


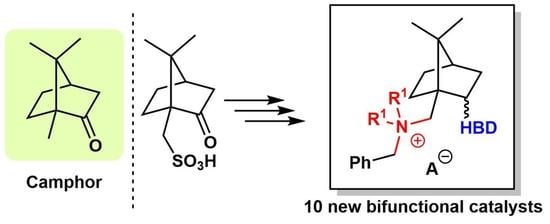
Synthesis and Catalytic Activity of Bifunctional Phase-Transfer Organocatalysts Based on Camphor
 ,
,  , ,
, ,  and
and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Structure Determination
2.3. Organocatalytic Activity
3. Materials and Methods
3.1. Reduction of (1S,4R,E)-1-[(Dimethylamino)methyl]-7,7-dimethylbicyclo[2.2.1]heptan-2-one oxime
3.1.1. (1S,2R,4R)-1-[(Dimethylamino)methyl]-7,7-dimethylbicyclo[2.2.1]heptan-2-amine (2a)
3.1.2. (1S,2S,4R)-1-[(Dimethylamino)methyl]-7,7-dimethylbicyclo[2.2.1]heptan-2-amine (1a)
3.2. Boc Protection of Chiral Amines—General Procedure 1 (GP1)
3.2.1. tert-Butyl {(1S,2S,4R)-1-[(dimethylamino)methyl]-7,7-dimethylbicyclo[2.2.1]heptan-2-yl}carbamate (3a)
3.2.2. tert-Butyl {(1S,2R,4R)-1-[(dimethylamino)methyl]-7,7-dimethylbicyclo[2.2.1]heptan-2-yl}carbamate (4a)
3.2.3. tert-Butyl [(1S,2S,4R)-7,7-dimethyl-1-(pyrrolidin-1-ylmethyl)bicyclo[2.2.1]heptan-2-yl}carbamate (3b)
3.3. Benzylation of Tertiary Amines—General Procedure 2 (GP2)
3.3.1. N-Benzyl-1-{(1S,2S,4R)-2-[(tert-butoxycarbonyl)amino]-7,7-dimethylbicyclo[2.2.1]heptan-1-yl}-N,N-dimethylmethanaminium Bromide (5a)
3.3.2. N-Benzyl-1-{(1S,2R,4R)-2-[(tert-butoxycarbonyl)amino]-7,7-dimethylbicyclo[2.2.1]heptan-1-yl}-N,N-dimethylmethanaminium Bromide (6a)
3.3.3. 1-Benzyl-1-({(1S,2S,4R)-2-[(tert-butoxycarbonyl)amino]-7,7-dimethylbicyclo[2.2.1]heptan-1-yl}methyl)pyrrolidin-1-ium Bromide (5b)
3.4. Boc Deprotection of Amines—General Procedure 3 (GP3)
3.4.1. (1S,2S,4R)-1-[(Benzyldimethylammonio)methyl]-7,7-dimethylbicyclo[2.2.1]heptan-2-aminium 2,2,2-Trifluoroacetate (7a)
3.4.2. (1S,2R,4R)-1-[(Benzyldimethylammonio)methyl]-7,7-dimethylbicyclo[2.2.1]heptan-2-aminium 2,2,2-Trifluoroacetate (8a)
3.4.3. Synthesis of 1-{[(1S,2S,4R)-2-Ammonio-7,7-dimethylbicyclo[2.2.1]heptan-1-yl]methyl}-1-benzylpyrrolidin-1-ium Iodide (7b)
3.5. Synthesis of Phase-Transfer Bifunctional Catalysts—General Procedure 4 (GP4)
3.6. Trifluoroacetate Anion Exchange—General Procedure 5 (GP5)
3.6.1. N-Benzyl-1-((1R,2R,4R)-2-{3-[3,5-bis(trifluoromethyl)phenyl]thioureido}-7,7-dimethylbicyclo[2.2.1]heptan-1-yl)-N,N-dimethylmethanaminium 2,2,2-Trifluoroacetate (I)
3.6.2. N-Benzyl-1-((1R,2R,4R)-2-{3-[3,5-bis(trifluoromethyl)phenyl]thioureido}-7,7-dimethylbicyclo[2.2.1]heptan-1-yl)-N,N-dimethylmethanaminium Iodide (II)
3.6.3. 1-Benzyl-1-[((1S,2S,4R)-2-{3-[3,5-bis(trifluoromethyl)phenyl]thioureido}-7,7-dimethylbicyclo[2.2.1]heptan-1-yl)methyl]yrrolidine-1-ium Iodide (III)
3.6.4. N-Benzyl-1-((1R,2S,4R)-2-{3-[3,5-bis(trifluoromethyl)phenyl]thioureido}-7,7-dimethylbicyclo[2.2.1]heptan-1-yl)-N,N-dimethylmethanaminium 2,2,2-Trifluoroacetate (IV)
3.6.5. N-Benzyl-1-((1R,2S,4R)-2-{3-[3,5-bis(trifluoromethyl)phenyl]thioureido}-7,7-dimethylbicyclo[2.2.1]heptan-1-yl)-N,N-dimethylmethanaminium Iodide (V)
3.6.6. N-Benzyl-1-[(1R,2S,4R)-7,7-dimethyl-2-(3-phenylthioureido)bicyclo[2.2.1]heptan-1-yl]-N,N-dimethylmethanaminium 2,2,2-Trifluoroacetate (VI)
3.6.7. N-Benzyl-1-[(1R,2S,4R)-7,7-dimethyl-2-(3-phenylureido)bicyclo[2.2.1]heptan-1-yl]-N,N-dimethylmethanaminium 2,2,2-Trifluoroacetate (VII)
3.6.8. N-Benzyl-1-[(1R,2S,4R)-7,7-dimethyl-2-(3-phenylureido)bicyclo[2.2.1]heptan-1-yl]-N,N-dimethylmethanaminium Iodide (VIII)
3.6.9. N-Benzyl-1-{(1R,2S,4R)-2-[(2-{[3,5-bis(trifluoromethyl)phenyl]amino}-3,4-dioxocyclobut-1-en-1-yl)amino]-7,7-dimethylbicyclo[2.2.1]heptan-1-yl}-N,N-dimethylmethanaminium 2,2,2-Trifluoroacetate (IX)
3.6.10. 1-Benzyl-1-{[(1S,2S,4R)-7,7-dimethyl-2-(3-phenylthioureido)bicyclo[2.2.1]heptan-1-yl]methyl}pyrrolidin-1-ium Iodide (X)
3.7. General Procedure for the α-Fluorination of β-Keto Ester 9
3.8. General Procedure for the α-Chlorination of β-Keto Ester 9
3.9. General Procedure for the α-Hydroxylation of β-Keto Ester 9
3.10. General Procedure for the Ring-Opening of Aryl-Aziridine 14 with β-Keto Ester 9
3.11. General Procedure for the Michael Addition of Glycine Schiff Base 16 with Methyl Acrylate (17)
3.12. X-ray Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Helder, R.; Hummelen, J.; Laane, R.; Wiering, J.; Wynberg, H. Catalytic asymmetric induction in oxidation reactions. The synthesis of optically active epoxides. Tetrahedron Lett. 1976, 17, 1831–1834. [Google Scholar] [CrossRef]
- Dolling, U.H.; Davis, P.; Grabowski, E.J.J. Efficient catalytic asymmetric alkylations. 1. Enantioselective synthesis of (+)-indacrinone via chiral phase-transfer catalysis. J. Am. Chem. Soc. 1984, 106, 446–447. [Google Scholar] [CrossRef]
- O’Donnell, M.J.; Bennett, W.D.; Wu, S. The stereoselective synthesis of α-amino acids by phase-transfer catalysis. J. Am. Chem. Soc. 1989, 111, 2353–2355. [Google Scholar] [CrossRef]
- O’Donnell, M.J. Catalytic Asymmetric Syntheses, 2nd ed.; Ojima, I., Ed.; Wiley-VCH: New York, NY, USA, 2000; pp. 727–755. [Google Scholar]
- Maruoka, K. (Ed.) Asymmetric Phase Transfer Catalysis; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Vitale, M.R.; Oudeyer, S.; Levacher, V.; Briere, J. Radical and Ion-Pairing Strategies in Asymmetric Organocatalysis; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Shirakawa, S.; Maruoka, K. Recent developments in asymmetric phase-transfer reactions. Angew. Chem. Int. Ed. 2013, 52, 4312–4348. [Google Scholar] [CrossRef] [PubMed]
- Waser, M.; Winter, M.; Mairhofer, C. (Thio)urea containing chiral ammonium salt catalysts. Chem. Rec. 2023, 23, e202200198. [Google Scholar] [CrossRef]
- Ooi, T.; Kameda, M.; Maruoka, K. Molecular Design of a C2-Symmetric Chiral Phase-Transfer Catalyst for Practical Asymmetric Synthesis of α-Amino Acids. J. Am. Chem. Soc. 1999, 121, 6519–6520. [Google Scholar] [CrossRef]
- Ooi, T.; Takeuchi, M.; Kameda, M.; Maruoka, K. Practical catalytic enantioselective synthesis of α,α-dialkyl-α-amino acids by chiral phase-transfer catalysis. J. Am. Chem. Soc. 2000, 122, 5228–5229. [Google Scholar] [CrossRef]
- Shibuguchi, T.; Fukuta, Y.; Akachi, Y.; Sekine, A.; Ohshima, T.; Shibasaki, M. Development of new asymmetric two-center catalysts in phase-transfer reactions. Tetrahedron Lett. 2002, 43, 9539–9543. [Google Scholar] [CrossRef]
- Waser, M.; Gratzer, K.; Herchl, R.; Müller, N. Design, synthesis, and application of tartaric acid derived N-spiro quaternary ammonium salts as chiral phase-transfer catalysts. Org. Biomol. Chem. 2012, 10, 251–254. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Chai, Z.; Zhao, G. Novel bifunctional thiourea–ammonium salt catalysts derived from amino acids: Application to highly enantio-and diastereoselective aza-Henry reaction. Tetrahedron 2013, 69, 5104–5111. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Zhang, J.-X.; Cao, D.-D.; Zhao, G. Enantioselective addition of thiols to imines catalyzed by thiourea–quaternary ammonium salts. ACS Catal. 2013, 3, 2218–2221. [Google Scholar] [CrossRef]
- Novacek, J.; Waser, M. Syntheses and Applications of (Thio) Urea-Containing Chiral Quaternary Ammonium Salt Catalysts. Eur. J. Org. Chem. 2014, 2014, 802–809. [Google Scholar] [CrossRef]
- Denmark, S.E.; Gould, N.D.; Wolf, L.M. A systematic investigation of quaternary ammonium ions as asymmetric phase-transfer catalysts. Synthesis of catalyst libraries and evaluation of catalyst activity. J. Org. Chem. 2011, 76, 4260–4336. [Google Scholar] [CrossRef]
- Wang, H. Chiral phase-transfer catalysts with hydrogen bond: A powerful tool in the asymmetric synthesis. Catalysts 2019, 9, 244. [Google Scholar] [CrossRef]
- Bernal, P.; Fernández, R.; Lassaletta, J.M. Organocatalytic asymmetric cyanosilylation of nitroalkenes. Chem. Eur. J. 2010, 16, 7714–7718. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liu, Y.; Sun, C.; Wei, Z.; Cao, J.; Liang, D.; Lin, Y.; Duan, H. Asymmetric phase-transfer catalysts bearing multiple hydrogen-bonding donors: Highly efficient catalysts for enantio- and diastereoselective nitro-Mannich reaction of amidosulfones. Org. Lett. 2014, 16, 6432–6435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, X.Y.; Zhou, Q.-L.; Sun, J. Chiral Camphor Derivatives as New Catalysts for Asymmetric Phase-Transfer Alkylation. Chin. J. Org. Chem. 2001, 19, 630–633. [Google Scholar] [CrossRef]
- Money, T. Remote functionalization of camphor: Application to natural product synthesis. Org. Synth. Theory Appl. 1996, 3, 1–83. [Google Scholar]
- Money, T. Camphor: A chiral starting material in natural product synthesis. Nat. Prod. Rep. 1985, 2, 253–289. [Google Scholar] [CrossRef] [PubMed]
- Holton, R.A.; Somoza, C.; Kim, H.-B.; Liang, F.; Biediger, R.J.; Boatman, P.D.; Shindo, M.; Smith, C.C.; Kim, S.; Nadizadeh, H. The Total Synthesis of Paclitaxel Starting with Camphor. ACS Symp. Ser. 1995, 583, 288–301. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Yang, Z.; Liu, J.J.; Ueno, H.; Nantermet, P.G.; Guy, R.K.; Claiborne, C.F.; Renaud, J.; Couladouros, E.A.; Paulvannan, K. Total synthesis of taxol. Nature 1994, 367, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Oppolzer, W. Camphor as a natural source of chirality in asymmetric synthesis. Pure Appl. Chem. 1990, 62, 1241–1250. [Google Scholar] [CrossRef]
- Kitamura, M.; Suga, S.; Kawai, K.; Noyori, R. Catalytic asymmetric induction. Highly enantioselective addition of dialkylzincs to aldehydes. J. Am. Chem. Soc. 1986, 108, 6071–6072. [Google Scholar] [CrossRef]
- Mahdy, A.-H.S.; Zayed, S.E.; Abo-Bakr, A.M.; Hassan, E.A. Camphor: Synthesis, reactions and uses as a potential moiety in the development of complexes and organocatalysts. Tetrahedron 2022, 121, 132913. [Google Scholar] [CrossRef]
- Grošelj, U. Camphor-Derivatives in Asymmetric Organocatalysis–Synthesis and Application. Curr. Org. Chem. 2015, 19, 2048–2074. [Google Scholar] [CrossRef]
- Ričko, S.; Požgan, F.; Štefane, B.; Svete, J.; Golobič, A.; Grošelj, U. Stereodivergent Synthesis of Camphor-Derived Diamines and Their Application as Thiourea Organocatalysts. Molecules 2020, 25, 2978. [Google Scholar] [CrossRef]
- Ričko, S.; Svete, J.; Štefane, B.; Perdih, A.; Golobič, A.; Meden, A.; Grošelj, U. 1,3-Diamine-Derived Bifunctional Organocatalyst Prepared from Camphor. Adv. Synth. Catal. 2016, 358, 3786–3796. [Google Scholar] [CrossRef]
- Ričko, S.; Meden, A.; Ivančič, A.; Perdih, A.; Štefane, B.; Svete, J.; Grošelj, U. Organocatalyzed Deracemization of Δ2-Pyrrolin-4-ones. Adv. Synth. Catal. 2017, 359, 2288–2296. [Google Scholar] [CrossRef]
- Xu, J.; Hu, Y.; Huang, D.; Wang, K.-H.; Xu, C.; Niua, T. Thiourea-Catalyzed Enantioselective Fluorination of β-Keto Esters. Adv. Synth. Catal. 2012, 354, 515–526. [Google Scholar] [CrossRef]
- Novacek, J.; Monkowius, U.; Himmelsbach, M.; Waser, M. Asymmetric α-chlorination of β-ketoesters using bifunctional ammonium salt catalysis. Monatsh. Chem. 2016, 147, 533–538. [Google Scholar] [CrossRef]
- Mairhofer, C.; Novacek, J.; Waser, M. Synergistic Ammonium (Hypo)Iodite/Imine Catalysis for the Asymmetric α-Hydroxylation of β-Ketoesters. Org. Lett. 2020, 22, 6138–6142. [Google Scholar] [CrossRef]
- Haider, V.; Kreuzer, V.; Tiffner, M.; Spingler, B.; Waser, M. Ammonium Salt-Catalyzed Ring-Opening of Aryl-Aziridines with β-Keto Esters. Eur. J. Org. Chem. 2020, 32, 5173–5177. [Google Scholar] [CrossRef] [PubMed]
- Tiffner, M.; Novacek, J.; Busillo, A.; Gratzer, K.; Massa, A.; Waser, M. Design of chiral urea-quaternary ammonium salt hybrid catalysts for asymmetric reactions of glycine Schiff bases. RSC Adv. 2015, 5, 78941–78949. [Google Scholar] [CrossRef]
- 37. In CrysAlis PRO; Agilent Technologies UK Ltd.: Oxfordshire, UK, 2011.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cristallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Synthesis, Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciber, L.; Požgan, F.; Brodnik, H.; Štefane, B.; Svete, J.; Waser, M.; Grošelj, U. Synthesis and Catalytic Activity of Bifunctional Phase-Transfer Organocatalysts Based on Camphor. Molecules 2023, 28, 1515. https://doi.org/10.3390/molecules28031515
Ciber L, Požgan F, Brodnik H, Štefane B, Svete J, Waser M, Grošelj U. Synthesis and Catalytic Activity of Bifunctional Phase-Transfer Organocatalysts Based on Camphor. Molecules. 2023; 28(3):1515. https://doi.org/10.3390/molecules28031515
Chicago/Turabian StyleCiber, Luka, Franc Požgan, Helena Brodnik, Bogdan Štefane, Jurij Svete, Mario Waser, and Uroš Grošelj. 2023. "Synthesis and Catalytic Activity of Bifunctional Phase-Transfer Organocatalysts Based on Camphor" Molecules 28, no. 3: 1515. https://doi.org/10.3390/molecules28031515
APA StyleCiber, L., Požgan, F., Brodnik, H., Štefane, B., Svete, J., Waser, M., & Grošelj, U. (2023). Synthesis and Catalytic Activity of Bifunctional Phase-Transfer Organocatalysts Based on Camphor. Molecules, 28(3), 1515. https://doi.org/10.3390/molecules28031515