Abstract
Herein we report a feasible study concerning the synthesis and the in vitro antimicrobial activity of some new homodrimane sesquiterpenoids with a benzimidazole unit. Based on some homodrimane carboxylic acids, on their acyl chlorides and intermediate monoamides, a series of seven N-homodrimenoyl-2-amino-1,3-benzimidazoles and 2-homodrimenyl-1,3-benzimidazoles was synthesized. The syntheses involved the decarboxylative cyclization and condensation of the said acids or acyl chlorides with o-phenylendiamine and 2-aminobenzimidazole, as well as the p-TsOH-mediated cyclodehydration of the said monoacylamides. The structures of the synthesized compounds have been fully confirmed, including by the X-ray diffraction. Their biological activities were evaluated on five species of fungi (Aspergillus niger, Fusarium solani, Penicillium chrysogenum, P. frequentans, and Alternaria alternata) and two strains of bacteria (Bacillus sp. and Pseudomonas aeruginosa). Compounds 7 and 20 showed higher antifungal (MIC = 0.064 and 0.05 μg/mL) and antibacterial (MIC = 0.05 and 0.032 μg/mL) activities compared to those of the standards: caspofungin (MIC = 0.32 μg/mL) and kanamycin (MIC = 2.0 μg/mL), and compounds 4, 10, 14, and 19 had moderate activities.
1. Introduction
In recent years, many countries have encountered microbial infections that are rapidly spreading, as they are becoming one of the most serious problems [1,2]. Those global trends stimulate the design of new molecular structures with antimicrobial properties, which could lead to new and effective medicinal preparations. Natural products have proven to be an important source of novel biologically active compounds because their natural origin implies biocompatibility, selective biological activity, and low toxicity. Drimane sesquiterpenoids are just such natural or synthetic compounds with wide spectra of applications in medicine, pharmaceuticals, cosmetics, and agriculture. Particular attention is paid to drimane sesquiterpenoids that exhibit certain biological properties, especially those with anticancer, antimicrobial, antifungal, antimalarial, antidiabetic, etc., activities [3,4,5,6,7,8,9].
On the other hand, many pharmaceuticals that mimic bioactive natural products are known to contain heterocycles. Among them are the benzimidazole derivatives that have found practical applications in various fields because they have numerous pharmacological activities such as antihypertensive, anticancer, antiviral, antidiabetic, antimicrobial, etc. [10].
The synthesis of molecules with a hybrid skeleton is often used in the design of drugs and especially in preparations with a promising biological activity. This approach is based on the combination of several pharmacophores which produce compounds with a combined skeleton and have a higher bioactivity than known drugs.
The synthesis of terpene—heterocyclic compounds has seen a vertiginous development in the last 10 years. A great number of molecular hybrids containing a terpene unit and one of the following heterocycles: diazine [11,12], 1,2,4-triazole and carbazole [13,14], azaheterocyclic [15,16], hydrazinecarbothioamide and 1,2,4-triazole [17], 1,3,4-oxadiazole and 1,3,4-thiadiazole [18], thiosemicarbazone and 1,3-thiazole [19], or benzothiazole [20], many of which showed excellent antifungal and/or antibacterial activity, were reported elsewhere.
The purpose of the present research was the development of original methods for the preparation of new terpene—heterocyclic derivatives based on the available natural diterpenoid—sclareol, and the designing of natural chiral molecules of interest to the pharmaceutical industry. Herein, we report the results of the synthesis of novel homodrimane sesquiterpenoids containing 2-substituted 1,3-benzimidazole and N-substituted 2-amino-1,3-benzimidazole, and their antimicrobial properties evaluation.
2. Results and Discussion
2.1. Synthesis and Characterization
Starting from sclareolide (1), carboxylic acids 2, 5, and 8 were obtained in five, six, and three steps, with overall yields of 81%, 62%, and 89%, respectively [13,21,22] (Scheme 1). The intermediate carboxylic acid 12 was obtained based on (-)-sclareol 11 in two steps with the overall yield of 75% [23].
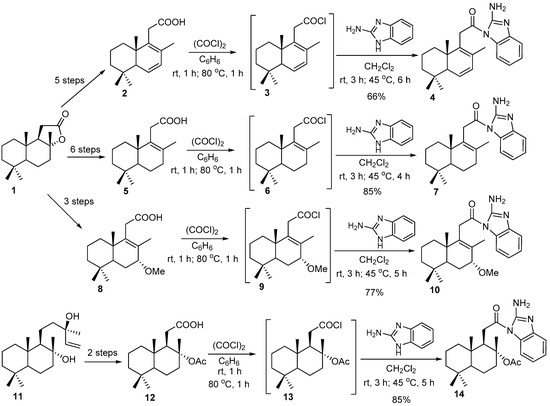
Scheme 1.
Synthesis of N-substituted 2-amino-1,3-aminobenzimidazoles 4, 7, 10, and 14 from carboxylic acids 2, 5, 8, 12, and 2-aminobenzimidazole.
In continuation of our previous work, a series of new N-homodrimenoyl-2-amino-1,3-benzimidazoles was prepared starting from the intermediate carboxylic acids 2, 5, 8, and 12, via their acyl chlorides 3, 6, 9, and 13 generated in situ. The desired N-substituted 2-amino-1,3-aminobenzimidazoles 4, 7, 10, and 14 were obtained, with yields between 66–85%, by acylation of 2-amino-1,3-aminobenzimidazole with the mentioned homodrimane acyl chlorides under the mentioned conditions [12] (Scheme 1).
According to the NMR spectra, the hybrids involved both heterocyclic and terpene units, and their accurate masses were confirmed by the high-resolution mass spectrometry (HRMS). All proton spectra of compounds 4, 7, 10, and 14 include the signals of aromatic protons in a range of 7.09–7.48 ppm, together with the signals specific for terpene units such as singlets of C8-bonded methyl groups at 1.54–1.69 ppm and doublets of C6- and C7-bonded protons at 3.88 and 5.95, or C7-bonded methoxy groups at 3.52 ppm, and broad singlets of amine protons in a range of 7.00–7.29 ppm. The structures of the reported N-substituted 2-amino-benzimidazoles were additionally confirmed by the 13C NMR spectra.
Several attempts to obtain desired benzimidazoles by the direct heterocyclization of acids 2, 5, 8, and 12 with o-phenylenediamine in the presence of 4N HCl [24], glacial AcOH [25] or BF3•OEt2 [26] gave no results. In the case of the treatment with triphenylphosphine and triethylamine [27], the monoacylated derivatives 15, 17, 19, and 20 were afforded in the yields depicted in Scheme 2. In the case of acids 2 and 5, diacylated derivatives 16 and 18 were also obtained, respectively.
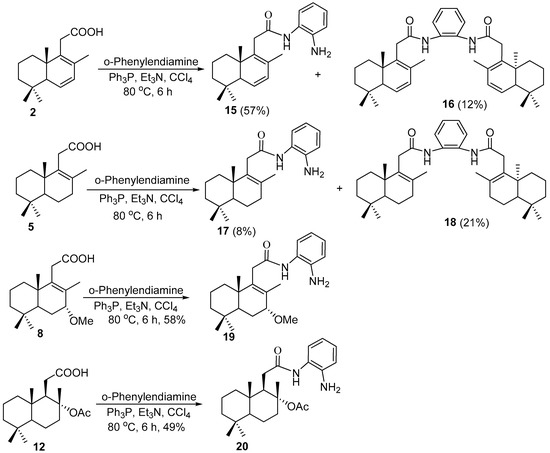
Scheme 2.
Synthesis of monoacilated precursors 15, 17, 19, and 20 from carboxylic acids 2, 5, 8, 12, and o-phenylendiamine.
The structures of the synthesized compounds were confirmed by the 1H, 13C, 15N, and 2D NMR spectroscopy and by the HRMS analysis, and finally, in the case of amide 20, by the single-crystal X-ray diffraction (XRD). The formation of compounds 15, 17, 19, and 20 was proven, first of all, by the presence of signals attributed to aromatic protons from a a common phenylene unit in a range of 6.74–7.29 ppm, and broad singlets of aminic and amidic protons in a range of 3.79–3.88 ppm and 7.55–8.31 ppm, respectively. In addition, some individual signals, such as a singlet corresponding to protons of C7-bonded methoxy group at 3.36 ppm, a singlet corresponding to protons of C8-bonded acethoxy group at 1.92 ppm, or a doublet of doublets of C6- and C7-bonded protons at 5.90 and 5.95 ppm, confirmed the presence of a terpene unit. Those structures were fully confirmed by the carbon spectral data.
The structural analysis of compound 16 by the 1H, 13C, 1H/1H COSY, and the 1H/13C HSQC NMR spectra suggested the presence of an isolated spin system: CH2CH2CH2 (C1 to C3) (Figure 1). In the 1H/13C HMBC spectrum, the correlations of H-C5,5′ with two sp2 hybridized carbons (C6,6′, δC 129.1 and C7,7′, δC 129.3) have confirmed the presence of the Δ6,7 double bond, which was also supported by the correlations of H3-C17,17′ with C7,7′.
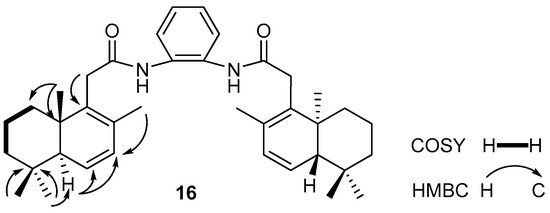
Figure 1.
Selected COSY and HMBC correlations for compound 16.
As mentioned above, the chemical composition and the crystal structure of compound 20 was confirmed via XRD. As shown in Figure 2, the asymmetric part of the unit cell consists of one molecular unit, which corresponds to that supposed on the base of the NMR spectra. There is no co-crystallized solvate molecule in the crystal. The values of the bond distances and angles are summarized in Table S1. The analysis of the crystal structure showed the presence of different fragments that are potential proton donors or proton acceptors, which creates premises for noncovalent intermolecular interactions. Therefore, the main structural motif is characterized as a 2D supramolecular layer assembled via the network of N−H∙∙∙O hydrogen bonding interactions, as shown in Figure 3.
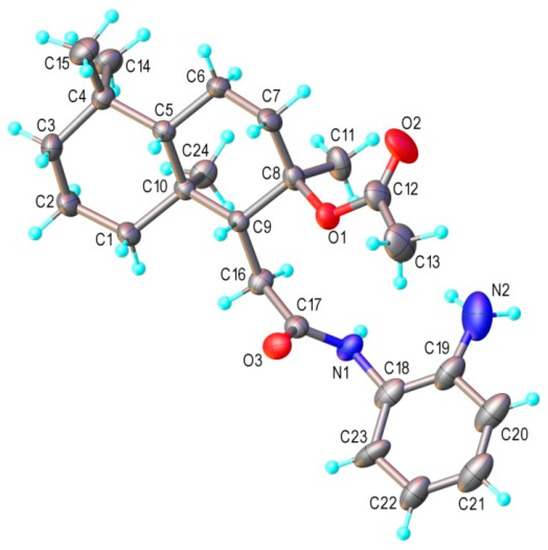
Figure 2.
X-ray molecular structure of compound 20, with atoms labeling and thermal ellipsoids at 50% level.
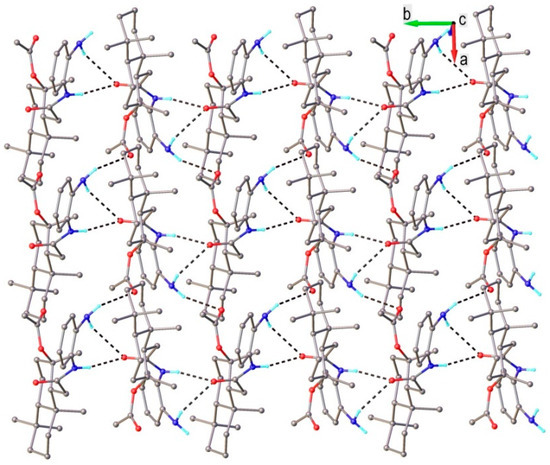
Figure 3.
Intermolecular hydrogen bonding in crystal 20 showing the formation of a 2D supramolecular network. Hydrogen bonds parameters: N1-H···O3 [N1-H 0.86 Å, H···O3(1 − x,−0.5 + y, 1 – z) 2.05 Å, N1···O3 2.892(4) Å, ∠N1HO3 164.6°]; N2-H···O3 [N2-H 0.87 Å, H···O3(−x,−0.5 + y, 1 – z) 2.46 Å, N2···O3 3.17(5) Å, ∠N2HO3 128.8°]; N2-H···O2 [N2-H 0.88 Å, H···O2(−x,−0.5 + y, 1 – z) 2.52 Å, N2···O2 3.20(2) Å, ∠N2HO2 134.0°].
In continuation, the cyclodehydration of the resulting monoacylamides 15, 17, 19, and 20 with p-TsOH in toluene [28] was performed. In the case of monoacylamides 15 and 19, 2-substituted benzimidazoles 21 and 22 were obtained (Scheme 3). The formation of the double unsaturated benzimidazole 22 from amides 15 and 19 can be explained by the elimination of the C7-methoxy group from compound 19 under acidic conditions, followed by the proton abstraction from the C5 position and, as result, isomerization of the Δ6–7 double bond into Δ5–6. Under similar conditions, monoacylamides 17 and 20 gave the same benzimidazole 23 (Scheme 3). The formation of the Δ8–9 benzimidazole 23 derivative from compound 20 is a result of the C8-acetoxy group elimination.
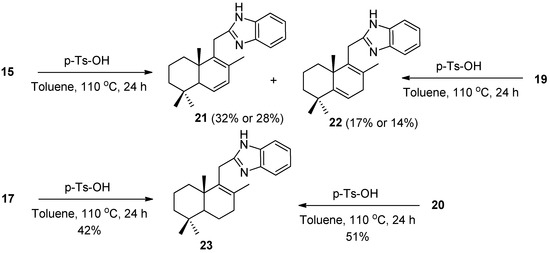
Scheme 3.
Synthesis of 2-substituted benzimidazoles 21–23 from monoacylamides 15, 17, 19, and 20.
According to the NMR spectra, the hybrid compounds 21–23 contained both heterocyclic and terpene units, and their accurate masses were confirmed by the HRMS analysis. The formation of the mentioned compounds was revealed, first of all, with the presence of the signals attributed to aromatic protons from a common 2-substituted-benzimidazole unit in a range of 7.18–7.24 ppm. Together with the signals specific for a terpene unit, such as singlets of C6- and C7-bonded protons at 5.83 and 5.97 ppm for compound 21, C6-bonded proton at 5.69 ppm for compound 22 and broad singlets of aminic protons at 8.79–9.00 ppm were obtained. The structures of the reported benzimidazoles were additionally confirmed by the 13C NMR spectra.
The NMR data of compound 22 have been assigned on the base of the 1D (1H, 13C, DEPT-135º) and 2D homo- (1H/13C HSQC, 1H/13C HMBC and 1H/1H COSY-45º) correlation spectra. An analysis of the 1H, 13C, 1H/1H COSY and 1H/13C HSQC NMR spectra suggested the presence of two isolated spin systems: CH2CH2CH2 (C1 to C3) and CHCH2 (C6 to C7) (Figure 4). The rearrangement of the double bond of compound 22 was established by a detailed analysis of its 1H/13C HMBC spectrum. Thus, the observed correlations of H3-C18 with two sp2 hybridized carbons (C5, δC 125.3 and C6, δC 120.1) were indicative of the Δ5,6 double bond localization. The position of a nitrogen atom was confirmed by the 1H/15N HMBC spectra and supported by the correlations of an H2-C11/N cross-peak (Figure 4).
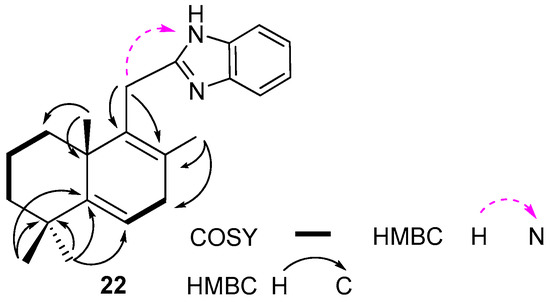
Figure 4.
Selected COSY and HMBC correlation for compound 22.
2.2. Antimicrobial Activity
All synthesized compounds were subjected to preliminary screening for their in vitro antifungal and antibacterial activities [29] against pure cultures of fungal species Aspergillus niger, Fusarium solani, Penicillium chrysogenum, Penicillium frequentans, and Alternaria alternata and both Gram-positive Bacillus sp. and Gram-negative Pseudomonas aeruginosa bacteria strains. The obtained minimum inhibitory concentration (MIC) values revealed that compounds 7 and 20 possess the highest antifungal (MIC 0.064 and 0.05 µg/mL, respectively,) and antibacterial (MIC 0.5 and 0.032 µg/mL, respectively,) activities, followed by compound 4 (MIC 1.6 and 4.0 µg/mL, respectively), which is comparable with the standards activity (Table 1, entries 1 and 5). Compounds 10, 14, and 19 have showed a moderate antifungal activity at MIC in a range from 0.80 to 1.16 µg/mL, and an antibacterial activity at MIC in a range from 3.90 to 6.0 µg/mL, vs the same standard (Table 1, entries 2–4). Compounds 15, 16, 17, 18, 21, 22, and 23 were found to be biologically inactive.

Table 1.
In vitro antifungal and antibacterial activities of compounds 4, 7, 10, 14, 19. and 20.
Based on the biological test data, it can be concluded that the activity of the reported compounds is largely determined by the nature of the heterocyclic unit. In this way, all N-homodrimenoyl-2-amino-1,3-benzimidazoles 4, 7, 10, and 14 were found to be active while 2-homodrimenyl-1,3-benzimidazoles 21–23 were inactive. Probably, the activity of 2-amino-1,3-benzimidazoles is primarily due to the presence of an unsubstituted amine group. This is also confirmed by compounds 19 and 20 which were produced by condensation with o-phenylenediamine on a single amine group and proved to be active, unlike bis-amides 16 and 18 which were inactive. However, it can be mentioned that the activity of the hybrid compounds depends also on the terpene unit; more precisely, it depends on the combination of the functional groups from the B cycle. It can be assumed that the presence of the C8-acetate group and of the Δ8–9 double bond in the molecules of compounds 20 and 7, in combination with the monosubstituted 2-amino-1,3-benzimidazolic or o-phenylenic fragment, make them the most active in this series. The presence of the Δ8–9 and Δ6–7 double bonds or the Δ8–9 bond and the C7-methoxy group in the molecules of compounds 4, 10, and 19 and of the C8-acetate group in compound 14, in combination with the 2-amino-1,3-benzimidazolic fragment or o-phenylenic fragment, influences the activity to a lesser extent. Compounds 15 and 17, which only contained a double bond Δ8–9 and Δ6–7 in combination with the monosubstituted o-phenylenic fragment, were inactive.
3. Materials and Methods
3.1. Synthesis and Characterization
The IR spectra were recorded on a Spectrum 100 FT-IR spectrometer (Perkin-Elmer, Shelton, CT, USA) using an ATR technique. The 1H, 13C, and 15N NMR (400, 100, and 40 MHz, respectively,) and COSY, 1H–13C HSQC, 1H–13C HMBC, DEPT, and 1H–15N HSQC, 1H–15N HMBC spectra were acquired on a Bruker Avance DRX 400 spectrometer (Bruker BioSpin, Rheinstetten, Germany) in CDCl3 (NMR spectra for all of the compounds are available online, see Supplementary Materials). The 1H NMR chemical shifts were reported relative to the residual solvent protons as internal standards (7.26 ppm). The solvent carbon atoms served as internal standard for the 13C NMR spectra (77.0 ppm). The 15N NMR spectra were obtained using MeNO2 (380.5 ppm) and urea (73.4 ppm) as internal standards. Optical rotations measurements were performed on a Jasco DIP-370 polarimeter (Rudolph Research Analytical, Hackettstown, NJ, USA) with a 10 cm microcell. Melting points were determined on a Boetius (VEB Analytik, DDR) hot stage apparatus and were not uncorrected. The run of reactions and the purity of products were examined by TLC on Merck silica gel 60 plates, eluent CH2Cl2, or a mixture of CH2Cl2–MeOH, 99:1; 49:1. Visualization was achieved by the treatment with conc. H2SO4 and heating at 80 °C or using an UV lamp (254 or 365 nm). All solvents were purified and dried by standard techniques prior to use.
Compounds 4, 7, 10, and 14 (General method).
The solution of one of the acids 2 (248 mg, 1 mmol), 5 (250 mg, 1 mmol), 8 (280 mg, 1 mmol), or 12 (310 mg, 1 mmol) dissolved in anhydrous C6H6 (5 mL) was treated with a solution of COCl2 (0.95 mL, 11 mmol) dissolved in C6H6 (2.5 mL). The reaction mixture was stirred at room temperature for 1 h and then refluxed for 1 h. The C6H6 and excess of COCl2 were removed at a reduced pressure on a rotary evaporator. Next, 2-aminobenzimidazole (225 mg, 1.5 mmol) was added to the solution of an acyl chloride 3, 6, 9, or 13 in CH2Cl2 (10 mL), and the resulting mixtures were stirred at r.t. for 3 h, then refluxed for 4–6 h. After cooling, the precipitates were filtered off, washed with CH2Cl2, and the filtrates were concentrated to dryness at a reduced pressure on a rotary evaporator. The crude reaction products were purified by silica gel flash chromatography (1 → 3% MeOH/CH2Cl2).
1-(2-Amino-1H-benzo[d]imidazol-1-yl)-2-((8aS)-2,5,5,8a-tetramethyl-4a,5,6,7,8,8a-hexahydronaphthalen-1-yl)ethanone 4. (239mg, 66%), colorless oil. −45.9 (c 3.4, CHCl3). IR spectrum, ν, cm−1: 729, 906, 1111, 1165, 1262, 1334, 1461, 1598, 1643, 1708, 2925, 3436. 1H NMR (400 MHz, CDCl3) δ 0.91 (3H, s, 10-CH3), 0.98 (3H, s, 4-CH3), 0.98 (3H, s, 4-CH3), 1.13–1.60 (6H, m, 3CH2), 1.69 (3H, s, 8-CH3), 2.22 (1H, t, J = 2.6 Hz, H-5), 3.72 (1H, d, J = 17.2 Hz, H-11), 3.81 (1H, d, J = 17.8 Hz, H-11), 5.88 (1H, dd, J= 9.3, 2.5 Hz, H-6), 5.95 (1H, dd, J= 9.6, 3.0 Hz, H-7), 7.10 (1H, td, J = 8.0, 1.1 Hz, H-Ar), 7.14 (1H, br.s, NH), 7.26 (1H, t, J = 8.0 Hz, H-Ar), 7.37 (1H, t, J = 2.3 Hz, H-Ar), 7.47 (1H, d, J = 8.2 Hz, H-Ar). 13C NMR (100 MHz, CDCl3) δ 15.3 (C-20), 18.3 (C-17), 18.8 (C-2), 22.7 (C-18), 32.3 (C-19), 33.0 (C-4), 34.7 (C-1), 36.9 (C-11), 38.4 (C-10), 40.7 (C-3), 52.3 (C-5), 113.2 (Ar), 117.0 (Ar), 120.6 (Ar), 125.0 (Ar), 128.7 (C-6), 129.0 (C-7), 129.6 (C-8), 129.7 (Ar), 134.5 (C-9), 142.8 (Ar), 155.0 (C=N), 173.1 (C-12). 15N NMR (40 MHz, CDCl3) δ 62, 196. HRMS (ESI) calculated for C23H29N3O [M + H]+, 363.23106. Found: 363.24188.
1-(2-Amino-1H-benzo[d]imidazol-1-yl)-2-((8aS)-2,5,5,8a-tetramethyl-3,4,4a,5,6,7,8,8a-octahydronaphthalen-1-yl) ethanone 7. (310 mg, 85%), mp 91–92 °C, 67.9 (c 2.7, CHCl3). IR spectrum, ν, cm−1: 688, 719, 753, 894, 1110, 1164, 1262, 1336, 1461, 1540, 1599, 1656, 1709, 2922, 3431. 1H NMR (400 MHz, CDCl3) δ 0.85 (3H, s, 4-CH3), 0.92 (3H, s, 4-CH3), 1.01 (3H, s, 10-CH3), 1.17–1.23 (2H, m, CH2), 1.33 (1H, dd, J = 12.5, 1.3 Hz, H-5), 1.38–1.50 (4H, m, 2CH2), 1.54 (3H, s, 8-CH3), 1.70–1.78 (2H, m, CH2), 2.10 (1H, dd, J = 18.2, 5.6 Hz, H-7), 2.20–2.29 (1H, m, H-7), 3.67 (1H, d, J = 12.2, H-11), 3.72 (1H, d, J = 17.8, H-11), 7.01 (2H, br.s, NH2), 7.11 (1H, t, J = 8.1 Hz, H-Ar), 7.26 (1H, t, J = 7.5 Hz, H-Ar), 7.37 (1H, d, J = 7.7 Hz, H-Ar), 7.48 (1H, d, J = 8.2 Hz, H-Ar). 13C NMR (100 MHz, CDCl3) δ 18.8 (C-2), 18.9 (C-6), 20.0 (C-17), 21.6 (C-18), 33.1 (C-19), 33.3 (C-4), 33.5 (C-7), 36.0 (C-1), 37.4 (C-11), 38.4 (C-10), 41.4 (C-3), 51.4 (C-5), 113.3, 117.0, 120.6), 124.7, 129.8, 142.9 (Ar), 131.7 (C-8), 132.4 (C-9), 154.9 (C=N), 173.3 (C-12). 15N NMR (40 MHz, CDCl3) δ 60, 191. HRMS (ESI) calculated for C23H31N3O [M + H]+, 365.24671. Found: 365.25388.
1-(2-Amino-1H-benzo[d]imidazol-1-yl)-2-((3R,8aS)-3-methoxy-2,5,5,8a-tetramethyl-3,4,4a,5,6,7,8,8a-octahydronaphthalen-1-yl)ethanone 10. (304 mg, 77%), mp 105–106 °C, 75.6 (c 1.4, CHCl3). IR spectrum, ν, cm−1: 739, 755, 1075, 1166, 1262, 1341, 1460, 1542, 1595, 1647, 1707, 2926, 3434. 1H NMR (400 MHz, CDCl3) δ 0.87 (3H, s, 10-CH3), 0.94 (3H, s, 4-CH3), 0.96 (3H, s, 4-CH3), 1.16–1.27 (2H, m, CH2), 1.39–1.52 (4H, m, 2CH2), 1.55 (1H, t, J = 3.4 Hz, H-5), 1.59 (1H, d, J = 1.6 Hz, H-6), 2.04 (1H, d, J = 2.0, H-6), 1.63 (3H, s, 8-CH3), 3.40 (3H, s, 7-OCH3), 3.38 (1H, d, J = 6.9 Hz, H-11), 3.52 (2H, d, J = 5.7 Hz, H-11 and H-7), 7.00 (2H, br.s, NH2), 7.09 (1H, dt, J = 7.7, 1.2 Hz, H-Ar), 7.25 (1H, dt, J = 7.9, 0.7 Hz, H-Ar), 7.37 (1H, dd, J = 7.9, 0.7 Hz, H-Ar), 7.43 (1H, d, J = 8.2 Hz, H-Ar). 13C NMR (100 MHz, CDCl3) δ 18.1 (C-17), 18.7 (C-2), 22.6 (C-6), 21.6 (C-18), 32.8 (C-19), 32.9 (C-4), 35.6 (C-1), 37.2 (C-11), 39.2 (C-10), 41.1 (C-3), 45.7 (7-OCH3), 56.8 (C-5), 78.8 (C-7), 113.1, 116.9, 120.6, 124.9, 129.5, 142.6 (Ar), 131.8 (C-8), 137.2 (C-9), 154.9 (C=N), 171.9 (C-12). 15N NMR (40 MHz, CDCl3) δ 65, 195. HRMS (ESI) calculated for C24H33N3O2 [M + H]+, 395.25728. Found: 395.26447.
(1R,2R,8aS)-1-(2-(2-amino-1H-benzo[d]imidazol-1-yl)-2-oxoethyl)-2,5,5,8a-tetramethyldecahydronaphthalen-2-yl acetate 14. (360 mg, 85%), colorless oil. −2.9 (c 7.2, CHCl3). IR spectrum, ν, cm−1: 728, 907, 1113, 1164, 1246, 1306, 1382, 1461, 1598, 1642, 1713, 2929, 3439. 1H NMR (400 MHz, CDCl3) δ 0.79 (3H, s, 10-CH3), 0.89 (3H, s, 4-CH3), 0.92 (3H, s, 4-CH3), 1.09–1.22 (3H, m, H-5, CH2), 1.29–1.44 (4H, m, 2CH2), 1.56 (3H, s, 8-CH3), 1.66–1.76 (2H, m, CH2), 1.78 (3H, s, 8-OCOCH3), 1.85 (1H, s, H-9), 2.73–2.80 (2H, m, H-7), 2.93 (1H, dd, J = 17.2, 4.8 Hz, H-11), 3.11 (1H, dd, J = 17.4, 4.3 Hz, H-11), 7.29 (2H, br.s, NH2), 7.10 (1H, td, J = 7.5, J = 1.4 Hz, H-Ar), 7.24 (1H, td, J = 7.6, 0.6 Hz, H-Ar), 7.34 (1H, d, J = 8.0 Hz, H-Ar), 7.45 (1H, d, J = 8.0 Hz, H-Ar). 13C NMR (100 MHz, CDCl3) δ 16.2 (C-17), 18.1 (C-2), 19.9 (C-6), 20.8 (C-18), 21.3 (8-OCOCH3), 33.1 (C-4), 33.2 (C-19), 38.7 (C-7), 38.8 (C-1), 34.5 (C-11), 38.7 (C-10), 41.6 (C-3), 53.3 (C-5), 55.3 (C-9), 86.2 (C-8), 113.1, 116.8, 120.7, 124.9, 129.6, 142.5 (Ar), 155.1 (C=N), 169.9 (8-OCOCH3), 174.6 (C-12). 15N NMR (40 MHz, CDCl3) δ 60, 190. HRMS (ESI) calculated for C25H35N3O3 [M + H]+, 425.26784. Found: 425.27448.
Compounds 15, 17, 19 and 20 (General method).
One of the acids 2 (248 mg, 1 mmol), 5 (250 mg, 1 mmol), 8 (280 mg, 1 mmol), or 12 (310 mg, 1 mmol) was added to an ice bath-cooled solution of Ph3P (786 mg, 3 mmol) and Et3N (0.16 mL, 1.2 mmol) dissolved in anhydrous CCl4 (7 mL). After 10 min of stirring, the solution of o-phenylendiamine (150 mg, 1.2 mmol) dissolved in anhydrous CCl4 (3 mL) was added, and the reaction mixture was refluxed under stirring for 6 h. The solvents were removed under a reduced pressure on a rotary evaporator to dryness, and the crude reaction products were subjected to silica gel flash column chromatography (CH2Cl2 →2% MeOH/CH2Cl2).
N-(2-aminophenyl)-2-((8aS)-2,5,5,8a-tetramethyl-4a,5,6,7,8,8a-hexahydronaphthalen-1-yl)acetamide 15. (182 mg, 57%), yellow oil. 0.79 (c 0.7, CHCl3). IR spectrum, ν, cm−1: 730, 907, 1033, 1370, 1455, 1503, 1591, 1654, 2926, 3268, 3354. 1H NMR (400 MHz, CDCl3) δ 0.88 (3H, s, 10-CH3), 0.97 (3H, s, 4-CH3), 0.99 (3H, s, 4-CH3), 1.12–1.64 (6H, m, 3CH2), 1.86 (3H, s, 8-CH3), 2.07 (1H, t, J = 2.5 Hz, H-5), 3.12 (1H, d, J = 17.2 Hz, H-11), 3.34 (1H, d, J = 17.2 Hz, H-11), 3.83 (2H, br.s,NH2), 5.90 (1H, dd, J = 9.5, 2.3 Hz, H-6), 5.95 (1H, dd, J = 9.6, 2.7 Hz, H-7), 6.78–6.82 (2H, m, H-Ar), 7.06 (1H, td, J = 7.6, 1.4 Hz, H-Ar), 7.14 (1H, dd, J = 8.1, 1.4 Hz, H-Ar), 7.67 (1H, br.s, NH). 13C NMR (100 MHz, CDCl3) δ 15.2 (C-20), 18.4 (C-17), 18.8 (C-2), 22.7 (C-18), 32.4 (C-19), 33.1 (C-4), 35.0 (C-1), 35.9 (C-11), 39.2 (C-10), 40.9 (C-3), 53.6 (C-5), 118.2, 119.4, 124.5, 124.7, 127.1, 140.8 (Ar), 128.9 (C-6), 129.1 (C-7), 130.0 (C-8), 138.2 (C-9), 169.8 (C-12). 15N NMR (40 MHz, CDCl3) δ 51, 126. HRMS (ESI) calculated for C22H30N2O [M + H]+, 338.23581. Found: 338.24301.
(S)-N,N′-(1,2-phenylene)bis(2-((8aS)-2,5,5,8a-tetramethyl-4a,5,6,7,8,8a-hexahydrona-phthalen-1-yl)acetamide) 16. (34 mg, 12%), yellow oil. −140.16 (c 1.7, CHCl3). IR spectrum, ν, cm−1: 728, 907, 1369, 1445, 1511, 1599, 1662, 2926, 3264. 1H NMR (400 MHz, CDCl3) δ 0.86 (6H, s, 10-CH3 and 10ʹ-CH3), 0.97 (6H, s, 4-CH3 and 4ʹ-CH3), 0.98 (6H, s, 4-CH3 and 4ʹ-CH3ʹ), 1.09–1.62 (12H, m, 3CH2), 1.80 (6H, s, 8-CH3 and 8ʹ-CH3), 2.18 (2H, t, J = 2.5, H-5 and H-5ʹ), 3.08 (2H, d, J = 16.9 Hz, H-11 and H-11ʹ), 3.25 (2H, d, J = 16.9 Hz, H-11 and H-11ʹ), 5.90 (2H, dd, J = 9.4, 2.1 Hz, H-6 and H-6ʹ), 5.94 (2H, dd, J = 9.7, 2.4 Hz, H-7 and H-7ʹ), 7.19 (4H, dd, J = 5.7, 3.4 Hz, H-Ar), 7.44 (4H, dd, J = 7.4, 3.4 Hz, H-Ar), 8.12 (2H, s, 2NH). 13C NMR (100 MHz, CDCl3) δ 15.2 (C-20 and C-20ʹ), 18.5 (C-2 and C-2ʹ), 18.8 (C-17 and C-17ʹ), 22.7 (C-18 and C-18ʹ), 32.4 (C-19 and C-19ʹ), 33.4 (C-4 and C-4ʹ), 35.0 (C-1 and C-1ʹ), 36.1 (C-11 and C-11ʹ), 39.2 (C-10 and C-10ʹ), 40.8 (C-3 and C-3ʹ), 52.9 (C-5 and C-5ʹ), 124.8, 126.2, 129.9 (Ar), 128.9 (C-6 and C-6ʹ), 129.1 (C-7 and C-7ʹ), 130.6 (C-8 and C-8ʹ), 137.5 (C-9 and C-9ʹ), 170.7 (C-12 and C-12ʹ). 15N NMR (40 MHz, CDCl3) δ 123. HRMS (ESI) calculated for C38H52N2O2 [M + H]–, 568.40288. Found: 568.39551.
N-(2-aminophenyl)-2-((8aS)-2,5,5,8a-tetramethyl-3,4,4a,5,6,7,8,8a-octahydronaphtha-len-1-yl)acetamide 17. (25 mg, 8%), yellow oil. 99.88 (c 0.4, CHCl3). IR spectrum, ν, cm−1: 738, 756, 1018, 1086, 1141, 1274, 1317, 1348, 1462, 1478, 1625, 1721, 2918, 3215. 1H NMR (400 MHz, CDCl3) δ 0.85 (3H, s, 4-CH3), 0.91 (3H, s, 4-CH3), 1.00 (3H, s, 10-CH3), 1.17–1.51 (7H, m, 3CH2, H-5), 1.55 (3H, s, 8-CH3), 1.61–1.75 (2H, m, CH2), 3.79 (2H, br.s. NH2), 2.04–2.29 (2H, m, H-7) 3.74 (1H, d, J = 19.1 Hz, H-11), 4.02 (1H, d, J = 19.1 Hz, H-11), 7.04 (1H, dd, J = 7.6, 1.1 Hz, H-Ar), 7.13 (1H, td, J = 7.8, 1.4 Hz, H-Ar), 7.19 (1H, td, J = 7.6, 1.3 Hz, H-Ar), 8.21 (1H, d, J = 7.4 Hz, H-Ar), 8.31 (1H, s, NH). 13C NMR (100 MHz, CDCl3) δ 18.9 (C-2), 18.9 (C-6), 19.8 (C-20), 20.1 (C-17), 21.7 (C-18), 33.1 (C-19), 33.3 (C-4), 33.7 (C-7), 36.0 (C-1), 36.2 (C-11), 38.4 (C-10), 41.5 (C-3), 51.4 (C-5), 108.8, 116.2, 122.8, 124.5, 127.6, 127.8 (Arʹ), 130.6 (C-8), 133.4 (C-9), 172.4 (C-12). 15N NMR (40 MHz, CDCl3) δ 52, 126. HRMS (ESI) calculated for C22H32N2O [M + H]–, 340.25146. Found: 340.19998.
(S)-N,N′-(1,2-phenylene)bis(2-((8aS)-2,5,5,8a-tetramethyl-3,4,4a,5,6,7,8,8a-octahydro-naphthalen-1-yl)acetamide) 18. (60 mg, 21%), yellow oil. 83.62 (c 3.6, CHCl3). IR spectrum, ν, cm−1: 751, 1043, 1161, 1295, 1376, 1443, 1511, 1599, 1663, 2865, 2923, 3264. 1H NMR (400 MHz, CDCl3) δ 0.86 (6H, s, 4-CH3 and 4ʹ-CH3), 0.93 (6H, s, 4-CH3 and 4ʹ-CH3), 1.00 (6H, s, 10-CH3 and 10ʹ-CH3), 1.16–1.62 (14H, m, 6CH2, H-5 and H-5ʹ), 1.67 (6H, s, 8-CH3 and 8ʹ-CH3), 1.70–1.81 (4H, m, 2CH2), 2.13–2.23 (4H, m, H-7 and H-7ʹ), 3.09 (2H, d, J = 17.6 Hz, H-11 and H-11ʹ), 3.23 (2H, d, J = 17.96 Hz, H-11 and H-11ʹ), 7.11 (4H, dd, J = 5.9, 3.5 Hz, H-Ar), 7.36 (4H, dd, J = 5.9, 3.2 Hz, H-Ar), 8.11 (2H, s, 2NH). 13C NMR (100 MHz, CDCl3) δ 18.9 (C-2 and C-2ʹ), 18.9 (C-6 and C-6ʹ), 20.1 (C-20 and C-20ʹ), 20.3 (C-17 and C-17ʹ), 21.7 (C-18 and C-18ʹ), 33.2 (C-19 and C-19ʹ), 33.4 (C-4 and C-4ʹ), 33.6 (C-7 and C-7ʹ), 36.3 (C-11 and C-11ʹ), 36.7 (C-1 and C-1ʹ), 38.9 (C-10 and C-10ʹ), 41.5 (C-3 and C-3ʹ), 51.8 (C-5 and C-5ʹ), 125.1, 126.1, 130.7 (Ar), 132.2 (C-8 and C-8ʹ), 135.4 (C-9 and C-9ʹ), 171.1 (C-12 and C-12ʹ). 15N NMR (40 MHz, CDCl3) δ 125. HRMS (ESI) calculated for C38H56N2O2 [M + H]–, 572.43418. Found: 572.42700.
N-(2-aminophenyl)-2-((3R,8aS)-3-methoxy-2,5,5,8a-tetramethyl-3,4,4a,5,6,7,8,8a-octa-hydronaphthalen-1-yl)acetamide 19. (204 mg, 58%), yellow oil. 84.78 (c 7.9, CHCl3). IR spectrum, ν, cm−1: 742, 1075, 1341, 1456, 1516, 1656, 2926, 3260, 3358. 1H NMR (400 MHz, CDCl3) δ 0.87 (3H, s, 10-CH3), 0.93 (3H, s, 4-CH3), 0.97 (3H, s, 4-CH3), 1.16–1.27 (3H, m, CH2), 1.41–1.63 (5H, m, H-5, 2CH2), 21.82 (3H, s, 8-CH3), 0.03 (1H, d, J = 14.2 Hz, CH2), 3.12 (1H, d, J = 17.8 Hz) and 3.25 (1H, d, J = 17.6 Hz, H-11), 3.37 (3H, s, 7-OCH3), 3.48 (1H, d, J = 3.6 Hz, H-7), 3.79 (2H,br.s. NH2), 6.74–6.79 (2H, m, H-Ar), 7.01 (1H, dt, J = 7.6, 1.2 Hz, H-Ar), 7.29 (1H, dd, J = 7.7, 1.1 Hz, H-Ar), 7.62 (1H, s, NH). 13C NMR (100 MHz, CDCl3) δ 18.4 (C-20), 18.5 (C-17), 18.7 (C-2), 21.6 (C-18), 22.4 (C-6), 32.8 (C-19), 32.9 (C-4), 35.5 (C-1), 36.6 (C-11), 39.7 (C-10), 41.2 (C-3), 45.9 (7-OCH3), 56.8 (C-5), 79.0 (C-7), 117.7, 119.2, 124.3, 126.6, 140.9 (Ar), 132.1 (C-8), 140.2 (C-9), 169.1 (C-12). 15N NMR (40 MHz, CDCl3) δ 51, 121. HRMS (ESI) calculated for C23H34N2O2 [M + H]–, 370.26230. Found: 370.25464.
(1R,2R,8aS)-1-(2-((2-aminophenyl)amino)-2-oxoethyl)-2,5,5,8a-tetramethyldecahydro-naphthalen-2-yl acetate 20. (187 mg, 49%), mp 188–189 °C, 11.09 (c 2.4, CHCl3). IR spectrum, ν, cm−1: 746, 1025, 1127, 1248, 1366, 1459, 1526, 1654, 1720, 2942, 3261, 3675. 1H NMR (400 MHz, CDCl3) δ 0.79 (3H, s, 10-CH3), 0.86 (6H, s, 4-CH3 and 4-CH3), 1.07–1.39 (5H, m, H-5, 2CH2), 1.41–1.44 (1H, m, CH2), 1.51 (3H, s, 8-CH3), 1,55–1.76 (4H, m, 2CH2), 1.92 (3H, s, 8-OCOCH3), 2.34 (H, s, H-9), 2.36 (1H, dd, J = 18.3, 6.7 Hz, H-11), 2.53 (1H, dd, J = 18.3, 6.8 Hz, H-11), 2.71 (1H, dt, J = 12.4, 3.1 Hz, H-7), 3.88 (2H, br.s., NH2), 6.77 (2H, t, J = 7.8 Hz, H-Ar), 7.03 (1H, td, J = 7.6, 1.1 H-Ar), 7.17 (1H, dd, J = 8.2, 1.3 Hz, H-Ar), 7.56 (1H, s, NH). 13C NMR (100 MHz, CDCl3) δ 15.6 (C-20), 18.2 (C-2), 19.9 (C-18 and C-6), 21.4 (8-OCOCH3), 23.0 (C-17), 33.1 (C-4), 33.2 (C-19), 33.6 (C-11), 38.7 (C-7), 39.2 (C-1 and C-10), 41.6 (C-3), 55.5 (C-5), 56.0 (C-9), 87.4 (C-8), 118.2, 119.4, 124.6, 124.8, 126.8, 140.5 (Ar), 170.3 (C-12), 172.5 (8-OCOCH3). 15N NMR (40 MHz, CDCl3) δ 53, 125. HRMS (ESI) calculated for C24H36N2O3 [M + H]–, 400.27259. Found: 400.19977.
Compounds 21–23 (General method).
A solution of monoacylated compounds 15 (168 mg, 0.5 mmol), 17 (170 mg, 0.5 mmol), 19 (185 mg, 0.5 mmol), or 20 (200 mg, 0.5 mmol) and para-toluenesulfonic acid (187 mg, 1 mmol) in toluene (3.5 mL) was stirred for 24 h at reflux. Then the solvent was evaporated, and the residue was diluted with CH2Cl2 (20 mL), washed with aqueuous NaHCO3 5%, dried over Na2SO4, and concentrated. The crude reaction products were purified by silica gel flash chromatography (CH2Cl2).
2-(((8aS)-2,5,5,8a-tetramethyl-4a,5,6,7,8,8a-hexahydronaphthalen-1-yl)methyl)-1H-benzo[d]imidazole 21. (51 mg, 32% or 44 mg, 28%), colorless oil. 6.70 (c 0.2, CHCl3). IR spectrum, ν, cm−1: 742, 1016, 1117, 1272, 1364, 1420, 1453, 1524, 1590, 1622, 1719, 2925, 3054. 1H NMR (400 MHz, CDCl3) δ 0.78 (6H, s, 4-CH3 and 4-CH3), 0.86 (3H, s, 10-CH3), 0.93–1.16 (4H, m, 2CH2), 1.52–1.62 (2H, m, CH2), 1.93 (3H, s, 8-CH3), 2.35 (1H, t, J = 3.4 Hz, H-5), 3.83 (1H, d, J = 16.7 Hz, H-11), 3.94 (1H, d, J = 15.6 Hz, H-11), 5.83 (1H, dd, J = 9.4, 6.1 Hz, H-6), 5.97 (1H, d, J = 9.6 Hz, H-7), 7.18–7.24 (4H, m, H-Ar), 8.79 (1H, br.s, NH). 13C NMR (100 MHz, CDCl3) δ 18.8 (C-20), 20.1 (C-2), 20.4 (C-17), 20.4 (C-18), 28.2 (C-11), 30.7 (C-19), 34.4 (C-4), 35.8 (C-1), 37.5 (C-10), 41.3 (C-3), 52.7 (C-5), 117.5, 122.1, 125.3, 127.6, 129.0, 132.1 (Ar), 128.2 (C-6), 129.2 (C-7), 129.2 (C-8), 134.3 (C-9), 153.5 (C=N, benzimidazole). 15N NMR (40 MHz, CDCl3) δ 130, 247. HRMS (ESI) calculated for C22H28N2 [M + H]+, 320.22525. Found: 320.23145.
(R)-2-((2,5,5,8a-tetramethyl-3,5,6,7,8,8a-hexahydronaphthalen-1-yl)methyl)-1H-benzo[d]imidazole 22. (27 mg, 17% or 22 mg, 14%), mp 188–189 °C, −138.9 (c 1.0, CHCl3). IR spectrum, ν, cm−1: 741, 1024, 1270, 1371, 1417, 1453, 1522, 1590, 1622, 2925, 3051. 1H NMR (400 MHz, CDCl3) δ 0.84 (3H, s, 4-CH3), 0.90 (3H, s, 4-CH3), 1.00 (3H, s, 10-CH3), 1.10–1.16 (1H, m, CH2), 1.55–1.79 (4H, m, 2CH2), 1.85 (3H, s, 8-CH3), 2.07–2.31 (3H, m, CH2), 3.90 (1H, d, J = 17.2 Hz, H-11), 4.04 (1H, d, J = 17.2 Hz, H-11), 5.69 (1H, t, J = 3.9 Hz, H-6), 7.19–7.23 (4H, m, H-Ar), 8.80 (1H, br.s, NH). 13C NMR (100 MHz, CDCl3) δ 20.7 (C-20), 20.8 (C-17), 23.0 (C-18), 23.3 (C-2), 24.5 (C-19), 27.5 (C-1), 29.1 (C-11), 29.9 (C-7), 32.4 (C-3), 34.5 (C-4), 37.8 (C-10), 125.3 (C-5), 117.1, 120.1, 121.9, 128.2, 129.0 (Ar), 120.1 (C-6), 133.6 (C-8), 139.8 (C-9), 154.2 (C=N, benzimidazole). 15N NMR (40 MHz, CDCl3) δ 134, 243. HRMS (ESI) calculated for C22H28N2 [M + H]+, 320.22525. Found: 320.23151.
2-(((8aS)-2,5,5,8a-tetramethyl-3,4,4a,5,6,7,8,8a-octahydronaphthalen-1-yl)methyl)-1H-benzo[d]imidazole 23. (80 mg, 42% or 97 mg, 51%), mp 175–176 °C, 69.29 (c 1.0, CHCl3). IR spectrum, ν, cm−1: 740, 1015, 1118, 1270, 1375, 1415, 1453, 1519, 1591, 1622, 2926, 3054. 1H NMR (400 MHz, CDCl3) δ 0.83 (3H, s, 4-CH3), 0.89 (3H, s, 4-CH3), 1.00 (3H, s, 10-CH3), 1.07–1.54 (7H, m, 3CH2, H-5), 1.68 (3H, s, 8-CH3), 1.73–1.77 (2H, m, CH2), 2.14–2.23 (2H, m, CH2), 3.70 (1H, d, J = 17.2 Hz, H-11), 3.77 (1H, d, J = 17.2 Hz, H-11), 7.16–7.22 (4H, m, H-Ar), 9.00 (1H, br.s, NH). 13C NMR (100 MHz, CDCl3) δ 18.7 (C-2), 18.9 (C-6), 19.9 (C-20), 20.3 (C-17), 21.6 (C-18), 28.1 (C-11), 33.2 (C-19), 33.3 (C-4), 33.6 (C-7), 36.6 (C-1), 39.0 (C-10), 41.5 (C-3), 52.1 (C-5), 118.9, 121.1, 122.0, 128.2, 129.1 (Ar), 130.7 (C-8), 136.5 (C-9), 154.5 (C=N, benzimidazole). 15N NMR (40 MHz, CDCl3) δ 139, 238. HRMS (ESI) calculated for C22H30N2 [M + H]+, 322.24090. Found: 322.25364.
3.2. X-ray Crystallography
Single-crystal XRD data were collected on an Oxford-Diffraction XCALIBUR Eos CCD diffractometer with graphite-monochromated Mo-Kα radiation. A single crystal was positioned at 46 mm from the detector, and 600 frames were measured each for 25 s over 1° scan width. The unit cell determination and data integration were carried out using the CrysAlisPro package from Oxford Diffraction [30]. A multiscan correction for absorption was applied. The structure was solved with the software SHELXT using the intrinsic phasing method and refined by the full-matrix least-squares method on F2 with SHELXL [31,32]. Olex2 was used as an interface to the SHELX programs [33]. Nonhydrogen atoms were refined anisotropically. Hydrogen atoms were added in idealized positions and refined using a riding model. The positional parameters of disordered atoms were refined in combination with PART and AFIXI restraints using an anisotropic model for non-H atoms. In the absence of a significant anomalous scattering, the absolute configuration of the structures could not be reliably determined, and therefore, Friedel pairs were merged and any references to the Flack parameter were removed. The molecular plots were obtained using the Olex2 program. Selected crystallographic data and structure refinement details for are provided in Table S2.
3.3. Antifungal and Antibacterial Activity Assay
Pure cultures of the fungi Aspergillus niger, Fusarium, Penicillium chrysogenum, Penicillium frequentans, and Alternaria alternata and bacteria Pseudomonas aeruginosa and Bacillus sp. were used as obtained from the American Type Culture Collection (ATCC). Suspensions of microorganisms in DMSO were prepared according to the direct colony method and the serial dilution procedure. The final concentration of the stock inoculum was 1·10–4 μg/mL. Both antifungal and antibacterial activities assays were performed by applying a mixture of a microorganism suspension and a solution of the target compound in a ratio 1:1 to Petri dishes with a solid medium: Merck Sabouraud agar or agar-agar. The DMSO did not have any inhibitory effect on the tested organisms.
4. Conclusions
A series of seven novel N-homodrimenoyl-2-amino-1,3-benzimidazoles and 2-homodrimenyl-1,3-benzimidazoles, four intermediate monoacylamides, and two bis-acylamides were designed, synthesized, and assessed as antimicrobial agents. Six of them showed high to moderate antifungal and antibacterial activities compared to those of the reference drugs.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28030933/s1, NMR spectra for all the compounds are available online. Table S1: Bond distances (Å) and angles (Å) for 20; Table S2: Selected crystallographic data refinement parameters for 20.
Author Contributions
Conceptualization, A.A.; experimental work, L.L., C.C. and S.B.; evaluated biological activity, N.V.; HRMS analysis, E.-I.G.; NMR data acquisition, A.B.; XRD analysis, S.S.; data analysis, A.C.; writing, review, and revision of the manuscript, A.C., A.A. and I.I.M.; funding acquisition and revision of the manuscript, V.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was financially supported by the National Agency for Research and Development (ANCD), Republic of Moldova, via the project PLANTERAS 20.80009.8007.03.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Grahman, P.L. An Introduction to Medicinal Chemistry, 2nd ed.; Oxford University Press: New York, NY, USA, 2001; p. 646. ISBN 9780198505334/978-0198505334. [Google Scholar]
- Greenwood, D.; Finch, R.; Davey, P.; Wilcox, M. Antimicrobial Chemotherapy, 5th ed.; Oxford University Press: New York, NY, USA, 2007; p. 504. ISBN 0198570163/978-0198570165. [Google Scholar]
- Fraga, B.M. Natural sesquiterpenoids. Nat. Prod. Rep. 2013, 30, 1226–1264. [Google Scholar] [CrossRef] [PubMed]
- Jansen, B.J.M.; De Groot, A. Occurrence, biological activity and synthesis of drimane sesquiterpenoids. Nat. Prod. Rep. 2004, 21, 449–477. [Google Scholar] [CrossRef] [PubMed]
- Allouche, N.; Apel, C.; Martin, M.-T.; Dumontet, V.; Guéritte, F.; Litaudon, M. Cytotoxic sesquiterpenoids from Winteraceae of Caledonian rainforest. Phytochemistry 2009, 70, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhu, T.; Cai, S.; Gu, Q.; Li, D. Drimane sesquiterpenoids from the mangrove-derived Fungus Aspergillus ustus. Chem. Pharm. Bull. 2011, 59, 762–766. [Google Scholar] [CrossRef] [PubMed]
- Lhinhatrakool, T.; Prabpai, S.; Kongsaeree, P.; Sutthivaiyakit, S. Antiplasmodial sesquiterpene alkaloids from the roots of Maytenus mekongensis. J. Nat. Prod. 2011, 74, 1386–1391. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Li, X.; Chen, W.; Xi, Z.; Sun, L. Three new sesquiterpenes from Tithonia diversifolia and their anti-hyperglycemic activity. Fitoterapia 2012, 83, 1590–1597. [Google Scholar] [CrossRef] [PubMed]
- Komala, I.; Ito, T.; Nagashima, F.; Yagi, Y.; Asakawa, Y. Cytotoxic, radical scavenging and antimicrobial activities of sesquiterpenoids from the Tahitian liverwort Mastigophora diclados (Brid.) Nees (Mastigophoraceae). J. Nat. Med. 2010, 64, 417–422. [Google Scholar] [CrossRef]
- Pathare, B.; Bansode, T. Review—Biological active benzimidazole derivatives. Results Chem. 2021, 3, 100200. [Google Scholar] [CrossRef]
- Kuchkova, K.; Aricu, A.; Barba, A.; Ungur, N.; Tuchilus, C.; Shova, S.; Zbancioc, G.; Mangalagiu, I.I. An Efficient and Straightforward Method to New Organic Compounds: Homodrimane Sesquiterpenoids with Diazine Units. Synlett 2013, 24, 697–700. [Google Scholar]
- Kuchkova, K.; Aricu, A.; Secara, E.; Barba, A.; Vlad, P.; Ungur, N.; Tuchilus, C.; Shova, S.; Zbancioc, G.; Mangalagiu, I.I. Design, Synthesis, and Antimicrobial Activity of Some Novel Homodrimane Sesquiterpenoids with Diazine Skeleton. Med. Chem. Res. 2014, 23, 1559–1568. [Google Scholar] [CrossRef]
- Kuchkova, K.I.; Arycu, A.N.; Sekara, E.S.; Barba, A.N.; Vlad, P.F.; Makaev, F.Z.; Mel’nik, E.; Kravtsov, V.K. Synthesis and Structure of Homodrimane Sesquiterpenoids Containing 1,2,4-Triazole and Carbazole Rings. Chem. Nat. Compd. 2015, 51, 684–688. [Google Scholar] [CrossRef]
- Duca, G.; Aricu, A.; Lungu, L.; Tenu, N.; Ciocarlan, A.; Gutu, Y.; Dragalin, I.; Barba, A. Synthesis of New Homodrimane Sesquiterpenoids Containing Diazine, 1,2,4-Triazole and Carbazole Rings. Chem. J. Mold. 2018, 13, 69–73. [Google Scholar] [CrossRef]
- Aricu, A.; Ciocarlan, A.; Lungu, L.; Barba, A.; Shova, S.; Zbancioc, G.; Mangalagiu, I.I.; D’Ambrosio, M.; Vornicu, N. Synthesis, Antibacterial, and Antifungal Activities of New Drimane Sesquiterpenoids with Azaheterocyclic Units. Med. Chem. Res. 2016, 25, 2316–2323. [Google Scholar] [CrossRef]
- Ciocarlan, A.; Aricu, A.; Lungu, L.; Edu, C.; Barba, A.; Shova, S.; Mangalagiu, I.I.; D’Ambrosio, M.; Nicolescu, A.; Deleanu, C.; et al. Synthesis of Novel Tetranorlabdane Derivatives with Unprecedented Carbon Skeleton. Synlett 2017, 28, 565–571. [Google Scholar] [CrossRef]
- Lungu, L.; Ciocarlan, A.; Barba, A.; Shova, S.; Pogrebnoi, S.; Mangalagiu, I.I.; Moldoveanu, C.; Vornicu, N.; D’Ambrosio, M.; Babak, M.V.; et al. Synthesis and Evaluation of Biological Activity of Homodrimane Sesquiterpenoids Bearing Hydrazinecarbothioamideor 1,2,4-Triazole Unit. Chem. Heterocycl. Compd. 2019, 55, 716–724. [Google Scholar] [CrossRef]
- Lungu, L.; Ciocarlan, A.; Smigon, C.; Ozer, I.; Shova, S.; Gutu, I.; Vornicu, N.; Mangalagiu, I.I.; D’Ambrosio, M.; Aricu, A. Synthesis and Evaluation of Biological Activity of Homodrimane Sesquiterpenoids Bearing 1,3,4-Oxadiazole or 1,3,4-Thiadiazole Units. Chem. Heterocycl. Compd. 2020, 56, 578–585. [Google Scholar] [CrossRef]
- Blaja, S.P.; Lungu, L.V.; Kuchkova, K.I.; Ciocarlan, A.G.; Barba, A.N.; Vornicu, N.; Aricu, A.N. Norlabdane Compounds Containing Thiosemicarbazone or 1,3-Thiazole Fragments: Synthesis and Antimicrobial Activity. Chem. Nat. Compd. 2021, 57, 101–110. [Google Scholar] [CrossRef]
- Lungu, L.; Cucicova, C.; Blaja, S.; Ciocarlan, A.; Dragalin, I.; Barba, A.; Vornicu, N.; Geana, E.-I.; Mangalagiu, I.I.; Aricu, A. Synthesis of Homodrimane Sesquiterpenoids Bearing1,3-Benzothiazole Unit and Their Antimicrobial Activity Evaluation. Molecules 2022, 27, 5082. [Google Scholar] [CrossRef]
- Ciocarlan, A.; Lungu, L.; Blaja, S.; Dragalin, I.; Aricu, A. The Use of Some Non-Conventional Methods in Chemistry of Bicyclohomofarnesenic Methyl Esters. Chem. J. Mold. 2020, 15, 69–77. [Google Scholar] [CrossRef]
- Aricu, A.N.; Kuchkova, K.I.; Barba, A.N.; Dragalin, I.P.; Shova, S.G.; Vornicu, N.; Gorincioi, E.K.; Secara, E.S.; Lungu, L.V.; Niculaua, M.; et al. Synthesis from Norambreinolide, Structure, and Antimicrobial Activity of Dihomodrimane Sesquiterpenoids with Azine, Hydrazide, and Dihydrazide Fragments. Chem. Nat. Compd. 2016, 52, 1029–1036. [Google Scholar] [CrossRef]
- Barrero, A.F.; Alvarez-Manzaneda, E.J.; Chahboun, R.; González Díaz, C.C. New Routes Toward Drimanes and nor-Drimanes from (-)-Sclareol. Synlett 2000, 11, 1561–1564. [Google Scholar]
- Mariappan, G.; Hazarika, R.; Alam, F.; Karki, R.; Patangia, U.; Nath, S. Synthesis and Biological Evaluation of 2-Substituted Benzimidazole Derivatives. Arab. J. Chem. 2015, 8, 715–719. [Google Scholar] [CrossRef]
- Leonard, J.T.; Jeyaseeli, L.; Shivakumar, R.; Gunasekaran, V. Synthesis, Antiimflamatory and Antibacterial Activities of 2-Hydroxyphenyl Benzimidazoles. Asian J. Chem. 2005, 17, 2674–2678. [Google Scholar]
- Sharma, M.C.; Kohli, D.V.; Sharma, S.; Sharma, A.D. Synthesis and Antihypertensive Activity of Some New Benzimidazolederivatives of 4’-(6-Methoxy-2-Substituted-Benzimidazole-1-ylmethyl)-biphenyl-2-carboxylic Acid in the Presences of BF3·OEt2. Pharm. Sinica 2010, 1, 104–115. [Google Scholar]
- Ge, F.; Wang, Z.; Wan, W.; Lua, W.; Hao, J. One-Pot Synthesis of 2-Trifluoromethyl and 2-Difluoromethyl Substituted Benzo-1,3-diazoles. Tetrahedron Lett. 2007, 48, 3251–3254. [Google Scholar] [CrossRef]
- Charton, J.; Girault-Mizzi, S.; Sergheraert, C. Conversion of Sterically Hindered Diacylated 1,2-Phenylenediamines into 2-Substituted Benzimidazoles. Chem. Pharm. Bull. 2005, 53, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Antimicrobial Susceptibility Testing (AST) Standards M07 and M100; National Committee on Clinical Laboratory Standards: Wayne, IL, USA, 2003.
- Rigaku Oxford Diffraction. CrysAlisPro Software System, Version 1.171.41.64; Rigaku Corporation: Oxford, UK, 2015. [Google Scholar]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).