



Multi-Analytical Approach to Characterize the Degradation of Different Types of Microplastics: Identification and Quantification of Released Organic Compounds


 ,
,  and
and 
Abstract
:1. Introduction
2. Results
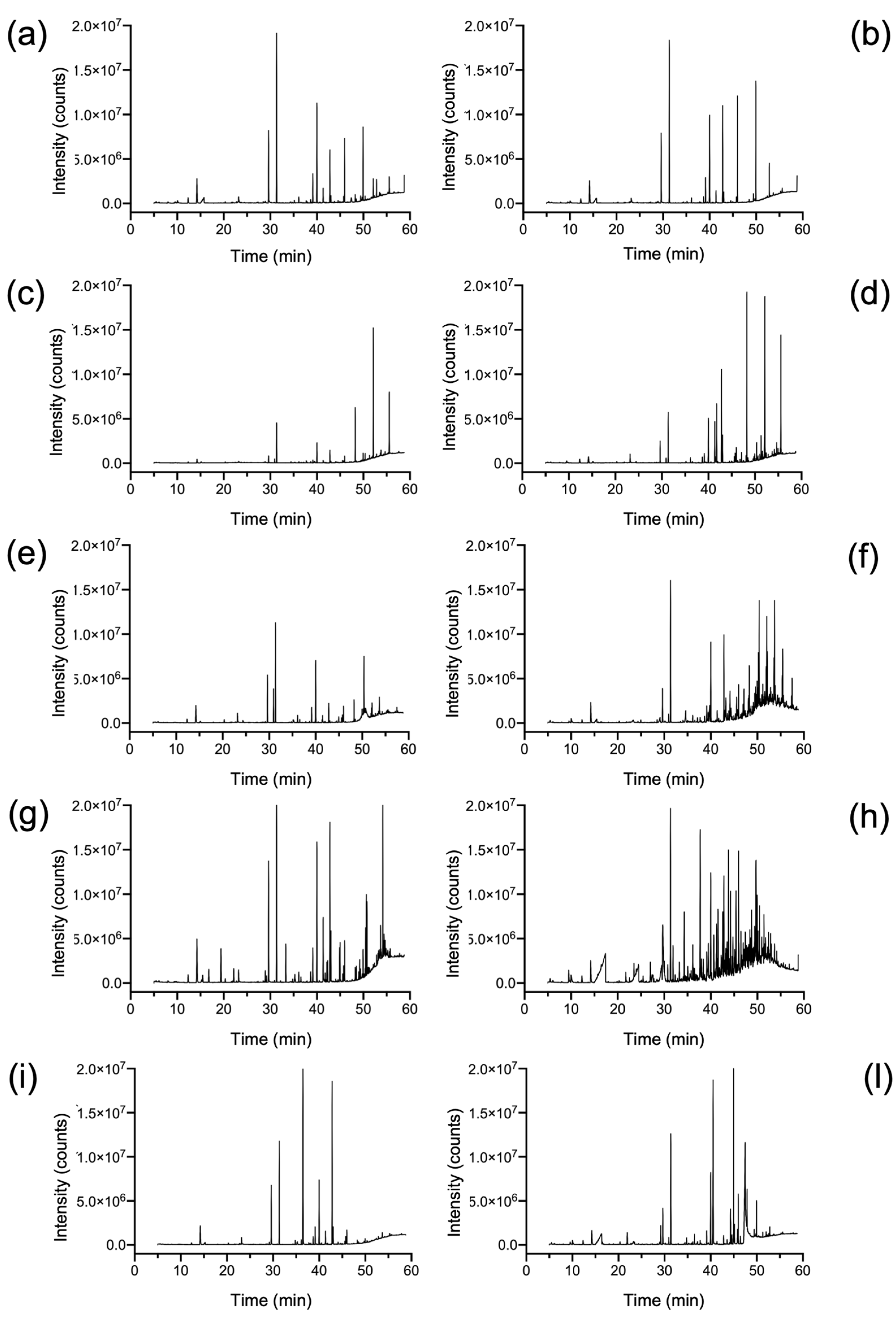
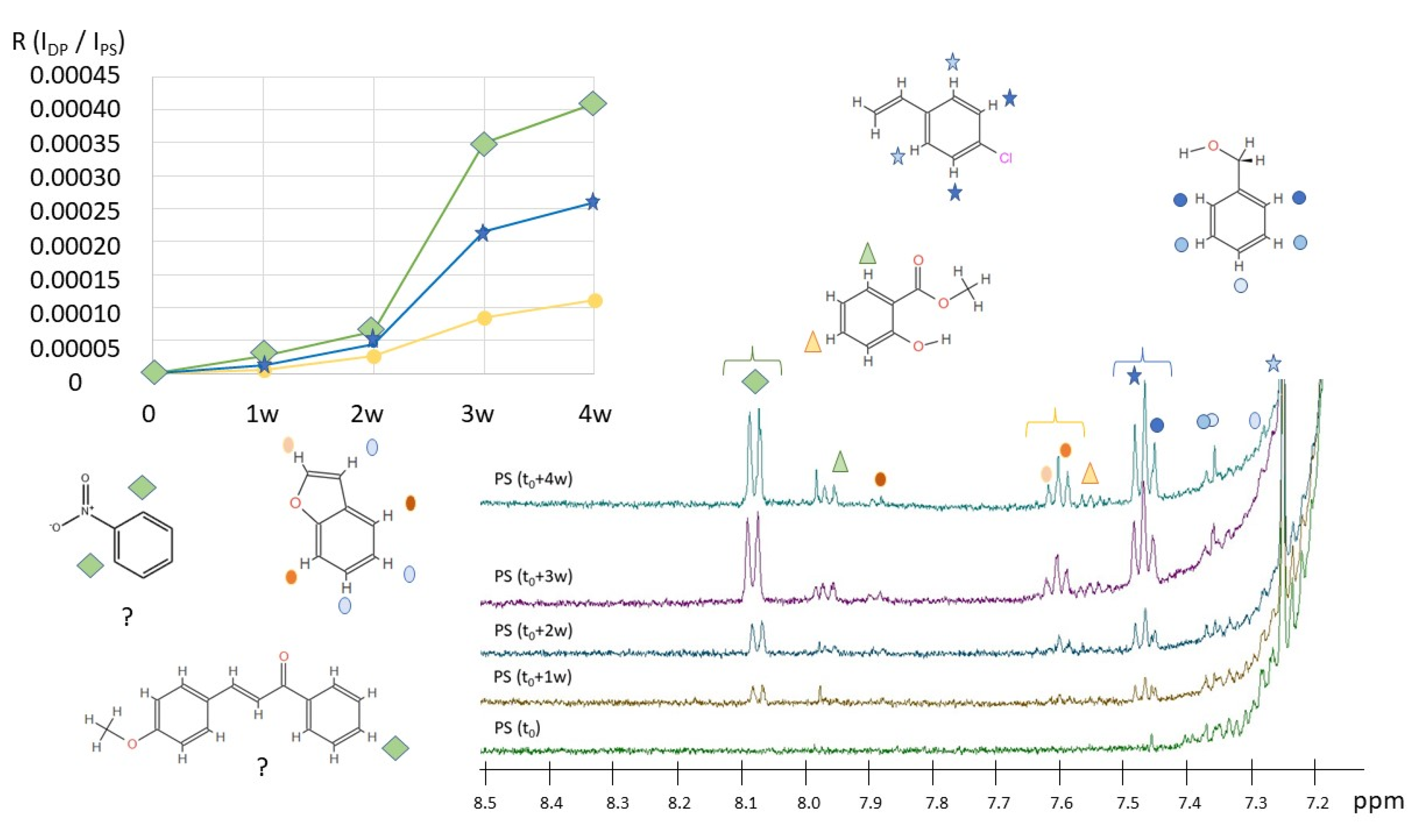
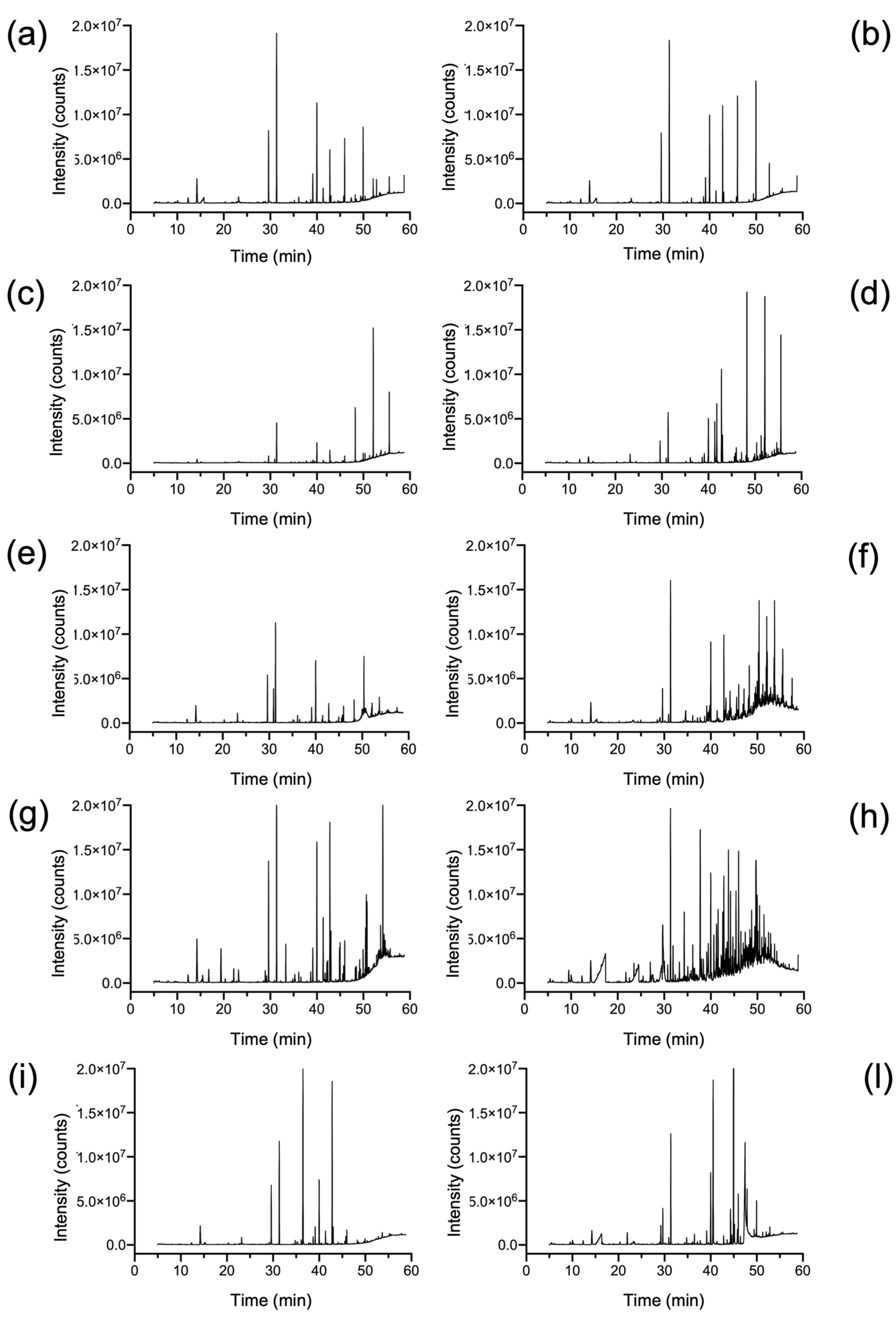
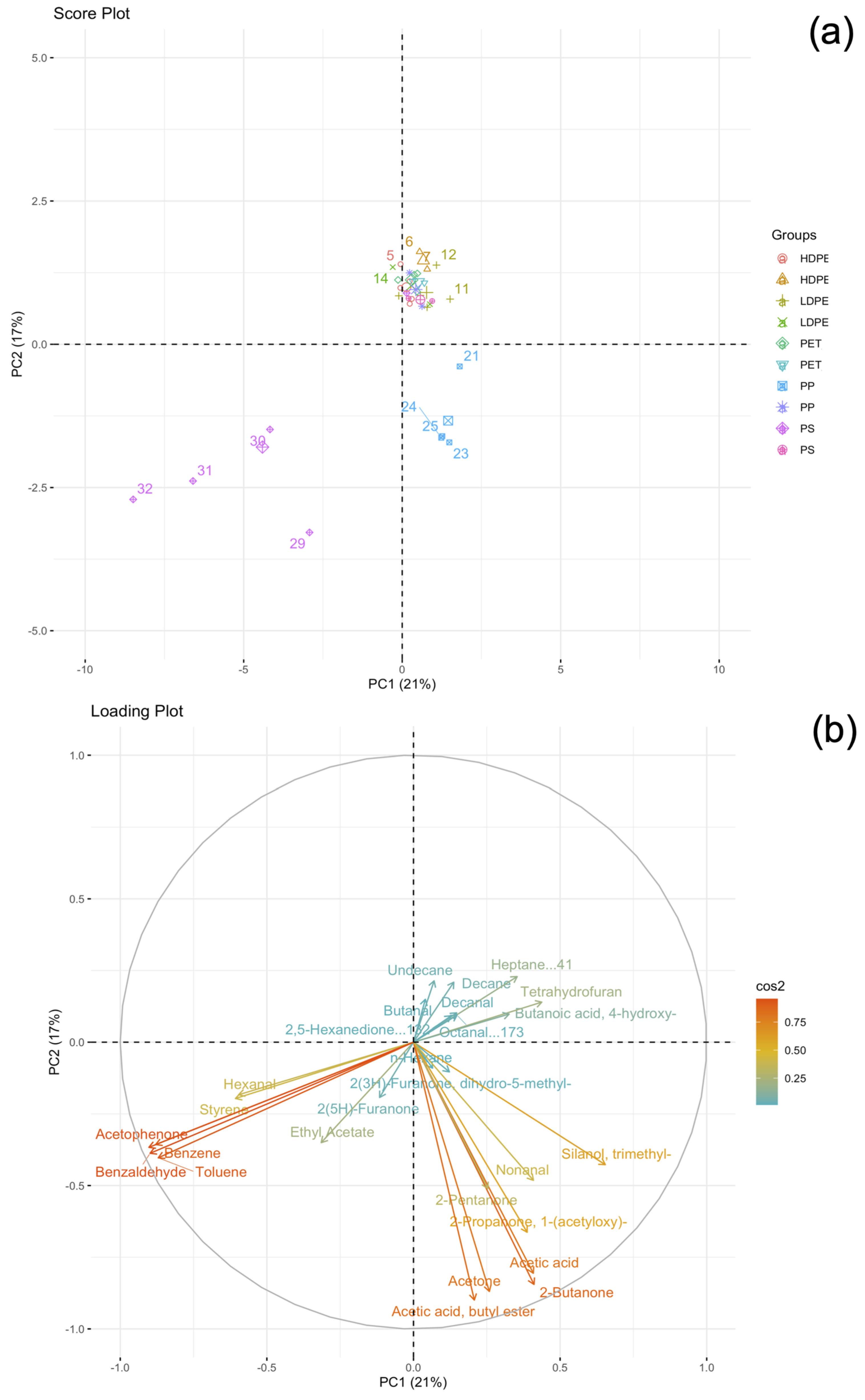
2.1. Qualitative and Quantitative Characterization of Polymer Materials with HiSorb-TD-GC-MS Analysis
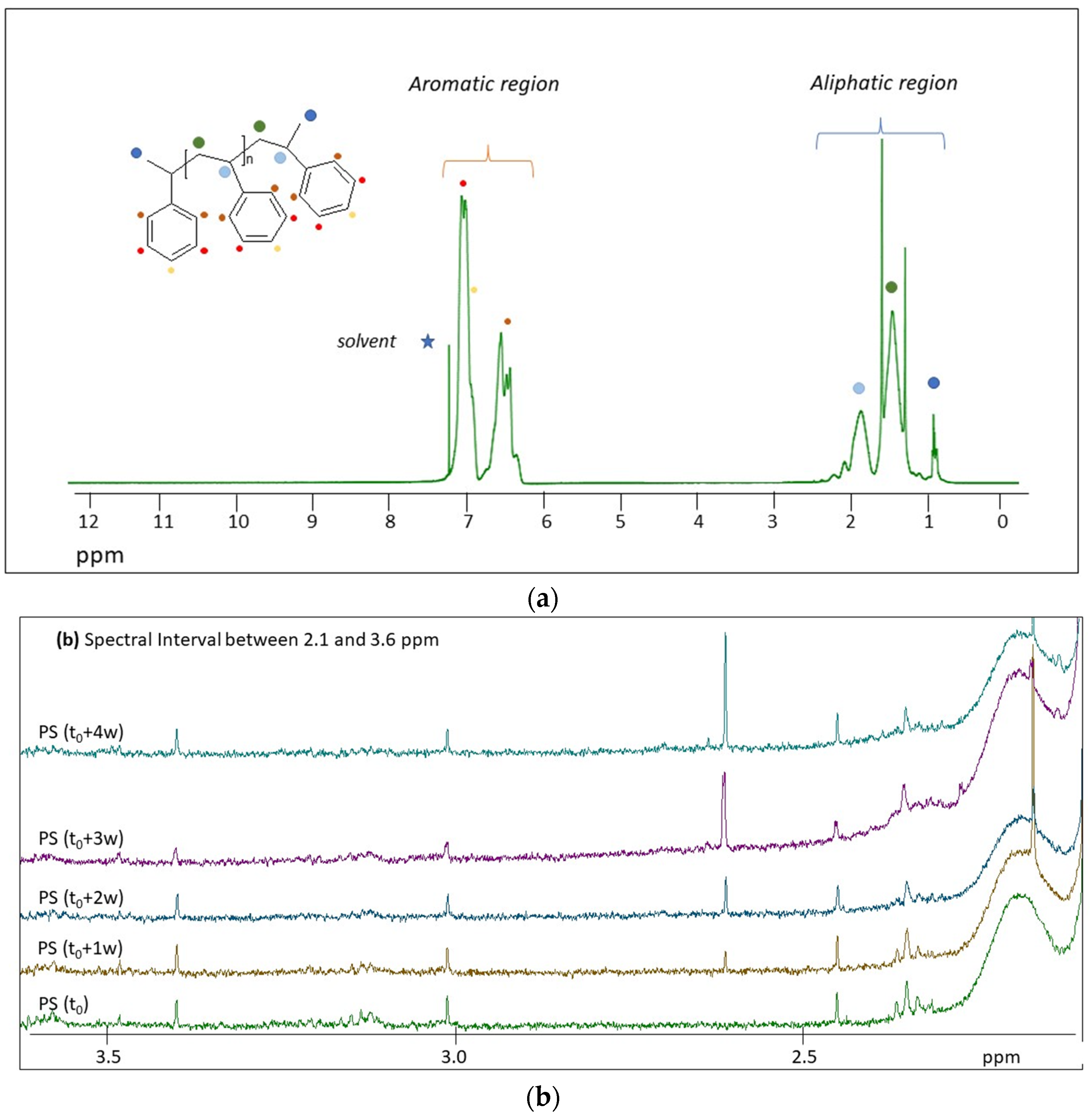
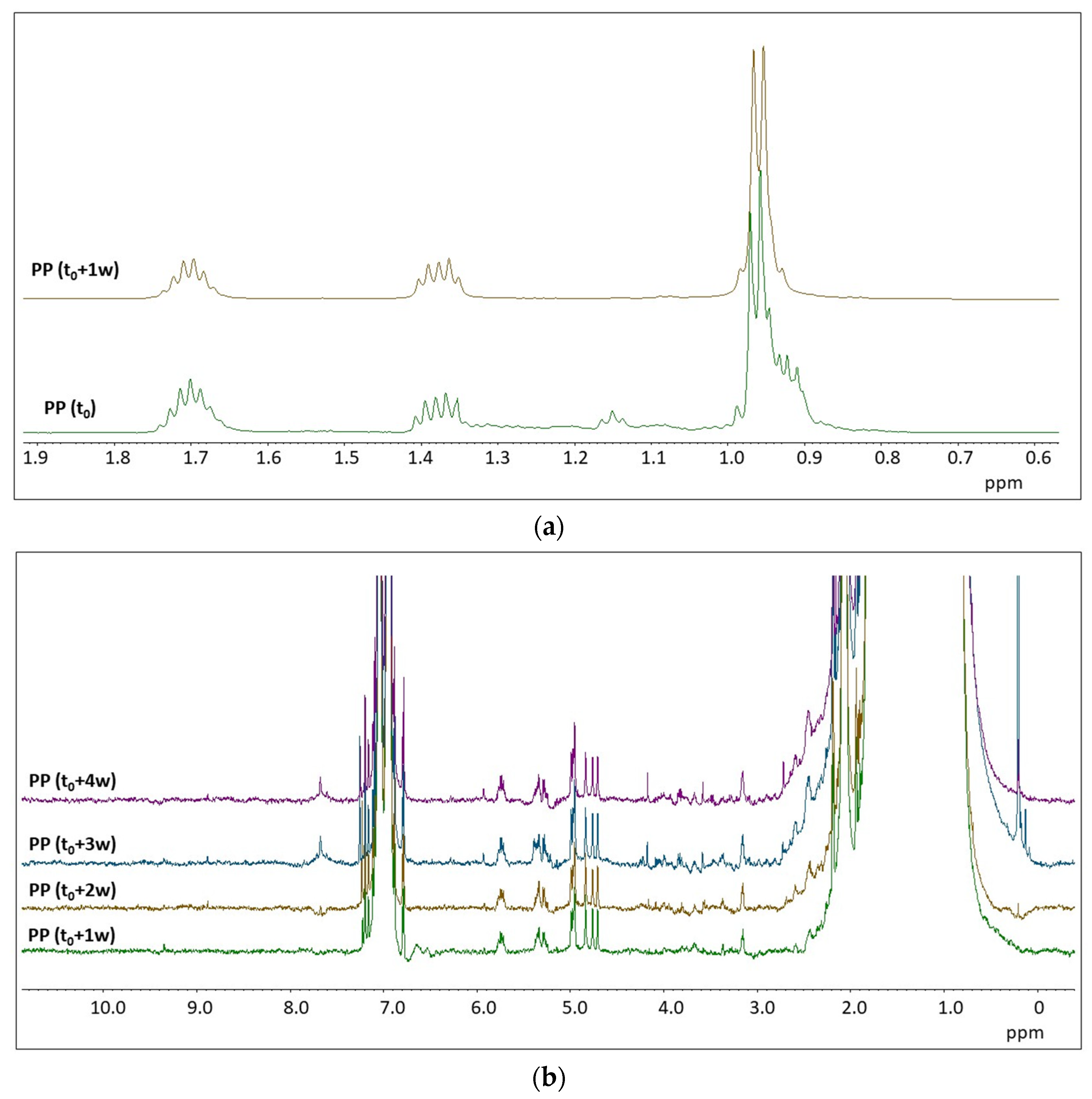
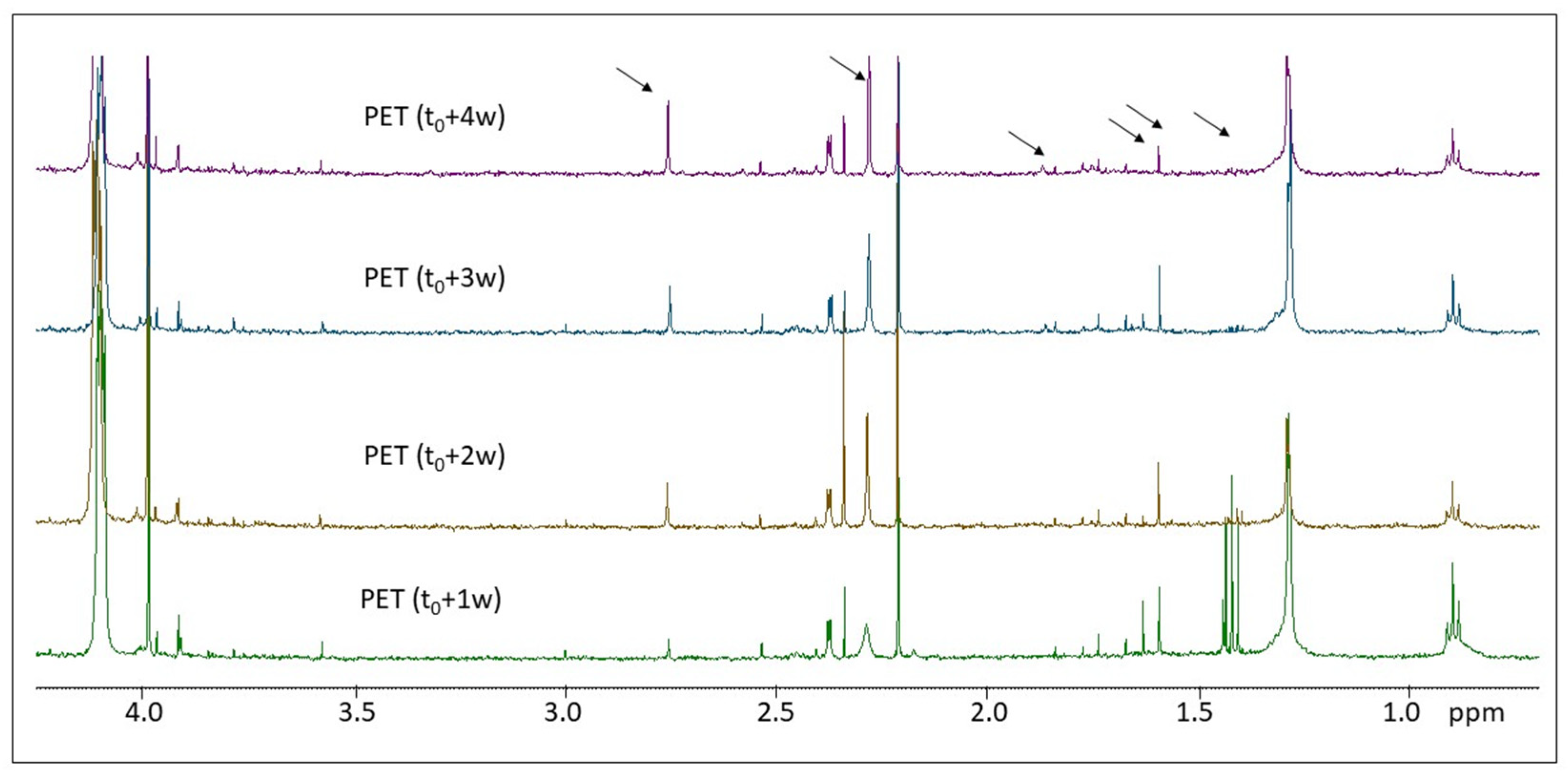
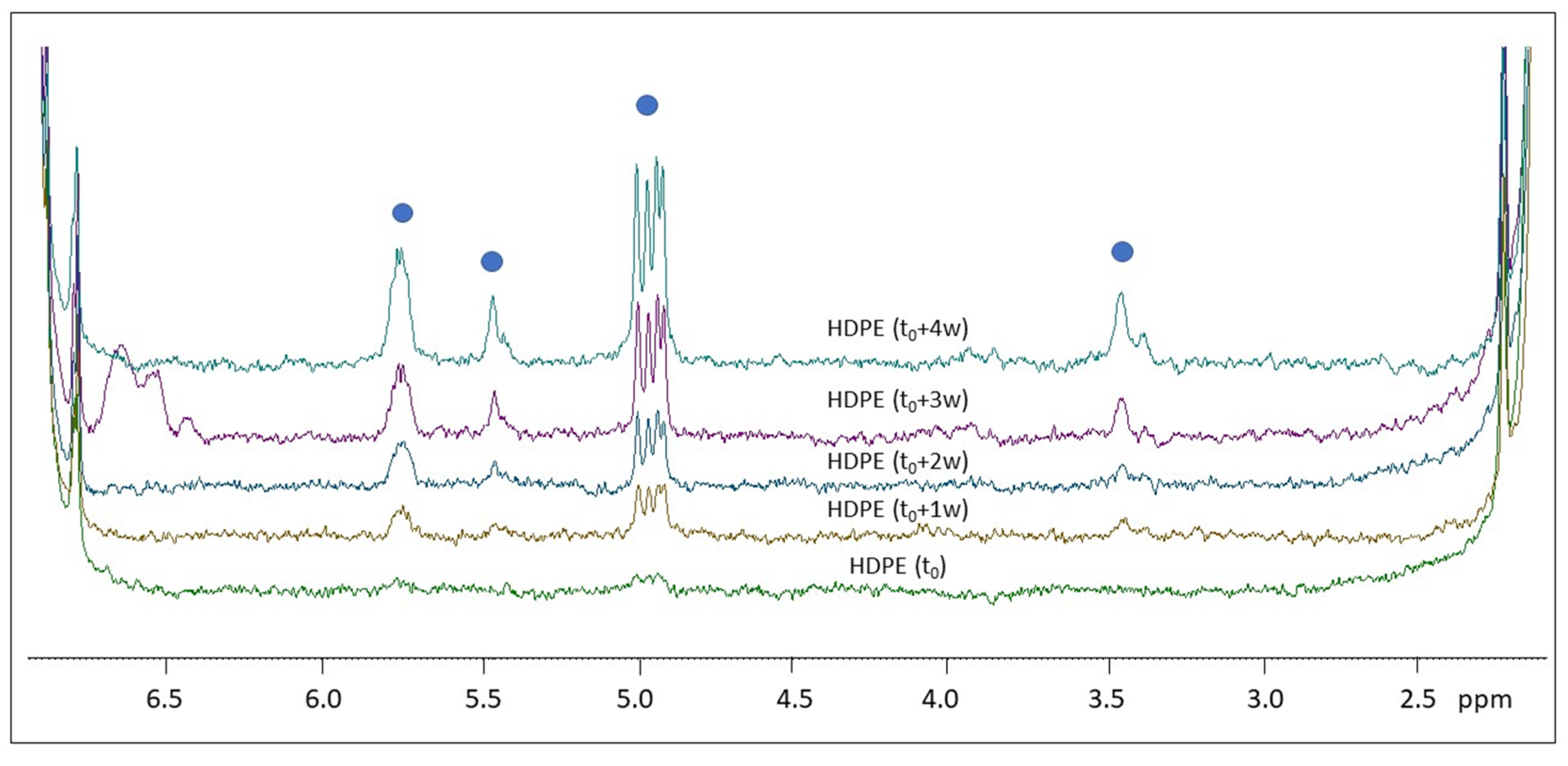
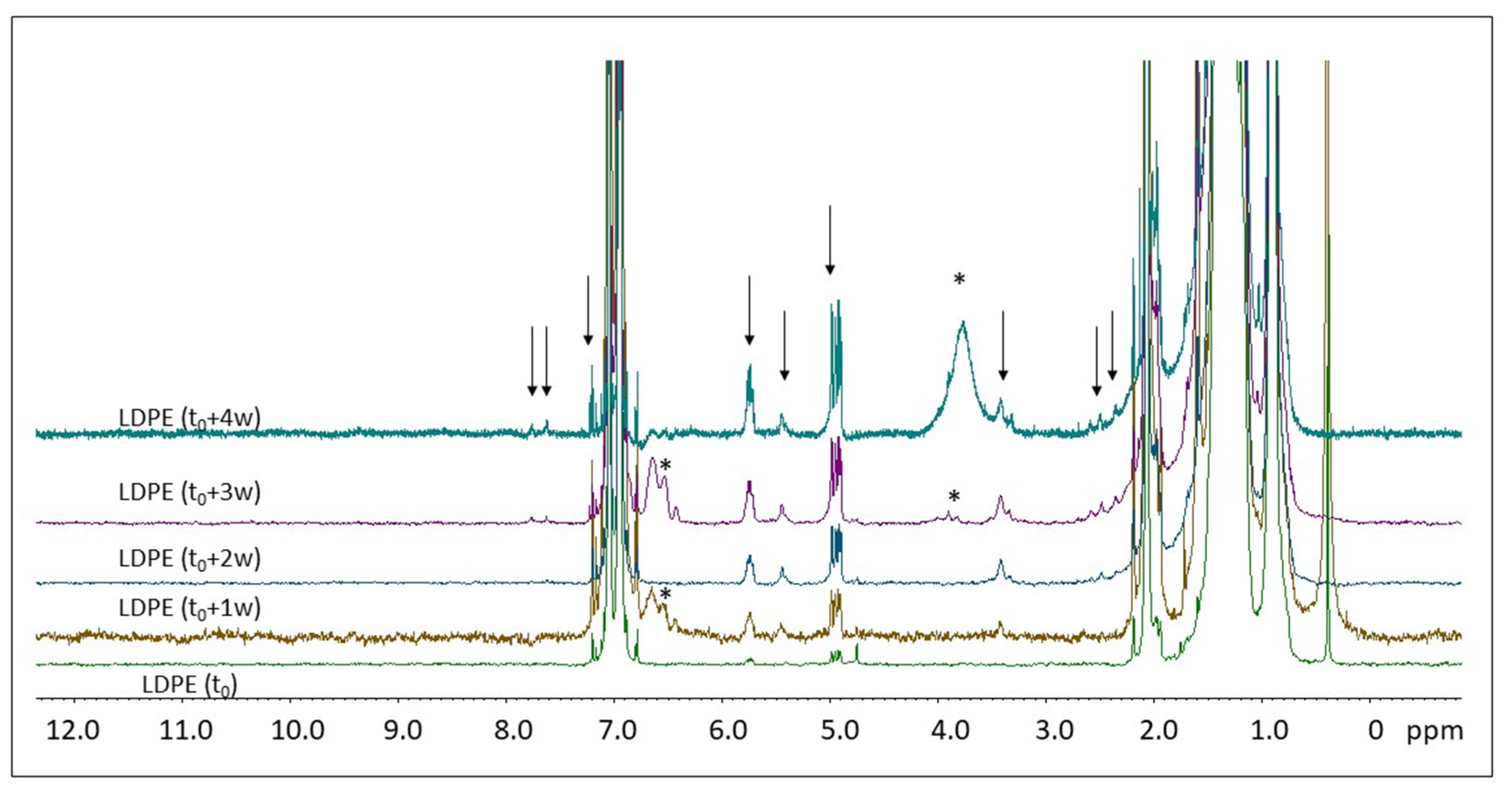
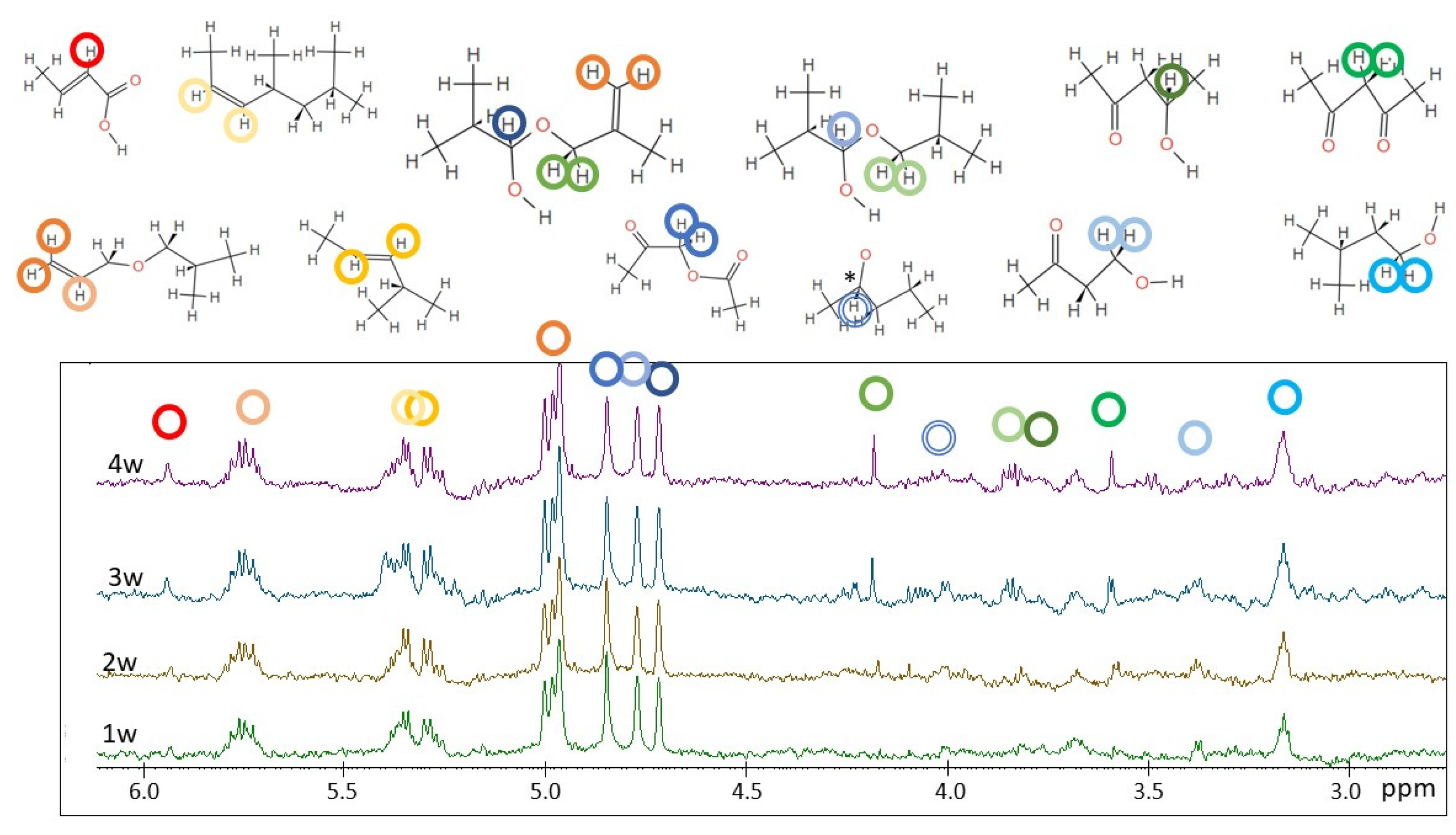
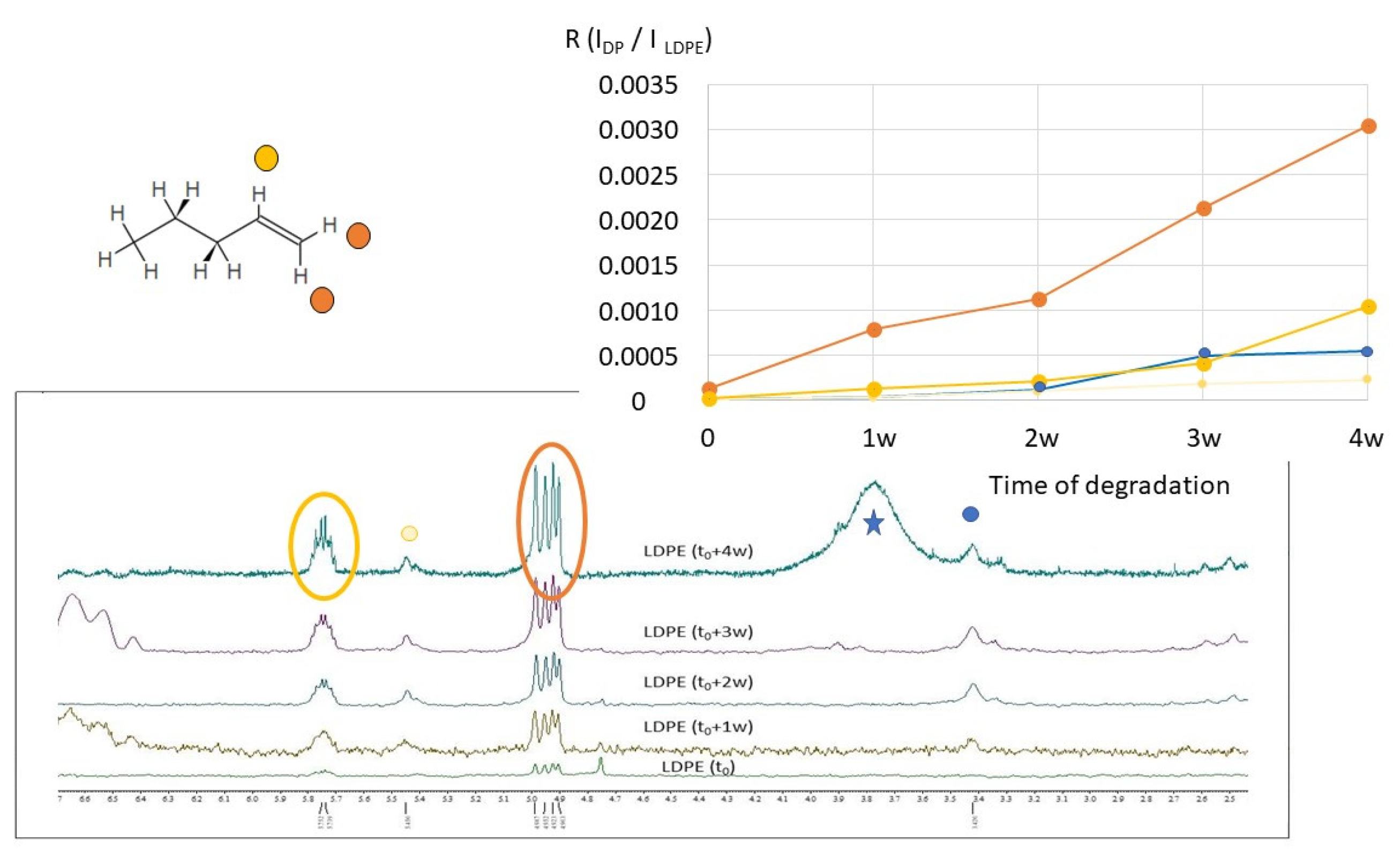
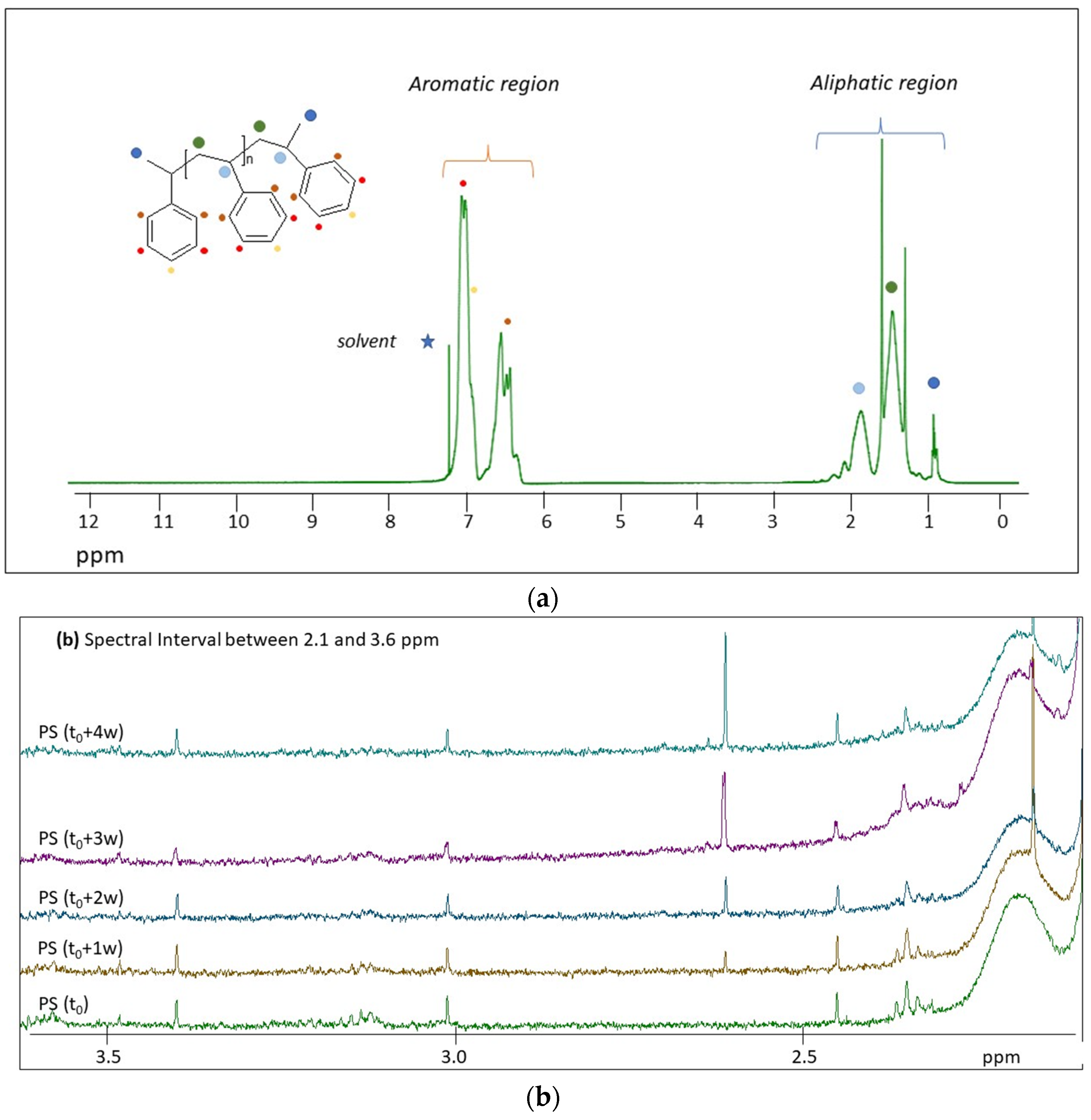
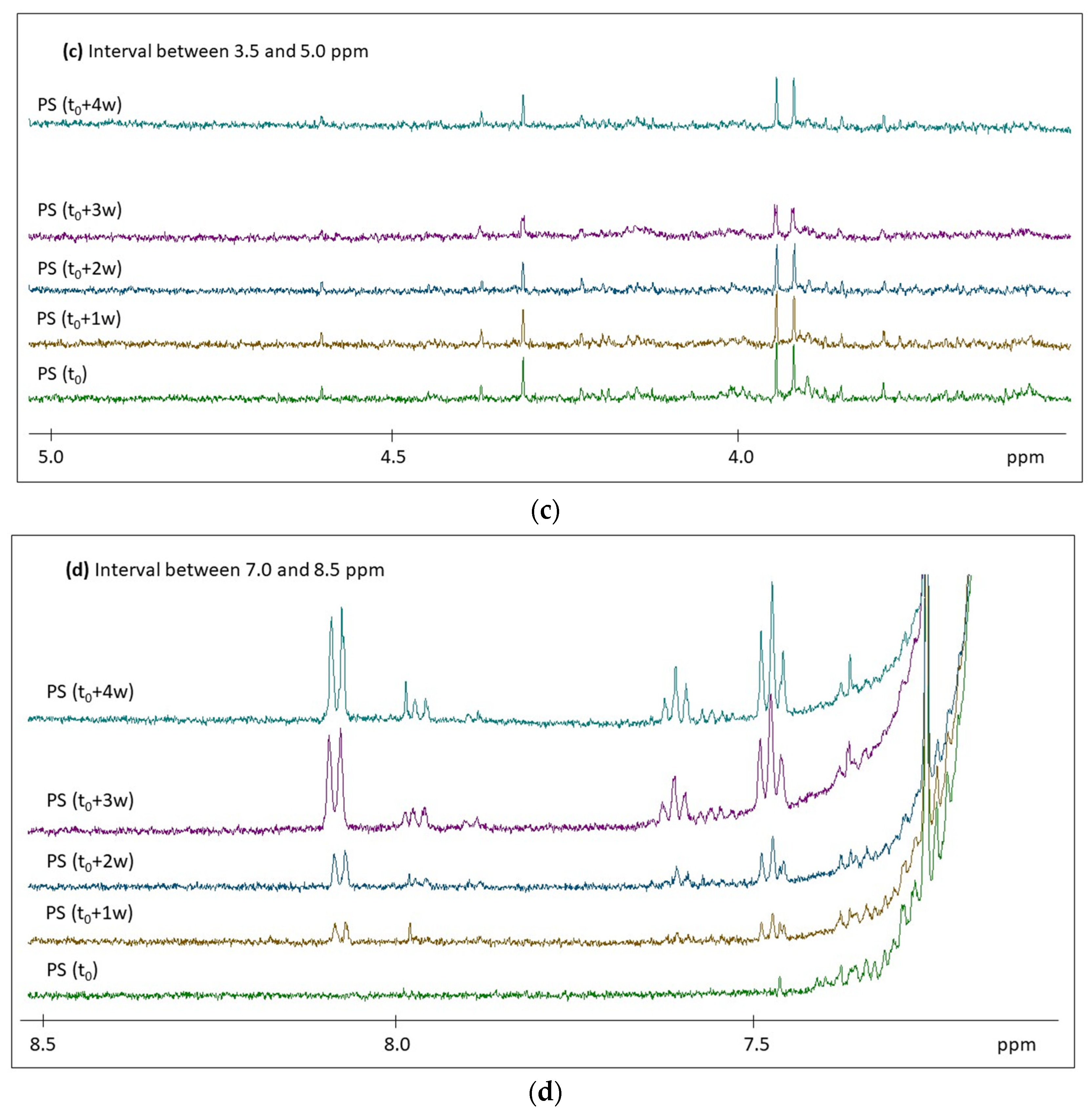
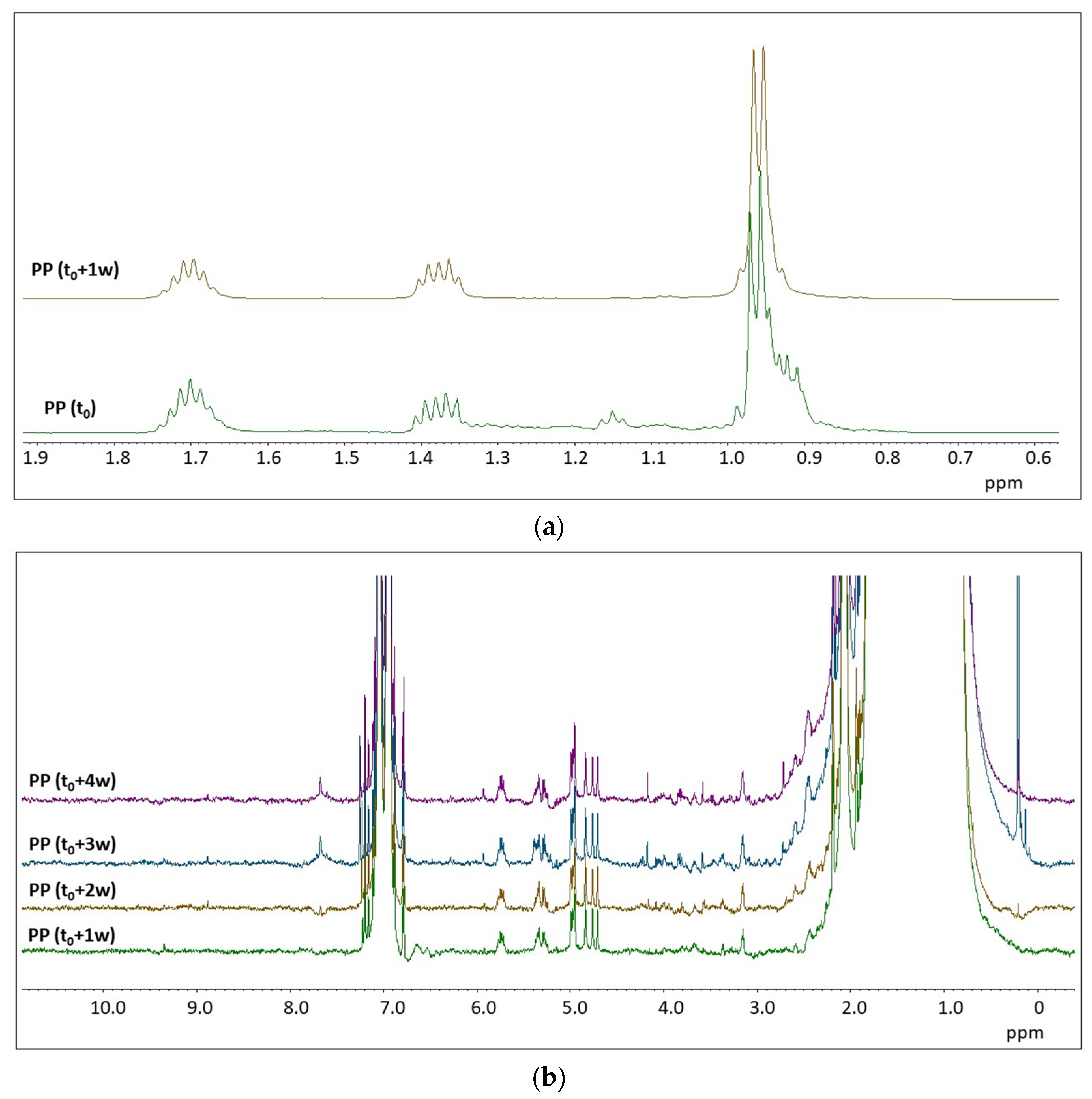
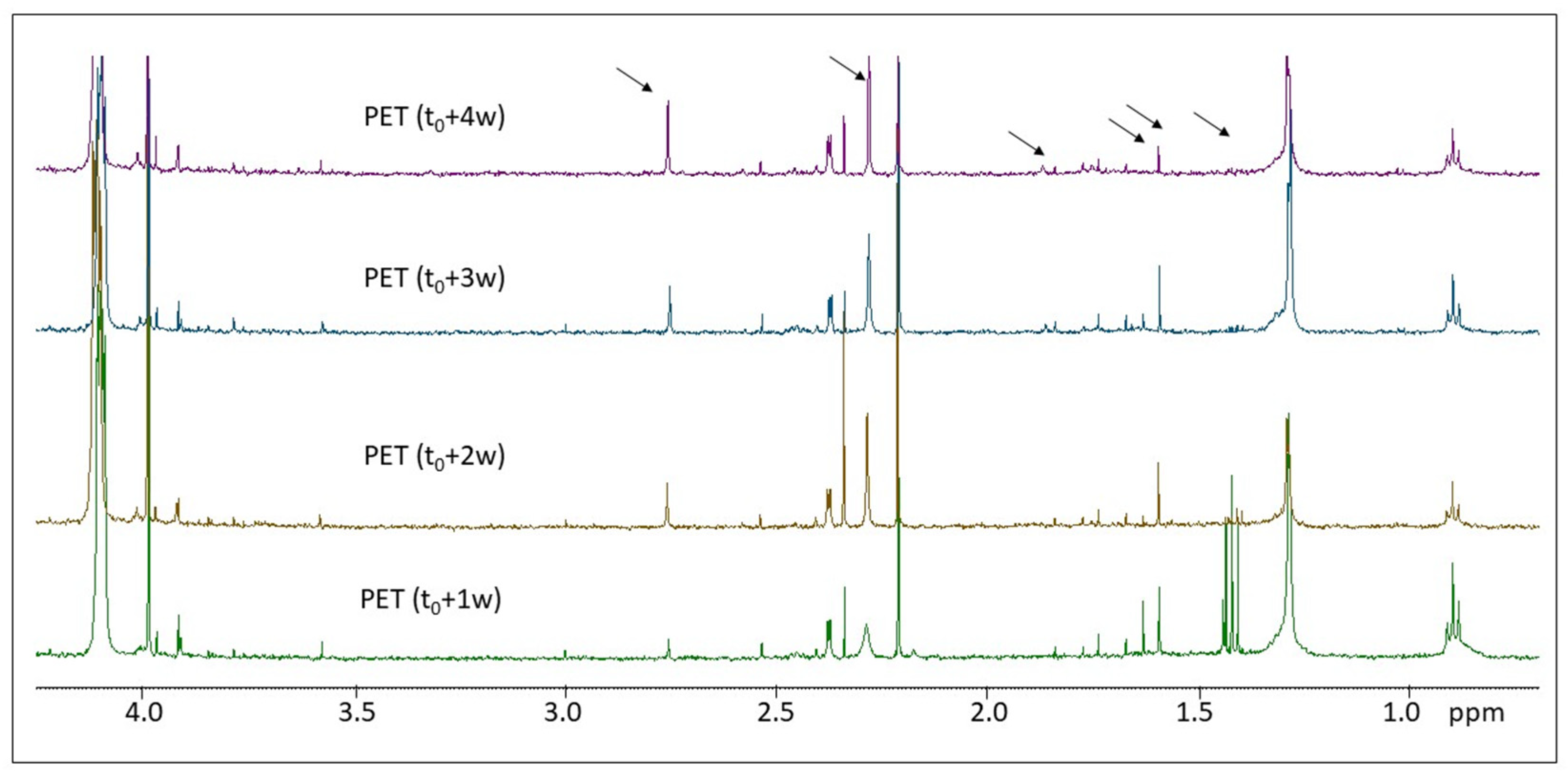
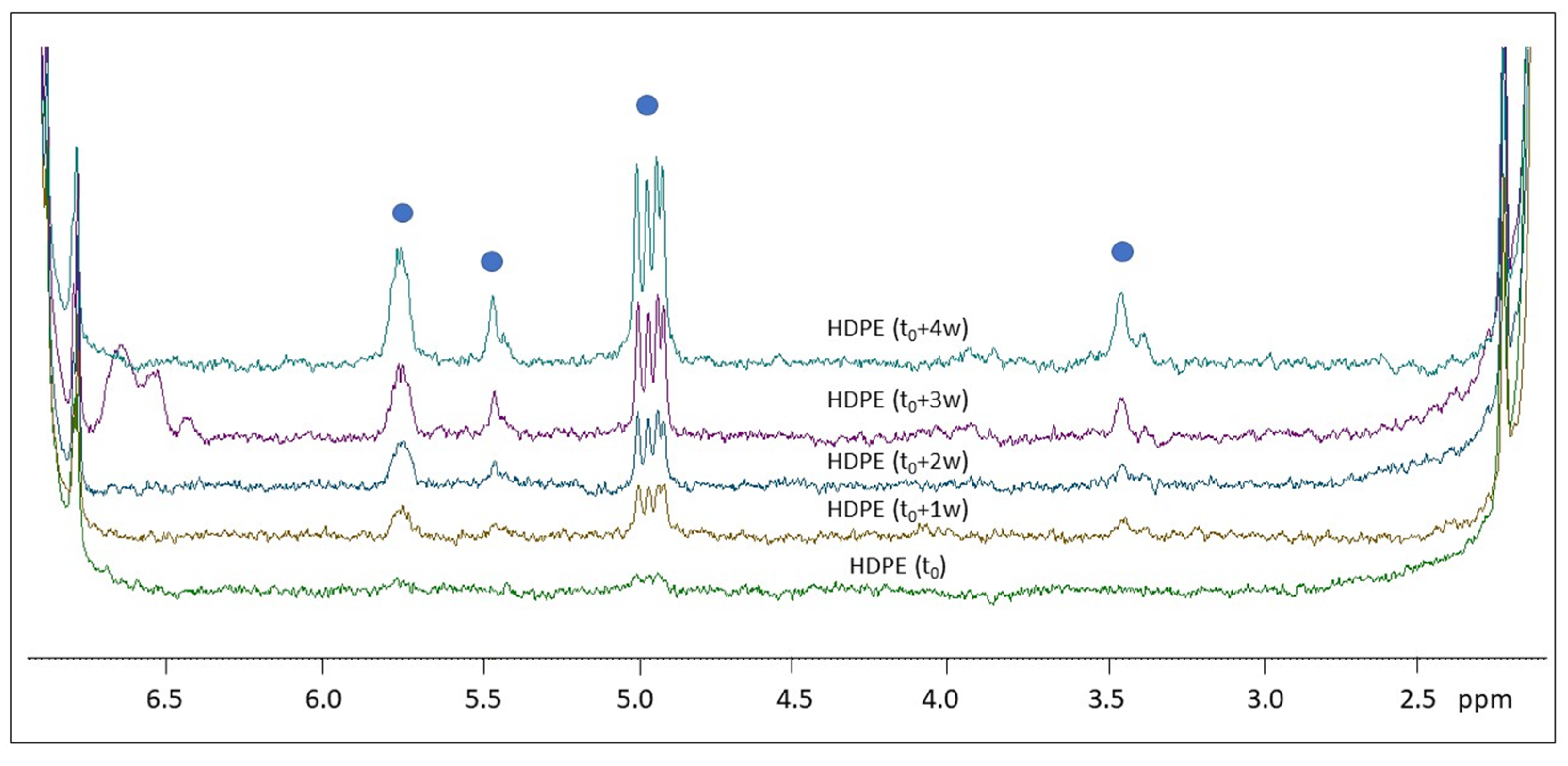
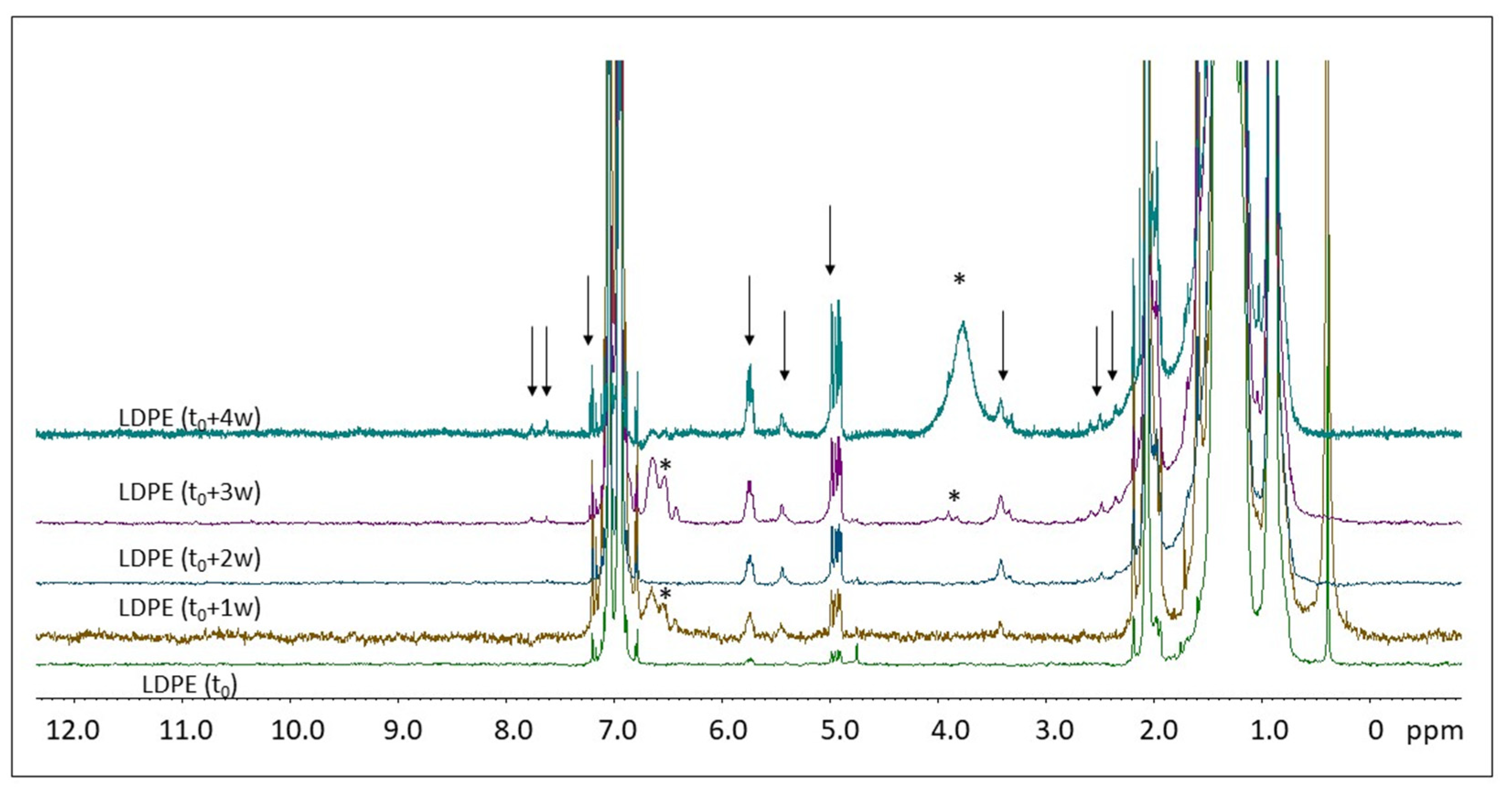
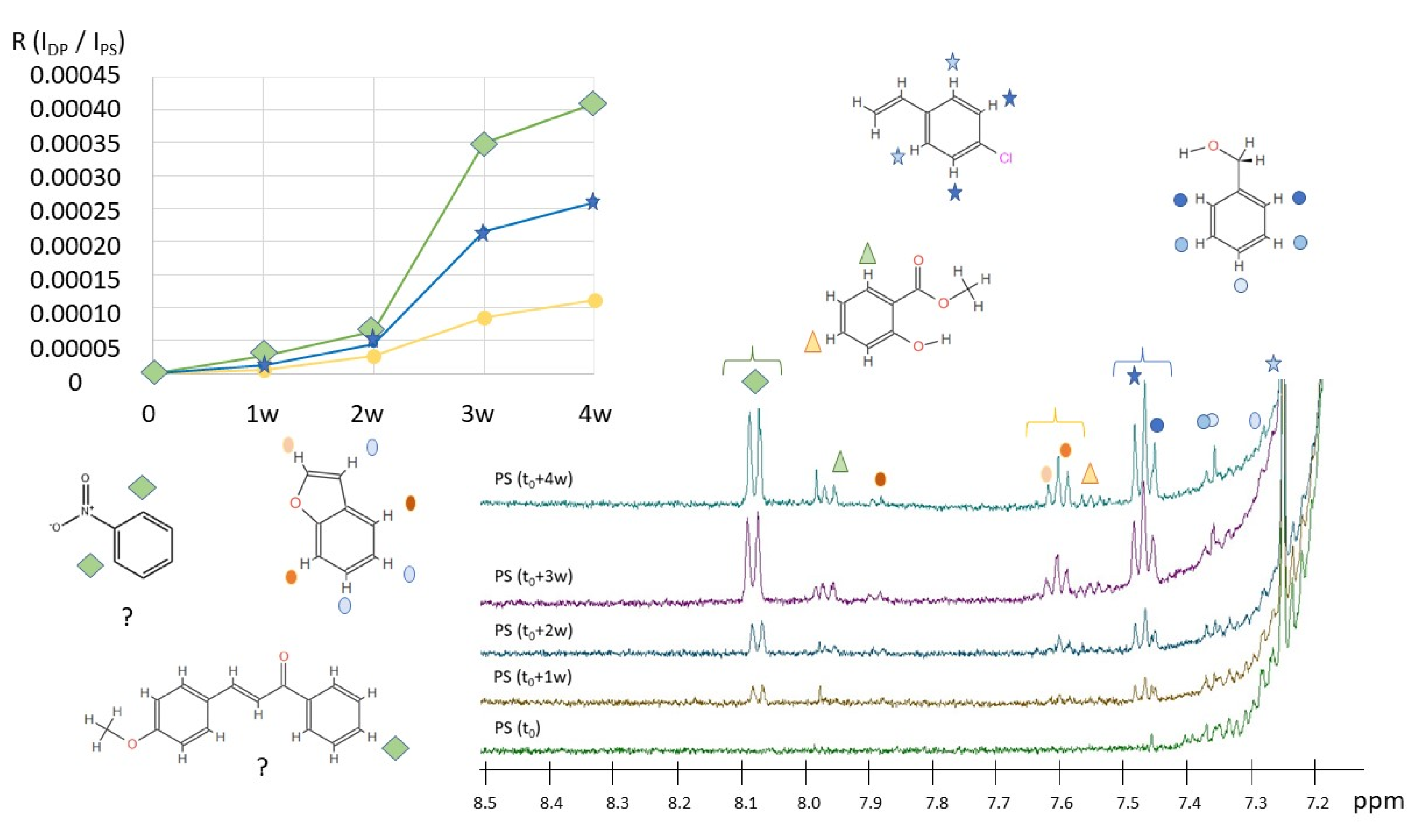
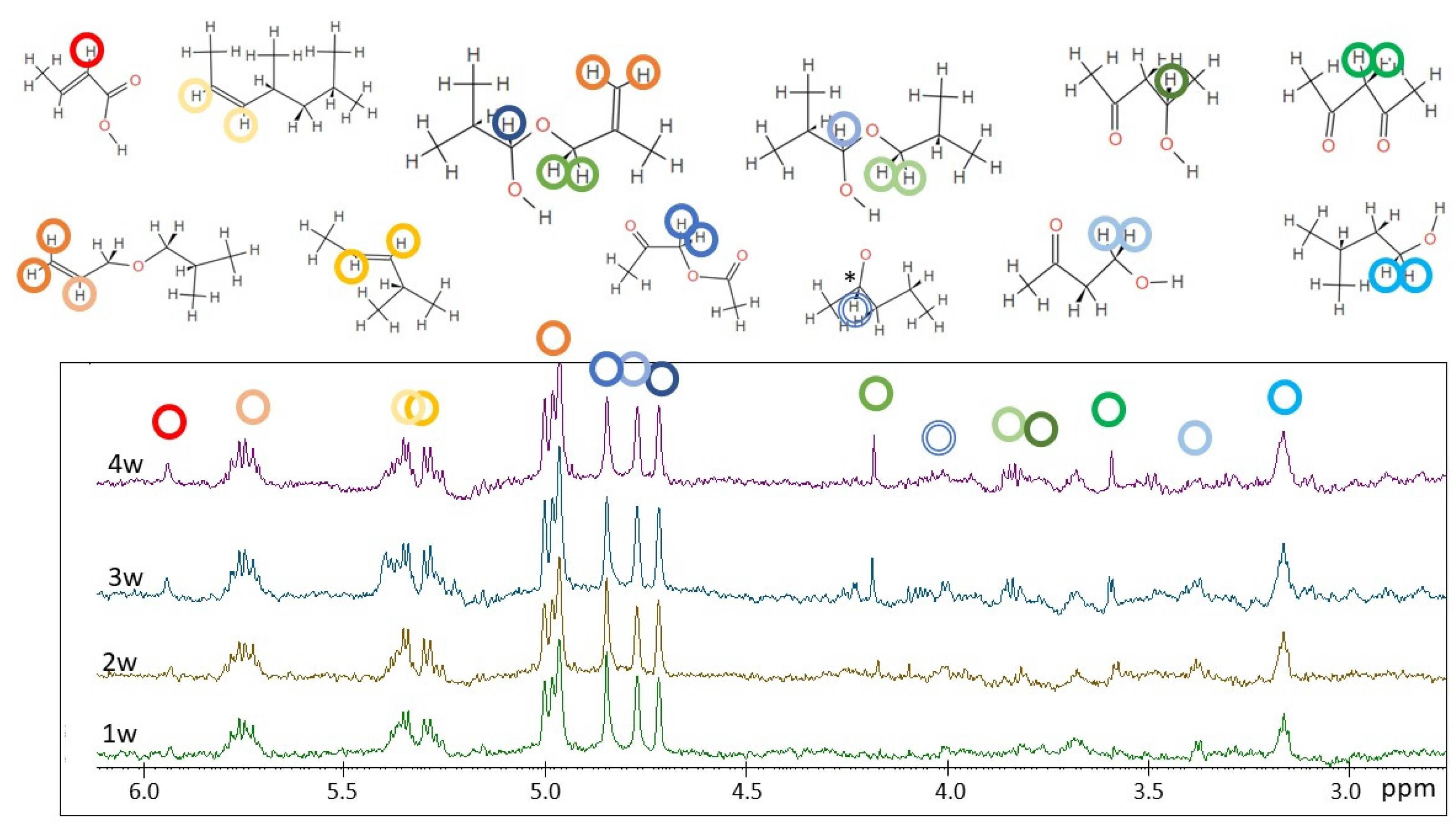
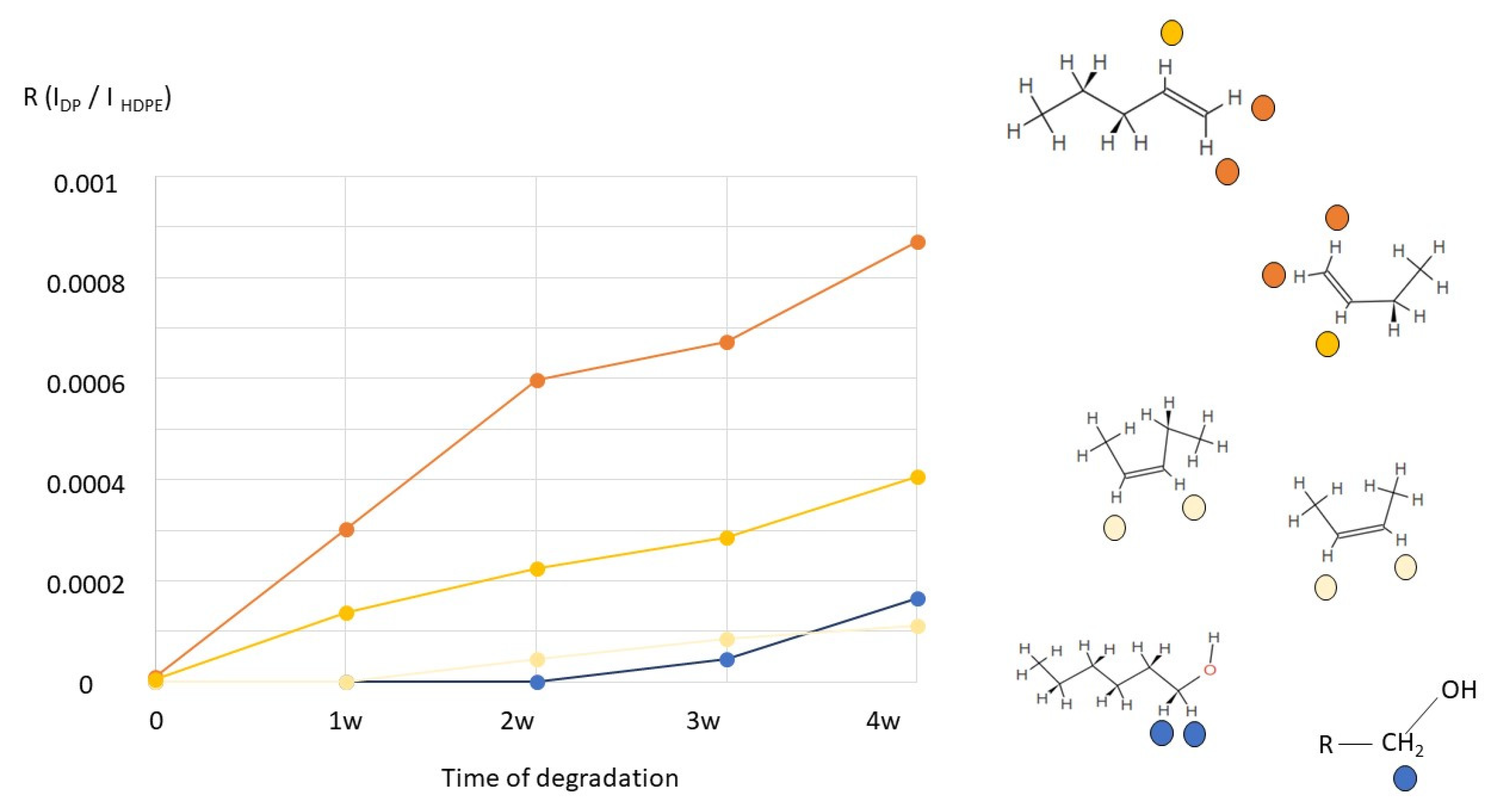
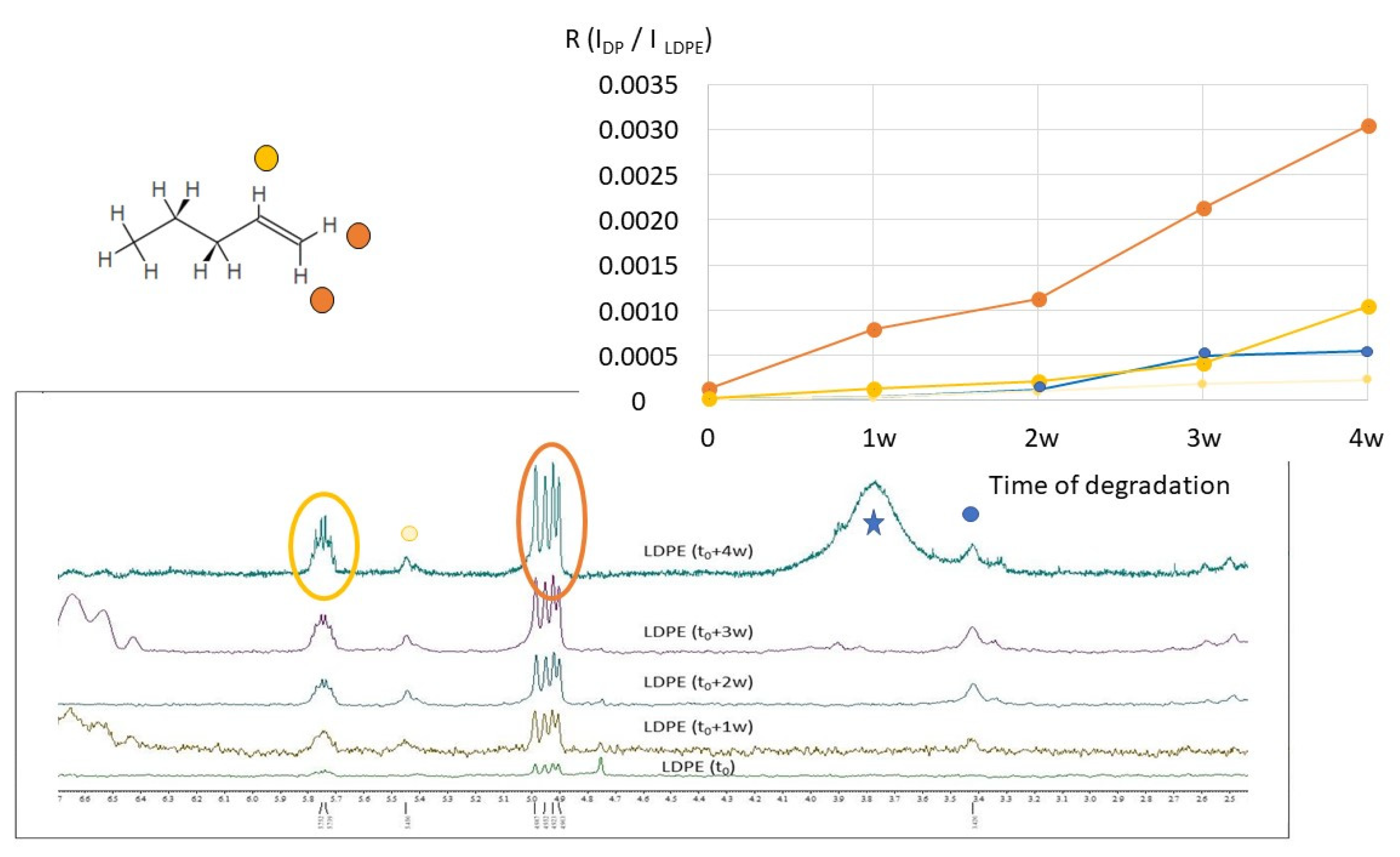
2.2. H NMR Spectroscopy of Polymer Microplastics during Degradation
3. Discussion
4. Materials and Methods
4.1. Chemicals and Materials
4.2. Samples and Artificial Aging Procedure
4.3. Samples Preparation for the NMR Analysis
4.4. Samples Preparation for the HiSorb-TD-GC-MS Analysis
4.5. Data Processing and Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Kundu, A.; Shetti, N.P.; Basu, S.; Reddy, K.R.; Nadagouda, M.N.; Aminabhavi, T.M. Identification and removal of micro- and nano-plastics: Efficient and cost-effective methods. Chem. Eng. J. 2021, 421, 129816. [Google Scholar]
- Ragusa, A.; Svelato, A.; Santacroce, C.; Catalano, P.; Notarstefano, V.; Carnevali, O.; Papa, F.; Rongioletti, M.C.A.; Baiocco, F.; Draghi, S.; et al. Plasticenta: First evidence of microplastics in human placenta. Environ. Int. 2021, 146, 106274. [Google Scholar]
- Leslie, H.A.; van Velzen, M.J.M.; Brandsma, S.H.; Vethaak, A.D.; Garcia-Vallejo, J.J.; Lamoree, M.H. Discovery and quantification of plastic particle pollution in human blood. Environ. Int. 2022, 163, 107199. [Google Scholar]
- Frias, J.P.G.L.; Nash, R. Microplastics: Finding a consensus on the definition. Mar. Pollut. Bull. 2019, 138, 145–147. [Google Scholar]
- Rocha-Santos, T.; Duarte, A.C. A critical overview of the analytical approaches to the occurrence, the fate and the behavior of microplastics in the environment. TrAC Trends Anal. Chem. 2015, 65, 47–53. [Google Scholar]
- Li, S.; Cai, M.; Liu, Y.; Wang, C.; Lv, K.; Chen, X. S-Scheme photocatalyst TaON/Bi2WO6 nanofibers with oxygen vacancies for efficient abatement of antibiotics and Cr(VI): Intermediate eco-toxicity analysis and mechanistic insights. Chin. J. Catal. 2022, 43, 2652–2664. [Google Scholar]
- Fu, Y.; Tan, M.; Guo, Z.; Hao, D.; Xu, Y.; Du, H.; Zhang, C.; Guo, J.; Li, Q.; Wang, Q. Fabrication of wide-spectra-responsive NA/NH2-MIL-125(Ti) with boosted activity for Cr(VI) reduction and antibacterial effects. Chem. Eng. J. 2023, 452, 139417. [Google Scholar]
- Xu, J.L.; Thomas, K.V.; Luo, Z.; Gowen, A.A. FTIR and Raman imaging for microplastics analysis: State of the art, challenges and prospects. TrAC Trends Anal. Chem. 2019, 119, 115629. [Google Scholar]
- Xue, Q.; Wang, N.; Lu, J.; Bai, H. Design of a confocal micro-Raman spectroscopy system and research on microplastics detection. Appl. Opt. 2021, 60, 8375–8383. [Google Scholar]
- La Nasa, J.; Biale, G.; Fabbri, D.; Modugno, F. A review on challenges and developments of analytical pyrolysis and other thermoanalytical techniques for the quali-quantitative determination of microplastics. J. Anal. Appl. Pyrolysis 2020, 149, 104841. [Google Scholar]
- Adhikari, S.; Kelkar, V.; Kumar, R.; Halden, R.U. Methods and challenges in the detection of microplastics and nanoplastics: A mini-review. Polym. Int. 2022, 71, 543–551. [Google Scholar]
- Castelvetro, V.; Corti, A.; Biale, G.; Ceccarini, A.; Degano, I.; La Nasa, J.; Lomonaco, T.; Manariti, A.; Manco, E.; Modugno, F.; et al. New methodologies for the detection, identification, and quantification of microplastics and their environmental degradation by-products. Environ. Sci. Pollut. Res. Int. 2021, 28, 46764. [Google Scholar]
- Lomonaco, T.; Manco, E.; Corti, A.; La Nasa, J.; Ghimenti, S.; Biagini, D.; Di Francesco, F.; Modugno, F.; Ceccarini, A.; Fuoco, R.; et al. Release of harmful volatile organic compounds (VOCs) from photo-degraded plastic debris: A neglected source of environmental pollution. J. Hazard. Mater. 2020, 394, 122596. [Google Scholar]
- La Nasa, J.; Lomonaco, T.; Manco, E.; Ceccarini, A.; Fuoco, R.; Corti, A.; Modugno, F.; Castelvetro, V.; Degano, I. Plastic breeze: Volatile organic compounds (VOCs) emitted by degrading macro- and microplastics analyzed by selected ion flow-tube mass spectrometry. Chemosphere 2021, 270, 128612. [Google Scholar]
- Saito, T.; Yamazaki, T.; Numata, M. Development of nuclear magnetic resonance as a tool of quantitative analysis for organic materials. Metrologia 2019, 56, 054002. [Google Scholar]
- Bharti, S.K.; Roy, R. Quantitative 1H NMR spectroscopy. TrAC Trends Anal. Chem. 2012, 35, 5–26. [Google Scholar]
- Simmler, C.; Napolitano, J.G.; McAlpine, J.B.; Chen, S.N.; Pauli, G.F. Universal quantitative NMR analysis of complex natural samples. Curr. Opin. Biotechnol. 2014, 25, 51–59. [Google Scholar]
- Choi, K.; Myoung, S.; Seo, Y.; Ahn, S. Quantitative NMR as a Versatile Tool for the Reference Material Preparation. Magnetochemistry 2021, 7, 15. [Google Scholar]
- Pauli, G.F.; Chen, S.N.; Simmler, C.; Lankin, D.C.; Gödecke, T.; Jaki, B.U.; Friesen, J.B.; McAlpine, J.B.; Napolitano, J.G. Importance of purity evaluation and the potential of quantitative 1H NMR as a purity assay. J. Med. Chem. 2014, 57, 9220–9231. [Google Scholar]
- Cullen, C.H.; Ray, G.J.; Szabo, C.M. A comparison of quantitative nuclear magnetic resonance methods: Internal, external, and electronic referencing. Magn. Reson. Chem. 2013, 51, 705–713. [Google Scholar]
- Peez, N.; Janiska, M.C.; Imhof, W. The first application of quantitative 1H NMR spectroscopy as a simple and fast method of identification and quantification of microplastic particles (PE, PET, and PS). Anal. Bioanal. Chem. 2019, 411, 823–833. [Google Scholar]
- Peez, N.; Imhof, W. Quantitative 1H-NMR spectroscopy as an efficient method for identification and quantification of PVC, ABS and PA microparticles. Analyst 2020, 145, 5363–5371. [Google Scholar]
- Peez, N.; Becker, J.; Ehlers, S.M.; Fritz, M.; Fischer, C.B.; Koop, J.H.E.; Winkelmann, C.; Imhof, W. Quantitative analysis of PET microplastics in environmental model samples using quantitative 1H-NMR spectroscopy: Validation of an optimized and consistent sample clean-up method. Anal. Bioanal. Chem. 2019, 411, 7409–7418. [Google Scholar]
- Corti, A.; Vinciguerra, V.; Iannilli, V.; Pietrelli, L.; Manariti, A.; Bianchi, S.; Petri, A.; Cifelli, M.; Domenici, V.; Castelvetro, V. Thorough Multianalytical Characterization and Quantification of Micro- and Nanoplastics from Bracciano Lake’s Sediments. Sustainability 2020, 12, 878. [Google Scholar]
- Peez, N.; Rinesch, T.; Kolz, J.; Imhof, W. Applicable and cost-efficient microplastic analysis by quantitative 1H-NMR spectroscopy using benchtop NMR and NoD methods. Magn. Reson. Chem. 2022, 60, 172–183. [Google Scholar]
- Mauel, A.; Pötzschner, B.; Meides, N.; Siegel, R.; Strohriegl, P.; Senker, J. Quantification of photooxidative defects in weathered microplastics using 13C multiCP NMR spectroscopy. RSC Adv. 2022, 12, 10875. [Google Scholar]
- Kumar, U.; Gaikwad, V.; Mayyas, M.; Bucknall, M.; Sahajwalla, V. Application of High-Resolution NMR and GC-MS to Study Hydrocarbon Oils Derived from Noncatalytic Thermal Transformation of e-Waste Plastics. ACS Omega 2018, 3, 9282–9289. [Google Scholar]
- Garcia-Torné, M.; Abad, E.; Almeida, D.; Llorca, M.; Farré, M. Assessment of Micro-and Nanoplastic Composition (Polymers and Additives) in the Gastrointestinal Tracts of Ebro River Fishes. Molecules 2023, 28, 239. [Google Scholar]
- Reza Bagheri, A.; Laforsch, C.; Greiner, A.; Agarwal, S.; Bagheri, A.R.; Greiner, A.; Agarwal, S.; Laforsch, C. Fate of So-Called Biodegradable Polymers in Seawater and Freshwater. Glob. Challenges 2017, 1, 1700048. [Google Scholar]
- Paço, A.; Duarte, K.; da Costa, J.P.; Santos, P.S.M.; Pereira, R.; Pereira, M.E.; Freitas, A.C.; Duarte, A.C.; Rocha-Santos, T.A.P. Biodegradation of polyethylene microplastics by the marine fungus Zalerion maritimum. Sci. Total Environ. 2017, 586, 10–15. [Google Scholar]
- Ceccarini, A.; Corti, A.; Erba, F.; Modugno, F.; La Nasa, J.; Bianchi, S.; Castelvetro, V. The Hidden Microplastics: New Insights and Figures from the Thorough Separation and Characterization of Microplastics and of Their Degradation Byproducts in Coastal Sediments. Environ. Sci. Technol. 2018, 52, 5634–5643. [Google Scholar]
- Strohriegl, P.; Senker, J.; Meides, N.; Menzel, T.; Poetzschner, B.; Löder, M.G.J.; Mansfeld, U.; Altstaedt, V. Reconstructing the environmental degradation of polystyrene by accelerated weathering. Environ. Sci. Technol. 2021, 55, 7930–7938. [Google Scholar]
- Busico, V.; Cipullo, R. Microstructure of polypropylene. Prog. Polym. Sci. 2001, 26, 443–533. [Google Scholar]
- Iedema, P.D.; Remerie, K.; Seegers, D.; McAuley, K.B. Tacticity Changes during Controlled Degradation of Polypropylene. Macromolecules 2021, 54, 8921–8935. [Google Scholar]
- Liu, P.; Qian, L.; Wang, H.; Zhan, X.; Lu, K.; Gu, C.; Gao, S. New Insights into the Aging Behavior of Microplastics Accelerated by Advanced Oxidation Processes. Environ. Sci. Technol. 2019, 53, 3579–3588. [Google Scholar]
- Chamas, A.; Moon, H.; Zheng, J.; Qiu, Y.; Tabassum, T.; Jang, J.H.; Abu-Omar, M.; Scott, S.L.; Suh, S. Degradation Rates of Plastics in the Environment. ACS Sustain. Chem. Eng. 2020, 8, 3494–3511. [Google Scholar]
- Zhang, K.; Hamidian, A.H.; Tubić, A.; Zhang, Y.; Fang, J.K.H.; Wu, C.; Lam, P.K.S. Understanding plastic degradation and microplastic formation in the environment: A review. Environ. Pollut. 2021, 274, 116554. [Google Scholar]
- Martínez De Ilarduya, A.; Muñoz-Guerra, S. Chemical Structure and Microstructure of Poly(alkylene terephthalate)s, their Copolyesters, and their Blends as Studied by NMR. Macromol. Chem. Phys. 2014, 215, 2138–2160. [Google Scholar]
- Brandolini, A.J.; Hills, D.D. NMR Spectra of Polymers and Polymer Additives. In NMR Spectra of Polymers and Polymer Additives; CRC Press: Boca Raton, FL, USA, 2000. [Google Scholar]
- Cheng, Z.; Mannion, D.T.; O’sullivan, M.G.; Miao, S.; Kerry, J.P.; Kilcawley, K.N. Comparison of Automated Extraction Techniques for Volatile Analysis of Whole Milk Powder. Foods 2021, 10, 2061. [Google Scholar]
- Jabarin, S.A.; Lofgren, E.A. Thermal stability of polyethylene terephthalate. Polym. Eng. Sci. 1984, 24, 1056–1063. [Google Scholar]
- Takac, M.J.-M.; Topic, V.D. FT-IR and NMR spectroscopic studies of salicylic acid derivatives. II. Comparison of 2-hydroxy-and 2,4-and 2,5-dihydroxy derivatives. Acta Pharm. 2004, 54, 177–191. [Google Scholar]
- Gehman, C.A.; Hantz, E.R.; Hamann, C.S. Distinguishing Vinylic and Aromatic 1H NMR Signals Using Selectively Deuterated Chalcones. J. Chem. Educ. 2021, 98, 2323–2332. [Google Scholar]
- Mc Naught, A.D.; Wilkinson, A. Compendium of Chemical Terminology. In The Gold Book; Iupac: Research Triangle Park, NC, USA, 2012; p. 1670. [Google Scholar]
- Lomonaco, T.; Salvo, P.; Ghimenti, S.; Biagini, D.; Vivaldi, F.; Bonini, A.; Fuoco, R.; Di Francesco, F. Stability of volatile organic compounds in sorbent tubes following SARS-CoV-2 inactivation procedures. J. Breath Res. 2021, 15, 037102. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plastic Material | Density (g/cm3) | Average Dimension of Plastic Samples (μm) |
|---|---|---|
| Low density polyethylene (LDPE) | 0.917 | 632 |
| High density polyethylene (HDPE) | 0.952 | 622 |
| Polyethylene terephthalate (PET) (About 5% crystallinity as provided) | 1.28 | 509 |
| Polypropylene (PP) | 0.900 | 857 |
| Polystyrene (PS) | 1.05 | 564 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giaganini, G.; Cifelli, M.; Biagini, D.; Ghimenti, S.; Corti, A.; Castelvetro, V.; Domenici, V.; Lomonaco, T. Multi-Analytical Approach to Characterize the Degradation of Different Types of Microplastics: Identification and Quantification of Released Organic Compounds. Molecules 2023, 28, 1382. https://doi.org/10.3390/molecules28031382
Giaganini G, Cifelli M, Biagini D, Ghimenti S, Corti A, Castelvetro V, Domenici V, Lomonaco T. Multi-Analytical Approach to Characterize the Degradation of Different Types of Microplastics: Identification and Quantification of Released Organic Compounds. Molecules. 2023; 28(3):1382. https://doi.org/10.3390/molecules28031382
Chicago/Turabian StyleGiaganini, Giulia, Mario Cifelli, Denise Biagini, Silvia Ghimenti, Andrea Corti, Valter Castelvetro, Valentina Domenici, and Tommaso Lomonaco. 2023. "Multi-Analytical Approach to Characterize the Degradation of Different Types of Microplastics: Identification and Quantification of Released Organic Compounds" Molecules 28, no. 3: 1382. https://doi.org/10.3390/molecules28031382
APA StyleGiaganini, G., Cifelli, M., Biagini, D., Ghimenti, S., Corti, A., Castelvetro, V., Domenici, V., & Lomonaco, T. (2023). Multi-Analytical Approach to Characterize the Degradation of Different Types of Microplastics: Identification and Quantification of Released Organic Compounds. Molecules, 28(3), 1382. https://doi.org/10.3390/molecules28031382