Abstract
It has been found that the addition of CH2CN− anion to the carbonyl group of acylethynylpyrroles, generated from acetonitrile and t-BuOK, results in the formation of acetylenic alcohols, which undergo unexpectedly easy (room temperature) decomposition to ethynylpyrroles and cyanomethylphenylketones (retro-Favorsky reaction). This finding allows a robust synthesis of ethynylpyrroles in up to 95% yields to be developed. Since acylethynylpyrroles became available, the strategy thus found makes ethynylpyrroles more accessible than earlier. The quantum-chemical calculations (B2PLYP/6-311G**//B3LYP/6-311G**+C-PCM/acetonitrile) confirm the thermodynamic preference of the decomposition of the intermediate acetylenic alcohols to free ethynylpyrroles rather than their potassium derivatives.
1. Introduction
Ethynylpyrroles are valuable building blocks in the synthesis of many natural and synthetic biologically active compounds, such as antibiotic roseophilin, a potent cytotoxic agent against K562 human erythroid leukemia cells [1] and alkaloid quinolactacide with insecticidal activity [2]. They are applied in the syntheses of inhibitors of EGFR tyrosine kinase, an important target for anticancer drug design [3], the HMG-CoA reductase inhibitors for the treatment of hypercholesterolemia, hyperlipoproteinemia, hyperlipidemia and atherosclerosis [4], selective dopamine D4 receptor ligands [5] and foldamers, synthetic receptors, modified for encapsulation of dihydrogenphosphate ions [6]. Pyrroles with terminal acetylenic substituents take part in the syntheses of both lipophilic and highly hydrophilic BODIPY dyes, which fluoresce with high quantum yields and have low cytotoxicity, which makes it possible to visualize cells [7].
These pyrroles are employed in the development of advanced materials capable of detecting various organic and inorganic targets, such as tetrahedral oxoanions (H2PO4− and SO42−) [8] and pyrophosphate anions [9].
Also, high-tech materials, including ultrasensitive fluorescent probes for glucopyranoside [10], photoswitchable materials [11,12,13], components of dye-sensitized solar cells [14], monomers for organic thin-film transistors [15], prospective for energy storage devices, electrochemically active photoluminescence films are based on terminal ethynylpyrroles [16].
In light of the previous, it is clear that the improvement of the synthesis of ethynylpyrroles is a challenge. Indeed, the approaches to the preparation of these functionalized pyrroles are mainly limited to the deprotection of substituted at the triple bond (usually with TMS/TIPS groups) ethynylpyrroles, the products of the reaction of halopyrroles with the corresponding terminal acetylenes (Sonogashira cross-coupling) [2,5,6,8,17,18,19]. However, in this case, this coupling has limitations, since many halogenated pyrroles, except for representatives with electron-withdrawing substituents, are neither readily available nor stable [20,21]. Variants of the cross-coupling, such as Negishi reaction of halopyrroles with ethynyl magnesium chloride or zinc bromide [22] or cross-coupling of (1-methylpyrrol-2-yl)lithium with fluoroacetylene [23], are used albeit less often. It should be especially emphasized that almost all ethynylpyrroles synthesized by the above methods lack the substituents at carbon atoms in the pyrrole ring, i.e., the assortment of accessible ethynylpyrrole remains small and need to be extended.
Among other methods are Corey–Fuchs reaction of pyrrole-2-carbaldehydes with CBr4 with further conversion of dibromoolefins to ethynylpyrroles under the action of bases [1,3,7,24,25] and flash vacuum pyrolysis (FVP) of cyclic and linear 2-alkenylpyrroles (750 °C), limited to a few examples [26,27,28,29] due to difficulties in hardware implementation and requirements for substrates. Base-catalyzed elimination of ketones from tertiary acetylenic alcohols (retro-Favorsky reaction), affording pyrroles with terminal acetylenic substituents [30], is a rarer approach to such acetylenes because they could decompose or polymerize at high temperatures (up to 180 °C) common for the realization of this synthesis.
The formation of ethynylpyrroles as a result of the deacylation of acylethynylpyrroles was mentioned in only a few cases [31,32], and their yield was insignificant (though alkynones without pyrrole substituents in the presence of alkali metal hydroxides undergo hydrolytic cleavage to form terminal acetylenes [33,34,35]). For instance, when benzoylethynylpyrrole was treated with NaOH in DMSO (45–50 °C, 4 h), debenzoylation was detected by 1H NMR in negligible extent [31] and 7-days keeping of trifluroacetyl ethynylpyrrole over Al2O3 led to the ethynylpyrrole in 24% yield [32]. Certainly, these results were not suitable for the preparative synthesis of ethynylpyrroles.
Recently [36], we have disclosed the reaction of acylethynylpyrroles 1 with MeCN and metal lithium affording pyrrolyl-cyanopyridines 2 in up 87% yield (Scheme 1).
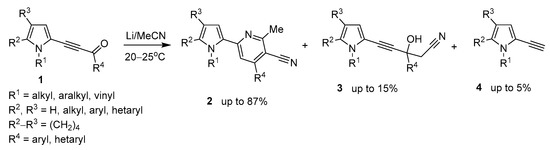
Scheme 1.
Previous work. Reaction of acylethynylpyrroles with Li/MeCN system to give pyrrolyl-cyanopyridines.
The synthesis was accompanied by the formation of propargyl alcohols 3 (up to 15%) and small amounts of ethynylpyrroles 4 (up to 5%). The propargyl alcohols 3 were proved to be intermediates in the synthesis of both pyridines and ethynylpyrroles.
These results served as a clue to develop a novel synthesis of ethynylpyrroles, provided we could manage to turn the above side process into a major reaction. Our further successful experiments confirmed this assumption. It appeared that if lithium metal is replaced by t-BuOK, the reaction is shifted almost completely to the formation of side ethynylpyrroles. The progress of this synthesis optimization is illustrated in Table 1, wherein the most representative results are presented. As a reference compound, 3-(1-benzyl-4,5,6,7-tetrahydro-1H-indol-2-yl)-1-(thiophen-2-yl)prop-2-yn-1-one (1a), was chosen believing that the optimal conditions, found for this pyrrolyl acetylenic ketone of higher complexity, will also be valid for the simpler congeners.
In this paper, we report the exceptionally mild decarbonylation of acylethynylpyrroles, readily available from the reaction of pyrroles with electrophilic haloacetylenes in the medium of solid oxides and metal salts [32,37,38,39,40], under the action of CH2CN− anion generated in situ in the system MeCN/t-BuOK.
2. Results and Discussion
As seen from Table 1, when the reaction was carried out by stirring acylethynylpyrrole 1a with 2 eq. n-BuLi in MeCN at room temperature under 1H NMR control, the isolated crude product contained 12% of the target ethynylpyrrole 4a (Table 1, Entry 1), i.e., the expected decarbonylation degree was noticeably increased. The major product, in this case, became tertiary propargylic alcohol 3a (content in the reaction mixture was 78%). Pyrrolylpyridine 2a, previously a major product [36], was also present in the reaction mixture but in a much smaller amount (10%). Almost the same results were obtained in the presence of 2 eq. of t-BuONa (Entry 3). But t-BuOLi turned out to be completely inactive in this reaction (Entry 2): the starting acylethynylpyrrole 1a, in this case, was almost returned from the reaction.
t-BuOK catalyzed the formation of ethynylpyrrole 4a much more actively: in the crude product obtained with one equivalent of this base, the content of the ethynylpyrrole 4a in the reaction mixture attained 66% (Entry 4). However, under these conditions, the conversion of the starting acylethynylpyrrole 1a was only 82%, but the content of pyrrolylpyridine 2a in the reaction mixture increased to 16%.
When 2 eq. t-BuOK were used, acylethynylpyrrole 1a reacted completely during the same time, and the content of ethynylpyrrole 4a in the reaction mixture became 90%. Pyrrolylpyridine 2a was also present as a by-product (10%) in the reaction mixture (Entry 5).
We found that it was possible to get rid of the pyridine almost completely (Entry 6 and 7) by carrying out the reaction in the mixed solvents (MeCN/THF or MeCN/DMSO in volume ratio 1:1). Thus, under these conditions, the reaction was excellently selective providing ethynylpyrrole 4a in ~80% isolated yield.

Table 1.
Optimization of the ethynylpyrrole 4a synthesis by decarbonylation of acylethynylpyrrole 1a a.
Table 1.
Optimization of the ethynylpyrrole 4a synthesis by decarbonylation of acylethynylpyrrole 1a a.
 | |||||
| Entry | Base, eq. | Content in the crude, % (1H NMR) | |||
|---|---|---|---|---|---|
| 1a | 2a | 3a | 4a | ||
| 1 | n-BuLi, 2 | traces | 10 | 78 | 12 |
| 2 | t-BuOLi, 2 | ~100 | traces | traces | traces |
| 3 | t-BuONa, 2 | traces | traces | 85 | 15 |
| 4 | t-BuOK, 1 | 18 | 16 | traces | 66 |
| 5 | t-BuOK, 2 | traces | 10 | traces | 90 |
| 6 b | t-BuOK, 2 | traces | traces | traces | ~100 d |
| 7 c | t-BuOK, 2 | traces | traces | traces | ~100 e |
a—reaction conditions: 0.5 mmol of 1a, acetonitrile (2.0 mL), 20–25 °C, nitrogen atmosphere. b—the reaction was carried out in the MeCN/THF (1:1) system. c—the reaction was carried out in the MeCN/DMSO (1:1) system. d—isolated yield 84%. e—isolated yield 82%.
Next, with the optimized reaction conditions (2 eq of t-BuOK, THF/MeCN, room temperature, 1 h) in hand, we have evaluated the scope of this reaction using benzoyl-, furoyl-, and thenoylethynylpyrroles with alkyl, aryl and hetaryl substituents at 4(4,5)-positions and methyl, benzyl, and vinyl moieties at the nitrogen atom of the pyrrole ring. Eventually, the series of earlier unknown ethynylpyrroles 4a–k were synthesized in good to excellent yields, the exception being pyrrole 4d (yield 36%) (Scheme 2).
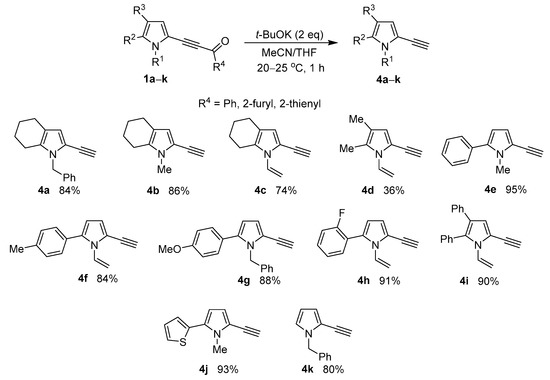
Scheme 2.
The scope of the acylethynylpyrroles 1a–k decarbonylation in the t-BuOK/MeCN/THF system.
The method proved to be extendable over indole compounds, as shown in the example of 3-benzoylethynylindole 5, which was transformed to the expected 1-methyl-3-ethynylindole 6 under the same conditions (Scheme 3).
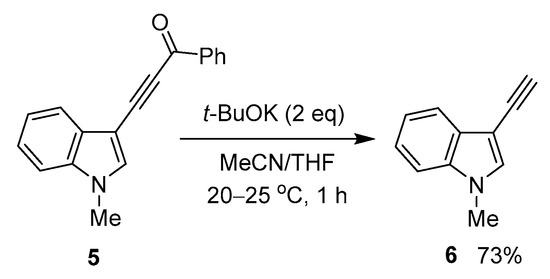
Scheme 3.
Reaction of 3-acylethynylindole 5 with t-BuOK.
Thus, this result shows that 3-ethynylindoles—valuable synthetic building blocks [41]—could be more accessible than previously due to the above-elaborated strategy. Noteworthy that the starting 3-acylethynylindoles can be easily prepared by the cross-coupling of the corresponding N-substituted indoles with acylbromoacetylenes in solid Al2O3 media [42].
Also, we have attempted to extend the synthesis of ethynylpyrroles over the furan series. For this, we have chosen menthofuran, a natural antioxidant component of peppermint oil [43]. It turned out that 2-benzoylethynylmenthofuran 7, which synthesis was previously described in [44], underwent similar decarbonylation under the above conditions to give the expected ethynyl derivative 8 in 80% yield (Scheme 4).
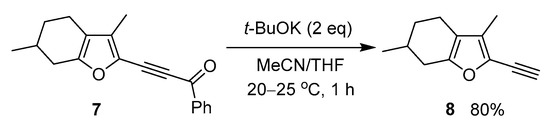
Scheme 4.
Reaction of 2-acylethynylmenthofuran 7 with t-BuOK.
The narrow range of the yields (74–95%) evidences that the structural effects on the synthesis efficiency are insignificant that are likely to result from the complex character of the process: (i) the formation of intermediate propargyl alcohols 3 and (ii) the decomposition of the latter. Besides, these steps are parallel to the formation of pyridine 2. Apart from these competing factors, the yields are influenced by the isolation procedure (chromatography on the SiO2), wherein a noticeable amount of the target products are lost (Table 1, cf. 1H NMR and isolated yields). Nevertheless, the following general trend in yields may be noted: alkyl substituents in the pyrrole ring slightly decrease the reaction efficiency compared to aromatic substituents (74–86% vs. 84–95%). That can be referred to as a higher acidophobicity of the alkyl pyrroles.
It is known that MeCN is easily deprotonated by the action of alkali metals to give acetonitrile dimers via the formation of an intermediate CH2CN− anion [45]. Also, it was reported that CH2CN− anion was added to ketones to form tertiary cyanomethyl alcohols [46,47,48,49]. Correspondingly, in the previous communication, we have shown that the intermediate propargyl alcohol 3 are actually adducts of acylethynylpyrroles and CH2CN− anion [36].
Although we failed to isolate propargyl alcohol 3a in the reaction mixture obtained in the presence of t-BuOK, the results produced with t-BuONa allowed us to assume that in the first case, the reaction also proceeded with the formation of the intermediate 3a, which was rapidly decomposed.
To verify this assumption, propargyl alcohols 3a,c,d,f, prepared from acylethynylpyrroles 1a,c,d,f and acetonitrile in the presence of t-BuONa according to the modified protocol (Scheme 5) [36], were rapidly and quantitatively converted in the presence of t-BuOK into the corresponding ethynylpyrroles 4a,c,d,f (Scheme 5).
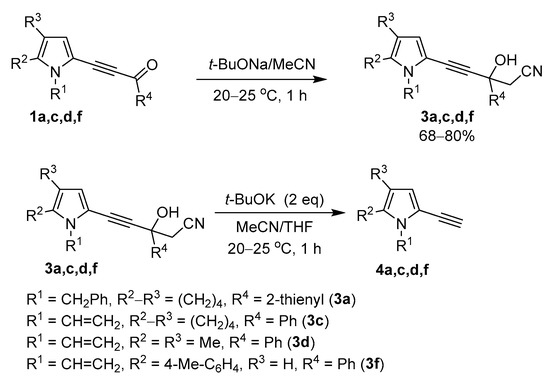
Scheme 5.
Synthesis and decomposition of propargyl alcohols 3a,c,d,f.
We performed the reaction in an NMR tube in deuterated acetonitrile. Immediate transformation of the characteristic signals of the protons of the benzoyl group at 8.16 ppm to protons of Ph-substituent occurs after the addition of t-BuOK to acylethynylpyrrole 1c solution, which corresponds to the formation of the intermediate acetylenic alcohol 3c (Scheme 3). Additionally, two nearly equal singlets at 6.30 (signal of H-3 of pyrrole ring in ethynylpyrrole 4c) and 6.44 ppm (signal of H-3 of pyrrole ring in intermediate alcohol 3c) appeared. The singlet at 6.44 ppm decreases rapidly and disappears after about 30 min of reaction. After 1 h reaction mixture contained only terminal alkyne 4c with a fully deuterated terminal acetylene position. Thus, the results confirm the proposed mechanism of the formation of ethynylpyrroles via intermediate acetylenic alcohol decomposition.
Cyanomethyl ketone (on the example of cyanomethyl-(2-thienyl)ketone 9a), a second product of the retro-Favorsky reaction, was detected (1H NMR) after acidification of the aqueous suspension received during the workup of the reaction mixture (Scheme 6).
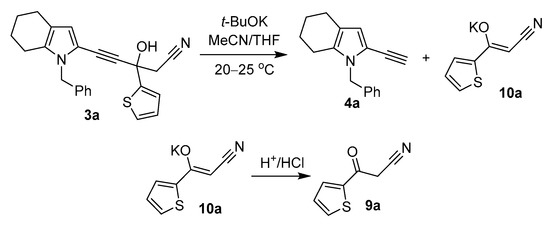
Scheme 6.
The decomposition of propargyl alcohol 3a.
Therefore, it is rigorously confirmed that in this reaction, ethynylpyrroles are the products of tertiary propargyl alcohol 3 decomposition, the retro-Favorsky reaction, which in this case occurs under extraordinarily mild (room temperature) conditions. Commonly this reaction requires a considerably higher temperature [120–140 °C (1 mm)] [30].
It could be emphasized that tertiary propargyl alcohols are one of the most attractive synthetic building blocks in organic synthesis [50,51,52,53,54,55,56,57,58]. This is primarily due to their bifunctionality (acetylene and hydroxyl functions), owing to which they can undergo cascade or multistage reactions with the formation of diverse compounds. In recent years, owing to the development of efficient methods for the synthesis of enantiomerically pure tertiary propargyl alcohols [59,60,61], interest in this class of compounds has increased significantly.
The tertiary propargyl alcohols here synthesized additionally contain one more synthetically valuable functional group (CN group and active C-H bond adjusted to nitrile function) and a pyrrole ring that significantly expands their potential for the design of novel functionalized compounds.
Despite the experimental evidence highlighting the mechanism of the cascade reaction studied, several mechanistic issues still need a quantum-chemical analysis. These issues mainly relate to the key stage of the synthesis, i.e., the t-BuOK-catalyzed decomposition of the intermediate propargyl alcohols 3. Here the following questions should be clarified: (i) are the intermediates 3 decompose to the corresponding ketones 9 and potassium derivatives 11 of ethynylpyrroles as so far usually considered or free ethynylpyrroles 4 and the corresponding potassium enolates 10 (Scheme 7) are formed? Although the Favorsky retro-reaction was synthetically thoroughly studied, this issue was never specially investigated. (ii) Is the experimentally observed formation of enolate from propargyl alcohols kinetically or thermodynamically controlled? (iii) Is the experimentally observed role of alkali metal cation, which fully controls the synthesis direction, an intrinsic (intramolecular) feature of the reaction, or is this influence of intermolecular solvation of the cations? (iv) What is the contribution of the solvent effect to the thermodynamics of this reaction?
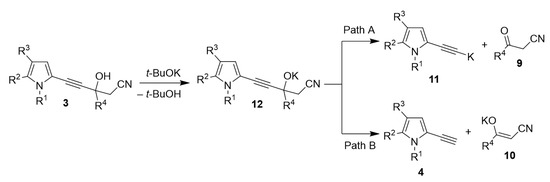
Scheme 7.
Possible ways of propargyl alcohols 3 decomposition.
To gain a clearer understanding of these mechanistic points, we have performed the quantum chemical calculations of the fundamental characteristics of the above reaction, the Gibbs free energy change, ∆G, using the DFT-based computational approach, which can be briefly referred to as B2PLYP/6-311G**//B3LYP/6-311G**+C-PCM/acetonitrile (see Supplementary Materials for details) and assuming R1 = Me, R2 = R3 = H, R4 = Ph in Scheme 5.
According to the results obtained, path A, i.e., formation of the metallated ethynylpyrroles and ketones (Scheme 5), is thermodynamically closed, whereas path B (Scheme 5), i.e., formation of the ethynylpyrrole and enolate, is thermodynamically opened (see SI for details). The calculations indicate that the decomposition of intermediate 3 proceeds via the formation of free ethynylpyrrole and potassium enolate. Also, these results evidence that path B is thermodynamically controlled. The ∆G values for path B calculated for Li, Na and K derivatives of propargyl alcohols 3 are −66.5, −78.6 and −88.3 kJ/mol, respectively. This explains experimental results according to which with the t-BuOLi, no products are formed (Table 1), while t-BuONa promotes the formation of sodium enolate, however stable under reaction conditions, and with t-BuOK, the decomposition of potassium enolate occurs. Thus, the effect of potassium alkali metal indeed has an intrinsic (intramolecular) character.
The computed O-Li, O-Na and O-K bond lengths in alkali metal derivatives of propargyl alcohols are 1.71, 2.08, and 2.43 Å, respectively, and in the corresponding enolates are 1.83, 2.16, 2.61 Å, respectively. These values correlate with the literature data: 1.95 Å (O-Li), 2.14–2.32 Å (O-Na), 2.60–2.80 Å (O-K) [62], respectively. The reported bond energies are 343, 255 and 238 kJ/mol [63]. From these results, it becomes clear why with t-BuOK, the pyridines 2 are not formed: the abstraction of a proton from the CHCN moiety would lead to dianionic-like species that are thermodynamically unfavorable. In the cases of Li- and Na-derivatives of propargyl alcohols, the negative charges on oxygen are smaller since they are tighter ion pairs, especially with lithium cation. Therefore, the reaction takes other directions: with Li cation, expectedly, pyridines are formed, and with t-BuONa, the propargyl alcohol decomposition slows down (Table 1, Entry 3).
The mechanism of ethynylpyrroles formation from potassium derivatives of propargyl alcohols (on the example of alcoholate 12, R1 = Me, R2 = R3 = H, R4 = Ph) likely represents an intramolecular process (Scheme 8) [36], involving the Csp-CH bond cleavage with simultaneous transfer of a proton from the CH bond.
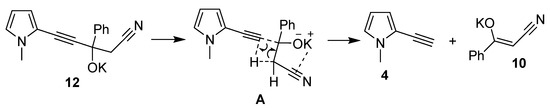
Scheme 8.
Ethynylpyrroles formation from potassium derivatives of propargyl alcohols.
This process is probably facilitated by the intramolecular interaction (coordination) between potassium cation and CN-bond (intermediate A). This is supported by the fact that the calculated K⋯N distance in the potassium derivative of propargyl alcohol (3.87 or 4.12 Å, depending on the molecular conformation, see Supplementary Materials) is smaller than the sum of the van der Waals radii of these atoms (4.2 Å). The above-mentioned two conformations are separated only by 0.8 kJ/mol. Since the latter value is well within the error margin of our computational scheme, they both can be considered legitimate propargyl alcohol equilibrium ground-state molecular structures (see Supplementary Materials for more details).
The ∆G values computed for the formation of ethynylpyrroles with the participation of the solvent (MeCN) and then without (gas phase) are close (−88.3 and −84.5 kJ/mol). This means that the contribution of the solvent effect is negligible.
The experiments with MeNO2 showed that in this solvent, the reaction did not proceed at all: the starting acylethynylpyrrole was recovered completely. In our previous work [36], we reported the reaction of benzoylethynylpyrrole 1a with isobutyronitrile and valeronitrile in the presence of lithium metal. In both cases, respective intermediate alcohols were isolated in 60 and 26% yields. In the presence of t-BuOK, both were readily transformed to corresponding ethynylpyrrole 4a.
3. Experimental Section
3.1. General Information
IR spectra were obtained on a “Bruker IFS-25” spectrometer (Bruker, Billerica, MA, USA) (KBr pellets or films in 400–4000 cm−1 region). 1H (400.13 MHz) and 13C (100.6 MHz) NMR spectra were recorded on a “Bruker Avance 400” instrument (Bruker, Billerica, MA, USA) in CDCl3. The assignment of signals in the 1H NMR spectra was made using COSY and NOESY experiments. Resonance signals of carbon atoms were assigned based on 1H-13C HSQC and 1H-13C HMBC experiments. The 1H chemical shifts (δ) were referenced to the residual solvent protons (7.26 ppm, CDCl3), and the 13C chemical shifts were expressed with respect to the deuterated solvent (77.16 ppm). Coupling constants in hertz (Hz) were measured from one-dimensional spectra, and multiplicities were abbreviated as follows: br (broad), s (singlet), d (doublet), t (triplet), and m (multiplet). The chemical shifts were recorded in ppm. The (C, H, N) microanalyses were performed on a Flash EA 1112 CHNS-O/MAS (CHN Analyzer) instrument (Thermo Finnigan, Italy). Sulfur was determined by complexometric titration with Chlorasenazo III. Fluorine content was determined on a SPECOL 11 (Carl Zeiss Jena, Germany) spectrophotometer. Melting points (uncorrected) were determined with SMP50 Stuart Automatic melting point (Cole-Palmer Ltd. Stone, Staffordshire, UK).
3.2. Synthesis of Ethynylpyrroles 4a–k, Ethynylindole 6, Ethynylfuran 8, General Procedure
Acylethynylpyrrole 1a–k, 3-acylethynylindole 5 or 2-acylethynylfuran 7 (1 mmol) was dissolved in dry THF/MeCN (1:1, 4 mL), and then t-BuOK (224 mg, 2 mmol) was added to reaction mixture under nitrogen. Reaction mixture was stirred at room temperature for 1 h while turning into an orange suspension. Then reaction mixture was diluted with cold (0–5 °C) water (30 mL) and extracted by cold (0–5 °C) n-hexane (3 × 10 mL). Combined extracts were washed with water (3 × 5 mL) and dried over Na2SO4. The residue, after removing solvent, was purified by flash chromatography (dried SiO2, n-hexane) to afford ethynylpyrrole 4a–k, ethynylindole 6 and ethynylfuran 8.
- 1-Benzyl-2-ethynyl-4,5,6,7-tetrahydro-1H-indole (4a). Yield: 197 mg (84%), colorless oil; 1H NMR (400.13 MHz, CDCl3): δ 7.37–7.24 (m, 3H, Hm,p, Ph), 7.13–7.08 (m, 2H, Ho, Ph), 6.37 (s, 1H, H-3, pyrrole), 5.14 (s, 2H, CH2-Ph), 3.35 (s, 1H, ≡CH), 2.54–2.49 (m, 2H, CH2-7), 2.43–2.38 (m, 2H, CH2-4), 1.82–1.68 (m, 4H, CH2-5, CH2-6); 13C NMR (100.6 MHz, CDCl3): δ 138.3, 130.9, 128.7 (2C), 127.3, 126.7 (2C), 118.0, 114.2, 99.7, 81.2, 77.0, 47.9, 23.5, 23.2, 23.1, 22.5; IR (KBr) 3287, 3087, 3063, 3030, 2928, 2849, 2097, 1495, 1457, 1388, 1357, 1301, 1130, 1077, 1029, 928, 795, 722, 696, 545, 457 cm−1; Anal. Calcd for C17H17N: C, 86.77; H, 7.28; N, 5.95%. Found: C, 86.47; H, 7.31; N, 6.14%.
- 2-Ethynyl-1-methyl-4,5,6,7-tetrahydro-1H-indole (4b). Yield: 137 mg (86%), white crystals, mp 53–54 °C; 1H NMR (400.13 MHz, CDCl3): δ 6.26 (s, 1H, H-3, pyrrole), 3.50 (s, 3H, NMe), 3.37 (s, 1H, ≡CH), 2.52–2.50 (m, 2H, CH2-7), 2.47–2.45 (m, 2H, CH2-4), 1.83–1.80 (m, 2H, CH2-5), 1.73–1.71 (m, 2H, CH2-6); 13C NMR (100.6 MHz, CDCl3,): δ 130.9, 117.4, 113.6, 112.6, 81.1, 76.9, 30.8, 23.6, 23.2, 23.0, 22.4; IR (film) 3288, 3100, 2929, 2847, 2097, 1570, 1462, 1442, 1386, 1302, 1130, 1055, 790, 667, 536 cm−1; Anal. Calcd for C11H13N: C, 82.97; H, 8.23; N, 8.80%. Found: C, 82.71; H, 8.44; N, 8.58%.
- 2-Ethynyl-1-vinyl-4,5,6,7-tetrahydro-1H-indole (4c). Yield: 127 mg (74%), colorless oil; 1H NMR (400.13 MHz, CDCl3): δ 6.97 (dd, J = 16.1, 9.4 Hz, 1H, Hx), 6.34 (s, 1H, H-3, pyrrole), 5.34 (d, J = 16.1 Hz, 1H, Ha), 4.83 (d, J = 9.4 Hz, 1H, Hb), 3.39 (s, 1H, ≡CH), 2.66–2.63 (m, 2H, CH2-7), 2.48–2.45 (m, 2H, CH2-4), 1.83–1.80 (m, 2H, CH2-5), 1.71–1.69 (m, 2H, CH2-6); 13C NMR (100.6 MHz, CDCl3): δ 130.5, 119.5, 116.8, 112.3, 102.1, 99.7, 82.1, 76.8, 24.2, 23.4, 23.1, 23.0; IR (film) 3292, 3128, 3049, 2932, 2849, 2099, 1643, 1577, 1483, 1438, 1387, 1324, 1294, 1136, 966, 871, 802, 669, 558 cm−1; Anal. Calcd for C12H13N: C, 84.17; H, 7.65; N, 8.18%. Found: C, 83.85; H, 7.81; N, 8.36%.
- 5-Ethynyl-2,3-dimethyl-1-vinyl-1H-pyrrole (4d). Yield: 52 mg (36%), colorless oil; 1H NMR (400.13 MHz, CDCl3): δ 6.91 (dd, J = 16.0, 9.2 Hz, 1H, Hx), 6.36 (s, 1H, H-3, pyrrole), 5.46 (d, J = 16.1 Hz, 1H, Ha), 4.94 (d, J = 9.2 Hz, 1H, Hb), 3.37 (s, 1H, ≡CH), 2.21 (s, 3H, Me), 1.99 (s, 3H, Me); 13C NMR (100.6 MHz, CDCl3): δ 130.8, 127.8, 119.0, 116.7, 111.6, 104.5, 81.7, 76.9, 11.4, 11.1; IR (film) 3291, 3106, 2920, 2866, 2099, 1643, 1483, 1432, 1392, 1335, 1310, 1162, 1113, 965, 879, 806, 671, 562 cm−1; Anal. Calcd for C10H11N: C, 82.72; H, 7.64; N, 9.65%. Found: C, 82.94; H, 7.49; N, 9.80%.
- 2-Ethynyl-1-methyl-5-phenyl-1H-pyrrole (4e). Yield: 172 mg (95%), colorless oil; 1H NMR (400.13 MHz, CDCl3): δ 7.42–7.34 (m, 5H, Ph), 6.55 (d, J = 3.8 Hz, 1H, H-3, pyrrole), 6.16 (d, J = 3.8 Hz, 1H, H-4, pyrrole), 3.69 (s, 3H, NMe), 3.44 (s, 1H, ≡CH); 13C NMR (100.6 MHz, CDCl3): δ 136.7, 132.9, 128.9 (2C), 128.6 (2C), 127.5, 116.1, 115.6, 108.6, 82.0, 76.5, 33.2; IR (film) 3287, 3106, 3060, 2948, 2102, 1602, 1498, 1457, 1390, 1324, 1234, 1155, 1074, 1028, 758, 698, 568 cm−1; Anal. Calcd for C13H11N: C, 86.15; H, 6.12; N, 7.73%. Found: C, 85.75; H, 5.86; N, 7.48%.
- 2-Ethynyl-5-(4-methylphenyl)-1-vinyl-1H-pyrrole (4f). Yield: 174 mg (84%), colorless oil; 1H NMR (400.13 MHz, CDCl3): δ 7.34–7.28 (m, 2H, Ho, Ph), 7.24–7.17 (m, 2H, Hm, Ph), 6.82 (dd, J = 15.9, 9.0 Hz, 1H, Hx), 6.63 (d, J = 3.8 Hz, 1H, H-3 pyrrole), 6.17 (d, J = 3.8 Hz, 1H, H-4, pyrrole), 5.53 (d, J = 15.9 Hz, 1H, Ha), 4.99 (d, J = 9.0 Hz, 1H, Hb), 3.43 (s, 1H, ≡CH), 2.38 (s, 3H, Me); 13C NMR (100.6 MHz, CDCl3): δ 137.6, 136.1, 131.1, 129.6, 129.2 (2C), 129.1 (2C), 118.5, 114.5, 109.9, 107.0, 82.5, 76.8, 21.3; IR (KBr) 3287, 3112, 3024, 2921, 2102, 1643, 1547, 1510, 1466, 1419, 1389, 1324, 1297, 1226, 1113, 963, 889, 822, 775, 672, 571, 500 cm−1; Anal. Calcd for C15H13N: C, 86.92; H, 6.32; N, 6.76%. Found: C, 86.68; H, 6.51; N, 6.85%.
- 1-Benzyl-2-ethynyl-5-(4-methoxyphenyl)-1H-pyrrole (4g). Yield: 253 mg (88%), white crystals; mp 92–93 °C; 1H NMR (400.13 MHz, CDCl3): δ 7.30–7.22 (m, 3H, Hm,p, Ph), 7.20–7.15 (m, 2H, Ho, Ph), 6.99–6.93 (m, 2H, Hm, Ph), 6.87–6.82 (m, 2H, Ho, Ph), 6.63 (d, J = 3.7 Hz, 1H, H-3 pyrrole), 6.16 (d, J = 3.7 Hz, 1H, H-4, pyrrole), 5.25 (s, 2H, CH2-Ph), 3.80 (s, 3H, MeO), 3.29 (s, 1H, ≡CH); 13C NMR (CDCl3, 100.6 MHz): δ 159.3, 138.8, 136.7, 130.4 (2C), 128.6 (2C), 127.2, 126.3 (2C), 125.3, 116.1, 115.5, 114.0 (2C), 108.8, 81.8, 76.6, 55.4, 48.9; IR (KBr) 3287, 3087, 3063, 3031, 2955, 2934, 2836, 2100, 1611, 1575, 1547, 1510, 1463, 1442, 1392, 1358, 1321, 1288, 1249, 1178, 1110, 1087, 1031, 977, 909, 836, 767, 731, 695, 575, 524, 459 cm−1; Anal. Calcd for C20H17NO: C, 83.59; H, 5.96; N, 4.87; O, 5.57%. Found: C, 83.31; H, 6.02; N, 5.02%.
- 2-Ethynyl-5-(2-fluorophenyl)-1-vinyl-1H-pyrrole (4h). Yield: 192 mg (91%), colorless oil; 1H NMR (400.13 MHz, CDCl3): δ 7.40–7.30 (m, 2H, Hm, Ph), 7.22–7.08 (m, 2H, Ho,p, Ph), 6.84 (dd, J = 15.9, 8.9 Hz, 1H, Hx), 6.66 (d, J = 3.7 Hz, 1H, H-3 pyrrole), 6.24 (d, J = 3.7 Hz, 1H, H-4, pyrrole), 5.34 (d, J = 15.9 Hz, 1H, Ha), 4.91 (d, J = 8.9 Hz, 1H, Hb), 3.45 (s, 1H, ≡CH); 13C NMR (100.6 MHz, CDCl3): δ 159.9 (d, J = 249.1 Hz, C-2, 2-FC6H4), 132.1 (d, J = 2.0 Hz, C-6, 2-FC6H4), 130.9, 130.1 (d, J = 8.2 Hz, C-4, 2-FC6H4), 129.2, 124.24 (d, J = 3.3 Hz, C-5, 2-FC6H4), 120.6 (d, J = 15.5 Hz, C-1, 2-FC6H4), 118.1, 116.1 (d, J = 22.0 Hz, C-3, 2-FC6H4), 115.2, 111.8, 106.4, 82.7, 76.4; IR (KBr) 3293, 3115, 3068, 2924, 2104, 1645, 1580, 1547, 1498, 1465, 1397, 1300, 1229, 1109, 963, 890, 817, 780, 759, 672, 577, 471 cm−1; Anal. Calcd for C14H10FN: C, 79.60; H, 4.77; F, 8.99; N, 6.63%. Found: C, 79.24; H, 4.96; F, 8.75; N, 6.39%.
- 5-Ethynyl-2,3-diphenyl-1-vinyl-1H-pyrrole (4i). Yield: 242 mg (90%), white crystals; mp 93–94 °C; 1H NMR (400.13 MHz, CDCl3): δ 7.39–7.34 (m, 3H, Ho,p, Ph), 7.31–7.26 (m, 2H, Ho, Ph), 7.21–7.15 (m, 2H, Hm, Ph), 7.15–7.09 (m, 3H, Hm,p, Ph), 6.84 (s, 1H, H-3 pyrrole), 6.71 (dd, J = 15.9, 9.2 Hz, 1H, Hx), 5.47 (d, J = 15.9 Hz, 1H, Ha), 4.91 (d, J = 9.2 Hz, 1H, Hb), 3.46 (s, 1H, ≡CH); 13C NMR (100.6 MHz, CDCl3): δ 135.1, 131.9, 131.8, 131.4 (2C), 130.8, 128.7 (2C), 128.3 (2C), 128.2 (3C), 126.1, 123.8, 118.6, 113.7, 106.5, 82.8, 76.5; IR (KBr) 3274, 3080, 3057, 2923, 2100, 1641, 1601, 1557, 1495, 1446, 1386, 1320, 1305, 1177, 1031, 964, 889, 800, 769, 699, 587, 522 cm−1; Anal. Calcd for C20H15N: C, 89.19; H, 5.61; N, 5.20%. Found: C, 88.89; H, 5.45; N, 5.34%.
- 2-Ethynyl-1-methyl-5-(thiophen-2-yl)-1H-pyrrole (4j). Yield: 174 mg (93%), colorless oil; 1H NMR (400.13 MHz, CDCl3): δ 7.32–7.28 (m, 1H, H-5, thiophene), 7.10–7.05 (m, 2H, H-3,4, thiophene), 6.51 (d, J = 3.9 Hz, 1H, H-3 pyrrole), 6.26 (d, J = 3.9 Hz, 1H, H-4, pyrrole), 3.76 (s, 3H, N-CH3), 3.43 (s, 1H, ≡CH); 13C NMR (100.6 MHz, CDCl3): δ 134.4, 129.2, 127.5, 125.8, 125.3, 116.6, 115.6, 109.7, 99.7, 82.2, 33.2; IR (KBr) 3288, 3106, 3074, 2944, 2922, 2101, 1445, 1417, 1395, 1345, 1314, 1201, 1034, 845, 766, 698, 570, 493 cm−1; Anal. Calcd for C11H9NS: C, 70.55; H, 4.84; N, 7.48; S, 17.12%. Found: C, 70.26; H, 4.69; N, 7.28; S, 16.82%.
- 1-Benzyl-2-ethynyl-1H-pyrrole (4k). Yield: 145 mg (80%), colorless oil; 1H NMR (400.13 MHz, CDCl3): δ 7.36–7.27 (m, 3H, Hm,p, Ph), 7.16–7.14 (m, 2H, Ho, Ph), 6.68–6.65 (m, 1H, H-3, pyrrole), 6.54–6.51 (m, 1H, H-5, pyrrole), 6.13–6.10 (m, 1H, H-4, pyrrole), 5.19 (s, 2H, CH2-Ph), 3.33 (s, 1H, ≡CH); 13C NMR (CDCl3, 100.6 MHz): δ 137.9, 128.8 (2C), 127.7, 127.3 (2C), 123.1, 116.0, 114.7, 108.7, 81.7, 76.0, 51.3; IR (KBr) 3288, 3106, 3064, 3031, 2925, 2853, 2103, 1495, 1466, 1455, 1435, 1300, 1018, 722, 694, 569, 522 cm−1; Anal. Calcd for C13H11N: C, 86.15; H, 6.12; N, 7.73%. Found: C, 85.84; H, 5.89; N, 7.45%.
- 3-Ethynyl-1-methyl-1H-indole (6). Yield: 113 mg (73%); Spectral characteristics are the same as previously published [64].
- 2-Ethynyl-3,6-dimethyl-4,5,6,7-tetrahydrobenzofuran (8). Yield: 139 mg (80%), colorless oil; 1H NMR (400.13 MHz, CDCl3): δ 3.55 (s, 1H, ≡CH), 2.67–2.62 (m, 1H, CH), 2.33–2.30 (m, 2H, CH2), 2.19–2.12 (m, 1H, CH), 2.00 (s, 3H, Me), 1.93–1.91 (m, 1H, CH), 1.85–1.81 (m, 1H, CH), 1.36–1.30 (m, 1H, CH), 1.07 (d, J = 6.7 Hz, 3H, CHMe); 13C NMR (100.6 MHz, CDCl3): δ 152.1, 131.5, 127.2, 118.4, 83.8, 74.7, 31.7, 31.2, 29.6, 21.5, 20.0, 9.0; IR (KBr) 3293, 2923, 2849, 2103, 1628, 1558, 1456, 1379, 1295, 1257, 1150, 1107, 1066, 1041, 774, 692 cm−1; Anal. Calcd for C12H14O: C, 82.72; H, 8.10; O, 9.18%. Found: C, 82.94; H, 7.88%.
3.3. Synthesis of Propargyl Alcohols 3a,c,d,f
Acylethynylpyrrole 1a,c,d,f (1 mmol) was dissolved in dry MeCN (4 mL), and then t-BuONa (192 mg, 2 mmol) was added to reaction mixture under nitrogen and reaction mixture was stirred at room temperature for 1 h. Then reaction mixture was diluted with water (30 mL) and extracted by diethyl ether (3 × 10 mL). Extracts were washed with water (3 × 5 mL) and dried over Na2SO4. The residue after removing solvents was fractionated by column chromatography (SiO2, n-hexane:diethyl ether, 10:1) to afford propargyl alcohol 3a,c,d,f.
- 5-(1-Benzyl-4,5,6,7-tetrahydro-1H-indol-2-yl)-3-hydroxy-3-(thiophen-2-yl)pent-4-ynenitrile (3a). Spectral characteristics are the same as previously published [36].
- 3-Hydroxy-3-phenyl-5-(1-vinyl-4,5,6,7-tetrahydro-1H-indol-2-yl)pent-4-ynenitrile (3c). Yield: 224 mg (71%), yellow oil; 1H NMR (400.13 MHz, CDCl3): δ 7.72–7.71 (m, 2H, Ho, Ph), 7.44–7.37 (m, 3H, Hm,p, Ph), 6.98 (dd, J = 15.9, 9.3 Hz, 1H, Hx), 6.40 (s, 1H, H-3, pyrrole), 5.34 (d, J = 15.9 Hz, 1H, Ha), 4.88 (d, J = 9.3 Hz, 1H, Hb), 3.03 (d, J = 4.8 Hz, 2H, CH2CN), 2.85 (s, 1H, OH), 2.67–2.65 (m, 2H, CH2-7), 2.49–2.47 (m, 2H, CH2-4), 1.83–1.81 (m, 2H, CH2-5), 1.74–1.73 (m, 2H, CH2-6); 13C NMR (CDCl3, 100.6 MHz): δ 141.7, 131.5, 130.4, 129.0, 128.8 (2C), 125.4 (2C), 119.9, 117.3, 116.4, 111.3, 103.2, 92.9, 81.4, 71.0, 35.7, 24.1, 23.3, 23.1, 23.0. IR (film) 3422, 3062, 3030, 2931, 2851, 2215, 1643, 1492, 1447, 1383, 1295, 1241, 1143, 1102, 1053, 968, 910, 805, 765, 733, 700, 646 cm−1; Anal. Calcd for C21H20N2O: C, 79.72; H, 6.37; N, 8.85; O, 5.06%. Found: C, 79.44; H, 6.20; N, 8.59%.
- 5-(4,5-Dimethyl-1-vinyl-1H-pyrrol-2-yl)-3-hydroxy-3-phenylpent-4-ynenitrile (3d). Yield: 197 mg (68%), yellow crystals, mp 101–102 °C; 1H NMR (400.13 MHz, CDCl3): δ 7.72–7.70 (m, 2H, Ho, Ph), 7.42–7.40 (m, 2H, Hm,p, Ph), 6.91 (dd, J = 15.9, 9.1 Hz, 1H, Hx), 6.41 (s, 1H, H-3, pyrrole), 5.45 (d, J = 15.9 Hz, 1H, Ha), 4.99 (d, J = 9.1 Hz, 1H, Hb), 3.02 (d, J = 5.1 Hz, 2H, CH2CN), 2.86 (s, 1H, OH), 2.22 (s, 3H, Me), 2.00 (s, 3H, Me); 13C NMR (100.6 MHz, CDCl3): δ 141.7, 130.6, 128.9, 128.7 (2C), 128.6, 125.4 (2C), 119.5, 117.0, 116.4, 110.6, 105.6, 92.6, 81.4, 70.9, 35.6, 11.3, 11.1; IR (KBr) 3422, 3062, 3030, 2921, 2215, 1643, 1493, 1449, 1392, 1357, 1304, 1172, 1100, 1049, 967, 910, 809, 765, 733, 700, 634 cm−1; Anal. Calcd for C19H18N2O: C, 78.59; H, 6.25; N, 9.65; O, 5.51%. Found: C, 78.22; H, 6.02; N, 9.42%.
- 3-Hydroxy-3-phenyl-5-(5-(4-methylphenyl)-1-vinyl-1H-pyrrol-2-yl)pent-4-ynenitrile (3f). Yield: 281 mg (80%), yellow oil; 1H NMR (CDCl3, 400 MHz): δ 7.74–7.72 (m, 2H, Ho, Ph), 7.45–7.39 (m, 2H, Hm,p, Ph), 7.31 (d, J = 7.9 Hz, 2H, Ho, C6H4), 7.21 (d, J = 7.9 Hz, 2H, Hm, C6H4), 6.83 (dd, J = 15.8, 8.9 Hz, 1H, Hx), 6.67 (d, J = 3.8 Hz, 1H, H-4, pyrrole), 6.22 (d, J = 3.8 Hz, 1H, H-3, pyrrole), 5.52 (d, J = 15.8 Hz, 1H, Ha), 5.05 (d, J = 8.9 Hz, 1H, Hb), 3.06 (d, J = 5.0 Hz, 2H, CH2CN), 2.85 (s, 1H, OH), 2.39 (s, 3H, Me); 13C NMR (CDCl3, 100.6 MHz): δ 141.6, 137.8, 136.8, 131.2, 129.4, 129.3 (2C), 129.1 (3C), 128.8 (2C), 125.4 (2C), 118.9, 116.3, 113.7, 110.1, 108.0, 93.1, 81.4, 71.0, 35.6, 21.4; IR (KBr) 3416, 3061, 3028, 2922, 2218, 1643, 1515, 1472, 1449, 1418, 1389, 1324, 1301, 1224, 1112, 1042, 964, 909, 823, 773, 733,701, 622, 503 cm−1, Anal. Calcd for C24H20N2O: C, 81.79; H, 5.72; N, 7.95; O, 4.54%. Found: C, 81.35; H, 5.60; N, 7.68%.
4. Conclusions
In conclusion, we have found efficient and extraordinarily easy (room temperature) access to ethynylpyrroles via decarbonylation of available acylethynylpyrroles. The reaction proceeds in the MeCN-THF/t-BuOK system via the addition of CH2CN− anion to the carbonyl group of acylethynylpyrroles followed by retro-Favorsky reaction of the intermediated propargylic alcohols. Thermodynamic aspects of the intermediate alcohol decomposition have been considered in the framework of B2PLYP/6-311G**//B3LYP/6-311G**+C-PCM/acetonitrile methodology. The substrate scope of the reaction includes benzoyl-, furoyl-, thenoylethynylpyrroles with alkyl, vinyl, aryl and hetaryl substituents at 1(4,5)-positions of the pyrrole ring, and methyl, benzyl, and vinyl moieties at the nitrogen atom, as well as acylethynyl derivatives of 1-methylindole and menthofuran.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28031389/s1. Synthesis of ethynylpyrroles 4a–k, ethynylindole 6, ethynylfuran 8 (S3–S7); Synthesis of propargyl alcohols 3a,c,d,f (S7–S8); Quantum chemical calculations details (S9–S12); 1H and 13C NMR spectra of synthesized compounds 3a,c,d,f, 4a–k, 6, 8 (S13–S44). References [36,64,65,66,67,68,69,70,71,72,73,74] are cited in the Supplementary Materials.
Author Contributions
Conceptualization, B.A.T.; investigation, D.N.T.; writing—review and editing, D.N.T., L.N.S. and B.A.T.; writing—original draft preparation, D.N.T. and L.N.S.; formal analysis, I.A.U.; calculation part, A.M.B. and A.B.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Education and Science of Russian Federation (State Registration No. 121021000199-6).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the Supplementary Material.
Acknowledgments
This work was supported by the research project plans in the State Register of the IPC RAS no. 121021000199-6. The authors acknowledge Baikal Analytical Center for the collective use of SB RAS for the equipment. A.B.T. gratefully acknowledges Grant No. FZZE-2020-0025 from the Ministry of Science and Higher Education of the Russian Federation. A.M.B. thanks the Irkutsk Supercomputer Center of SB RAS for providing computational resources of the HPC-cluster “Akademik V. M. Matrosov”.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of compounds 1a–k, 5, and 7 are available from the authors.
References
- Bitar, A.Y.; Frontier, A.J. Formal Synthesis of (±)-Roseophilin. Org. Lett. 2009, 11, 49–52. [Google Scholar] [CrossRef]
- Saito, K.; Yoshida, M.; Uekusa, H.; Doi, T. Facile Synthesis of Pyrrolyl 4-Quinolinone Alkaloid Quinolactacide by 9-AJ-Catalyzed Tandem Acyl Transfer–Cyclization of o-Alkynoylaniline Derivatives. ACS Omega 2017, 2, 4370–4381. [Google Scholar] [CrossRef]
- Kitano, Y.; Suzuki, T.; Kawahara, E.; Yamazaki, T. Synthesis and inhibitory activity of 4-alkynyl and 4-alkenylquinazolines: Identification of new scaffolds for potent EGFR tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 5863–5867. [Google Scholar] [CrossRef]
- Thottathil, J.K.; Li, W.S. Process for the Preparation of 4-phosphinyl-3-Keto-Carboxylate and 4-Phosphonyl-3-Keto-Carboxylate Intermediates Useful in the Preparation of Phosphorus Containing HMG-CoA Reductase Inhibitors. U.S. Patent US5298625A, 29 March 1994. [Google Scholar]
- Haubmann, C.; Hübner, H.; Gmeiner, P. Piperidinylpyrroles: Design, synthesis and binding properties of novel and selective dopamine D4 receptor ligands. Bioorg. Med. Chem. Lett. 1999, 9, 3143–3146. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Lee, H.; Lee, S.; Jeon, H.-G.; Jeong, K.-S. Encapsulation of dihydrogenphosphate ions as a cyclic dimer to the cavities of site-specifically modified indolocarbazole-pyridine foldamers. Org. Chem. Front. 2019, 6, 299–303. [Google Scholar] [CrossRef]
- Guérin, C.; Jean-Gérard, L.; Octobre, G.; Pascal, S.; Maury, O.; Pilet, G.; Ledoux, A.; Andrioletti, B. Bis-triazolyl BODIPYs: A simple dye with strong red-light emission. RSC Adv. 2015, 5, 76342–76345. [Google Scholar] [CrossRef]
- Lim, J.Y.C.; Beer, P.D. A pyrrole-containing cleft-type halogen bonding receptor for oxoanion recognition and sensing in aqueous solvent media. New J. Chem. 2018, 42, 10472–10475. [Google Scholar] [CrossRef]
- Sessler, J.L.; Cai, J.; Gong, H.-Y.; Yang, X.; Arambula, J.F.; Hay, B.P. A Pyrrolyl-Based Triazolophane: A Macrocyclic Receptor with CH and NH Donor Groups That Exhibits a Preference for Pyrophosphate Anions. J. Am. Chem. Soc. 2010, 132, 14058–14060. [Google Scholar] [CrossRef]
- Liao, J.-H.; Chen, C.-T.; Chou, H.-C.; Cheng, C.-C.; Chou, P.-T.; Fang, J.-M.; Slanina, Z.; Chow, T.J. 2,7-Bis(1H-pyrrol-2-yl)ethynyl-1,8naphthyridine: An Ultrasensitive Fluorescent Probe for Glucopyranoside. Org. Lett. 2002, 4, 3107–3110. [Google Scholar] [CrossRef]
- Li, Y.; Zuo, Z.; Liu, H.; Li, Y. Highly-conductive carbon material and low-temperature preparation method thereof. Chinese Patent CN108298516A, 20 July 2018. [Google Scholar]
- Heynderickx, A.; Mohamed Kaou, A.; Moustrou, C.; Samat, A.; Guglielmetti, R. Synthesis and photochromic behaviour of new dipyrrolylperfluorocyclopentenes. New J. Chem. 2003, 27, 1425–1432. [Google Scholar] [CrossRef]
- Tanaka, Y.; Ishisaka, T.; Koike, T.; Akita, M. Synthesis and properties of diiron complexes with heteroaromatic linkers: An approach for modulation of organometallic molecular wire. Polyhedron 2015, 86, 105–110. [Google Scholar] [CrossRef]
- Cheema, H.; Baumann, A.; Loya, E.K.; Brogdon, P.; McNamara, L.E.; Carpenter, C.A.; Hammer, N.I.; Mathew, S.; Risko, C.; Delcamp, J.H. Near-Infrared-Absorbing Indolizine-Porphyrin Push–Pull Dye for Dye-Sensitized Solar Cells. ACS Appl. Mater. Interfaces 2019, 11, 16474–16489. [Google Scholar] [CrossRef] [PubMed]
- Debnath, S.; Singh, S.; Bedi, A.; Krishnamoorthy, K.; Zade, S.S. Site-selective synthesis and characterization of BODIPY–acetylene copolymers and their transistor properties. J. Polym. Sci. A Polym. Chem. 2016, 54, 1978–1986. [Google Scholar] [CrossRef]
- Yasuda, T.; Imase, T.; Nakamura, Y.; Yamamoto, T. New Alternative Donor−Acceptor Arranged Poly(Aryleneethynylene)s and Their Related Compounds Composed of Five-Membered Electron-Accepting 1,3,4-Thiadiazole, 1,2,4-Triazole, or 3,4-Dinitrothiophene Units: Synthesis, Packing Structure, and Optical Properties. Macromolecules 2005, 38, 4687–4697. [Google Scholar] [CrossRef]
- Martire, D.O.; Jux, N.; Aramendia, P.F.; Martin Negri, R.; Lex, J.; Braslavsky, S.E.; Schaffner, K.; Vogel, E. Photophysics and photochemistry of 22π and 26π acetylene-cumulene porphyrinoids. J. Am. Chem. Soc. 1992, 114, 9969–9978. [Google Scholar] [CrossRef]
- Tu, B.; Ghosh, B.; Lightner, D.A. Novel Linear Tetrapyrroles: Hydrogen Bonding in Diacetylenic Bilirubins. Mon. Für Chem. Chem. Mon. 2004, 135, 519–541. [Google Scholar] [CrossRef]
- Rana, A.; Lee, S.; Kim, D.; Panda, P.K. β-Octamethoxy-Substituted 22π and 26π Stretched Porphycenes: Synthesis, Characterization, Photodynamics, and Nonlinear Optical Studies. Chem. Eur. J. 2015, 21, 12129–12135. [Google Scholar] [CrossRef]
- Blomquist, A.T.; Wasserman, H.H. Organic Chemistry: A Series of Monographs. In Organic Chemistry: A Series of Monographs; Jones, R.A., Bean, G.P., Eds.; Academic Press: Cambridge, MA, USA, 1977; Volume 34, pp. 129–140. [Google Scholar]
- Gossauer, A. Die Chemie der Pyrrole; Springer: Berlin/Heidelberg, Germany, 2013; Volume 15. [Google Scholar]
- Negishi, E.; Xu, C.; Tan, Z.; Kotora, M. Direct Synthesis of Heteroarylethynes via Palladium-catalyzed Coupling of Heteroaryl Halides with Ethynylzinc Halides. Its Application to an Efficient Synthesis of a Thiophenelactone from Chamaemelum nobile L. Heterocycles 1997, 46, 209–214. [Google Scholar] [CrossRef]
- Sauvêtre, R.; Normant, J.F. Une nouvelle preparation du fluoroacetylene—Sa reaction avec les organometalliques. Synthese d’alcynes et d’enynes divers. Tetrahedron Lett. 1982, 23, 4325–4328. [Google Scholar] [CrossRef]
- Tietze, L.F.; Kettschau, G.; Heitmann, K. Synthesis of N-Protected 2-Hydroxymethylpyrroles and Transformation into Acyclic Oligomers. Synthesis 1996, 1996, 851–857. [Google Scholar] [CrossRef]
- Morri, A.K.; Thummala, Y.; Doddi, V.R. The Dual Role of 1,8-Diazabicyclo [5.4.0]undec-7-ene (DBU) in the Synthesis of Terminal Aryl- and Styryl-Acetylenes via Umpolung Reactivity. Org. Lett. 2015, 17, 4640–4643. [Google Scholar] [CrossRef] [PubMed]
- Wentrup, C.; Winter, H.-W. A General and Facile Synthesis of Aryl- and Hetero-arylacetylenes. Angew. Chem. Int. Ed. 1978, 17, 609–610. [Google Scholar] [CrossRef]
- Benzies, D.W.M.; Fresneda, P.M.; Jones, R.A.; McNab, H. Flash vacuum pyrolysis of 5-(indol-2- and -3-ylmethylene)-2,2-dimethyl-1,3-dioxane-4,6-diones. J. Am. Chem. Soc. Perkin Trans. 1986, 1, 1651–1654. [Google Scholar] [CrossRef]
- Comer, M.C.; Despinoy, X.L.M.; Gould, R.O.; McNab, H.; Parsons, S. Synthesis and unexpectedly facile dimerisation of 1-methoxycarbonylpyrrolizin-3-one. Chem. Commun. 1996, 9, 1083–1084. [Google Scholar] [CrossRef]
- Despinoy, X.L.M.; McNab, H. 1-Methoxycarbonylpyrrolizin-3-one and related compounds. Org. Biomol. Chem. 2009, 7, 2187–2194. [Google Scholar] [CrossRef]
- Vasilevskii, S.F.; Sundukova, T.A.; Shvartsberg, M.S.; Kotlyarevskii, I.L. Synthesis of acetylenyl-N-methylpyrroles. Izv. Akad. Nauk. SSSR Seriya Khimicheskaya 1980, 8, 1346–1350. [Google Scholar] [CrossRef]
- Sobenina, L.N.; Tomilin, D.N.; Gotsko, M.D.; Ushakov, I.A.; Mikhaleva, A.I.; Trofimov, B.A. From 4,5,6,7-tetrahydroindoles to 3- or 5-(4,5,6,7-tetrahydroindol-2-yl)isoxazoles in two steps: A regioselective switch between 3- and 5-isomers. Tetrahedron 2014, 70, 5168–5174. [Google Scholar] [CrossRef]
- Tomilin, D.N.; Gotsko, M.D.; Sobenina, L.N.; Ushakov, I.A.; Afonin, A.V.; Soshnikov, D.Y.; Trofimov, A.B.; Koldobsky, A.B.; Trofimov, B.A. N-Vinyl-2-(trifluoroacetylethynyl)pyrroles and E-2-(1-bromo-2-trifluoroacetylethenyl)pyrroles: Cross-coupling vs. addition during CH-functionalization of pyrroles with bromotrifluoroacetylacetylene in solid Al2O3 medium. H-bonding control. J. Fluorine Chem. 2016, 186, 1–6. [Google Scholar] [CrossRef]
- Vereshchagin, L.I.; Kirillova, L.P.; Buzilova, S.R. Unsaturated carbonyl-containing compounds. 18. Alkaline cleavage of alpha-acetylenic ketones. Zh. Org. Khim. 1975, 11, 292. [Google Scholar]
- Fedenok, L.G.; Shvartsberg, M.S. A method for the preparation of terminal acetylenes. Bull. Acad. Sci. USSR Div. Chem. Sci. 1990, 39, 2376–2377. [Google Scholar] [CrossRef]
- Trofimov, B.A.; Sobenina, L.N.; Mikhaleva, A.I.; Ushakov, I.A.; Vakul’skaya, T.I.; Stepanova, Z.V.; Toryashinova, D.-S.D.; Mal’kina, A.G.; Elokhina, V.N. N- and C-Vinylation of Pyrroles with Disubstituted Activated Acetylenes. Synthesis 2003, 2003, 1272–1278. [Google Scholar] [CrossRef]
- Tomilin, D.N.; Sobenina, L.N.; Saliy, I.V.; Ushakov, I.A.; Belogolova, A.M.; Trofimov, B.A. Substituted pyrrolyl-cyanopyridines on the platform of acylethynylpyrroles via their 1:2 annulation with acetonitrile under the action of lithium metal. New J. Chem. 2022, 46, 13149–13155. [Google Scholar] [CrossRef]
- Trofimov, B.A.; Stepanova, Z.V.; Sobenina, L.N.; Mikhaleva, A.I.; Ushakov, I.A. Ethynylation of pyrroles with 1-acyl-2-bromoacetylenes on alumina: A formal ‘inverse Sonogashira coupling’. Tetrahedron Lett. 2004, 45, 6513–6516. [Google Scholar] [CrossRef]
- Trofimov, B.A.; Sobenina, L.N. Targets in Heterocyclic Systems.; Attanasi, O.A., Spinelli, D., Eds.; Società Chimica Italiana: Roma, Italy, 2009; Volume 13, pp. 92–119. [Google Scholar]
- Sobenina, L.N.; Tomilin, D.N.; Petrova, O.V.; Gulia, N.; Osowska, K.; Szafert, S.; Mikhaleva, A.I.; Trofimov, B.A. Cross-coupling of 4,5,6,7-tetrahydroindole with functionalized haloacetylenes on active surfaces of metal oxides and salts. Russ. J. Org. Chem. 2010, 46, 1373–1377. [Google Scholar] [CrossRef]
- Sobenina, L.N.; Trofimov, B.A. Recent Strides in the Transition Metal-Free Cross-Coupling of Haloacetylenes with Electron-Rich Heterocycles in Solid Media. Molecules 2020, 25, 2490. [Google Scholar] [CrossRef]
- Tarshits, D.L.; Przhiyalgovskaya, N.M.; Buyanov, V.N.; Tarasov, S.Y. Ethynylindoles and their derivatives. Methods of synthesis and chemical transformations (review). Chem. Heterocycl. Compd. 2009, 45, 501–523. [Google Scholar] [CrossRef]
- Sobenina, L.N.; Demenev, A.P.; Mikhaleva, A.I.; Ushakov, I.A.; Vasil’tsov, A.M.; Ivanov, A.V.; Trofimov, B.A. Ethynylation of indoles with 1-benzoyl-2-bromoacetylene on Al2O3. Tetrahedron Lett. 2006, 47, 7139–7141. [Google Scholar] [CrossRef]
- Tisserand, R.; Young, R. 12—Cancer and the immune system. In Essential Oil Safety, 2nd ed.; Tisserand, R., Young, R., Eds.; Churchill Livingstone: St. Louis, MO, USA, 2014; pp. 165–186. [Google Scholar]
- Sobenina, L.N.; Tomilin, D.N.; Gotsko, M.D.; Ushakov, I.A.; Trofimov, B.A. Transition metal-free cross-coupling of furan ring with haloacetylenes. Tetrahedron 2018, 74, 1565–1570. [Google Scholar] [CrossRef]
- Uchida, A.; Doyama, A.; Matsuda, S. Reaction of Acetonitrile with Carboxylic Esters. Bull. Chem. Soc. Jpn. 1970, 43, 963–965. [Google Scholar] [CrossRef]
- Ko, E.-Y.; Lim, C.-H.; Chung, K.-H. Additions of Acetonitrile and Chloroform to Aromatic Aldehydes in the Presence of Tetrabutylammonium Fluoride. Bull. Korean Chem. Soc. 2006, 27, 432–434. [Google Scholar] [CrossRef]
- Xiao, S.; Chen, C.; Li, H.; Lin, K.; Zhou, W. A Novel and Practical Synthesis of Ramelteon. Org. Process Res. Dev. 2015, 19, 373–377. [Google Scholar] [CrossRef]
- Yu, Y.; Li, G.; Jiang, L.; Zu, L. An Indoxyl-Based Strategy for the Synthesis of Indolines and Indolenines. Angew. Chem. Int. Ed. 2015, 54, 12627–12631. [Google Scholar] [CrossRef] [PubMed]
- Hoff, B.H. Acetonitrile as a Building Block and Reactant. Synthesis 2018, 50, 2824–2852. [Google Scholar] [CrossRef]
- Engel, D.A.; Dudley, G.B. The Meyer–Schuster rearrangement for the synthesis of α,β-unsaturated carbonyl compounds. Org. Biomol. Chem. 2009, 7, 4149–4158. [Google Scholar] [CrossRef]
- Wang, L.-X.; Tang, Y.-L. Cycloisomerization of Pyridine-Substituted Propargylic Alcohols or Esters To Construct Indolizines and Indolizinones. Eur. J. Org. Chem. 2017, 2017, 2207–2213. [Google Scholar] [CrossRef]
- Roy, R.; Saha, S. Scope and advances in the catalytic propargylic substitution reaction. RSC Adv. 2018, 8, 31129–31193. [Google Scholar] [CrossRef]
- Qian, H.; Huang, D.; Bi, Y.; Yan, G. 2-Propargyl Alcohols in Organic Synthesis. Adv. Synth. Catal. 2019, 361, 3240–3280. [Google Scholar] [CrossRef]
- Kumar, G.R.; Rajesh, M.; Lin, S.; Liu, S. Propargylic Alcohols as Coupling Partners in Transition-Metal-Catalyzed Arene C−H Activation. Adv. Synth. Catal. 2020, 362, 5238–5256. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Liu, Y.-L.; Chen, L. Tandem Annulations of Propargylic Alcohols to Indole Derivatives. Adv. Synth. Catal. 2020, 362, 5170–5195. [Google Scholar] [CrossRef]
- Du, S.; Zhou, A.-X.; Yang, R.; Song, X.-R.; Xiao, Q. Recent advances in the direct transformation of propargylic alcohols to allenes. Org. Chem. Front. 2021, 8, 6760–6782. [Google Scholar] [CrossRef]
- Song, X.-R.; Yang, R.; Xiao, Q. Recent Advances in the Synthesis of Heterocyclics via Cascade Cyclization of Propargylic Alcohols. Adv. Synth. Catal. 2021, 363, 852–876. [Google Scholar] [CrossRef]
- Bai, J.-F.; Tang, J.; Gao, X.; Jiang, Z.-J.; Tang, B.; Chen, J.; Gao, Z. Regioselective Cycloaddition and Substitution Reaction of Tertiary Propargylic Alcohols and Heteroareneboronic Acids via Acid Catalysis. Org. Lett. 2022, 24, 4507–4512. [Google Scholar] [CrossRef] [PubMed]
- Noda, H.; Kumagai, N.; Shibasaki, M. Catalytic Asymmetric Synthesis of α-Trifluoromethylated Carbinols: A Case Study of Tertiary Propargylic Alcohols. Asian J. Org. Chem. 2018, 7, 599–612. [Google Scholar] [CrossRef]
- Kobayashi, D.; Miura, M.; Toriyama, M.; Motohashi, S. Stereoselective synthesis of secondary and tertiary propargylic alcohols induced by a chiral sulfoxide auxiliary. Tetrahedron Lett. 2019, 60, 120–123. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, W.; Wu, P.; Huang, C.; Zheng, Y.; Zheng, W.-F.; Qian, H.; Ma, S. Chiral tertiary propargylic alcohols via Pd-catalyzed carboxylative kinetic resolution. Org. Chem. Front. 2020, 7, 3907–3911. [Google Scholar] [CrossRef]
- Bradley, D.C.; Mehrotra, R.C.; Rothwell, I.P.; Singh, A. 4—X-Ray Crystal Structures of Alkoxo Metal Compounds. In Alkoxo and Aryloxo Derivatives of Metals; Bradley, D.C., Mehrotra, R.C., Rothwell, I.P., Singh, A., Eds.; Academic Press: London, UK, 2001; pp. 229–382. [Google Scholar]
- Dean, J.A.; Lange, N.A. Lange’s Handbook of Chemistry; McGraw-Hill: New York, NY, USA, 1999. [Google Scholar]
- Gupton, J.T.; Telang, N.; Gazzo, D.F.; Barelli, P.J.; Lescalleet, K.E.; Fagan, J.W.; Mills, B.J.; Finzel, K.L.; Kanters, R.P.F.; Crocker, K.R.; et al. Preparation of indole containing building blocks for the regiospecific construction of indole appended pyrazoles and pyrroles. Tetrahedron 2013, 69, 5829–5840. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Vitkovskaya, N.M.; Orel, V.B.; Absalyamov, D.Z.; Trofimov, B.A. Self-Assembly of N-Phenyl-2,5-dimethylpyrrole from Acetylene and Aniline in KOH/DMSO and KOBut/DMSO Superbase Systems: A Quantum-Chemical Insight. J. Org. Chem. 2020, 85, 10617–10627. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Petersson, G.A. Complete Basis Set Models for Chemical Reactivity: From the Helium Atom to Enzyme Kinetics. In Quantum-Mechanical Prediction of Thermochemical Data; Cioslowski, J., Ed.; Springer: Dordrecht, The Netherlands, 2001; pp. 99–130. [Google Scholar]
- Montgomery, J.A., Jr.; Frisch, M.J.; Ochterski, J.W.; Petersson, G.A. A complete basis set model chemistry. VII. Use of the minimum population localization method. J. Chem. Phys. 2000, 112, 6532–6542. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).