design of Amorphous Solid Dispersions. Pharmaceutics 2019, 11, 202. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Harada, T.; Yoshihashi, Y.; Yonemochi, E.; Terada, K.; Moriyama, H. Correlation between Glass-Forming Ability and Fragility of Pharmaceutical Compounds. J. Phys. Chem. B 2015, 119, 4873–4880. [Google Scholar] [CrossRef] [PubMed]
- Van Duong, T.; Lüdeker, D.; Van Bockstal, P.-J.; De Beer, T.; Van Humbeeck, J.; Van den Mooter, G. Polymorphism of Indomethacin in Semicrystalline Dispersions: Formation, Transformation, and Segregation. Mol. Pharm. 2018, 15, 1037–1051. [Google Scholar] [CrossRef] [PubMed]
- Surwase, S.A.; Boetker, J.; Saville, D.; Boyd, B.; Gordon, K.; Peltonen, L.; Strachan, C.J. Indomethacin: New Polymorphs of an Old Drug. Mol. Pharm. 2013, 10, 4472–4480. [Google Scholar] [CrossRef] [PubMed]
- Alonzo, D.E.; Zhang, G.G.Z.; Zhou, D.; Gao, Y.; Taylor, L.S. Understanding the Behavior of Amorphous Pharmaceutical Systems during Dissolution. Pharm. Res. 2010, 27, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Erdemir, D.; Myerson, A.S. Crystal Polymorphism in Chemical Process Development. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 259–280. [Google Scholar] [CrossRef]
- Musumeci, D.; Hasebe, M.; Yu, L. Crystallization of Organic Glasses: How Does Liquid Flow Damage Surface Crystal Growth? Cryst. Growth Des. 2016, 16, 2931–2936. [Google Scholar] [CrossRef]
- Hasebe, M.; Musumeci, D.; Powell, C.T.; Cai, T.; Gunn, E.; Zhu, L.; Yu, L. Fast Surface Crystal Growth on Molecular Glasses and Its Termination by the Onset of Fluidity. J. Phys. Chem. B 2014, 118, 7638–7646. [Google Scholar] [CrossRef]
- Newman, A.; Zografi, G. WhatWe Need to Know about Solid-State Isothermal Crystallization of Organic Molecules from the Amorphous State below the Glass Transition Temperature. Mol. Pharm. 2020, 17, 1761–1777. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Brian, C.W.; Yu, L. Fast Surface Diffusion of Amorphous o-Terphenyl and Its Competition with Viscous Flow in Surface Evolution. J. Phys. Chem. B 2015, 119, 5071–5078. [Google Scholar] [CrossRef]
- Andronis, V.; Zografi, G. Crystal nucleation and growth of indomethacin polymorphs from the amorphous state. J. Non-Cryst. Solids 2000, 271, 236–248. [Google Scholar] [CrossRef]
- Ueda, H.; Ida, Y.; Kadota, K.; Tozuka, Y. Raman mapping for kinetic analysis of crystallization of amorphous drug based on distributional images. Int. J. Pharm. 2014, 462, 115–122. [Google Scholar] [CrossRef]
- Tool, Q. Relation between inelastic deformability and thermal expansion of glass in its annealing range. J. Am. Ceram. Soc. 1946, 29, 240. [Google Scholar] [CrossRef]
- Naraynaswamy, S. A model of structural relaxation in glass. J. Am. Ceram. Soc. 1971, 54, 491. [Google Scholar] [CrossRef]
- Moynihan, T.; Easteal, A.J.; DeBolt, M.A.; Tucker, J. Dependence of the fictive temperature of glass on cooling rate. J. Am. Ceram. Soc. 1976, 59, 12. [Google Scholar] [CrossRef]
- Šesták, J. Thermophysical Properties of Solids, Their Measurements and Theoretical Analysis; Elsevier: Amsterdam, The Netherlands, 1984. [Google Scholar]
- Svoboda, R. Crystallization of glasses—When to use the Johnson-Mehl-Avrami kinetics? J. Eur. Ceram. Soc. 2021, 41, 7862–7867. [Google Scholar] [CrossRef]
- Johnson, W.A.; Mehl, K.F. Reaction kinetics in processes of nucleation and growth. Trans. Am. Inst. Min. (Metall) Eng. 1939, 135, 416–442. [Google Scholar]
- Avrami, M. Kinetics of phase change I–general theory. J. Chem. Phys. 1939, 7, 1103–1112. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of phase change. II–transformation-time relations for random distribution of nuclei. J. Chem. Phys. 1940, 7, 212–224. [Google Scholar] [CrossRef]
- Avrami, M. Granulation, phase change, and microstructure—kinetics of phase change III. J. Chem. Phys. 1941, 7, 177–184. [Google Scholar] [CrossRef]
- Svoboda, R.; Chovanec, J.; Slang, S.; Beneš, L.; Konrád, P. Single-curve multivariate kinetic analysis: Application to the crystallization of commercial Fe-Si-Cr-B amorphous alloys. J. Alloys Compd. 2021, 889, 161672. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| daver/µm | 50–125 | 250–300 | ||
| q+/°C·min−1 | 0.5 | 20 | 0.5 | 20 |
| Ec/kJ·mol−1 | 98 | 98 | 68 | 68 |
| log(A/s) | 12.1551 | 11.6612 | 7.6702 | 7.3840 |
| MAC | 0.8235 | 0.4249 | 0.7781 | 0.5538 |
| NAC | 0.4209 | 0.9941 | 0.8679 | 0.8719 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Svoboda, R.; Koutná, N.; Košťálová, D.; Krbal, M.; Komersová, A. Indomethacin: Effect of Diffusionless Crystal Growth on Thermal Stability during Long-Term Storage. Molecules 2023, 28, 1568. https://doi.org/10.3390/molecules28041568
Svoboda R, Koutná N, Košťálová D, Krbal M, Komersová A. Indomethacin: Effect of Diffusionless Crystal Growth on Thermal Stability during Long-Term Storage. Molecules. 2023; 28(4):1568. https://doi.org/10.3390/molecules28041568
Chicago/Turabian StyleSvoboda, Roman, Nicola Koutná, Daniela Košťálová, Miloš Krbal, and Alena Komersová. 2023. "Indomethacin: Effect of Diffusionless Crystal Growth on Thermal Stability during Long-Term Storage" Molecules 28, no. 4: 1568. https://doi.org/10.3390/molecules28041568
APA StyleSvoboda, R., Koutná, N., Košťálová, D., Krbal, M., & Komersová, A. (2023). Indomethacin: Effect of Diffusionless Crystal Growth on Thermal Stability during Long-Term Storage. Molecules, 28(4), 1568. https://doi.org/10.3390/molecules28041568