
Synthesis and Antiproliferative Activity of Steroidal Diaryl Ethers
 , , and
, , and
Abstract
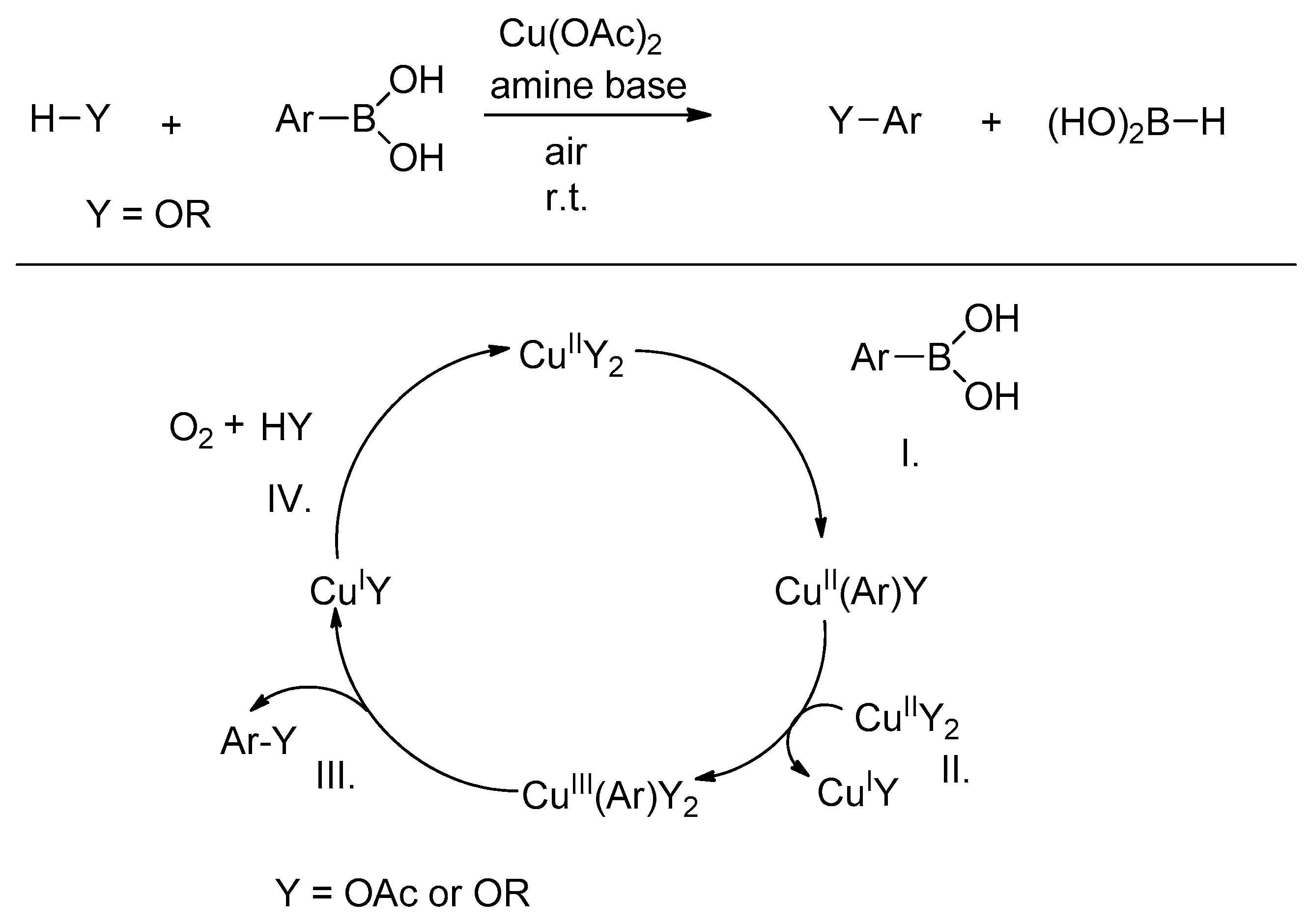
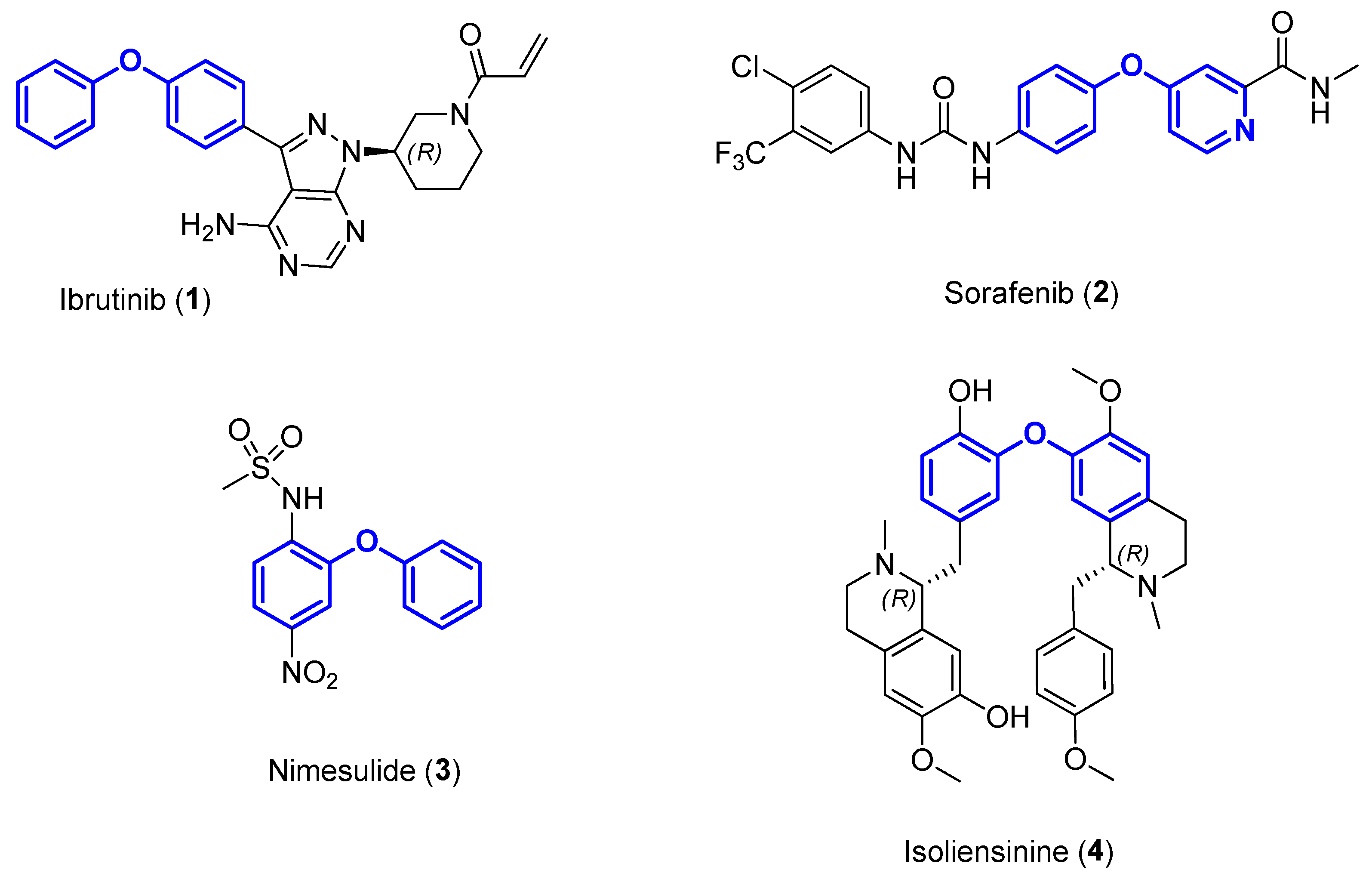
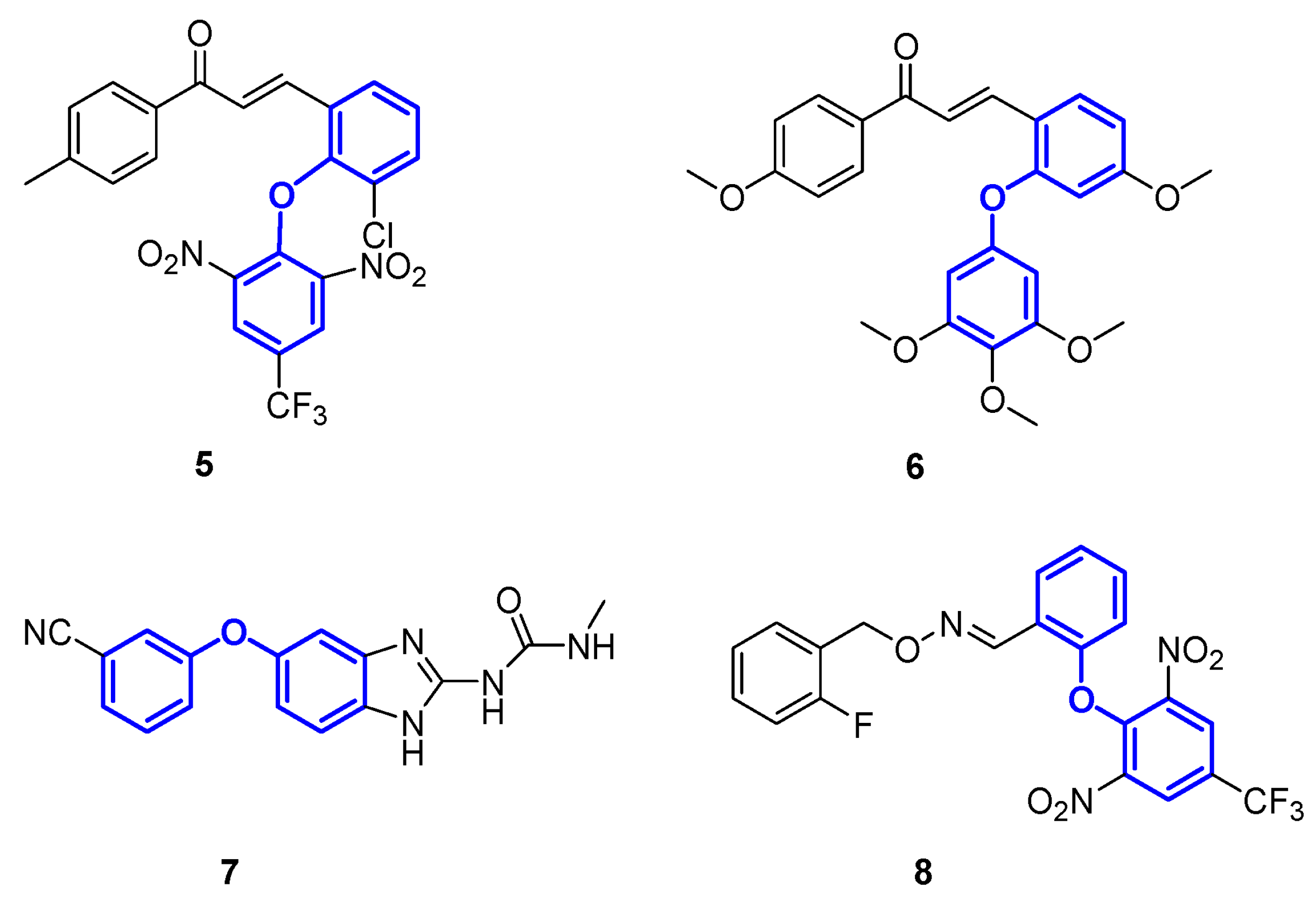
1. Introduction
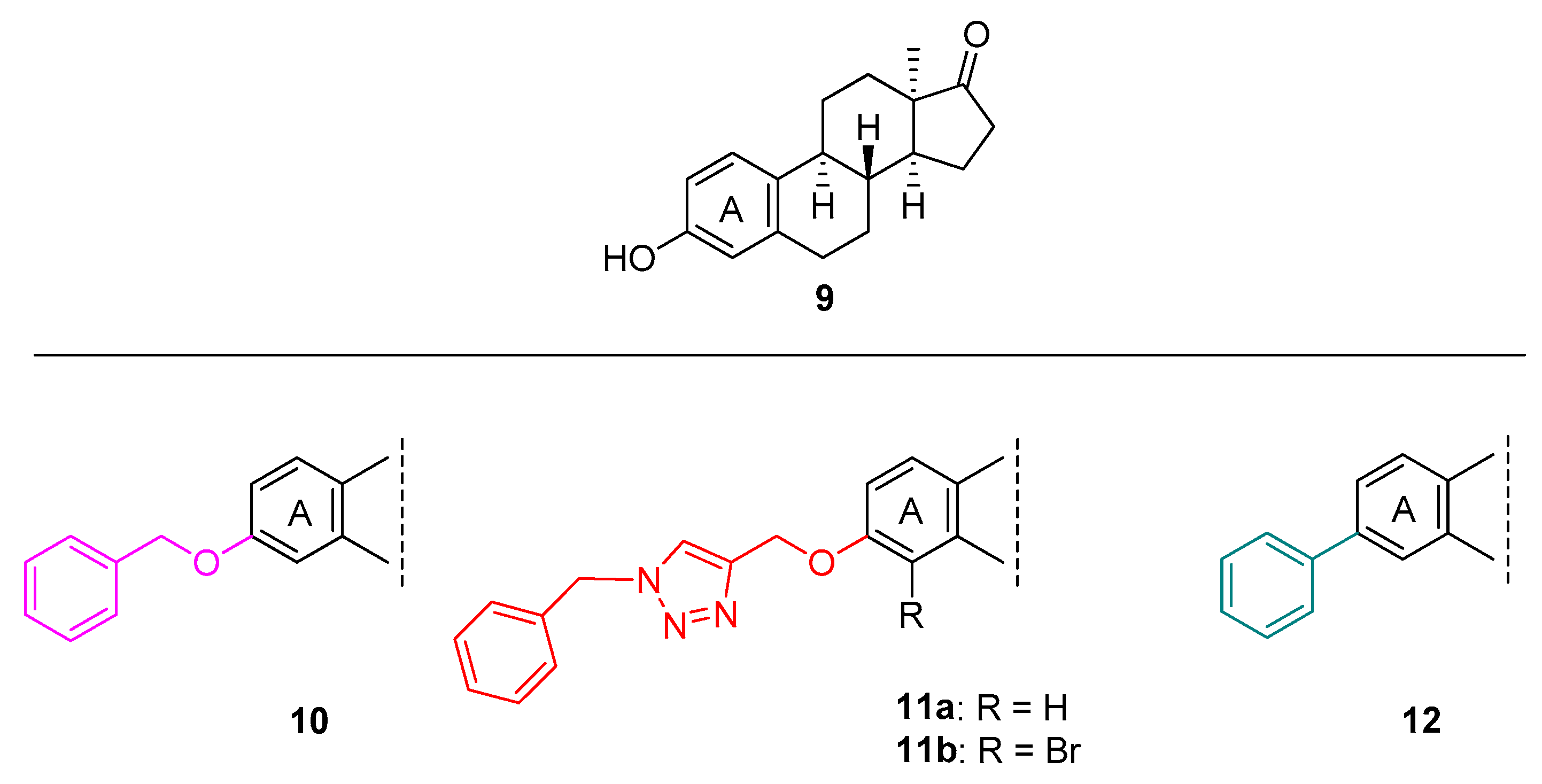
2. Results and Discussion
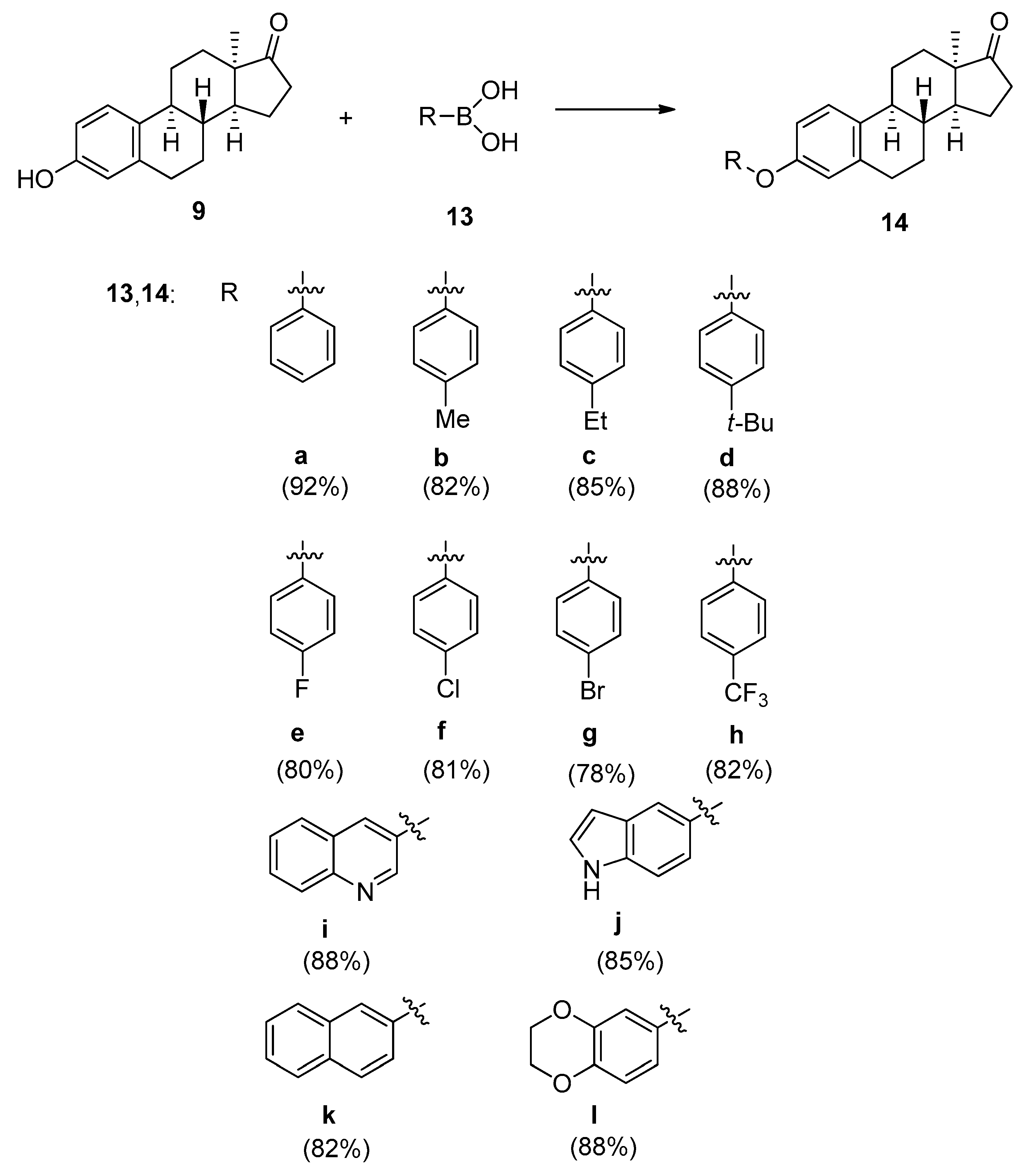
2.1. Chemistry
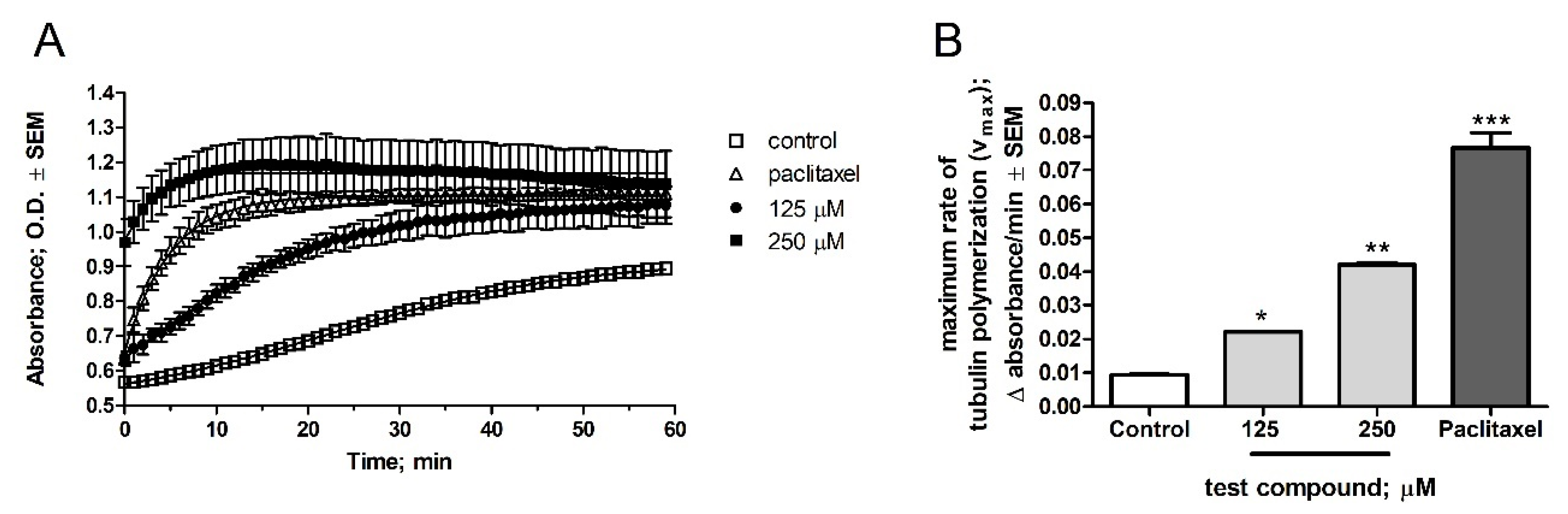
2.2. Pharmacology
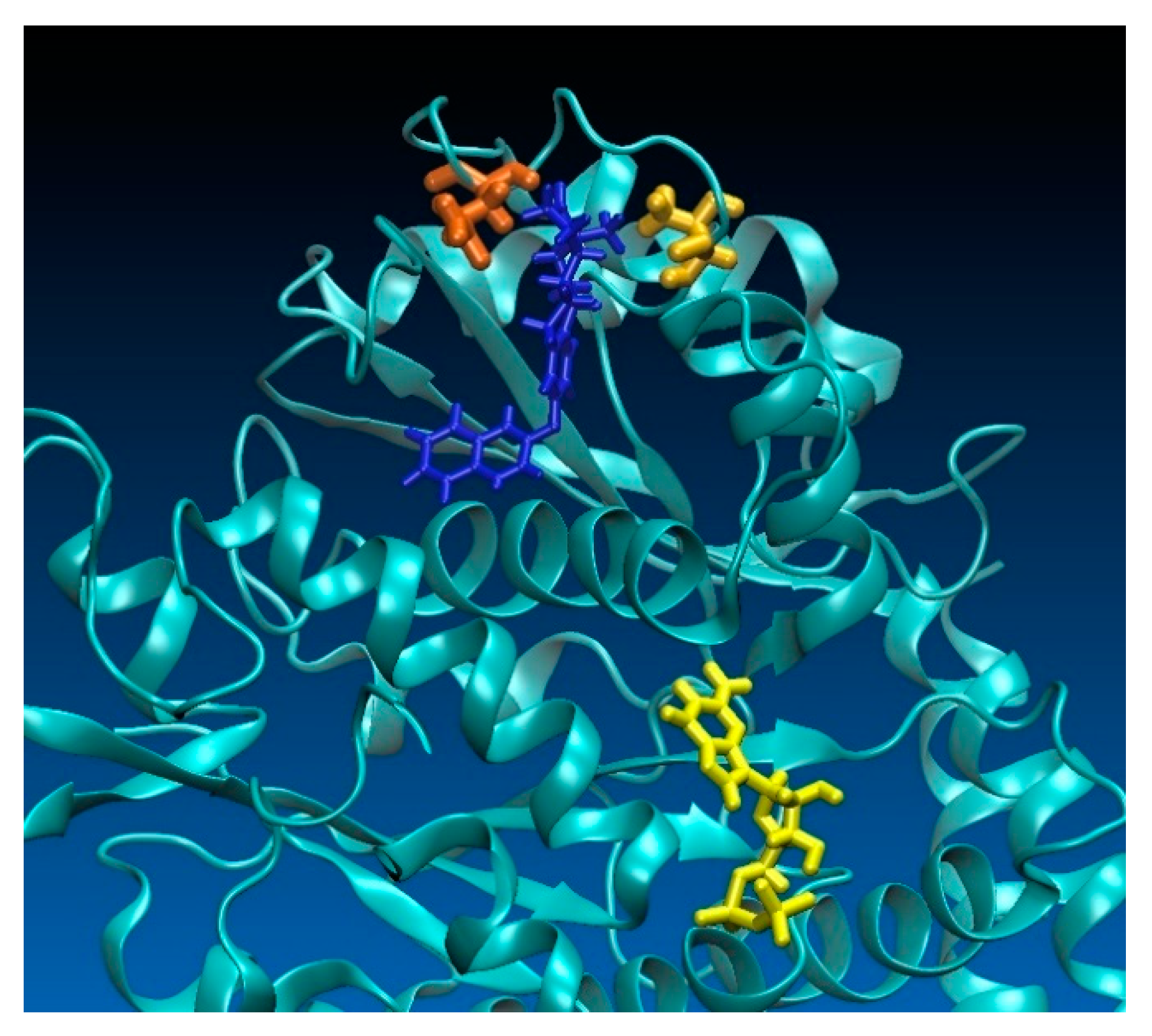
2.3. Computational Investigations
3. Materials and Methods
3.1. Chemistry
3.2. General Procedure for the Synthesis of 3-aryloxy-13α-estra-1,3,5(10)-triene-17-ones
4. Determination of Antiproliferative Activities
5. Tubulin Polymerization Assay
6. Computational Simulations
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Chen, J.Q.; Li, J.H.; Dong, Z.B. A Review on the Latest Progress of Chan-Lam Coupling Reaction. Adv. Synth. Catal. 2020, 362, 3311–3331. [Google Scholar] [CrossRef]
- Pal, T.; Lahiri, G.K.; Maiti, D. Copper in Efficient Synthesis of Aromatic Heterocycles with Single heteroatom. Eur. J. Org. Chem. 2020, 44, 6859–6869. [Google Scholar] [CrossRef]
- De Nino, A.; Maiuolo, L.; Costanzo, P.; Algieri, V.; Jiritano, A.; Olivito, F.; Tallarida, M.A. Recent Progress in Catalytic Synthesis of 1,2,3-Triazoles. Catalysts 2021, 11, 1120. [Google Scholar] [CrossRef]
- Yadav, P.; Bhalla, A. Recent Advances in Green Synthesis of Functionalized Quinolines of Medicinal Impact (2018–Present). ChemistrySelect 2022, 7, e202201721. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, H.; Shi, W.; Lei, A.W. Bond Formations between Two Nucleophiles: Transition Metal Catalyzed Oxidative Cross-Coupling Reactions. Chem. Rev. 2011, 111, 1780–1824. [Google Scholar] [CrossRef]
- Mousseau, J.J.; Charette, A.B. Direct Functionalization Processes: A Journey from Palladium to Copper to Iron to Nickel to Metal-Free Coupling Reactions. Acc. Chem. Res. 2013, 46, 412–424. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, X.; Macmillan, D.W.C. Decarboxylative sp3 C–N coupling via dual copper and photoredox catalysis. Nature 2018, 559, 83–88. [Google Scholar] [CrossRef]
- Kong, D.; Moon, P.J.; Bsharat, O.; Lundgren, R.J. Direct Catalytic Decarboxylative Amination of Aryl Acetic Acids. Angew. Chem. Int. Ed. 2020, 59, 1313–1319. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, K.; Zhen, H.Y.; Ding, R.C.; Yin, F.J.; Wang, N.; Li, Y.; Cheng, B.; Wang, H.F.; Zhai, H.B. A One-Pot Synthesis of Dibenzofurans from 6-Diazo-2-cyclohexenones. Org. Lett. 2015, 17, 5744–5747. [Google Scholar] [CrossRef]
- Zhang, C.; Shi, Y.L.; Zhang, L.Y.; Yuan, D.P.; Ban, M.T.; Zheng, J.Y.; Liu, D.H.; Guo, S.N.; Cui, D.M. NaOH-promoted reaction of 1;1-dihaloalkenes and 1H-azoles: Synthesis of dihetaryl substituted alkenes. New J. Chem. 2018, 42, 17732–17739. [Google Scholar] [CrossRef]
- Chan, D.M.T.; Monaco, K.L.; Wang, R.P.; Winters, M.P. New N- and O-arylations with phenylboronic acids and cupric acetate. Tetrahedron Lett. 1998, 39, 2933–2936. [Google Scholar] [CrossRef]
- Evans, D.A.; Katz, J.L.; West, T.R. Synthesis of diaryl ethers through the copper-promoted arylation of phenols with arylboronic acids. An expedient synthesis of thyroxine. Tetrahedron Lett. 1998, 39, 2937–2940. [Google Scholar] [CrossRef]
- Doyle, M.G.; Lundgren, R.J. Oxidative cross-coupling processes inspired by the Chan-Lam reaction. ChemCommun 2021, 57, 2724. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Xiong, H.; Yang, J.F.; Zhu, X.L.; Qu, R.Y.; Yang, G.F. Diaryl Ether: A Privileged Scaffold for Drug and Agrochemical Discovery. J. Agric. Food Chem. 2020, 68, 9839–9877. [Google Scholar] [CrossRef]
- Bedos-Belval, F.; Rouch, A.; Vanucci-Bacque, C.; Baltas, M. Diaryl ether derivatives as anticancer agents—A review. MedChemComm 2012, 3, 1356–1372. [Google Scholar] [CrossRef]
- Pitsinos, E.N.; Vidali, V.P.; Couladouros, E.A. Diaryl ether formation in the synthesis of natural products. Eur. J. Org. Chem. 2011, 2011, 1207–1222. [Google Scholar] [CrossRef]
- Pan, Z.Y.; Scheerens, H.; Li, S.J.; Schultz, B.E.; Sprengeler, P.A.; Burrill, L.C.; Mendonca, R.V.; Sweeney, M.D.; Scott, K.C.K.; Grothaus, P.G.; et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem 2007, 2, 58–61. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Gupta, N.; Wish, J.B. Hypoxia-inducible factor prolyl hydroxylase inhibitors: A potential new treatment for anemia in patients with CKD. Am. J. Kidney Dis. 2017, 69, 815–826. [Google Scholar] [CrossRef]
- Rainsford, K.D. Nimesulide—A multifactorial approach to inflammation and pain: Scientific and clinical consensus. Curr. Med. Res. Opin. 2006, 22, 1161–1170. [Google Scholar] [CrossRef]
- Shu, G.W.; Yue, L.; Zhao, W.H.; Xu, C.; Yang, J.; Wang, S.B.; Yang, X.Z. Isoliensinine; a bioactive alkaloid derived from embryos of Nelumbo nucifera; induces hepatocellular carcinoma cell apoptosis through suppression of NF-κB signaling. J. Agric. Food Chem. 2015, 63, 8793–8803. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, R.; Che, X.F.; Yamaguchi, T.; Ushiyama, M.; Zheng, C.L.; Okumura, H.; Takeda, Y.; Shibayama, Y.; Nakamura, K.; Jeung, H.C.; et al. Cepharanthine potently enhances the sensitivity of anticancer agents in K562 cells. Cancer Sci. 2005, 96, 372–376. [Google Scholar] [CrossRef]
- Meng, Z.P.; Li, T.; Ma, X.X.; Wang, X.Q.; Van Ness, C.; Gan, Y.C.; Zhou, H.; Tang, J.F.; Lou, G.Y.; Wang, Y.F.; et al. Berbamine inhibits the growth of liver cancer cells and cancer-initiating cells by targeting Ca2+/calmodulin-dependent protein kinase II. Mol. Cancer Ther. 2013, 12, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- da Silva, A.; Maciel, D.; Freitas, V.P.; Conserva, G.A.A.; Alexandre, T.R.; Purisco, S.U.; Tempone, A.G.; Melhem, M.S.C.; Kato, M.J.; Guimaraes, E.F.; et al. Bioactivity-guided isolation of laevicarpin; an antitrypanosomal and anticryptococcal lactam from Piper laevicarpu (Piperaceae). Fitoterapia 2016, 111, 24–28. [Google Scholar] [CrossRef]
- Hucke, O.; Coulombe, R.; Bonneau, P.; Bertrand-Laperle, M.; Brochu, C.; Gillard, J.; Joly, M.A.; Landry, S.; Lepage, O.; Llinas-Brunet, M.; et al. Molecular dynamics simulations and structure-based rational design lead to allosteric HCV NS5B polymerase thumb pocket 2 inhibitor with picomolar cellular replicon potency. J. Med. Chem. 2014, 57, 1932–1943. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, P.L.; Coulombe, R.; Duan, J.M.; Fazal, G.; Godbout, C.; Hucke, O.; Jakalian, A.; Joly, M.A.; Lepage, O.; Llinas-Brunet, M.; et al. Structure-based design of novel HCV NS5B thumb pocket 2 allosteric inhibitors with submicromolar gt1 replicon potency: Discovery of a quinazolinone chemotype. Bioorg. Med. Chem. Lett. 2013, 23, 4132–4140. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.H.; Wang, Z.L.; Yang, J.Z.; Yang, T.; Pi, W.Y.; Ang, W.; Lin, Y.N.; Liu, Y.Y.; Li, Z.C.; Luo, Y.F.; et al. Design, synthesis and evaluation of novel molecules with a diphenyl ether nucleus as potential antitubercular agents. Bioorg. Med. Chem. Lett. 2012, 22, 954–957. [Google Scholar] [CrossRef]
- Phainuphong, P.; Rukachaisirikul, V.; Phongpaichit, S.; Preedanon, S.; Sakayaroj, J. Diphenyl ethers and indanones from the soil-derived fungus Aspergillus unguis PSU-RSPG204. Tetrahedron 2017, 73, 5920–5925. [Google Scholar] [CrossRef]
- Luemmen, P.; Kunz, K.; Greul, J.; Guth, O.; Hartmann, B.; Ilg, K.; Moradi, W.A.; Seitz, T.; Mansfield, D.; Vors, J.P.; et al. Pesticide Phenyloxy Substituted Phenylamidine Derivatives. U.S. Patent Application 8,183,296 B2, 22 May 2012. [Google Scholar]
- HRAC (Herbicide Resistance Action Committee). Available online: http://www.hracglobal.com (accessed on 17 May 2020).
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.; Eastman, A. Microtubule destabilising agents: Far more than just antimitotic anticancer drugs. Br. J. Clin. Pharmacol. 2017, 83, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Naaz, F.; Haider, M.R.; Shafi, S.; Yar, M.S. Anti-tubulin agents of natural origin: Targeting taxol; vinca; and colchicine binding domains. Eur. J. Med. Chem. 2019, 171, 310–331. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.N.; Zheng, L.L.; Wang, D.; Liang, X.X.; Gao, F.; Zhou, X.L. Recent advances in microtubule-stabilizing agents. Eur. J. Med. Chem. 2018, 143, 806. [Google Scholar] [CrossRef] [PubMed]
- Field, J.J.; Dıaz, J.F.; Miller, J.H. The binding sites of microtubule stabilizing agents. Chem. Biol. 2013, 20, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sun, H.; Xu, S.; Zhu, Z.; Xu, J. Tubulin inhibitors targeting the colchicine binding site: A perspective of privileged structures. Future Med. Chem. 2017, 9, 1765–1794. [Google Scholar] [CrossRef]
- Beale, T.M.; Allwood, D.M.; Bender, A.; Bond, P.J.; Brenton, J.D.; Charnock-Jones, D.S.; Ley, S.V.; Myers, R.M.; Shearman, J.W.; Temple, J.; et al. A-ring dihalogenation increases the cellular activity of combretastatin templated tetrazoles. ACS Med. Chem. Lett. 2012, 3, 177–181. [Google Scholar] [CrossRef]
- Butenandt, A.; Wolff, A.; Karlson, P. Uber Lumi-oestron. Chem. Ber. 1941, 74, 1308–1312. [Google Scholar] [CrossRef]
- Yaremenko, F.G.; Khvat, A.V. A new one-pot synthesis of 17-oxo-13α-steroids of the androstane series from their 13β-analogues. Mendeleev Commun. 1994, 187, 187–188. [Google Scholar] [CrossRef]
- Ayan, D.; Roy, J.; Maltais, R.; Poirier, D. Impact of estradiol structural modifications (18-methyl and/or 17-hydroxy inversion of configuration) on the in vitro and in vivo estrogenic activity. J. Steroid Biochem. Mol. Biol. 2011, 127, 324–330. [Google Scholar] [CrossRef]
- Schonecker, B.; Lange, C.; Kotteritzsch, M.; Gunther, W.; Weston, J.; Anders, E.; Gorls, H. Conformational design for 13α-steroids. J. Org. Chem. 2000, 65, 5487–5497. [Google Scholar] [CrossRef] [PubMed]
- Szabo, J.; Pataki, Z.; Wolfling, J.; Schneider, G.; Bózsity, N.; Minorics, R.; Zupkó, I.; Mernyák, E. Synthesis and biological evaluation of 13a-estrone derivatives as potential antiproliferative agents. Steroids 2016, 113, 14–21. [Google Scholar] [CrossRef]
- Bacsa, I.; Herman, B.E.; Jojart, R.; Herman, K.S.; Wölfling, J.; Schneider, G.; Varga, M.; Tömböly, C.; Rižner, T.L.; Szécsi, M.; et al. Synthesis and structure–activity relationships of 2- and/or 4-halogenated 13β- and 13α-estrone derivatives as enzyme inhibitors of estrogen biosynthesis. J. Enzym. Inhib. Med. Chem. 2018, 33, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Jojart, R.; Pecsy, S.; Keglevich, G.; Szécsi, M.; Rigó, R.; Özvegy-Laczka, C.; Kecskeméti, G.; Mernyák, E. Pd-catalyzed microwave-assisted synthesis of phosphonated 13α-estrones as potential OATP2B1; 17β-HSD1 and/or STS inhibitors. Beilstein J. Org. Chem. 2018, 14, 2838–2845. [Google Scholar] [CrossRef] [PubMed]
- Sinreih, M.; Jójárt, R.; Kele, Z.; Büdefeld, T.; Paragi, G.; Mernyák, E.; Rižner, T.L. Synthesis and evaluation of AKR1C inhibitory properties of A-ring halogenated oestrone derivatives. J. Enzym. Inhib. Med. Chem. 2021, 36, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Jójárt, R.; Laczkó-Rigó, R.; Klement, M.; Kőhl, G.; Kecskeméti, G.; Özvegy-Laczka, C.; Mernyák, E. Design, synthesis and biological evaluation of novel estrone phosphonates as high affinity organic anion-transporting polypeptide 2B1 (OATP2B1) inhibitors. Bioorg. Chem. 2021, 112, 104914–104925. [Google Scholar] [CrossRef]
- Traj, P.; Abdolkhaliq, A.H.; Németh, A.; Dajcs, S.T.; Tömösi, F.; Lanisnik-Rizner, T.; Zupkó, I.; Mernyák, E. Transition metal-catalysed A-ring C-H activations and C(sp2)-C(sp2) couplings in the 13α-oestrone series and in vitro evaluation of antiproliferative properties. J. Enzym. Inhib. Med. Chem. 2021, 36, 895–902. [Google Scholar] [CrossRef]
- Jójárt, R.; Tahaei, S.A.S.; Trungel-Nagy, P.; Kele, Z.; Minorics, R.; Paragi, G.; Zupkó, I.; Mernyák, E. Synthesis and evaluation of anticancer activities of 2- or 4-substituted 3-(N-benzyltriazolylmethyl)-13α-oestrone derivatives. J. Enzym. Inhib. Med. Chem. 2021, 36, 58–67. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Lee, Y.K.; Lim, J.; Yoon, S.Y.; Joo, J.C.; Park, S.J.; Park, Y.J. Promotion of cell death in cisplatin-resistant ovarian cancer cells through KDM1BDCLRE1B modulation. Int. J. Mol. Sci. 2019, 20, 2443. [Google Scholar] [CrossRef]
- Schelz, Z.; Ocsovszki, I.; Bozsity, N.; Hohmann, J.; Zupkó, I. Antiproliferative effects of various furanoacridones isolated from Ruta graveolens on human breast cancer cell lines. Anticancer Res. 2016, 36, 2751–2758. [Google Scholar]
- Chavez, K.J.; Garimella, S.V.; Lipkowitz, S. Triple negative breast cancer cell lines: One tool in the search for better treatment of triple negative breast cancer. Breast Dis. 2010, 32, 35–48. [Google Scholar] [CrossRef]
- Anders, C.K.; Zagar, T.M.; Carey, L.A. The management of earlystage and metastatic triple-negative breast cancer: A review. Hematol. Oncol. Clin. N. Am. 2013, 27, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Suba, Z. Triple-negative breast cancer risk in women is defined by the defect of estrogen signaling: Preventive and therapeutic implications. Onco Targets Ther. 2014, 7, 147–164. [Google Scholar] [CrossRef]
- Goodman, A. HPV testing as a screen for cervical cancer. BMJ 2015, 350, h2372. [Google Scholar] [CrossRef] [PubMed]
- Ghittoni, R.; Accardi, R.; Chiocca, S.; Tommasino, M. Role of human papillomaviruses in carcinogenesis. Ecancermedicalscience 2015, 9, 526. [Google Scholar] [CrossRef] [PubMed]
- Kellogg, E.H.; Hejab, N.M.; Howes, S.; Northcote, P.; Miller, J.H.; Diaz, J.F.; Downing, K.H.; Nogales, E. Insights into the Distinct Mechanisms of Action of Taxane and Non-Taxane Microtubule Stabilizers from Cryo-EM Structures. J. Mol. Biol. 2017, 429, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2021-3: Protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY, USA, 2021; Impact, Schrödinger, LLC, New York, NY; Prime, Schrödinger, LLC, New York, NY, USA, 2021.
- Maestro Schrödinger Release 2021-3: Maestro, Schrödinger, LLC, New York, NY, USA, 2021.
- Schrödinger Release 2021-3: Glide, Schrödinger, LLC, New York, NY, USA, 2021.
- Schrödinger Release 2021-3: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, USA, 2021. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, USA, 2021.
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.; Dahlgren, M.; Russell, E.; Von Bargen, C.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antiproliferative Activities of Newly Synthesized Diaryl Ethers (14a–l) | |||||||
|---|---|---|---|---|---|---|---|
| Compd. | Conc. (μM) | Inhibition (%) ± SEM (Calculated IC50 Value; μM) | |||||
| MCF-7 | MDA-MB-231 | HeLa | SiHa | A2780 | NIH/3T3 | ||
| 14a | 10 | 37.52 ± 2.46 | – * | 60.24 ± 0.58 | – | 23.00 ± 1.32 | - |
| 30 | 81.42 ± 1.50 (10.12) | 25.30 ± 1.82 | 92.02 ± 1.38 (5.53) | – | 58.48 ± 1.03 (23.81) | 38.75 ± 1.21 | |
| 14b | 10 | – | – | 56.69 ± 1.29 | – | 39.64 ± 1.84 | – |
| 30 | 29.28 ± 1.26 | 33.34 ± 1.75 | 83.29 ± 1.34 (7.99) | – | 62.15 ± 1.15 (16.67) | 43.34 ± 2.17 | |
| 14c | 10 | 43.99 ± 1.77 | – | 62.38 ± 1.19 | 27.24 ± 2.60 | – | – |
| 30 | 58.64 ± 1.08 | 33.16 ± 0.95 | 88.14 ± 0.79 | 43.55 ± 0.86 | 45.08 ± 1.75 | 21.99 ± 2.03 | |
| (16.44) | (5.13) | ||||||
| 14d | 10 | 24.75 ± 0.74 | - | 65.28 ± 1.02 | - | 30.84 ± 2.24 | – |
| 30 | 30.77 ± 1.67 | 32.93 ± 1.88 | 77.06 ± 0.93 (7.11) | 41.14 ± 2.38 | 56.98 ± 1.07 (23.65) | 24.07 ± 1.67 | |
| 14e | 10 | 49.02 ± 0.85 | – | 30.14 ± 3.05 | – | – | – |
| 30 | 60.22 ± 1.68 (13.28) | – | 37.78 ± 3.86 | – | 46.50 ± 2.43 | – | |
| 14f | 10 | - | - | 60.72 ± 1.24 | – | 34.93 ± 2.85 | – |
| 30 | 37.51 ± 1.44 | 30.28 ± 2.24 | 75.40 ± 1.42 (5.78) | 38.91 ± 1.69 | 51.48 ± 1.93 (26.17) | – | |
| 14g | 10 | 48.82 ± 2.18 | – | 65.97 ± 0.61 | 20.31 ± 3.10 | – | – |
| 30 | 59.91 ± 1.73 (11.98) | 28.28 ± 1.39 | 80.70 ± 0.76 (5.21) | 38.42 ± 1.30 | 39.58 ± 2.60 | 30.92 ± 2.15 | |
| 14h | 10 | 35.08 ± 0.69 | 27.06 ± 2.93 | 67.46 ± 0.46 | 31.80 ± 2.77 | 36.13 ± 1.39 | – |
| 30 | 45.42 ± 3.30 | 39.67 ± 2.39 | 80.74 ± 0.58 (6.90) | 45.55 ± 2.63 | 52.60 ± 1.00 (23.47) | 45.47 ± 0.28 | |
| 14i | 10 | 49.68 ± 2.46 | 24.07 ± 2.47 | 76.18 ± 1.49 | 22.46 ± 2.36 | 48.66 ± 1.90 | 35.48 ± 1.33 |
| 30 | 74.67 ± 0.51 (8.52) | 58.28 ± 1.39 (22.95) | 90.11 ± 0.80 (3.98) | 48.33 ± 1.27 | 71.21 ± 1.35 (11.54) | 49.87 ± 0.89 | |
| 14j | 10 | – | – | 50.99 ± 2.21 | – | – | – |
| 30 | 42.25 ± 1.33 | – | 71.63 ± 1.45 (9.60) | – | 28.02 ± 3.57 | – | |
| 14k | 10 | – | 29.61 ± 3.01 | 73.86 ± 1.48 | 28.80 ± 2.10 | 42.23 ± 0.71 | – |
| 30 | 43.11 ± 2.035 | 41.87 ± 0.85 | 77.08 ± 1.34 (5.52) | 54.33 ± 0.83 (23.90) | 51.25 ± 1.43 (25.25) | 24.15 ± 0.60 | |
| 14l | 10 | 29.08 ± 1.88 | 20.72 ± 1.33 | 61.30 ± 0.98 | 20.79 ± 3.34 | 48.44 ± 0.49 | – |
| 30 | 63.02 ± 1.09 (18.91) | 24.10 ± 3.15 | 74.01 ± 0.80 (9.16) | 38.58 ± 2.61 | 69.17 ± 0.50 (11.47) | 46.16 ± 2.11 – | |
| cisplatin | 10 | 53.03 ± 2.29 | 20.75 ± 0.81 | 42.61 ± 2.33 | 60.98 ± 0.92 | 83.57 ± 1.21 | 76.74 ± 1.26 |
| 30 | 86.90 ± 1.24 | 74.47 ± 1.20 | 99.93 ± 0.26 | 88.95 ± 0.53 | 95.02 ± 0.28 | 96.90 ± 0.25 | |
| (5.78) | (19.13) | (12.43) | (4.29) | (1.30) | (4.73) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács, É.; Ali, H.; Minorics, R.; Traj, P.; Resch, V.; Paragi, G.; Bruszel, B.; Zupkó, I.; Mernyák, E. Synthesis and Antiproliferative Activity of Steroidal Diaryl Ethers. Molecules 2023, 28, 1196. https://doi.org/10.3390/molecules28031196
Kovács É, Ali H, Minorics R, Traj P, Resch V, Paragi G, Bruszel B, Zupkó I, Mernyák E. Synthesis and Antiproliferative Activity of Steroidal Diaryl Ethers. Molecules. 2023; 28(3):1196. https://doi.org/10.3390/molecules28031196
Chicago/Turabian StyleKovács, Édua, Hazhmat Ali, Renáta Minorics, Péter Traj, Vivien Resch, Gábor Paragi, Bella Bruszel, István Zupkó, and Erzsébet Mernyák. 2023. "Synthesis and Antiproliferative Activity of Steroidal Diaryl Ethers" Molecules 28, no. 3: 1196. https://doi.org/10.3390/molecules28031196
APA StyleKovács, É., Ali, H., Minorics, R., Traj, P., Resch, V., Paragi, G., Bruszel, B., Zupkó, I., & Mernyák, E. (2023). Synthesis and Antiproliferative Activity of Steroidal Diaryl Ethers. Molecules, 28(3), 1196. https://doi.org/10.3390/molecules28031196