
Synthetic Studies on Tetracyclic Diquinane Lycopodium Alkaloids Magellanine, Magellaninone and Paniculatine


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
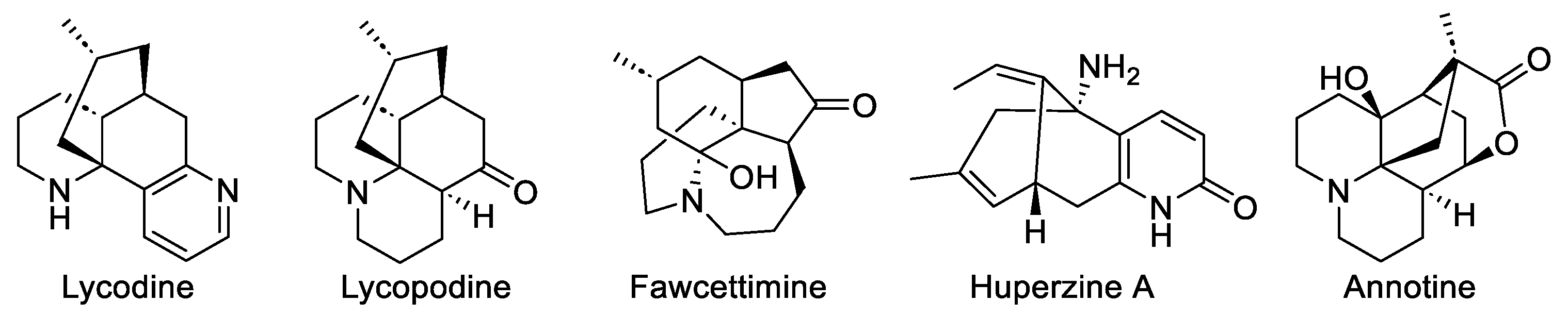
1. Introduction
2. Early Synthetic Efforts from 1986–1993
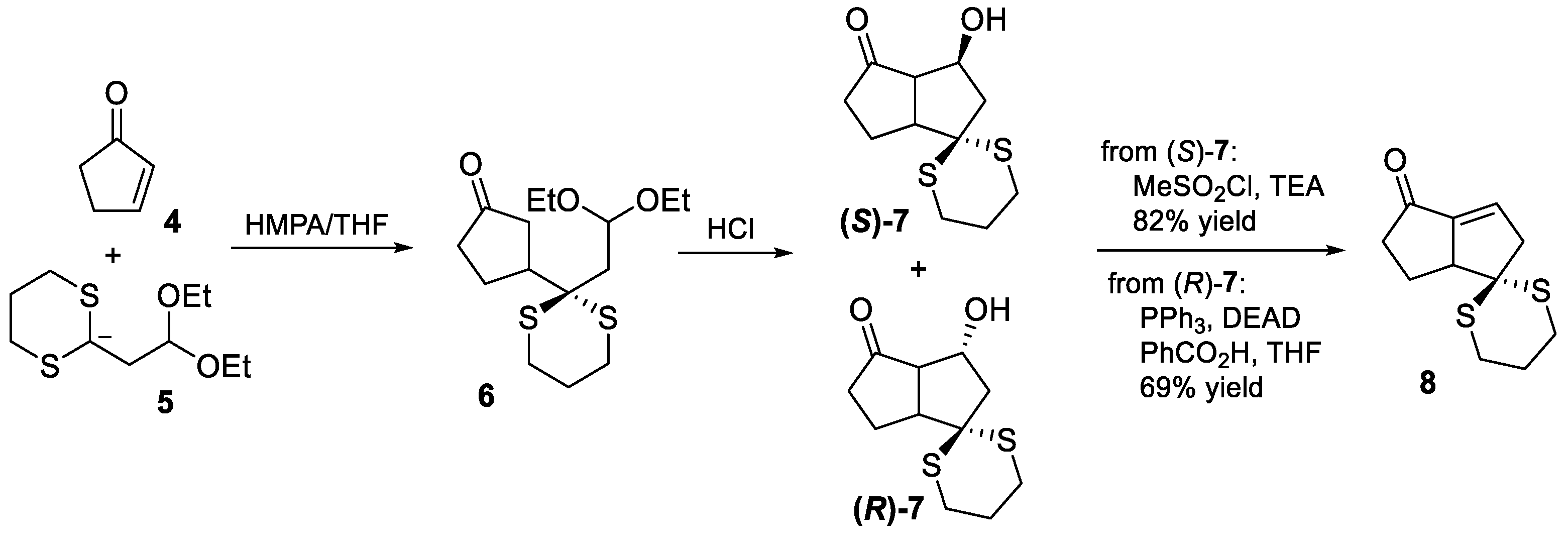
2.1. Paquette’s 1986 Synthesis
2.2. Overman’s 1989 Synthesis
2.3. Mehta’s 1987 and 1990 Syntheses
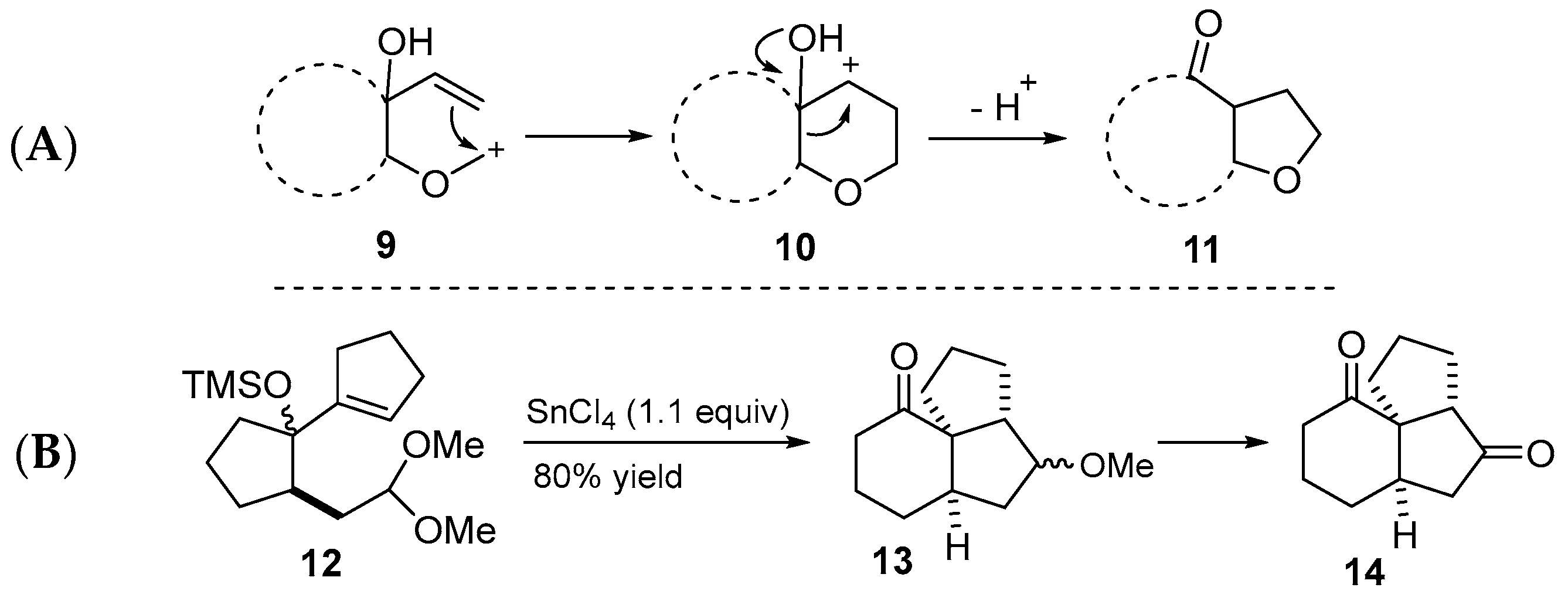
2.4. Crimmins’ 1993 Synthesis
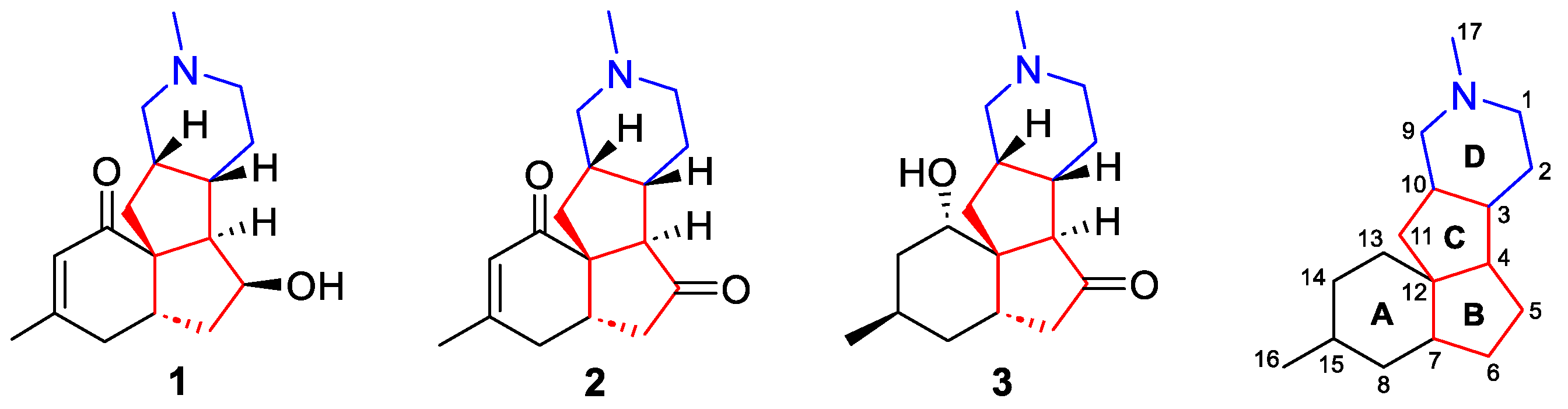
3. Total Synthesis of Magellanine, Magellaninone, and Paniculatine (1–3)
3.1. Overman’s 1993 Synthesis of (–)-Magellanine and (+)-Magellaninone
3.2. Paquette’s 1993/1994 Synthesis of (–)-Magellanine and (+)-Magellaninone
3.3. Sha’s 1999 Synthesis of (+)-Paniculatine
3.4. Liao’s 2002 Synthesis of (±)-Magellanine
3.5. Ishizaki’s 2005 Synthesis of (±)-Magellanine
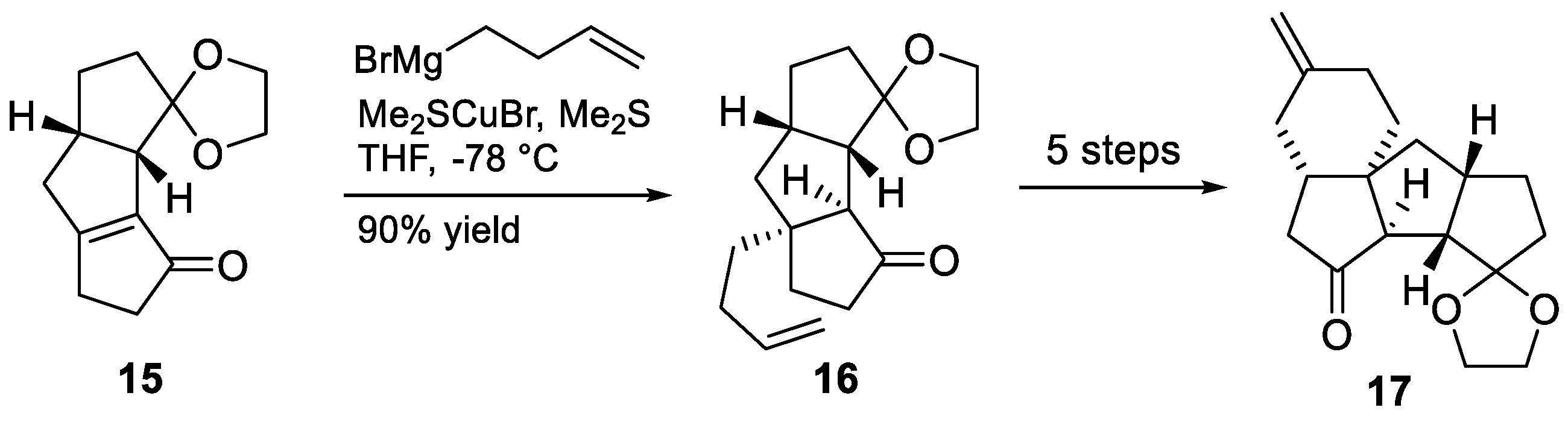
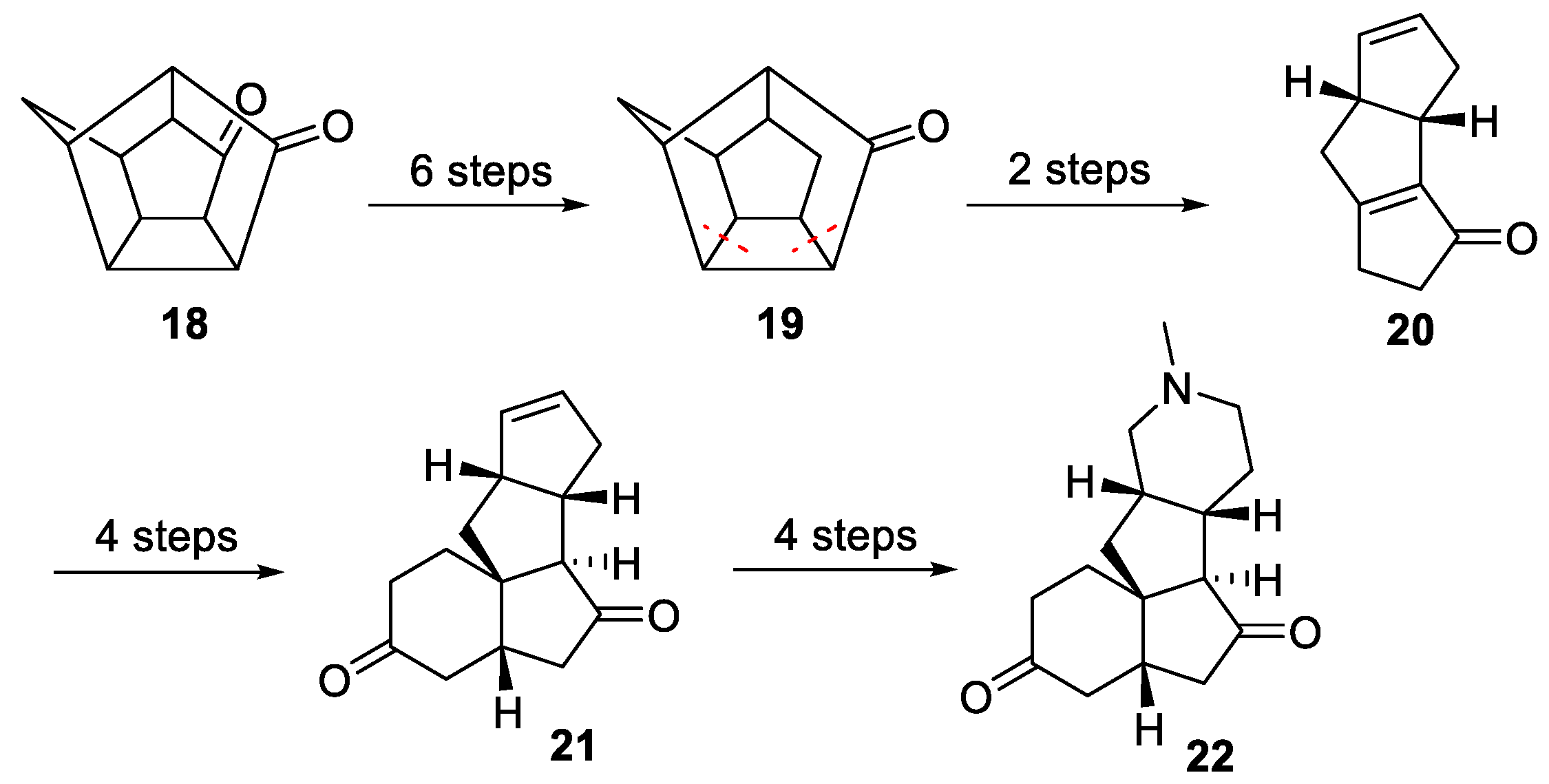
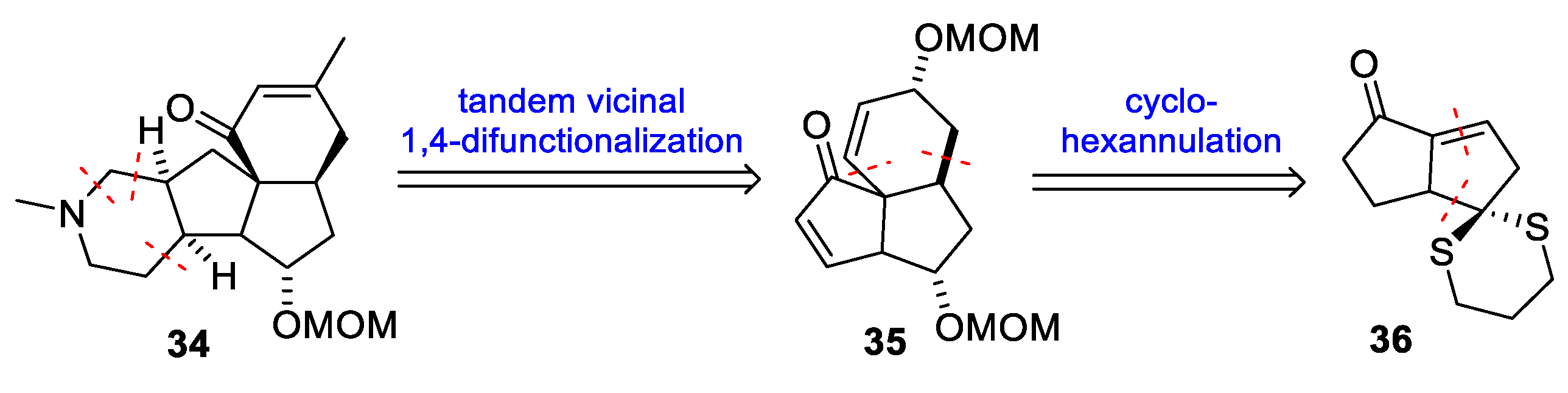
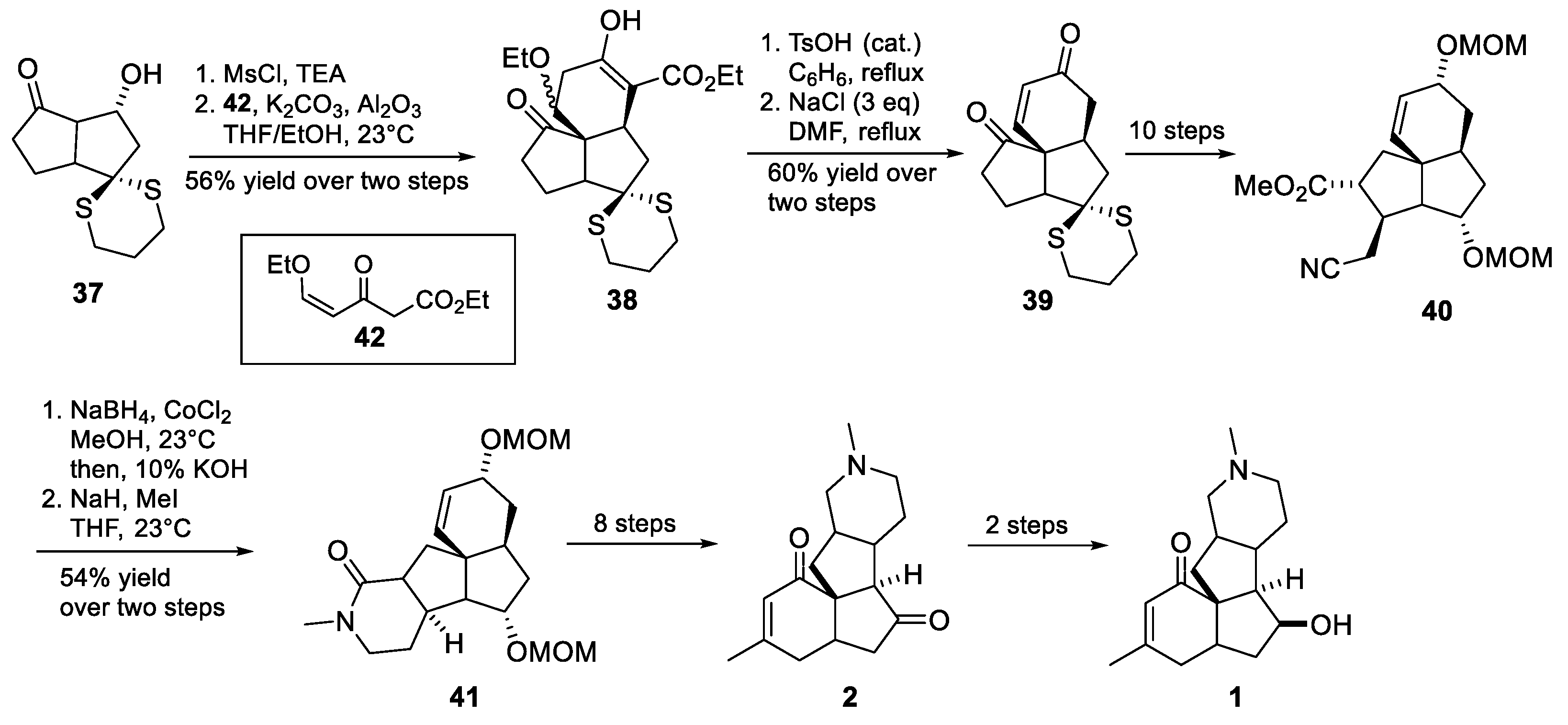
3.6. Mukai’s 2007 Synthesis of 1–3
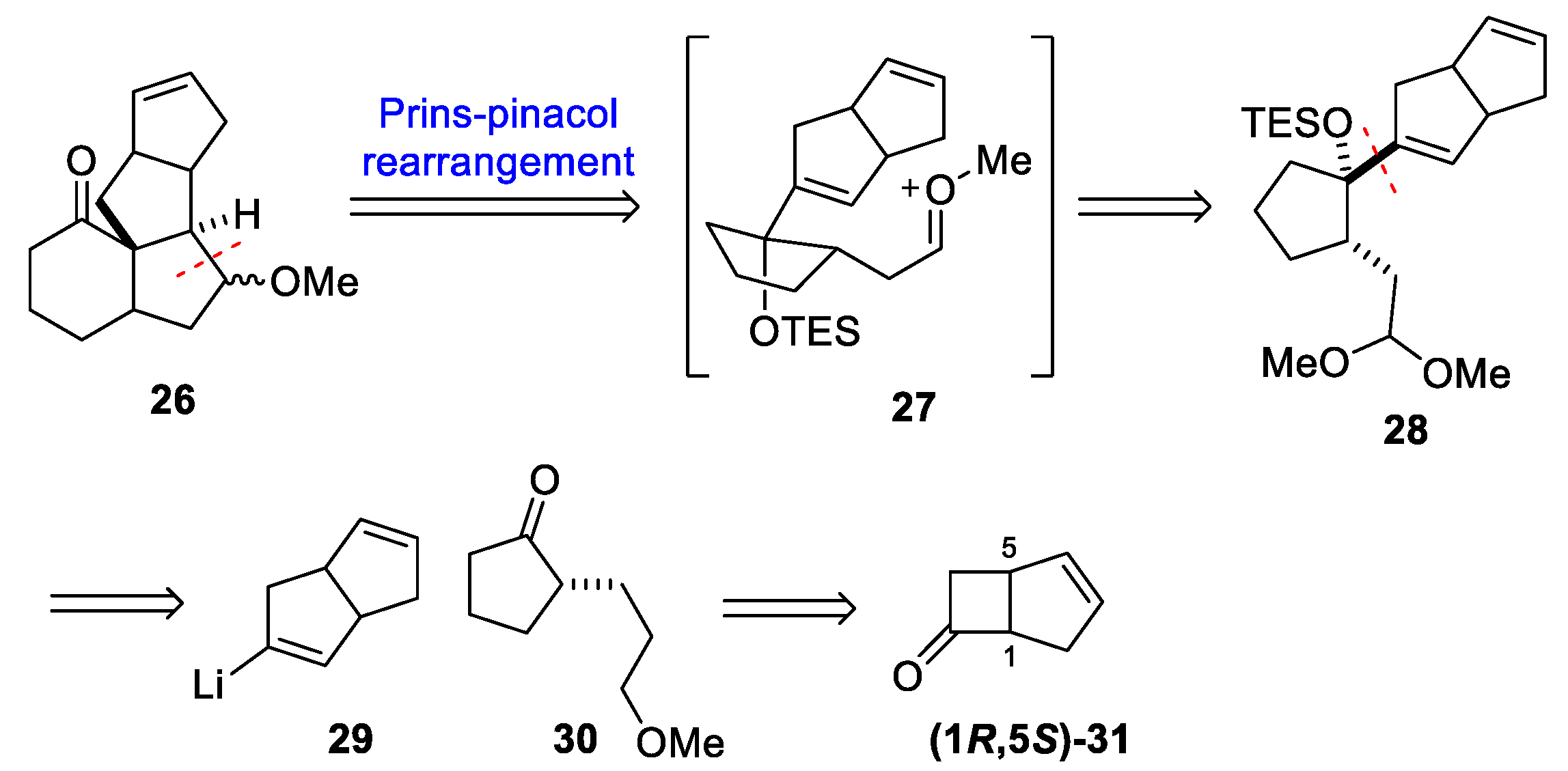
3.7. Yang’s 2014 Synthesis of 1–3
3.8. Yan’s 2015 Synthesis of 1–3
3.9. Qiu’s 2019 Synthesis of (+)-Paniculatine
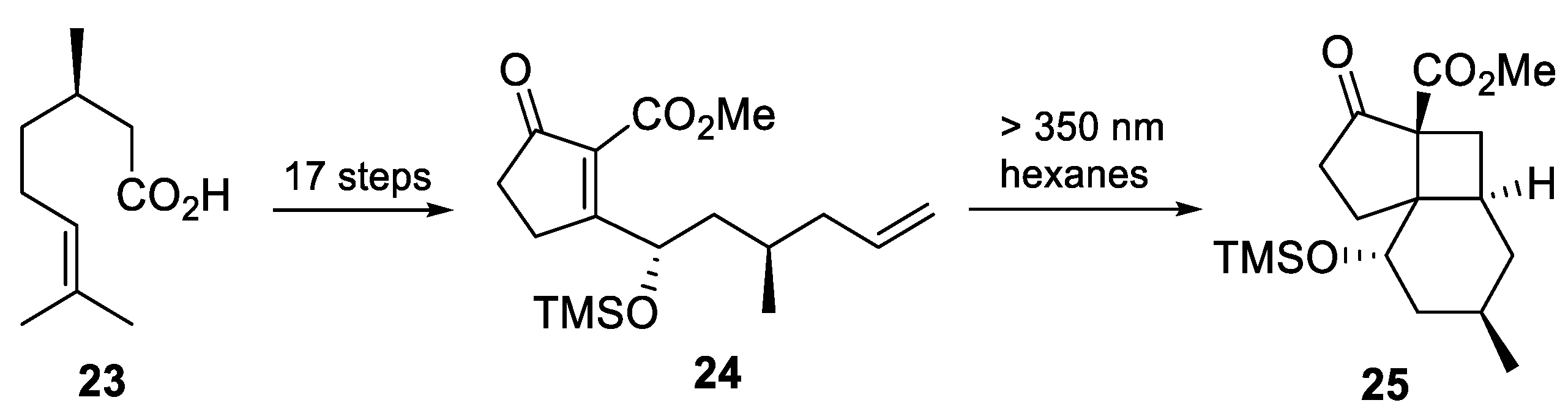
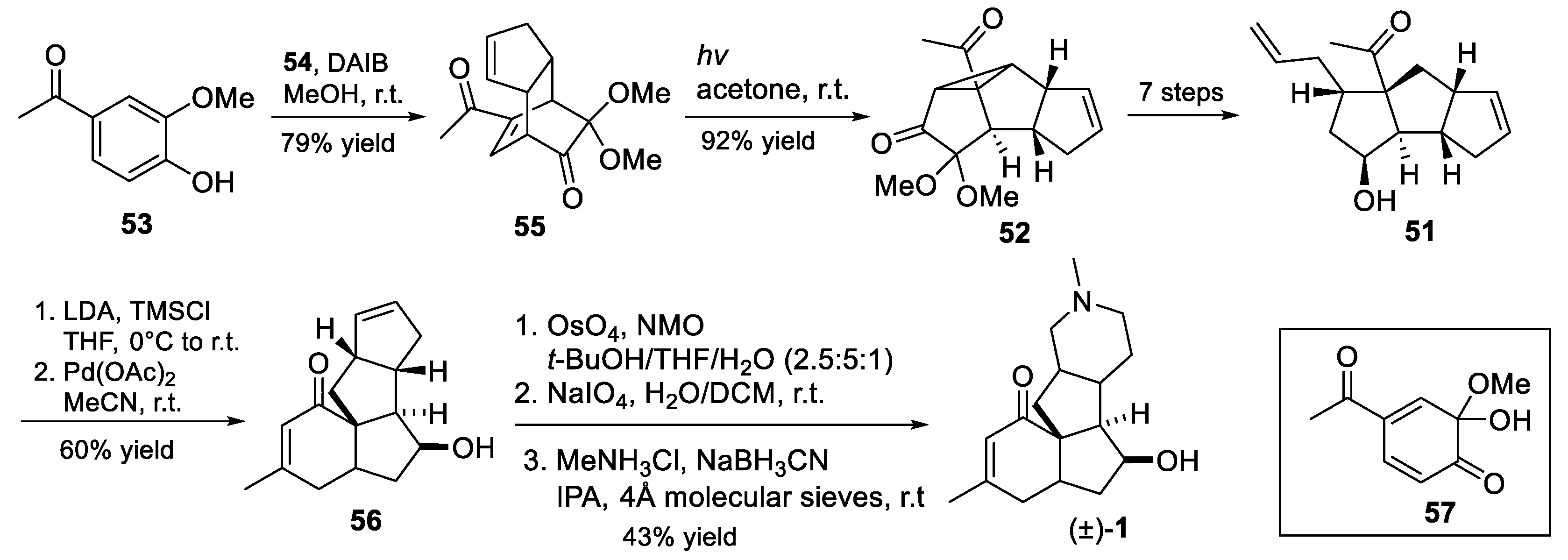
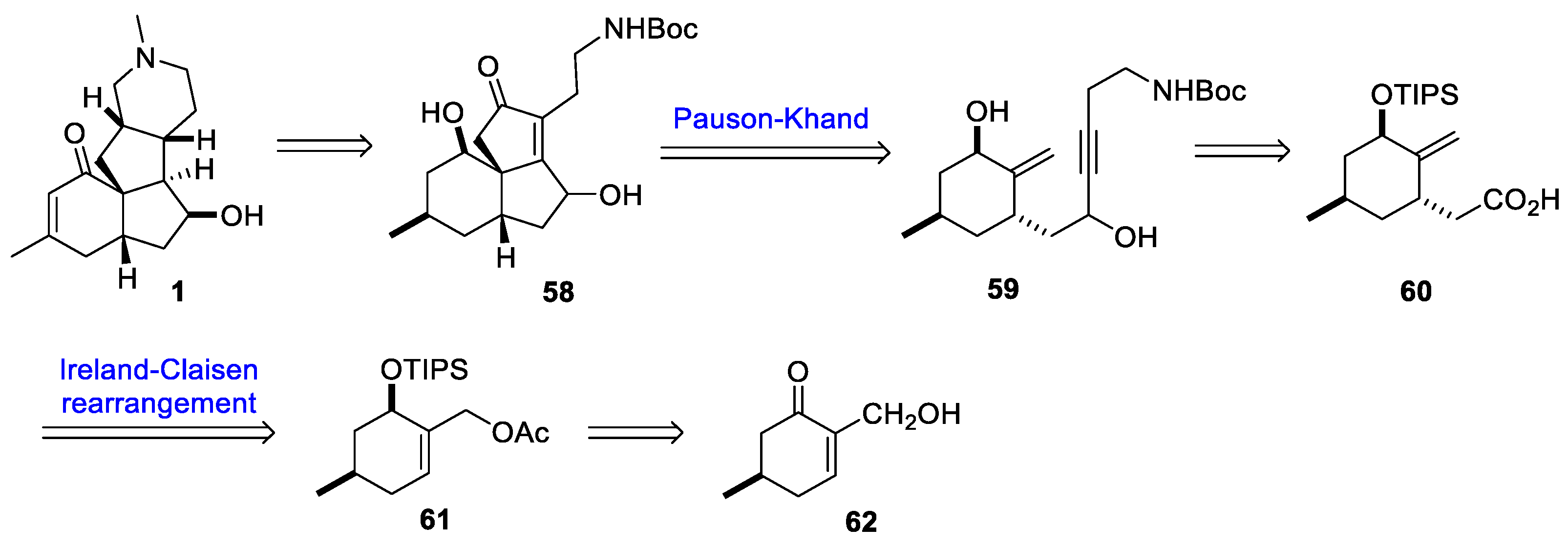
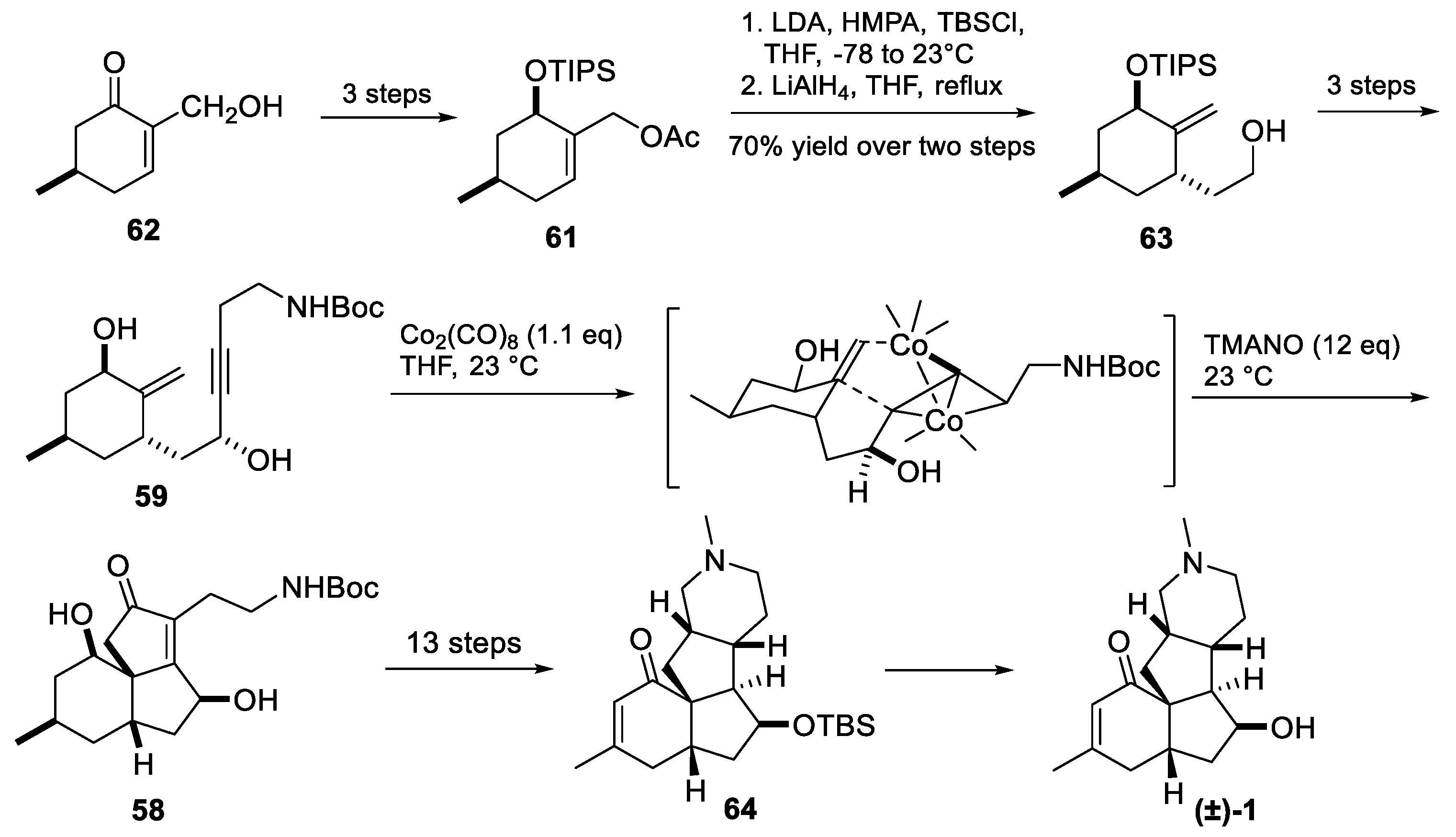
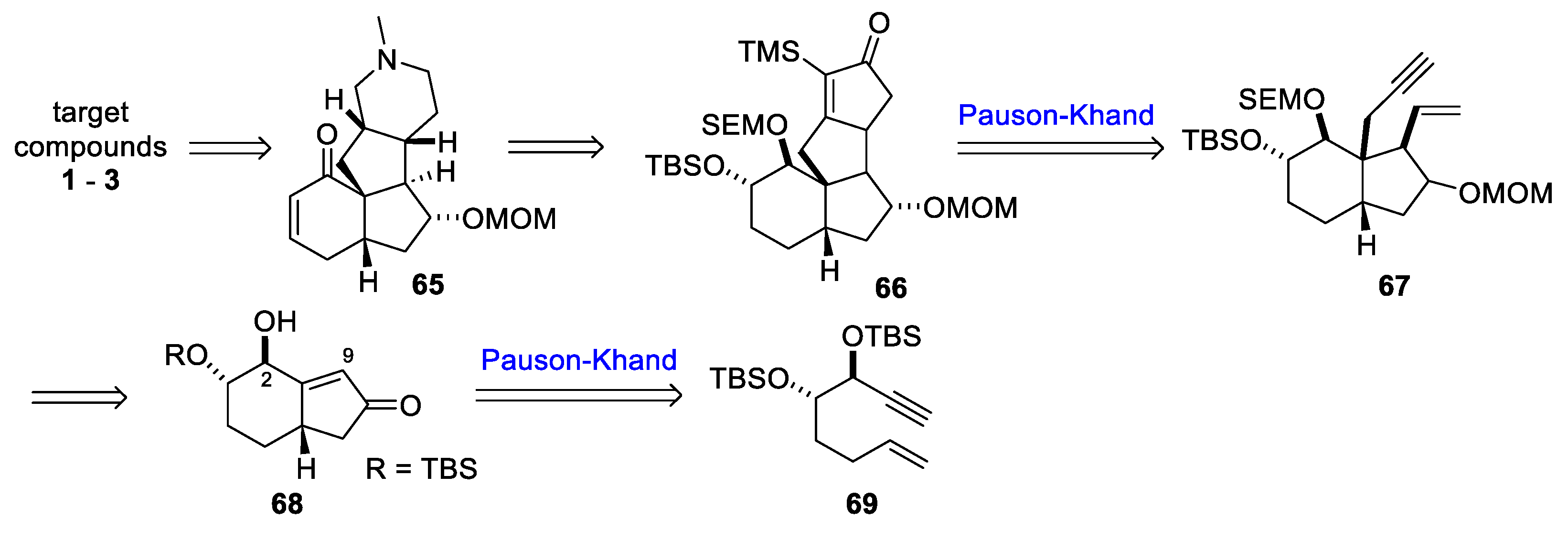
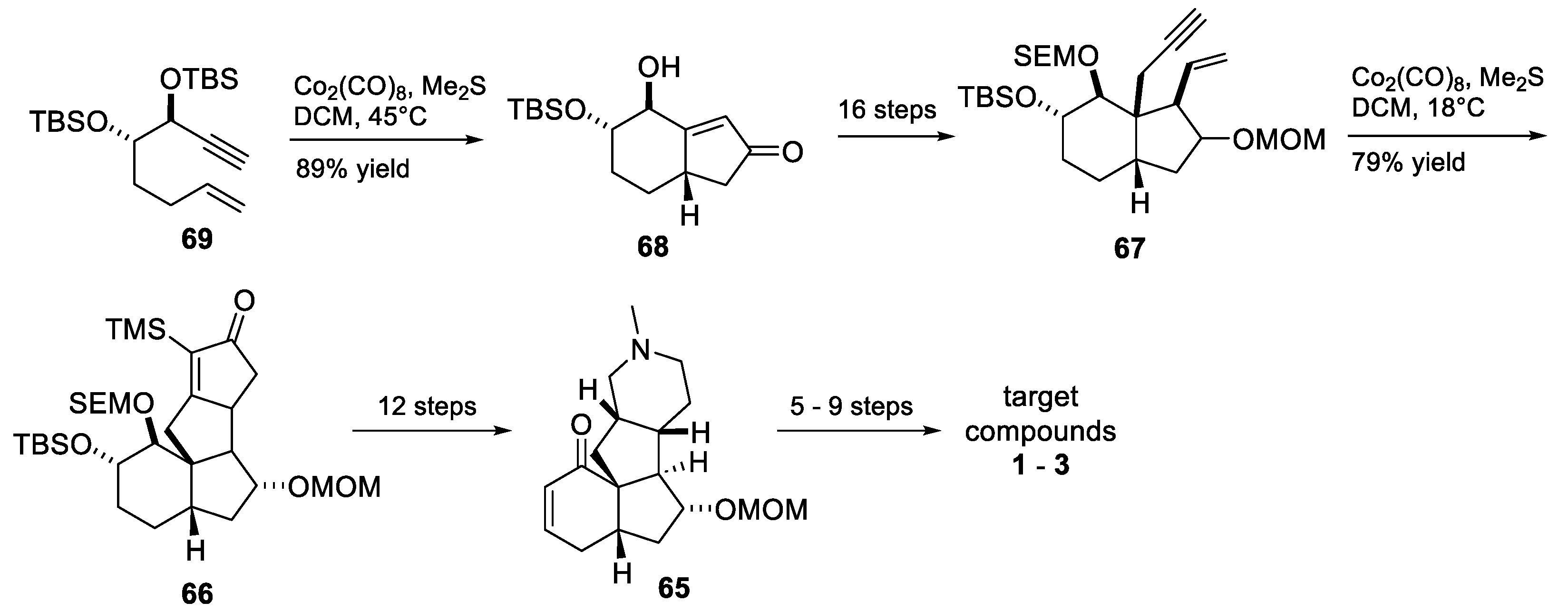
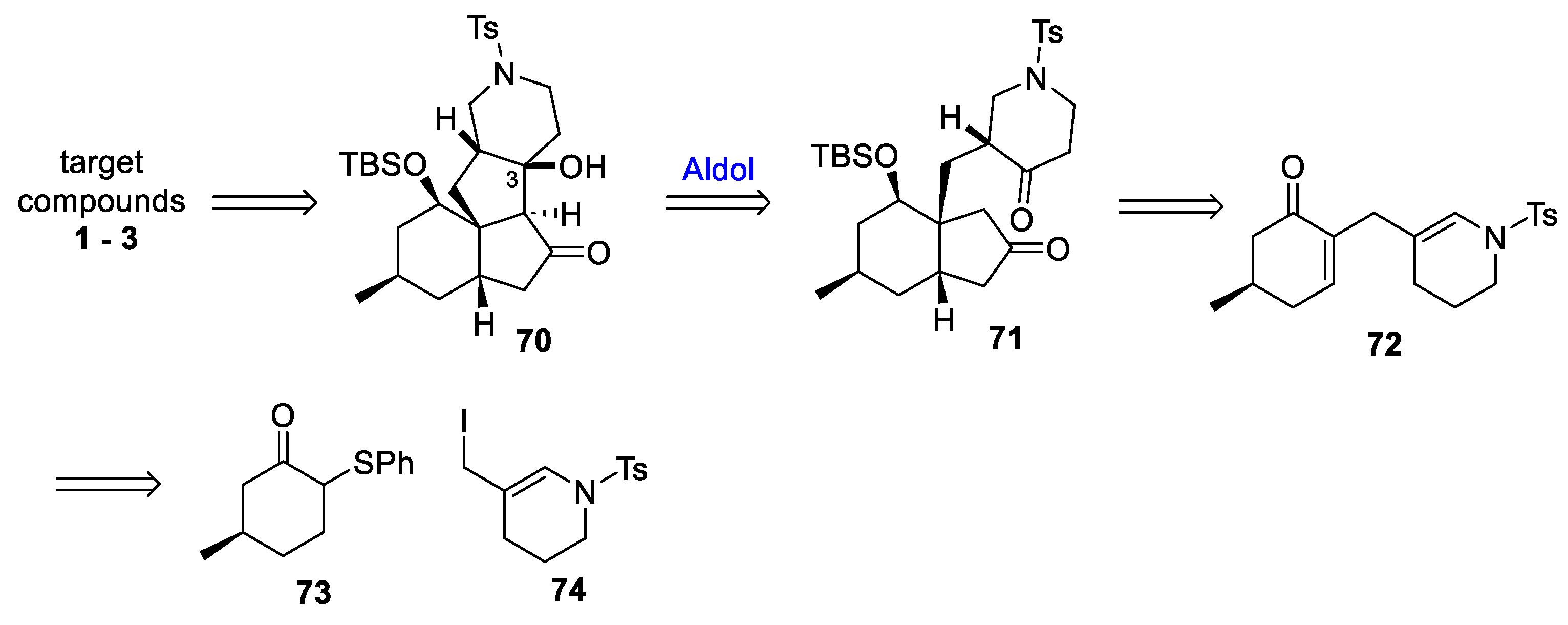
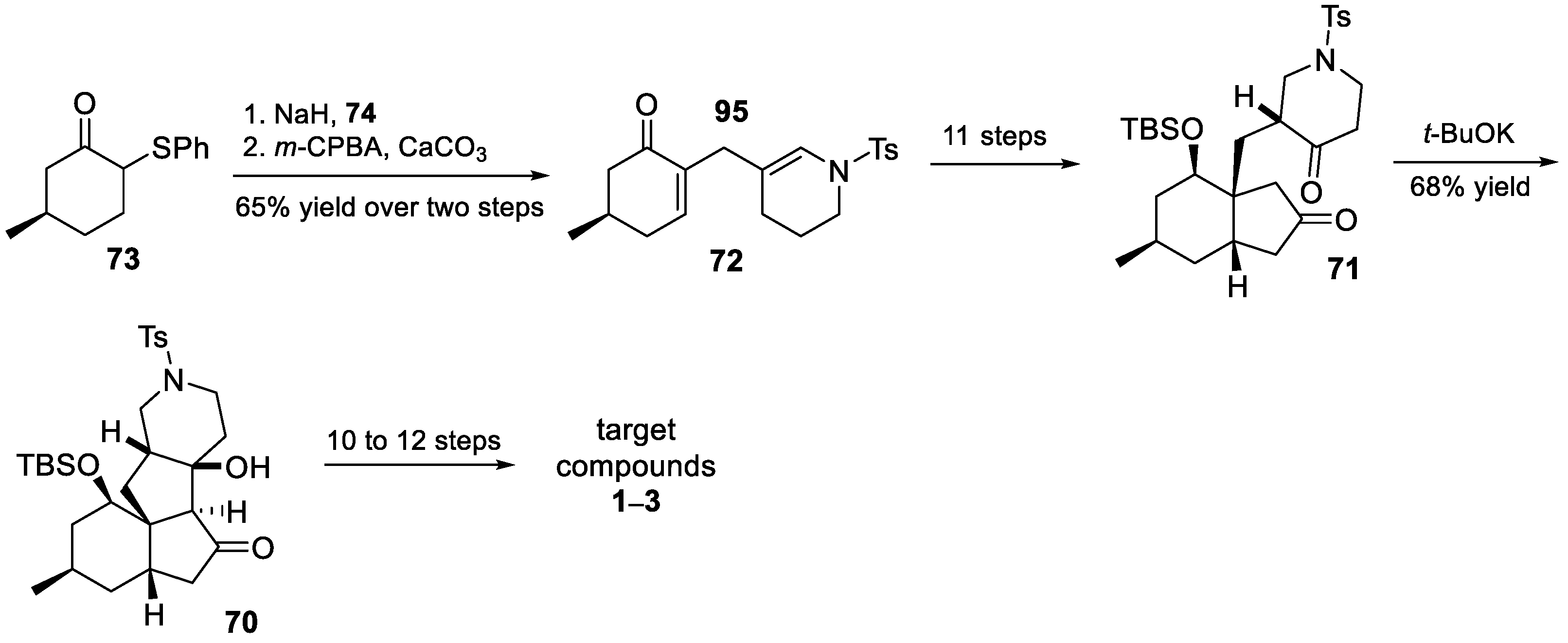
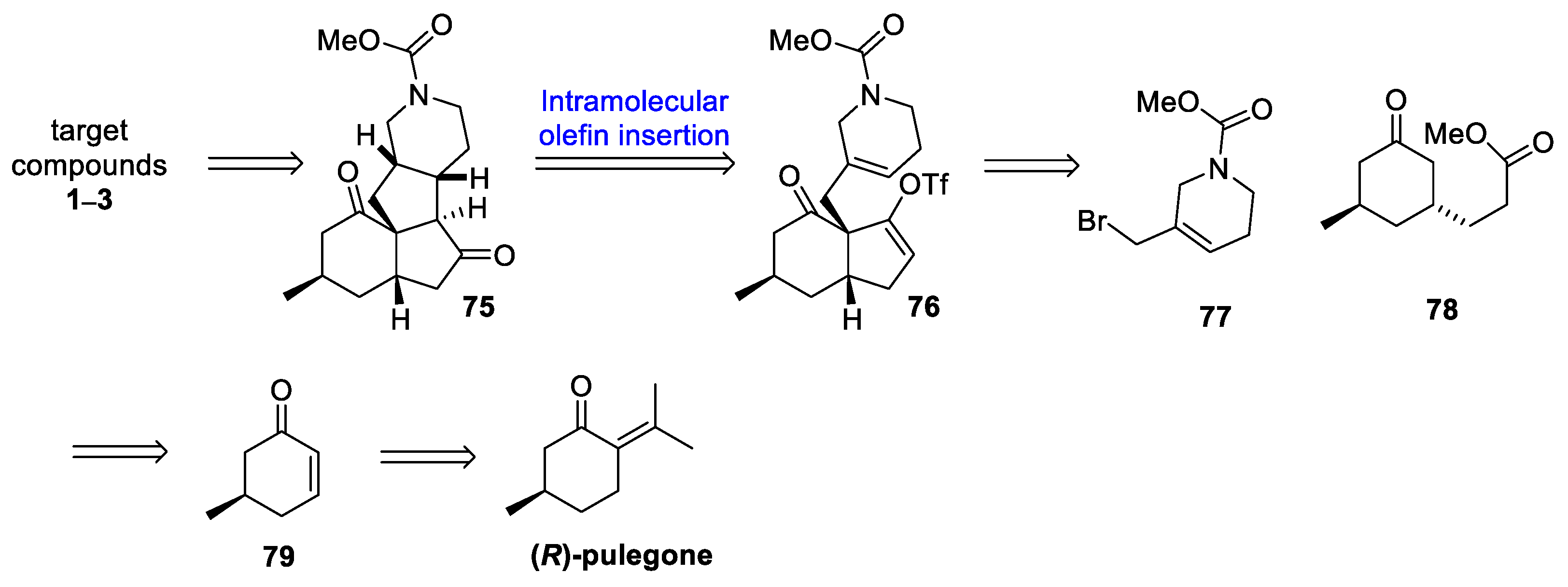
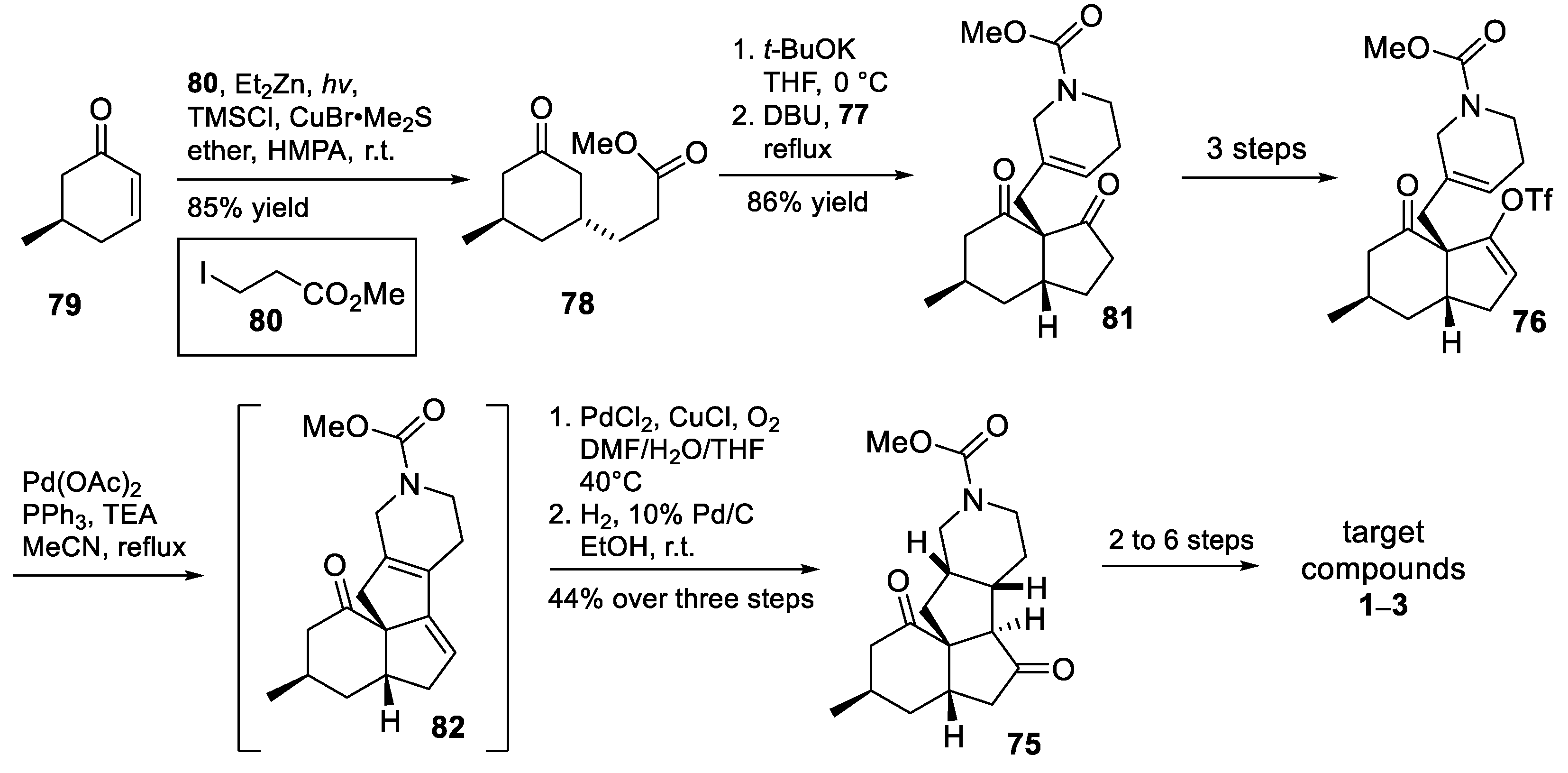
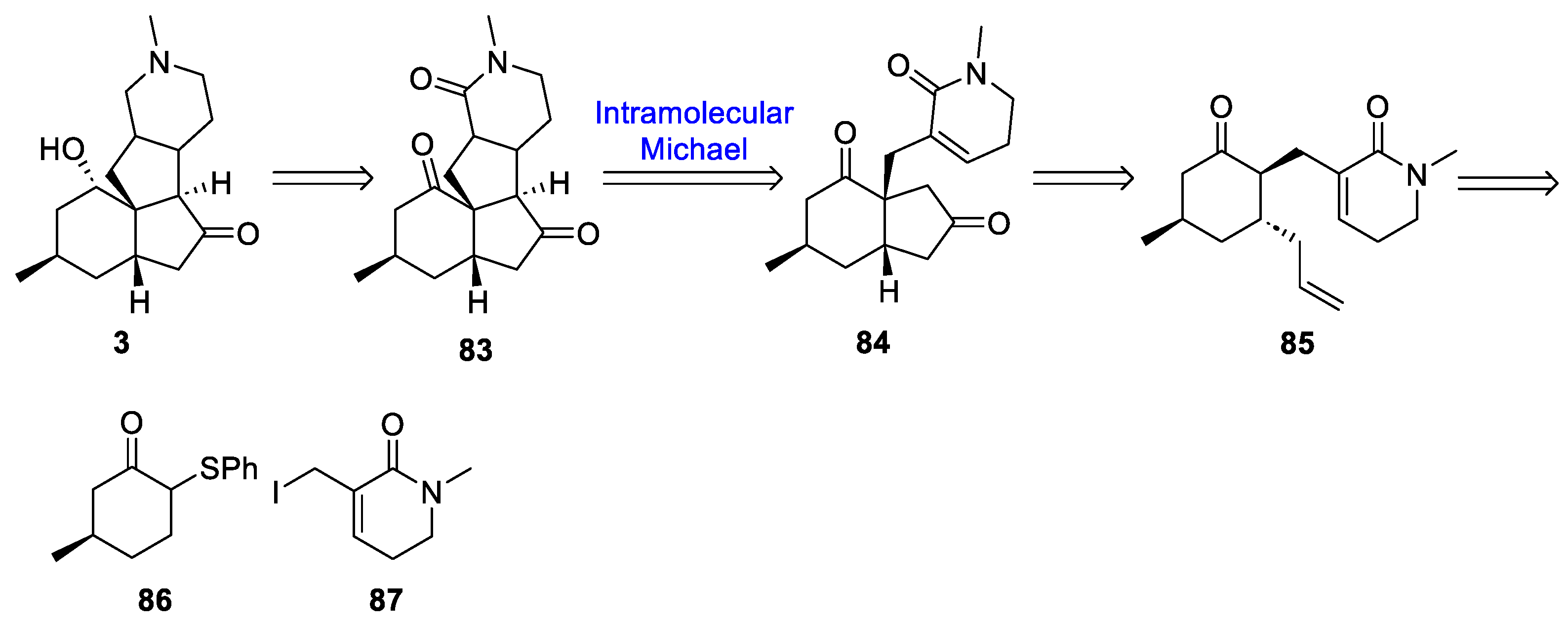
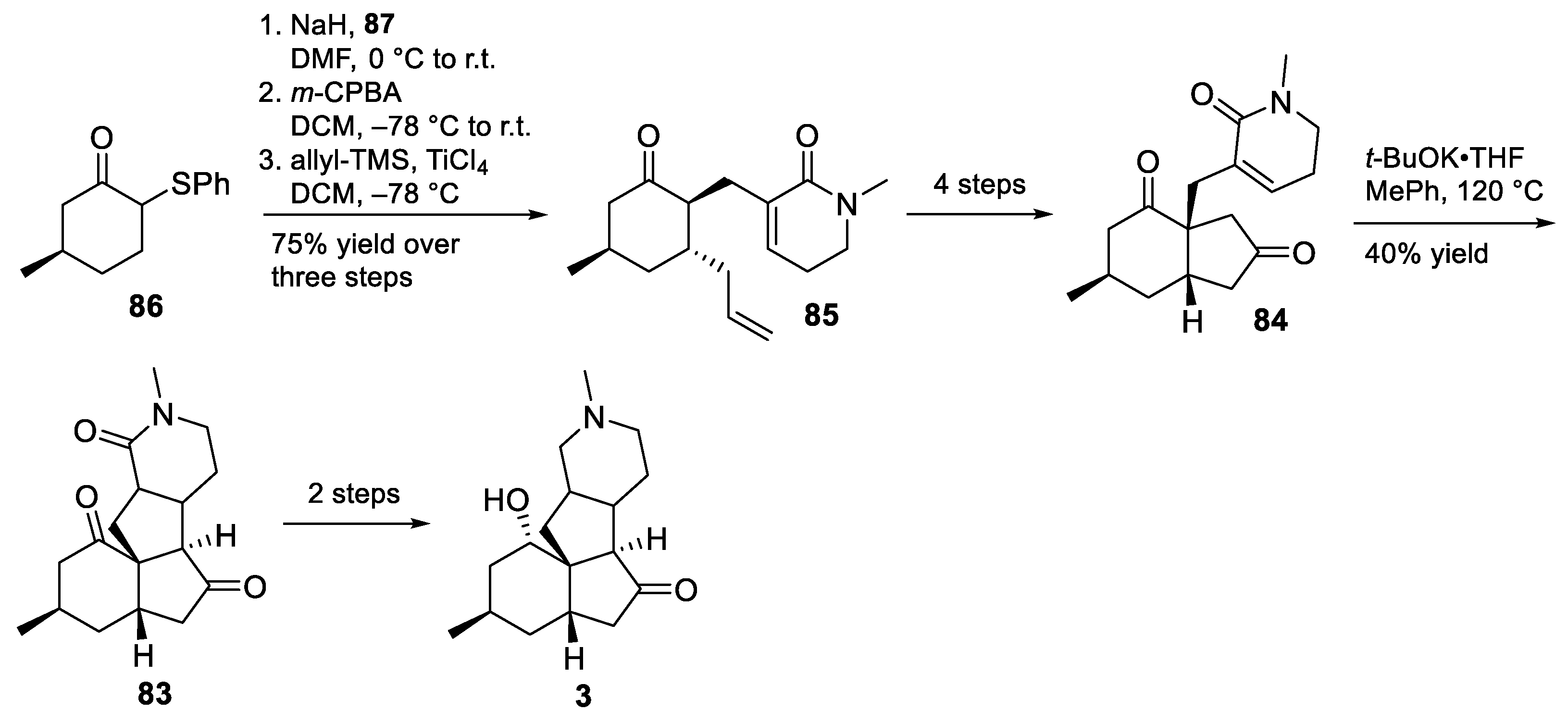
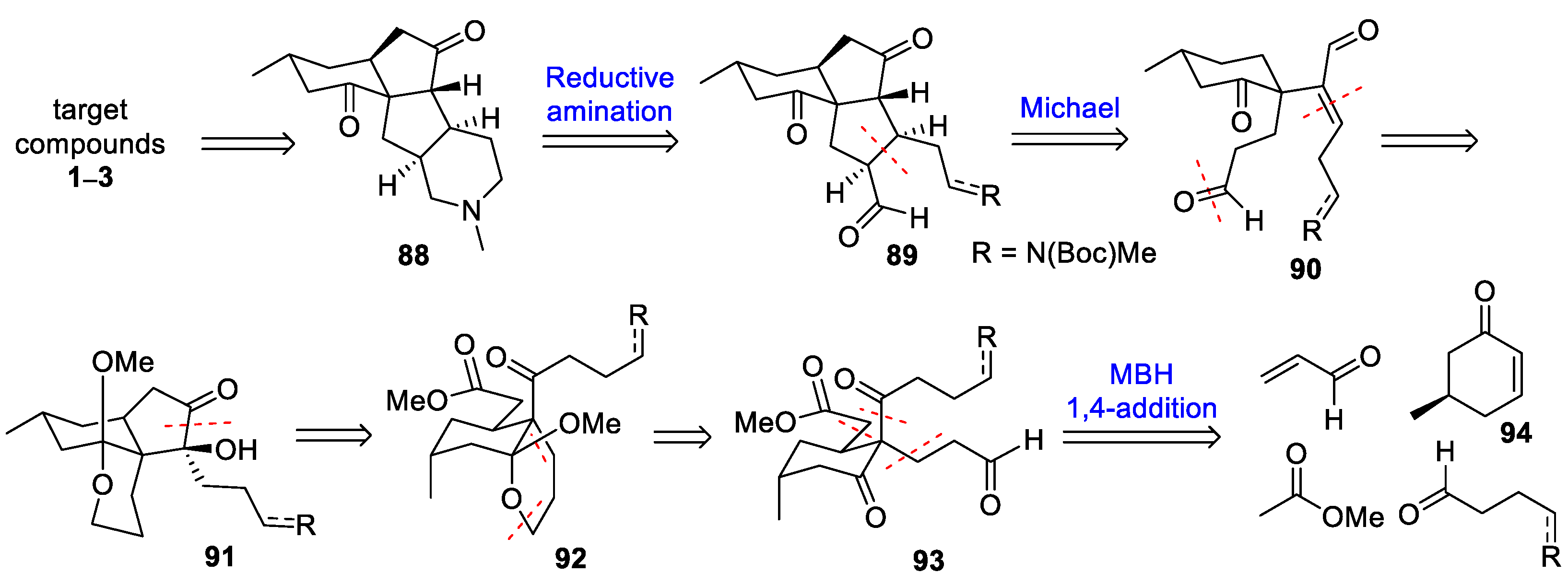
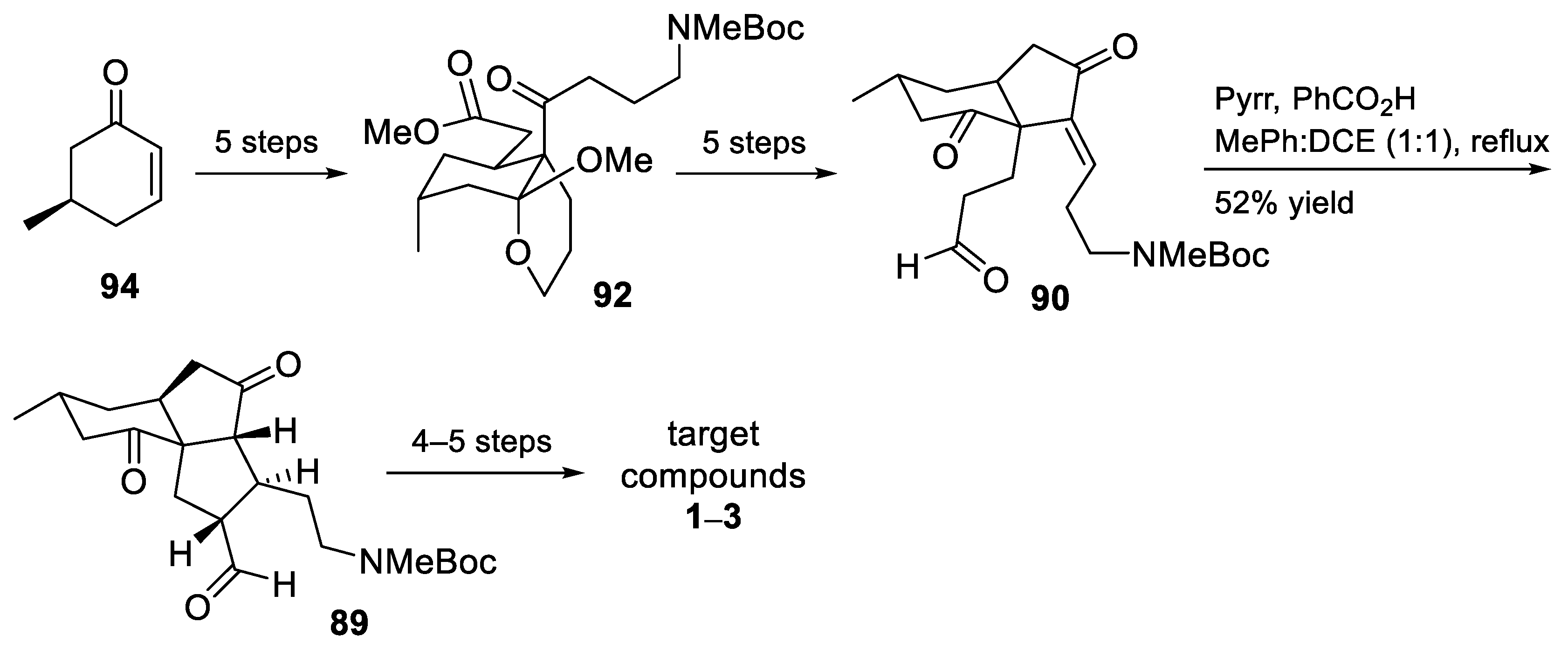
3.10. Yao’s 2022 Synthesis of 1–3
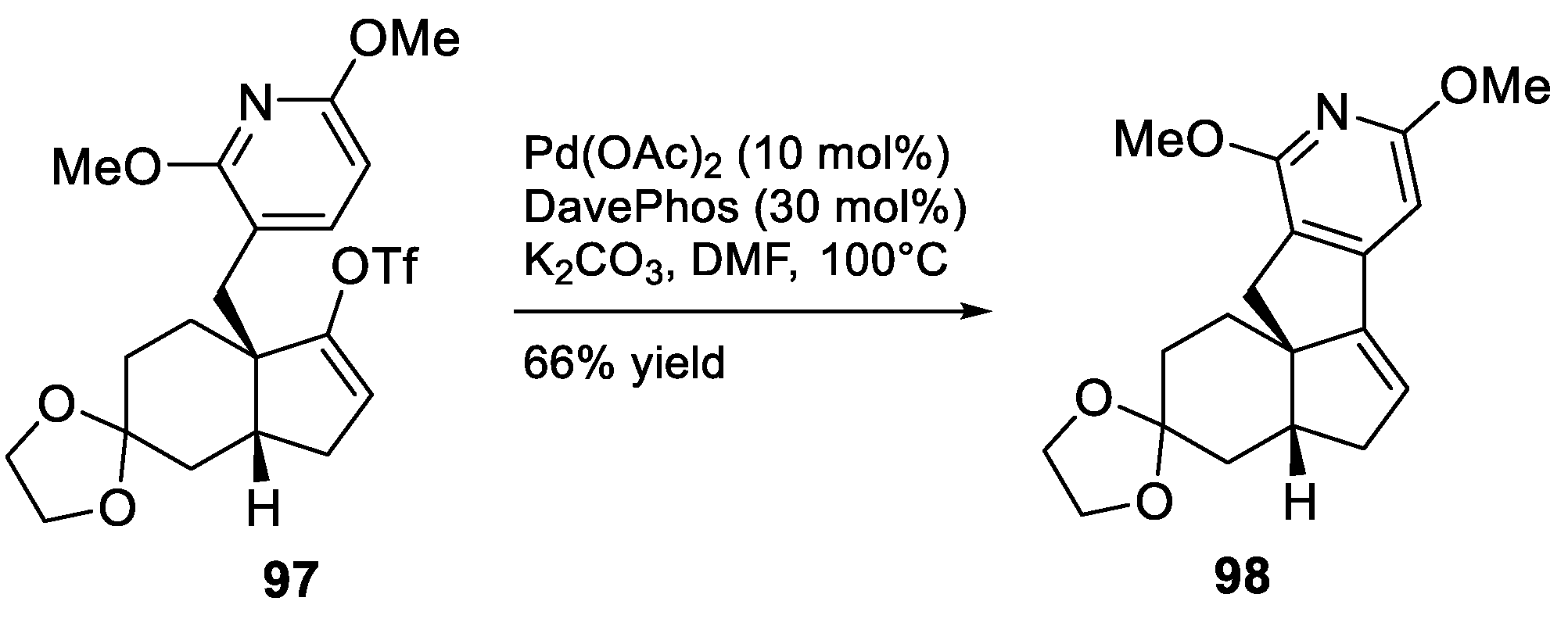
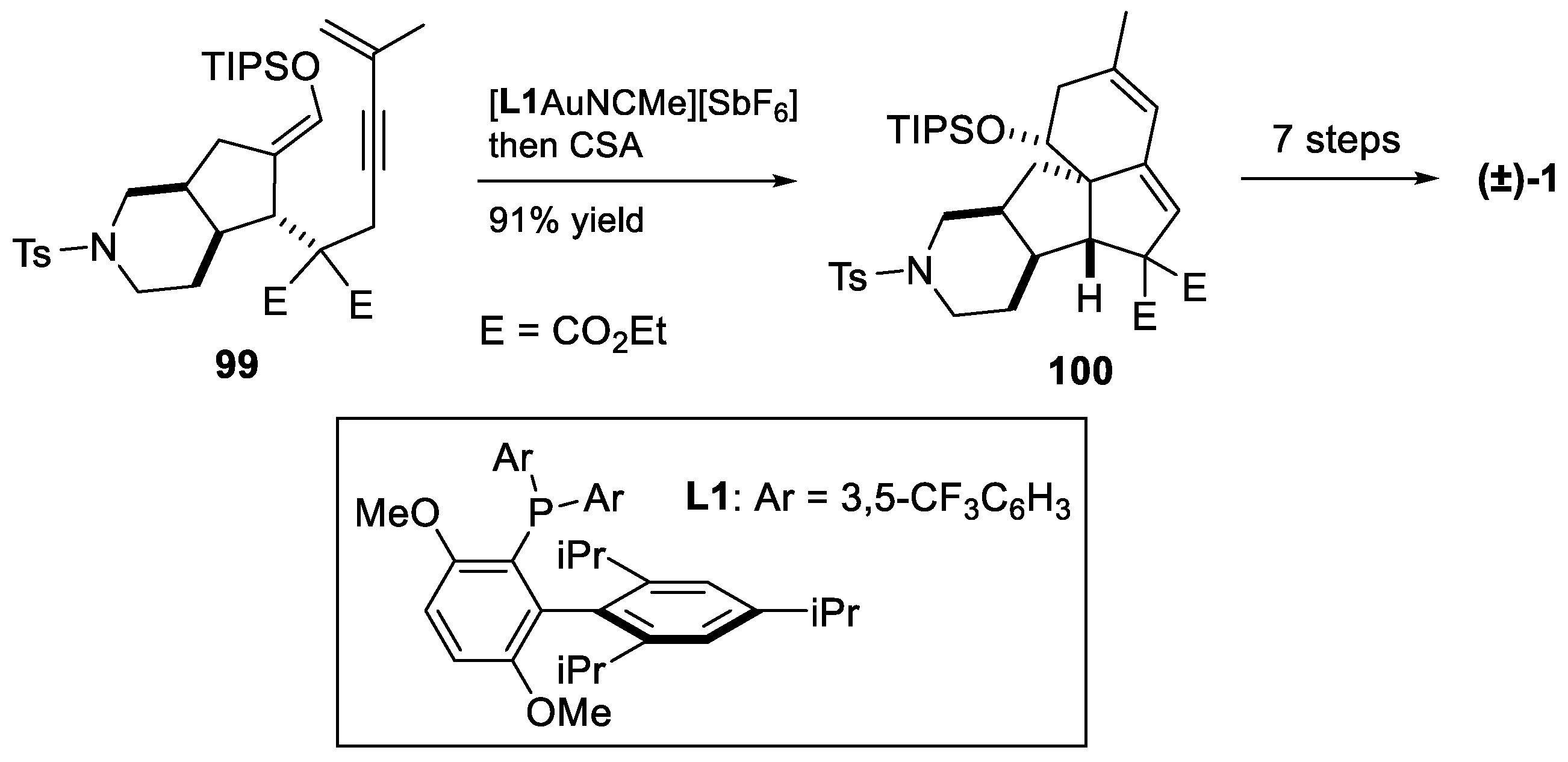
4. Other Notable Syntheses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ma, X.; Gang, D.R. The Lycopodium Alkaloids. Nat. Prod. Rep. 2004, 21, 752. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.P. Recently Isolated Lycodine-Type Lycopodium Alkaloids and Their Total Synthesis: A Review. Future J. Pharm. Sci. 2020, 6, 99. [Google Scholar] [CrossRef]
- Kobayashi, J.; Morita, H. The Lycopodium Alkaloids. In The Alkaloids: Chemistry and Biology; Elsevier: Amsterdam, The Netherlands, 2005; Volume 61, pp. 1–57. [Google Scholar] [CrossRef]
- Hardardottir, I.; Olafsdottir, E.S.; Freysdottir, J. Dendritic Cells Matured in the Presence of the Lycopodium Alkaloid Annotine Direct T Cell Responses toward a Th2/Treg Phenotype. Phytomedicine 2015, 22, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Hu, X.; Ao, H.; Ma, H.; Gao, Z.; Liu, F.; Kong, D.; Bao, Z.; Yu, Z. The Neurovascular Protective Effects of Huperzine A on D-Galactose-Induced Inflammatory Damage in the Rat Hippocampus. Gerontology 2014, 60, 424–439. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.X.; Zhu, X.Q.; Chen, Y.; Wu, G.C.; Wang, J. Huperzine A Inhibits CCL2 Production in Experimental Autoimmune Encephalomyelitis Mice and in Cultured Astrocyte. Int. J. Immunopathol. Pharmacol. 2013, 26, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, W.; Fujiwara, Y.; Kosuge, Y.; Monthakantirat, O.; Fujikawa, K.; Watthana, S.; Kitanaka, S.; Makino, T.; Ishiuchi, K. Phlenumdines D and E, New Lycopodium Alkaloids from Phlegmariurus Nummulariifolius, and Their Regulatory Effects on Macrophage Differentiation during Tumor Development. Phytochem. Lett. 2019, 29, 98–103. [Google Scholar] [CrossRef]
- Castillo, M.; Loyola, L.A.; Morales, G.; Singh, I.; Calvo, C.; Rolland, H.L.; MacLean, D.B. The Alkaloids of L. Magellanicum and the Structure of Magellanine. Can. J. Chem. 1976, 54, 2893–2899. [Google Scholar] [CrossRef]
- Castillo, M.; Morales, G.; Loyola, L.A.; Singh, I.; Calvo, C.; Holland, H.L.; MacLean, D.B. Paniculatine. A New Alkaloid from L. paniculatum Desvaux. Can. J. Chem. 1975, 53, 2513–2514. [Google Scholar] [CrossRef]
- Castillo, M.; Morales, G.; Loyola, L.A.; Singh, I.; Calvo, C.; Holland, H.L.; MacLean, D.B. The Alkaloids of L. Paniculatum and the Structure of Paniculatine. Can. J. Chem. 1976, 54, 2900–2908. [Google Scholar] [CrossRef]
- Loyola, L.A.; Morales, G.; Castillo, M. Alkaloids of Lycopodium Magellanicum. Phytochemistry 1979, 18, 1721–1723. [Google Scholar] [CrossRef]
- Lei, S. Synthetic Studies towards the Lycopodium Alkaloid Paniculatine. RSC Adv. 2013, 3, 11014. [Google Scholar] [CrossRef]
- Lindsay, V.N.G.; Murphy, R.A.; Sarpong, R. Effect of Protic Additives in Cu-Catalysed Asymmetric Diels–Alder Cycloadditions of Doubly Activated Dienophiles: Towards the Synthesis of Magellanine-Type Lycopodium Alkaloids. Chem. Commun. 2017, 53, 10291–10294. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, M.; Niimi, Y.; Hoshino, O. Synthetic Studies on Lycopodium Alkaloid, Magellanine: Stereoselective Construction of Functionallized Angular Tricyclic Skeletons by Intramolecular Pauson–Khand Reaction. Chem. Lett. 2001, 30, 546–547. [Google Scholar] [CrossRef]
- St. Laurent, D.R.; Paquette, L.A. Cyclopentannulation with a 1,3-Dicarbonyl Dipole Equivalent. Synthesis of Bicyclo[3.3.0]Oct-1(5)-Ene-2,6-Dione. J. Org. Chem. 1986, 51, 3861–3864. [Google Scholar] [CrossRef]
- Seebach, D.; Corey, E.J. Generation and Synthetic Applications of 2-Lithio-1,3-Dithianes. J. Org. Chem. 1975, 40, 231–237. [Google Scholar] [CrossRef]
- Rosen, T.; Taschner, M.J.; Thomas, J.A.; Heathcock, C.H. Synthetic and Biological Studies of Compactin and Related Compounds. 3. Synthesis of the Hexalin Portion of Compactin. J. Org. Chem. 1985, 50, 1190–1201. [Google Scholar] [CrossRef]
- Mitsunobu, O. The Use of Diethyl Azodicarboxylate and Triphenylphosphine in Synthesis and Transformation of Natural Products. Synthesis 1981, 1981, 1–28. [Google Scholar] [CrossRef]
- Agosta, W.C.; Wolff, S. Strain in a Bicyclo[3.3.0]Oct-1-Ene. Preparation of 5,8-Dimethylbicyclo[3.3.0]Oct-8-En-2-One and Related Compounds. J. Org. Chem. 1975, 40, 1699–1701. [Google Scholar] [CrossRef]
- Herrinton, P.H.; Hopkins, M.H.; Mishra, P.; Brown, M.J.; Overman, L.E. Ring-Enlarging Furan Annulations. J. Org. Chem. 1987, 52, 3711–3712. [Google Scholar] [CrossRef]
- Hopkins, M.H.; Overman, L.E. Stereocontrolled Preparation of Tetrahydrofurans by Acid-Catalyzed Rearrangement of Allylic Acetals. J. Am. Chem. Soc. 1987, 109, 4748–4749. [Google Scholar] [CrossRef]
- Hirst, G.C.; Howard, P.N.; Overman, L.E. Stereocontrolled Construction of Carbocyclic Rings by Sequential Cationic Cyclization-Pinacol Rearrangements. J. Am. Chem. Soc. 1989, 111, 1514–1515. [Google Scholar] [CrossRef]
- Mehta, G.; Rao, K.S. Model Studies towards Crinipellin Diterpenes and Paniculatine-Type Lycopodium Alkaloids from a Common Triquinane Precursor. J. Chem. Soc. Chem. Commun. 1987, 1987, 1578. [Google Scholar] [CrossRef]
- Tsuji, J.; Shimizu, I.; Yamamoto, K. Convenient General Synthetic Method for 1,4- and 1,5-Diketones by Palladium Catalyzed Oxidation of α-Allyl and α-3-Butenyl Ketones. Tetrahedron Lett. 1976, 17, 2975–2976. [Google Scholar] [CrossRef]
- Mehta, G.; Sreenivasa Reddy, M. A Strategy for the Construction of Novel Tetracyclic Lycopodium Alkaloids of Paniculatine- and Magellanine-Type. Tetrahedron Lett. 1990, 31, 2039–2042. [Google Scholar] [CrossRef]
- Mehta, G.; Srikrishna, A.; Reddy, A.V.; Nair, M.S. A Novel, Versatile Synthetic Approach to Linearly Fused Tricyclopentanoids via Photo-Thermal Olefin Metathesis. Tetrahedron 1981, 37, 4543–4559. [Google Scholar] [CrossRef]
- Crimmins, M.T.; Watson, P.S. Stereoselective Intramolecular Enone-Olefin Photocycloadditions of 1,7-Dienes: Model Studies on the Synthesis of Lycopodium Alkaloids. Tetrahedron Lett. 1993, 34, 199–202. [Google Scholar] [CrossRef]
- Crimmins, M.T. Synthetic Applications of Intramolecular Enone-Olefin Photocycloadditions. Chem. Rev. 1988, 88, 1453–1473. [Google Scholar] [CrossRef]
- Winkler, J.D.; Hershberger, P.M.; Springer, J.P. A Stereoselective Synthesis of the Azaspiboundecane Ring System of (−)-Histrionicotoxin from (+)-Glutamic Acid. Tetrahedron Lett. 1986, 27, 5177–5180. [Google Scholar] [CrossRef]
- Pirrung, M.C.; Thomson, S.A. Intramolecular [2+2] Photocycloaddition of Enone-Acetals. Tetrahedron Lett. 1986, 27, 2703–2706. [Google Scholar] [CrossRef]
- Hirst, G.C.; Johnson, T.O.; Overman, L.E. First Total Synthesis of Lycopodium Alkaloids of the Magellanane Group. Enantioselective Total Syntheses of (-)-Magellanine and (+)-Magellaninone. J. Am. Chem. Soc. 1993, 115, 2992–2993. [Google Scholar] [CrossRef]
- Abraham, W.D.; Bhupathy, M.; Cohen, T. Carbenoid Type Base Induced Ring Expansion of the Adducts of Cyclic Ketones with Bis(Phenylthio)Methyllithium. Tetrahedron Lett. 1987, 28, 2203–2206. [Google Scholar] [CrossRef]
- Cohen, T.; Kuhn, D.; Falck, J.R. Application of Copper(I)-Induced Thiophenoxide Removal to Ring Expansions or Chain Extensions of Aldehydes and Ketones. Detection, Isolation, and Independent Preparation of the Alpha-Epoxy Thioether Intermediate. J. Am. Chem. Soc. 1975, 97, 4749–4751. [Google Scholar] [CrossRef]
- Seyferth, D. Vinyl Derivatives of the Metals. II. The Cleavage of Vinyltin Compounds by the Halogens and by Protonic Acids. J. Am. Chem. Soc. 1957, 79, 2133–2136. [Google Scholar] [CrossRef]
- Pappo, R.; Allen, D., Jr.; Lemieux, R.; Johnson, W. Notes—Osmium Tetroxide-Catalyzed Periodate Oxidation of Olefinic Bonds. J. Org. Chem. 1956, 21, 478–479. [Google Scholar] [CrossRef]
- Danheiser, R.L.; Morin, J.M.; Salaski, E.J. Efficient Total Synthesis of (.+-.)-Anatoxin a. J. Am. Chem. Soc. 1985, 107, 8066–8073. [Google Scholar] [CrossRef]
- Ito, Y.; Hirao, T.; Saegusa, T. Synthesis of.Alpha.,.Beta.-Unsaturated Carbonyl Compounds by Palladium(II)-Catalyzed Dehydrosilylation of Silyl Enol Ethers. J. Org. Chem. 1978, 43, 1011–1013. [Google Scholar] [CrossRef]
- Paquette, L.A.; Friedrich, D.; Pinard, E.; Williams, J.P.; St. Laurent, D.; Roden, B.A. Total Synthesis of the Tetracyclic Diquinane Lycopodium Alkaloids Magellanine and Magellaninone. J. Am. Chem. Soc. 1993, 115, 4377–4378. [Google Scholar] [CrossRef]
- Williams, J.P.; Friedrich, D.; Pinard, E.; Roden, B.A.; Paquette, L.A.; St. Laurent, D.R. Total Synthesis of the Lycopodium Alkaloids Magellanine and Magellaninone by Three-Fold Annulation of 2-Cyclopentenone. J. Am. Chem. Soc. 1994, 116, 4689–4696. [Google Scholar] [CrossRef]
- Chapdelaine, M.J.; Hulce, M. Tandem Vicinal Difunctionalization: β-Addition to α,β-Unsaturated Carbonyl Substrates Followed by α-Functionalization. In Organic Reactions; John Wiley & Sons, Inc., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1990; pp. 225–653. ISBN 978-0-471-26418-7. [Google Scholar]
- Nazarov, I.N. Obshch. Khim. 1953, 23, 1703.
- Collins, D.; Tomkins, C. Acid-Catalysed Isomerization of Ethyl 5-Hydroxy-7a-Methyl-1-Oxo-2,6,7,7a Tetrahydro-1H-Indene-4-Carboxylate. Aust. J. Chem. 1977, 30, 443. [Google Scholar] [CrossRef]
- Chang, S.-J.; McNally, D.; Shary-Tehrany, S.; Hickey, S.M.J.; Boyd, R.H. Heats of Combustion and Strain Energies of Bicyclo[n.m.O]Alkanes. J. Am. Chem. Soc. 1970, 92, 3109–3118. [Google Scholar] [CrossRef]
- Barrett, J.W.; Linstead, R.P. 138. Fused Carbon Rings. Part X. Some Fundamental Properties of the 0 : 3 : 3-Bicyclooctane Ring. J. Chem. Soc. Resumed 1936, 1936, 611–616. [Google Scholar] [CrossRef]
- Krapcho, A.P. Synthetic Applications of Dealkoxycarbonylations of Malonate Esters, β-Keto Esters, α-Cyano Esters and Related Compounds in Dipolar Aprotic Media—Part I. Synthesis 1982, 1982, 805–822. [Google Scholar] [CrossRef]
- Satoh, T.; Suzuki, S.; Suzuki, Y.; Miyaji, Y.; Imai, Z. Reduction of Organic Compounds with Sodium Borohydride-Transition Metal Salt Systems. Tetrahedron Lett. 1969, 10, 4555–4558. [Google Scholar] [CrossRef]
- Harayama, T.; Ohtani, M.; Oki, M.; Inubushi, Y. Total Synthesis of the Lycopodium Alkaloid Dl-Serratinine. Chem. Pharm. Bull. (Tokyo) 1975, 23, 1511–1515. [Google Scholar] [CrossRef]
- Heinzman, S.W.; Ganem, B. Mechanism of Sodium Borohydride-Cobaltous Chloride Reductions. J. Am. Chem. Soc. 1982, 104, 6801–6802. [Google Scholar] [CrossRef]
- Hughes, D.L. The Mitsunobu Reaction. In Organic Reactions; John Wiley & Sons, Inc., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1992; pp. 335–656. ISBN 978-0-471-26418-7. [Google Scholar]
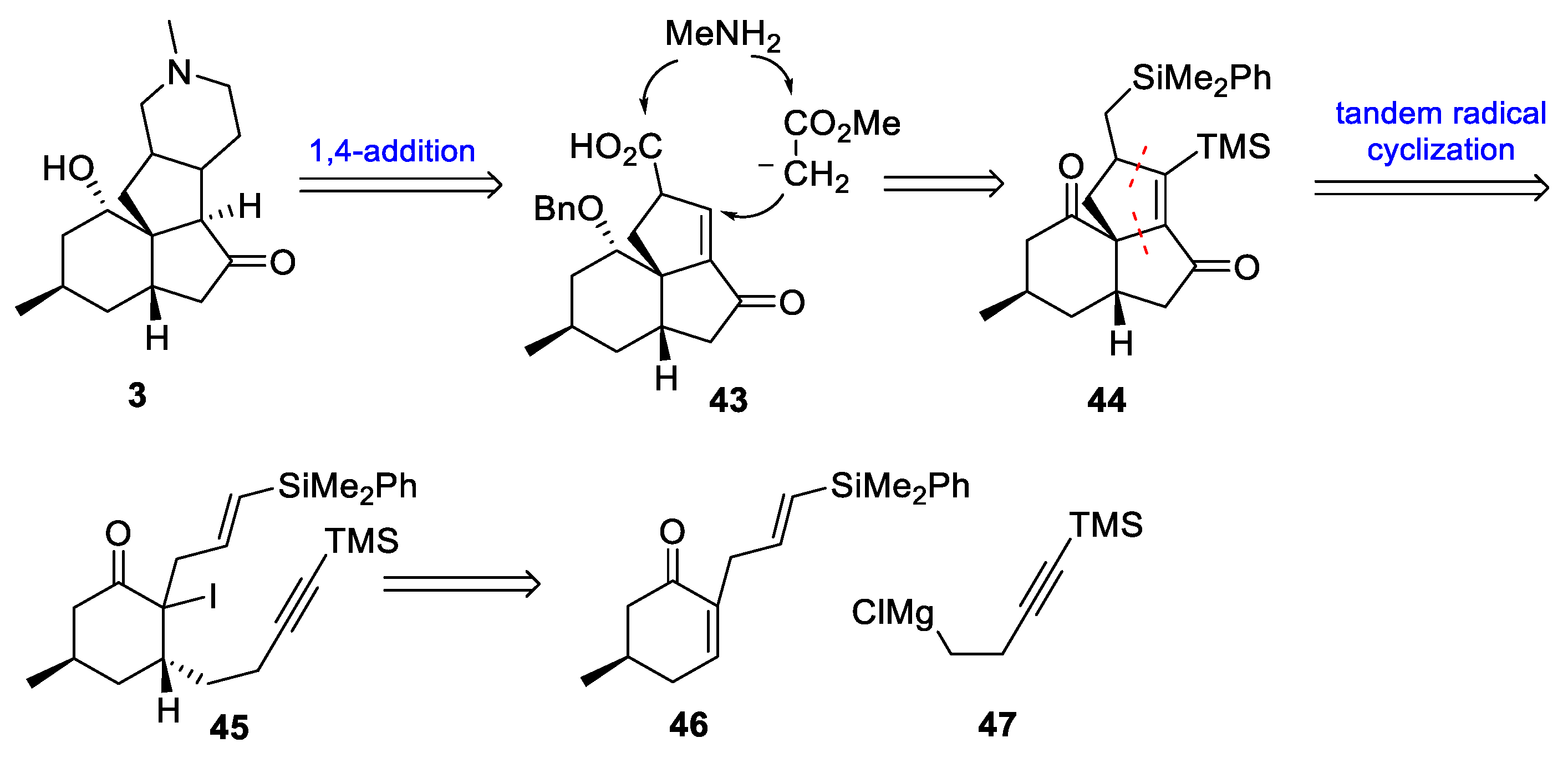
- Sha, C.-K.; Lee, F.-K.; Chang, C.-J. Tandem Radical Cyclizations Initiated with α-Carbonyl Radicals: First Total Synthesis of (+)-Paniculatine. J. Am. Chem. Soc. 1999, 121, 9875–9876. [Google Scholar] [CrossRef]
- Chin-Kang, S.; Tsong-Shin, J.; Deh-Chi, W. Intramolecular Radical Cyclization of Silylacetylenic or Olefinic α-Iodo Ketones: Application to the Total Synthesis of (±)-Modhephene. Tetrahedron Lett. 1990, 31, 3745–3748. [Google Scholar] [CrossRef]
- Sha, C.-K.; Chiu, R.-T.; Yang, C.-F.; Yao, N.-T.; Tseng, W.-H.; Liao, F.-L.; Wang, S.-L. Total Synthesis of (−)-Dendrobine via α-Carbonyl Radical Cyclization. J. Am. Chem. Soc. 1997, 119, 4130–4135. [Google Scholar] [CrossRef]
- Sha, C.-K.; Santhosh, K.C.; Lih, S.-H. α-Carbonyl Radical Cyclization Approach toward Angular Triquinanes: Total Synthesis of Enantiomerically Pure (−)-5-Oxosilphiperfol-6-Ene. J. Org. Chem. 1998, 63, 2699–2704. [Google Scholar] [CrossRef]
- Allinger, N.L.; Riew, C.K. The Stereochemistry of the Michael Addition Reaction. The Synthesis of 3,5-Dimethylcyclohexanone. Tetrahedron Lett. 1966, 7, 1269–1272. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Achille, B.; Simoneta, B.; Gian Piero, P.; Vinicio, Z. 2,3a,5,6,7,7a-Hexahydro-3h,4h-Benzothiophene-3,4-Dione and Cyclopenta [b]-Tetrahydrothiophene-3,4-Dione Enolate Anions as Synthetic Equivalents to Cyclohex-2-Enone and Cyclopent-2-Enone c-2-Carbanions. Tetrahedron Lett. 1984, 25, 4291–4294. [Google Scholar] [CrossRef]
- Caine, D.; Procter, K.; Cassell, R.A. A Facile Synthesis of (-)-R-5-Methyl-2-Cyclohexen-1-One and Related 2-Substituted Enones from (+)-Pulegone. J. Org. Chem. 1984, 49, 2647–2648. [Google Scholar] [CrossRef]
- Grieco, P.A.; Cooke, R.J.; Henry, K.J.; VanderRoest, J.M. Lithium Perchlorate Catalyzed Conjugate Addition of O-Silylated Ketene Acetals to Hindered α,β-Unsaturated Carbonyl Compounds at Atmospheric Pressure. Tetrahedron Lett. 1991, 32, 4665–4668. [Google Scholar] [CrossRef]
- Liao, C.-C. Concise and Efficient Total Synthesis of Lycopodium Alkaloid Magellanine. Angew. Chem. Int. Ed. 2002, 41, 4090. [Google Scholar]
- Hsu, D.-S.; Rao, P.D.; Liao, C.-C. Short and Efficient Route to Substituted Linear Triquinanes from 2-Methoxyphenols. Chem. Commun. 1998, 17, 1795–1796. [Google Scholar] [CrossRef]
- Givens, R.S.; Oettle, W.F. Mechanistic Studies in Organic Photochemistry. IV. Mechanism of the Photorearrangement of Bicyclo[2.2.2]Octadienones to Tricyclo[3.3.0.0.2.8]Octen-3-Ones. J. Am. Chem. Soc. 1971, 93, 3963–3968. [Google Scholar] [CrossRef]
- Givens, R.S.; Oettle, W.F.; Coffin, R.L.; Carlson, R.G. Mechanistic Studies in Organic Photochemistry. III. Photochemistry of Bicyclo[2.2.2]Octenone and Benzobicyclo[2.2.2]Octadiene. J. Am. Chem. Soc. 1971, 93, 3957–3962. [Google Scholar] [CrossRef]
- Lee, T.-H.; Rao, P.D.; Liao, C.-C. Photochemistry of Bicyclo[2.2.2]Octenones: An Uncommon Oxidative Decarbonylation. Chem. Commun. 1999, 9, 801–802. [Google Scholar] [CrossRef]
- VanRheenen, V.; Kelly, R.C.; Cha, D.Y. An Improved Catalytic OsO4 Oxidation of Olefins to -1,2-Glycols Using Tertiary Amine Oxides as the Oxidant. Tetrahedron Lett. 1976, 17, 1973–1976. [Google Scholar] [CrossRef]
- Borch, R.F.; Bernstein, M.D.; Durst, H.D. Cyanohydridoborate Anion as a Selective Reducing Agent. J. Am. Chem. Soc. 1971, 93, 2897–2904. [Google Scholar] [CrossRef]
- Ishizaki, M.; Niimi, Y.; Hoshino, O.; Hara, H.; Takahashi, T. A Formal Total Synthesis of Lycopodium Alkaloid, (±)-Magellanine, by Using the Intramolecular Pauson Khand Reaction. Tetrahedron 2005, 61, 4053–4065. [Google Scholar] [CrossRef]
- Ishizaki, M.; Hoshino, O. Pauson-Khand Reaction of Enynes Bearing Exo-Olefin and Its Application to Total Synthesis of Lycopodium Alkaloid, Magellanine. J. Synth. Org. Chem. Jpn. 2003, 61, 1166–1175. [Google Scholar] [CrossRef]
- Ishizaki, M.; Masamoto, M.; Hoshino, O.; Nishitani, K.; Hara, H. Intramolecular Pauson-Khand Reaction of 3-Alkynyl-1-Alkyl- Idenecycles: A Convenient Synthesis of [5.n.1.01,5] Tricyclic Compounds. Heterocycles 2004, 63, 1359. [Google Scholar] [CrossRef]
- Ishizaki, M.; Zyo, M.; Kasama, Y.; Niimi, Y.; Hoshino, O.; Nishitani, K.; Hara, H. Investigation of the Intermolecular Pauson-Khand Reaction of Various 1-Alkynes with Cyclic Exo-Methylene Compounds. Heterocycles 2003, 60, 2259. [Google Scholar] [CrossRef]
- Ishizaki, M.; Masamoto, M.; Hoshino, O. Facile Synthesis of [5.n.1.01,5]- Tricycles Mediated by Intra- Molecular Pauson-Khand Reaction of 3-Alkynyl-1-Alkylidenecyclic Compounds. Heterocycles 2002, 57, 1409. [Google Scholar] [CrossRef]
- Ishizaki, M.; Kasama, Y.; Zyo, M.; Niimi, Y.; Hoshino, O. A Facile Synthesis of Spiro-Cyclopentenones from Various 1-Alkynes and Cyclic Exo-Methylene Compounds by Intermolecular Pauson-Khand Reaction. Heterocycles 2001, 55, 1439. [Google Scholar] [CrossRef]
- Ishizaki, M.; Iwahara, K.; Kyoumura, K.; Hoshino, O. An Effective Synthesis of Angular Tricyclic Compounds Having Two Contiguous Quaternary Centers by Intramolecular Pauson-Khand Reaction of Exocyclic Enynes. Synlett 1999, 1999, 587–589. [Google Scholar] [CrossRef]
- Ishizaki, M.; Iwahara, K.; Niimi, Y.; Satoh, H.; Hoshino, O. Investigation of the Synthesis of Angular Tricyclic Compounds by Intramolecular Pauson–Khand Reaction of Exo- and Endo-Cyclic Enynes. Tetrahedron 2001, 57, 2729–2738. [Google Scholar] [CrossRef]
- Ishizaki, M.; Niimi, Y.; Hoshino, O. Formal Total Synthesis of (±)-Magellanine. Tetrahedron Lett. 2003, 44, 6029–6031. [Google Scholar] [CrossRef]
- Rezgui, F.; El Gaied, M.M. DMAP-Catalyzed Hydroxymethylation of 2-Cyclohexenones in Aqueous Medium through Baylis-Hillman Reaction. Tetrahedron Lett. 1998, 39, 5965–5966. [Google Scholar] [CrossRef]
- Jeong, N.; Chung, Y.K.; Lee, B.Y.; Lee, S.H.; Yoo, S.-E. A Dramatic Acceleration of the Pauson-Khand Reaction by Trimethylamine N -Oxide. Synlett 1991, 1991, 204–206. [Google Scholar] [CrossRef]
- Kozaka, T.; Miyakoshi, N.; Mukai, C. Stereoselective Total Syntheses of Three Lycopodium Alkaloids, (−)-Magellanine, (+)-Magellaninone, and (+)-Paniculatine, Based on Two Pauson−Khand Reactions. J. Org. Chem. 2007, 72, 10147–10154. [Google Scholar] [CrossRef] [PubMed]
- Mukai, C. A Highly Diastereoselective Construction of Optically Active Bicyclo[3.3.0]Octenone Derivatives from L-Ascorbic Acid via Pauson-Khand Reaction. Tetrahedron Lett. 1995, 36, 5761–5764. [Google Scholar] [CrossRef]
- Mukai, C.; Uchiyama, M.; Hanaoka, M. Bis(Cyclopentadienyl)Tetracarbonyldimolybdenum-Alkyne Complexes Mediated [2 + 2 + 1] Cycloaddition: The Formation of 3-Substituted Cyclopentenone Derivatives. J. Chem. Soc. Chem. Commun. 1992, 1992, 1014. [Google Scholar] [CrossRef]
- Mukai, C.; Kim, J.S.; Uchiyama, M.; Sakamoto, S.; Hanaoka, M. Stereoselective Construction of Optically Active Bicyclo[3.3.0]Octenone Derivatives Based on the Pauson–Khand Reaction. J. Chem. Soc. Perkin 1 1998, 17, 2903–2916. [Google Scholar] [CrossRef]
- Mukai, C.; Kim, J.S.; Uchiyama, M.; Hanaoka, M. Diastereocomplementary Construction of Optically Active Bicyclo[4.3.0]Nonenone Skeleton Based on Pauson-Khand Reaction. Tetrahedron Lett. 1998, 39, 7909–7912. [Google Scholar] [CrossRef]
- Mukai, C.; Kim, J.S.; Sonobe, H.; Hanaoka, M. Stereocomplementary Construction of Optically Active Bicyclo[4.3.0]Nonenone Derivatives. J. Org. Chem. 1999, 64, 6822–6832. [Google Scholar] [CrossRef]
- Mukai, C.; Sonobe, H.; Kim, J.S.; Hanaoka, M. Pauson−Khand Reaction of Optically Active 6,7-Bis( Tert -Butyldimethylsiloxy)Non-1-En-8-Ynes. J. Org. Chem. 2000, 65, 6654–6659. [Google Scholar] [CrossRef]
- Mukai, C.; Kobayashi, M.; Kim, I.J.; Hanaoka, M. Total Synthesis of (±)-Ceratopicanol. Tetrahedron 2002, 58, 5225–5230. [Google Scholar] [CrossRef]
- Mukai, C.; Kozaka, T.; Suzuki, Y.; Kim, I.J. Trimethylgermyl Group in Pauson–Khand Reaction. Tetrahedron Lett. 2002, 43, 8575–8578. [Google Scholar] [CrossRef]
- Mukai, C.; Kozaka, T.; Suzuki, Y.; Kim, I.J. New Entry to the Pauson–Khand Reaction: Trimethylgermyl Group at the Triple Bond Terminus as a Latent Functional Group. Tetrahedron 2004, 60, 2497–2507. [Google Scholar] [CrossRef]
- Nomura, I.; Mukai, C. Studies on the Total Synthesis of Streptazolin and Its Related Natural Products: First Total Synthesis of (±)-8α-Hydroxystreptazolone. J. Org. Chem. 2004, 69, 1803–1812. [Google Scholar] [CrossRef]
- Nomura, I.; Mukai, C. Total Synthesis of (±)-8α-Hydroxystreptazolone. Org. Lett. 2002, 4, 4301–4304. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, T.; Yamada, M.; Yamaguchi, M.; Nishizawa, M. The Intra- and Intermolecular Pauson-Khand Reaction Promoted by Alkyl Methyl Sulfides. Synlett 1999, 1999, 771–773. [Google Scholar] [CrossRef]
- Salom-Roig, X.; Dénès, F.; Renaud, P. Radical Cyclization of Haloacetals: The Ueno-Stork Reaction. Synthesis 2004, 2004, 1903–1928. [Google Scholar] [CrossRef]
- Jiang, S.-Z.; Lei, T.; Wei, K.; Yang, Y.-R. Collective Total Synthesis of Tetracyclic Diquinane Lycopodium Alkaloids (+)-Paniculatine, (−)-Magellanine, (+)-Magellaninone and Analogues Thereof. Org. Lett. 2014, 16, 5612–5615. [Google Scholar] [CrossRef] [PubMed]
- Shenvi, R.A.; O’Malley, D.P.; Baran, P.S. Chemoselectivity: The Mother of Invention in Total Synthesis. Acc. Chem. Res. 2009, 42, 530–541. [Google Scholar] [CrossRef]
- Lin, K.-W.; Ananthan, B.; Tseng, S.-F.; Yan, T.-H. Exceedingly Concise and Elegant Synthesis of (+)-Paniculatine, (−)-Magellanine, and (+)-Magellaninone. Org. Lett. 2015, 17, 3938–3940. [Google Scholar] [CrossRef]
- Charette, A.B.; Beauchemin, A.; Marcoux, J.-F. Photoinduced Synthesis of Diorganozinc and Organozinc Iodide Reagents. J. Am. Chem. Soc. 1998, 120, 5114–5115. [Google Scholar] [CrossRef]
- Nakamura, E.; Kuwajima, I. Copper-Catalyzed Acylation and Conjugate Addition of Zinc Homoenolate. Synthesis of 4- and 5-Oxo Esters. J. Am. Chem. Soc. 1984, 106, 3368–3370. [Google Scholar] [CrossRef]
- Tsuji, J.; Nagashima, H.; Hori, K. Regioselective Oxidation of Internal Olefins Bearing Neighboring Oxygen Functions by Means of Palladium Catalysts. Preparation of β-Alkoxy or Acetoxy Ketones from Allyl and Homoallyl Ethers or Esters. Tetrahedron Lett. 1982, 23, 2679–2682. [Google Scholar] [CrossRef]
- Liu, J.; Chen, S.; Li, N.; Qiu, F.G. A Concise Total Synthesis of (+)-Paniculatine. Adv. Synth. Catal. 2019, 361, 3514–3517. [Google Scholar] [CrossRef]
- Smietana, M.; Gouverneur, V.; Mioskowski, C. An Improved Synthesis of Iodohydrins from Alkenes. Tetrahedron Lett. 2000, 41, 193–195. [Google Scholar] [CrossRef]
- Huang, B.-B.; Lei, K.; Zhong, L.-R.; Yang, X.; Yao, Z.-J. Unified Total Synthesis of Tetracyclic Diquinane Lycopodium Alkaloids (+)-Paniculatine, (−)-Magellanine, and (+)-Magellaninone. J. Org. Chem. 2022, 87, 8685–8696. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.M.; Zhang, L.-D.; Tan, R.X.; Yao, Z.-J. Protecting Group-Free Total Synthesis of (−)-Lannotinidine B. J. Am. Chem. Soc. 2012, 134, 12323–12325. [Google Scholar] [CrossRef]
- Zhong, L.-R.; Huang, B.-B.; Yang, X.-L.; Wang, S.; Yao, Z.-J. Concise Unified Access to (−)-8-Deoxy-13-Dehydroserratinine, (+)-Fawcettimine, (+)-Fawcettidine, and (−)-8-Deoxyserratinine Using a Direct Intramolecular Reductive Coupling. Org. Lett. 2021, 23, 3578–3583. [Google Scholar] [CrossRef]
- Zhong, L.; Yang, Y.; Huang, B.; Yao, Z. An Eight-step Total Synthesis of Pyrroloquinolone-type Lycopodium Alkaloid via a Tandem Annulation Approach. Chin. J. Chem. 2020, 38, 1560–1564. [Google Scholar] [CrossRef]
- Sibi, M.P.; Asano, Y. Enantioselective Conjugate Addition of Organomagnesium Amides to Enamidomalonates: Synthesis of Either Enantiomer of β-Amino Acid Derivatives. J. Am. Chem. Soc. 2001, 123, 9708–9709. [Google Scholar] [CrossRef]
- Schinzer, D. Conjugate Addition Reactions in Organic Synthesis. Angew. Chem. 1993, 105, 1738–1739. [Google Scholar] [CrossRef]
- Seayad, J.; List, B. Asymmetric Organocatalysis. Org. Biomol. Chem. 2005, 3, 719. [Google Scholar] [CrossRef] [PubMed]
- Dalko, P.I.; Moisan, L. In the Golden Age of Organocatalysis. Angew. Chem. Int. Ed. 2004, 43, 5138–5175. [Google Scholar] [CrossRef] [PubMed]
- List, B. Proline-Catalyzed Asymmetric Reactions. Tetrahedron 2002, 58, 5573–5590. [Google Scholar] [CrossRef]
- Jarvo, E.R.; Miller, S.J. Amino Acids and Peptides as Asymmetric Organocatalysts. Tetrahedron 2002, 58, 2481–2495. [Google Scholar] [CrossRef]
- Sandham, D.A.; Meyers, A.I. Chiral Bicyclic Lactams–an Asymmetric Synthesis of the Framework of the Lycopodium Alkaloid Magellanine Containing All Six Adjacent Stereogenic Centres. J Chem Soc Chem Commun 1995, 1995, 2511–2512. [Google Scholar] [CrossRef]
- 1Akiba, K.; Nishihara, Y.; Wada, M. Regioselective Synthesis of 4-(2-Oxoalkyl)Pyridines via 1,4-Dihydro-Pyridine Derivatives Using Silyl Enol Ethers and Pyridinium Salts. Tetrahedron Lett. 1983, 24, 5269–5272. [Google Scholar] [CrossRef]
- Comins, D.L.; Brown, J.D. Regioselective Addition of Titanium Enolates to 1-Acylpyridinium Salts. A Convenient Synthesis of 4-(2-Oxoalkyl)Pyridines. Tetrahedron Lett. 1984, 25, 3297–3300. [Google Scholar] [CrossRef]
- Comins, D.L.; Brown, J.D. Additon of Metallo Enolates to 1-Acylpyridinium Salts. A Short Synthesis of (±)-Lupinine. Tetrahedron Lett. 1986, 27, 2219–2222. [Google Scholar] [CrossRef]
- Courtois, G.; Al-Arnaout, A.; Miginiac, L. Addition Regioselective d’organometalliques α-Insatures Ou α-Fonctionnels Au Chlorure de n-Ethoxycarbonylpyridinium: Synthese de Dihydro-1,2 (Ou -1,4) Pyridines 2-(Ou 4-) Substituees. Tetrahedron Lett. 1985, 26, 1027–1030. [Google Scholar] [CrossRef]
- Evans, D.A.; Rieger, D.L.; Bilodeau, M.T.; Urpi, F. Stereoselective Aldol Reactions of Chlorotitanium Enolates. An Efficient Method for the Assemblage of Polypropionate-Related Synthons. J. Am. Chem. Soc. 1991, 113, 1047–1049. [Google Scholar] [CrossRef]
- Murphy, R.A.; Sarpong, R. Direct Methoxypyridine Functionalization Approach to Magellanine-Type Lycopodium Alkaloids. Org. Lett. 2012, 14, 632–635. [Google Scholar] [CrossRef] [PubMed]
- García-Cuadrado, D.; de Mendoza, P.; Braga, A.A.C.; Maseras, F.; Echavarren, A.M. Proton-Abstraction Mechanism in the Palladium-Catalyzed Intramolecular Arylation: Substituent Effects. J. Am. Chem. Soc. 2007, 129, 6880–6886. [Google Scholar] [CrossRef] [PubMed]
- García-Cuadrado, D.; Braga, A.A.C.; Maseras, F.; Echavarren, A.M. Proton Abstraction Mechanism for the Palladium-Catalyzed Intramolecular Arylation. J. Am. Chem. Soc. 2006, 128, 1066–1067. [Google Scholar] [CrossRef] [PubMed]
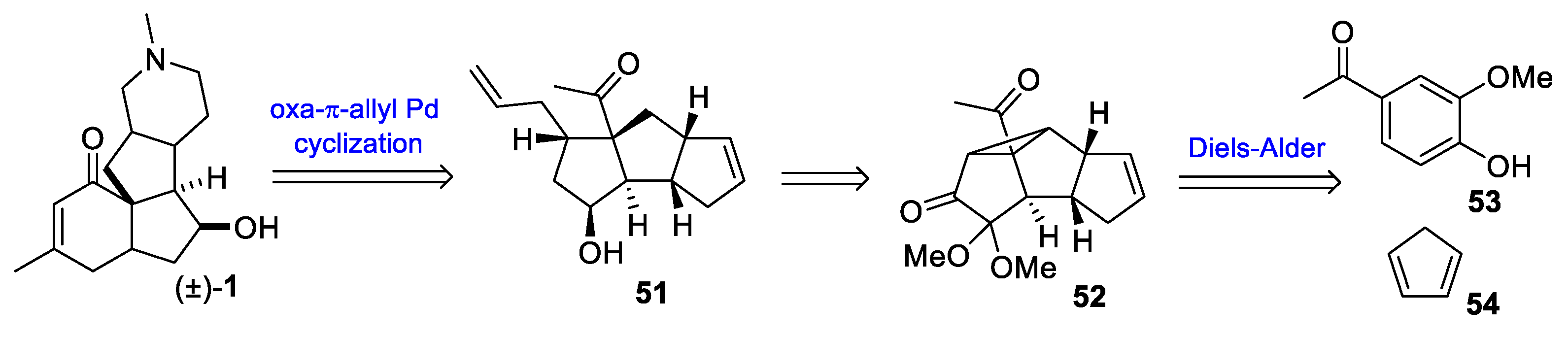
- McGee, P.; Bétournay, G.; Barabé, F.; Barriault, L. A 11-Steps Total Synthesis of Magellanine through a Gold(I)-Catalyzed Dehydro Diels-Alder Reaction. Angew. Chem. Int. Ed. 2017, 56, 6280–6283. [Google Scholar] [CrossRef]
- Barabé, F.; Levesque, P.; Korobkov, I.; Barriault, L. Synthesis of Fused Carbocycles via a Selective 6-Endo-Dig Gold(I)-Catalyzed Carbocyclization. Org. Lett. 2011, 13, 5580–5583. [Google Scholar] [CrossRef]






























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saito, T.; Awad, J.M.; Zhang, W. Synthetic Studies on Tetracyclic Diquinane Lycopodium Alkaloids Magellanine, Magellaninone and Paniculatine. Molecules 2023, 28, 1501. https://doi.org/10.3390/molecules28031501
Saito T, Awad JM, Zhang W. Synthetic Studies on Tetracyclic Diquinane Lycopodium Alkaloids Magellanine, Magellaninone and Paniculatine. Molecules. 2023; 28(3):1501. https://doi.org/10.3390/molecules28031501
Chicago/Turabian StyleSaito, Takeru, John Mark Awad, and Wei Zhang. 2023. "Synthetic Studies on Tetracyclic Diquinane Lycopodium Alkaloids Magellanine, Magellaninone and Paniculatine" Molecules 28, no. 3: 1501. https://doi.org/10.3390/molecules28031501
APA StyleSaito, T., Awad, J. M., & Zhang, W. (2023). Synthetic Studies on Tetracyclic Diquinane Lycopodium Alkaloids Magellanine, Magellaninone and Paniculatine. Molecules, 28(3), 1501. https://doi.org/10.3390/molecules28031501