
Structural and Mechanistic Basis for the Inactivation of Human Ornithine Aminotransferase by (3S,4S)-3-Amino-4-fluorocyclopentenecarboxylic Acid

 , , , , and
, , , , and
Abstract
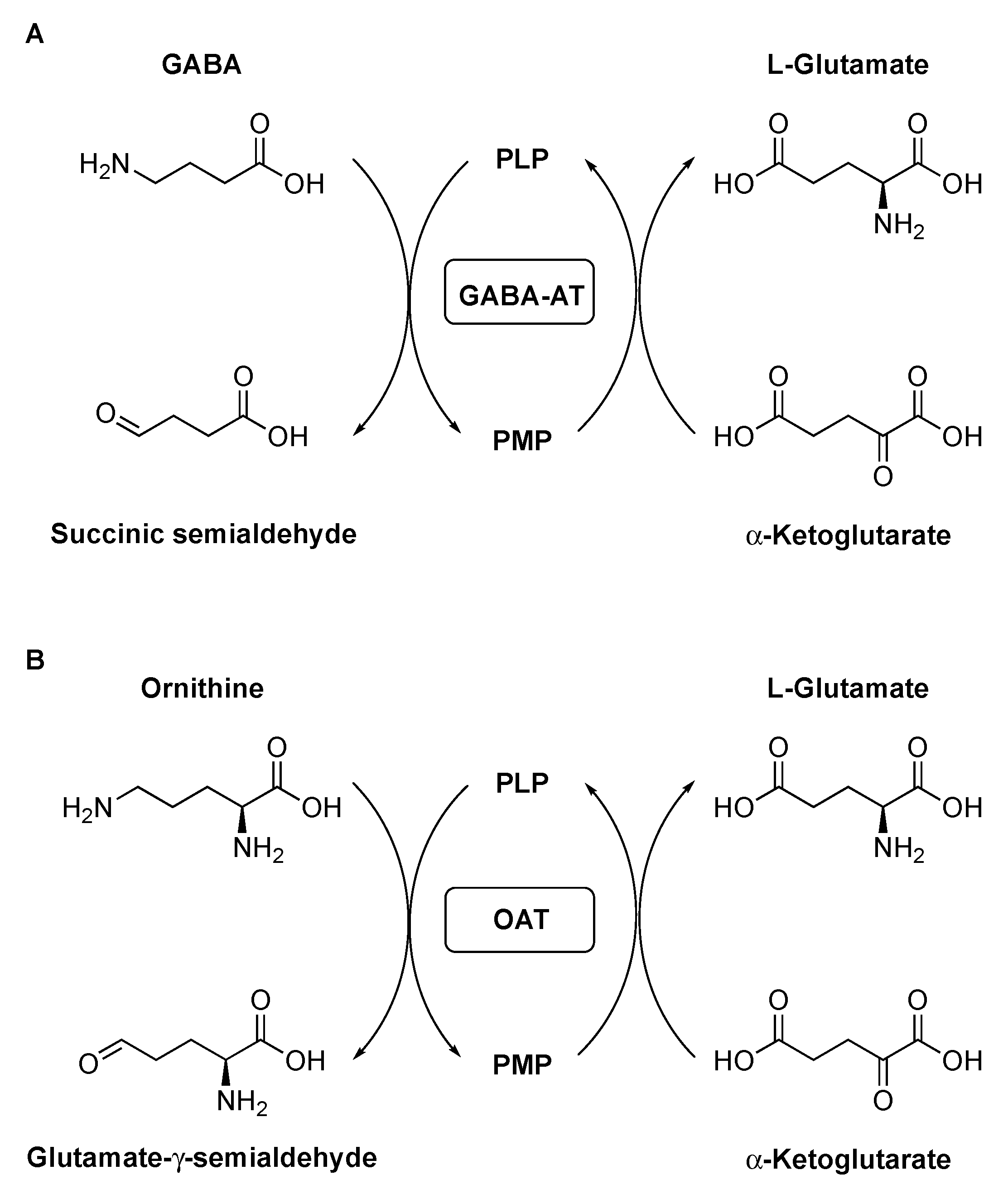
1. Introduction
2. Results and Discussion
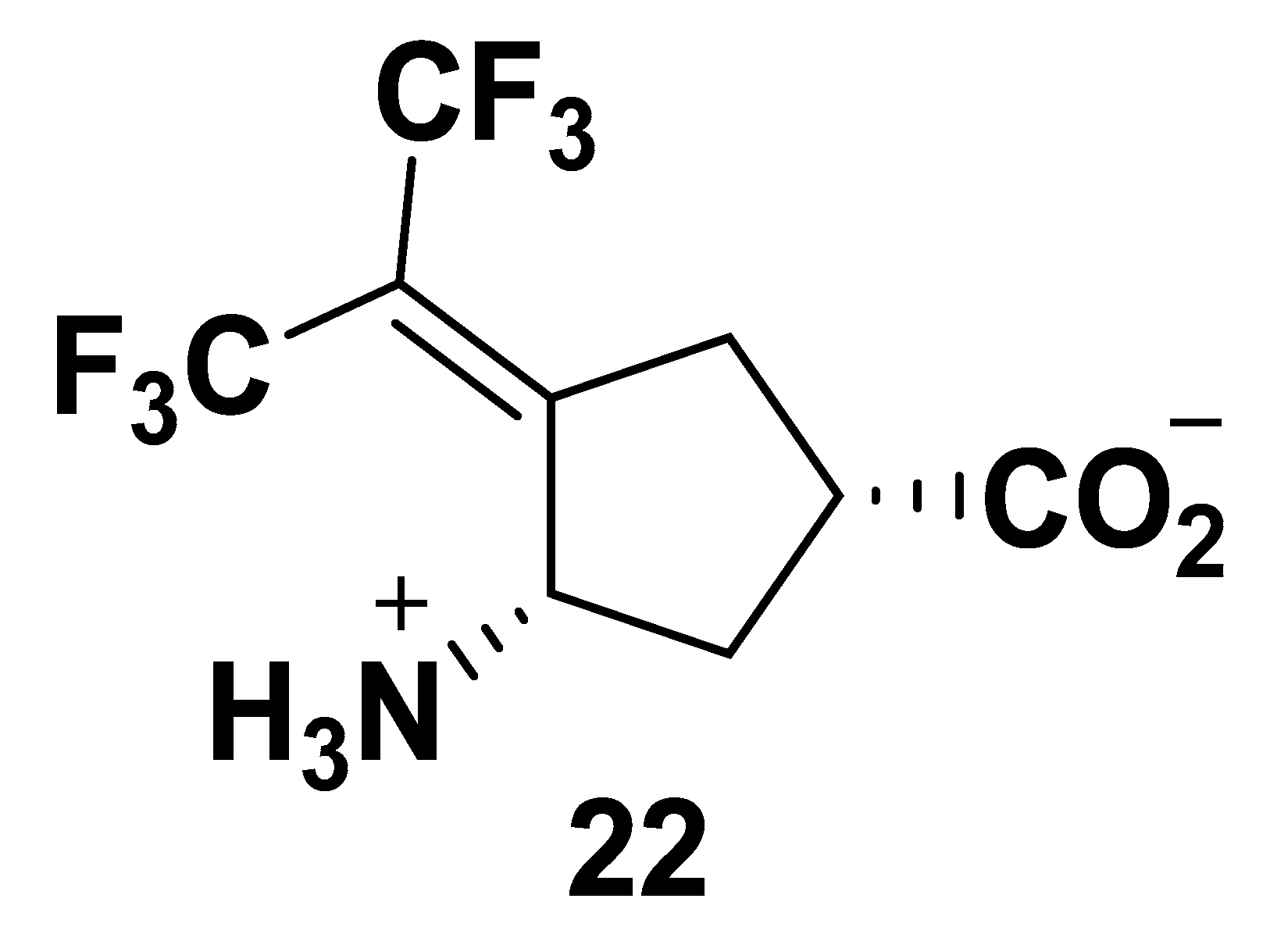
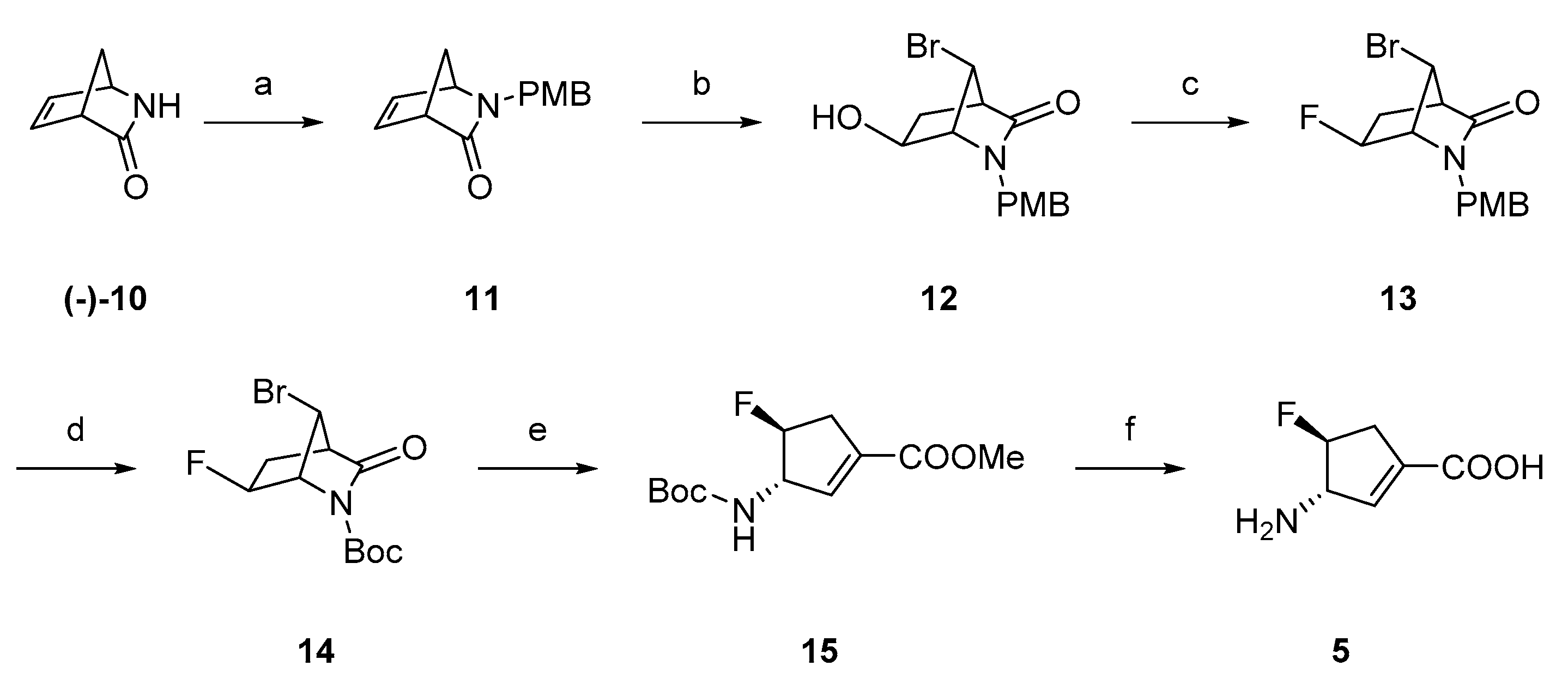
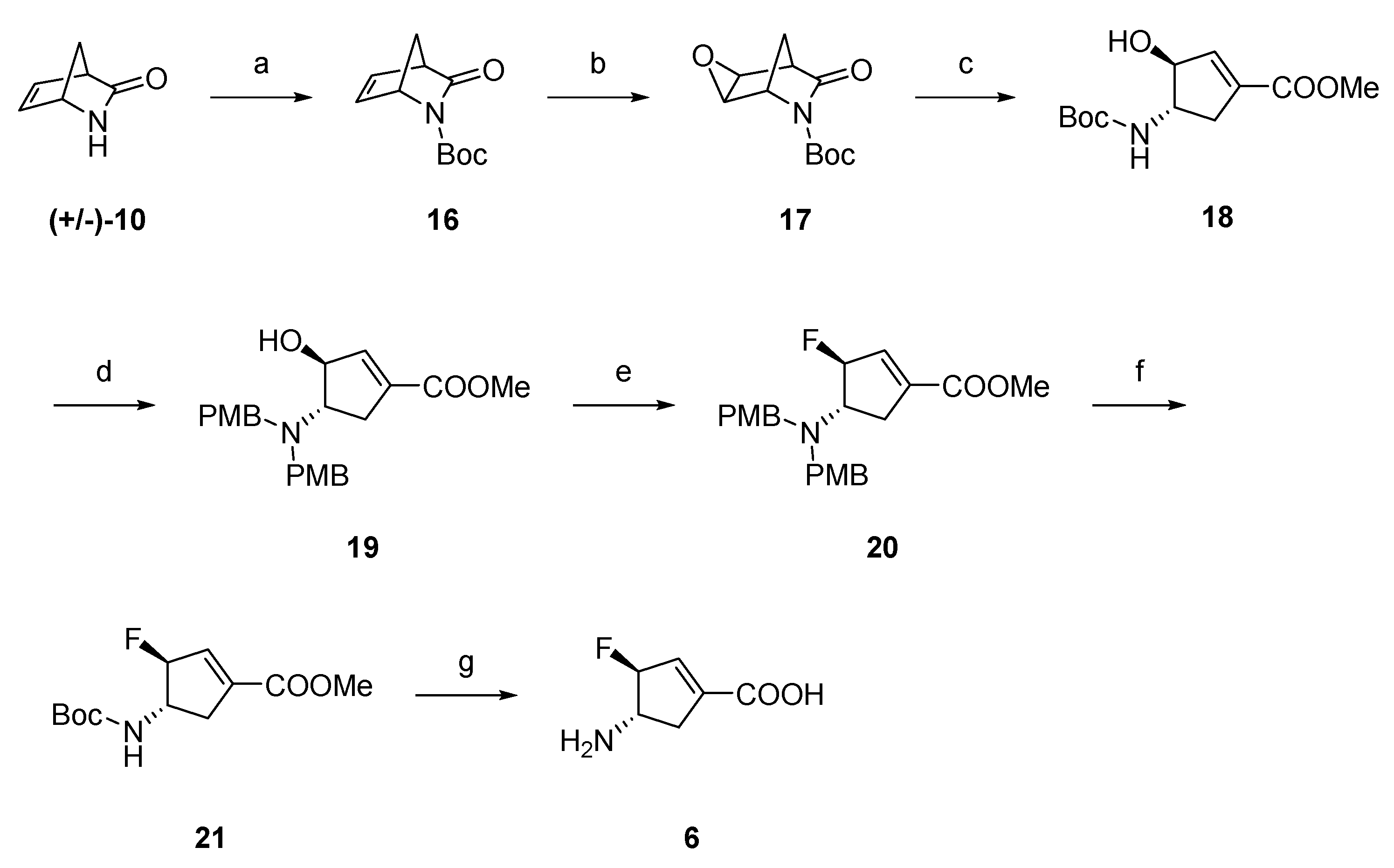
2.1. Synthesis of Monofluorinated Cyclopentene Analogs 5 and 6
2.2. The Significance of the Conjugated Alkene of 5
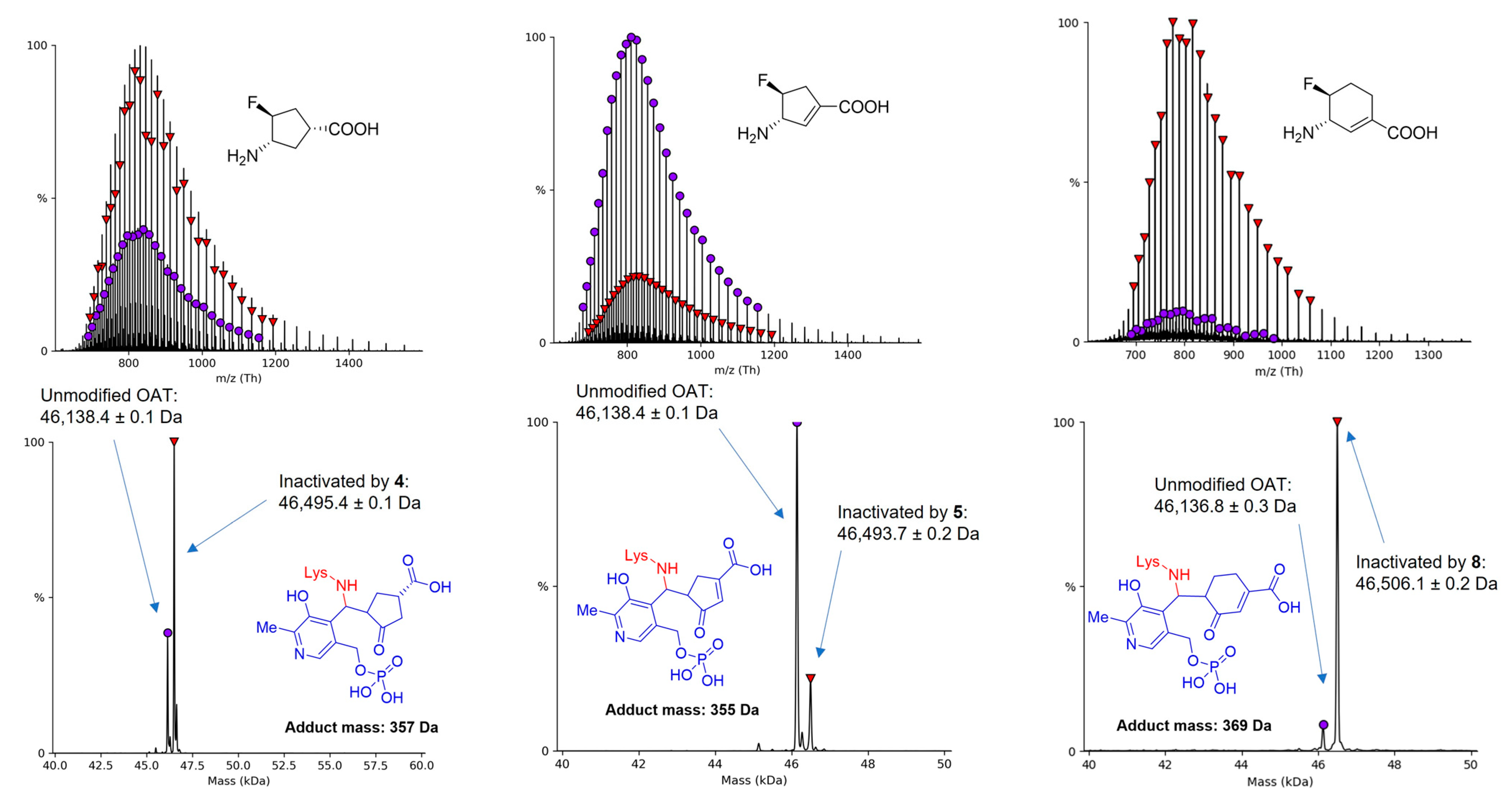
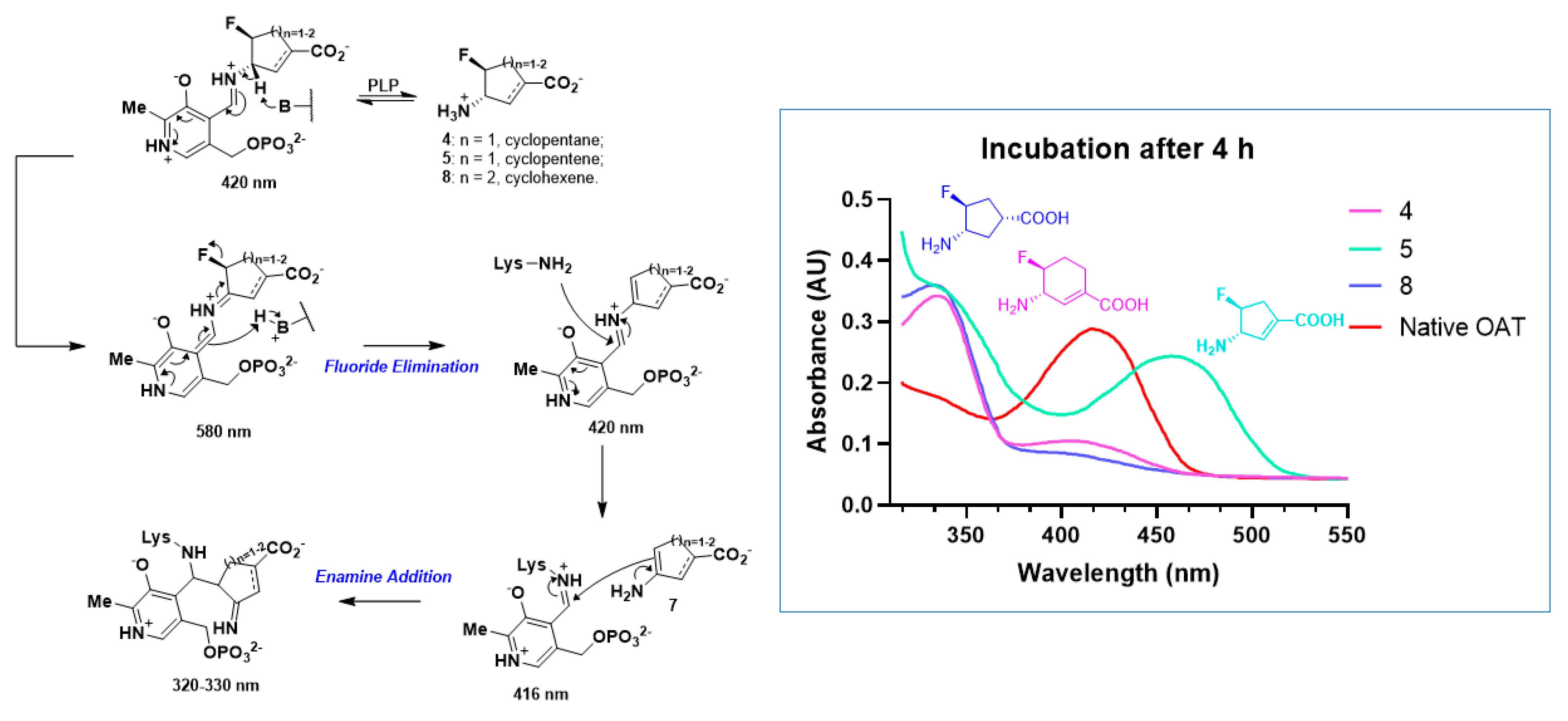
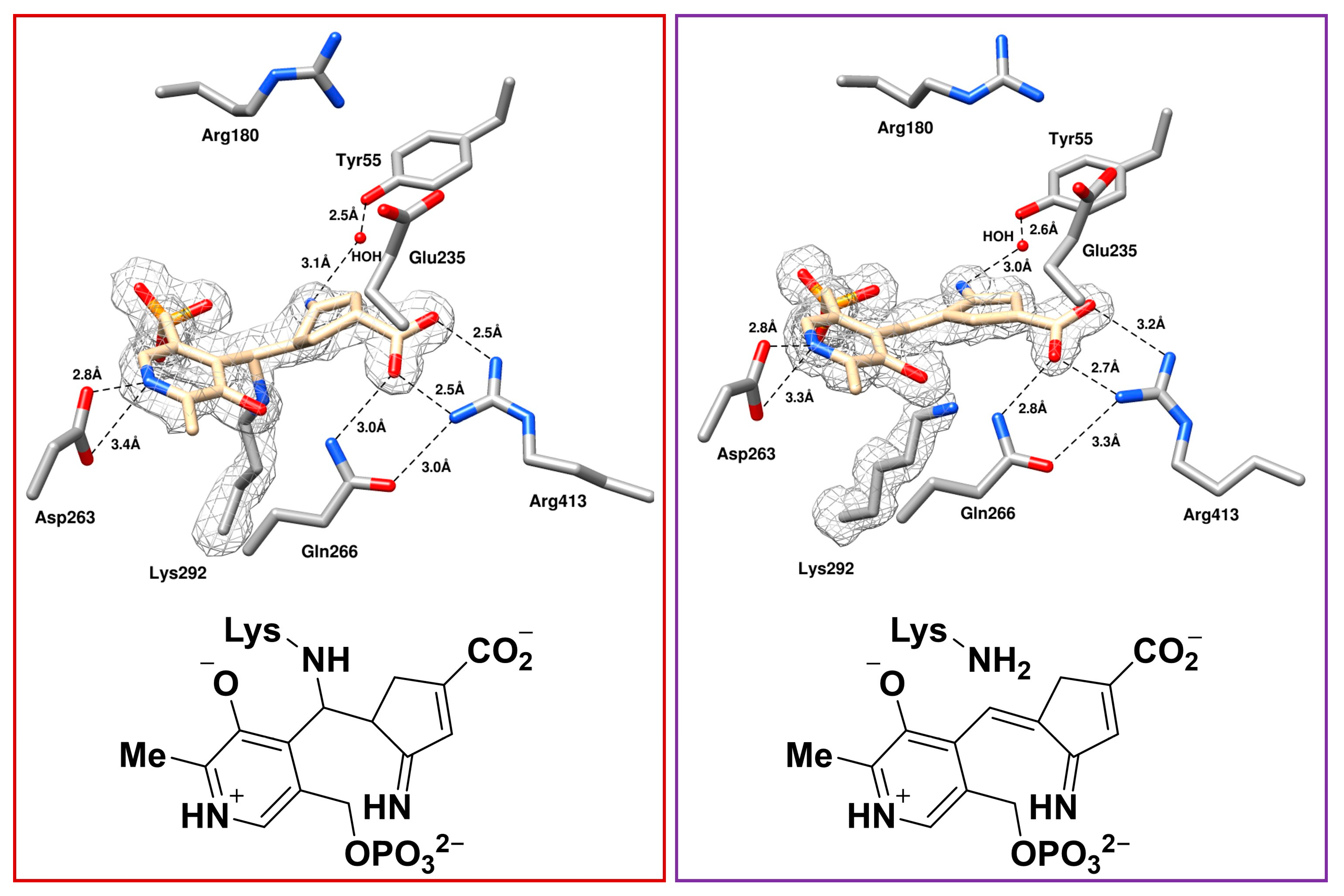
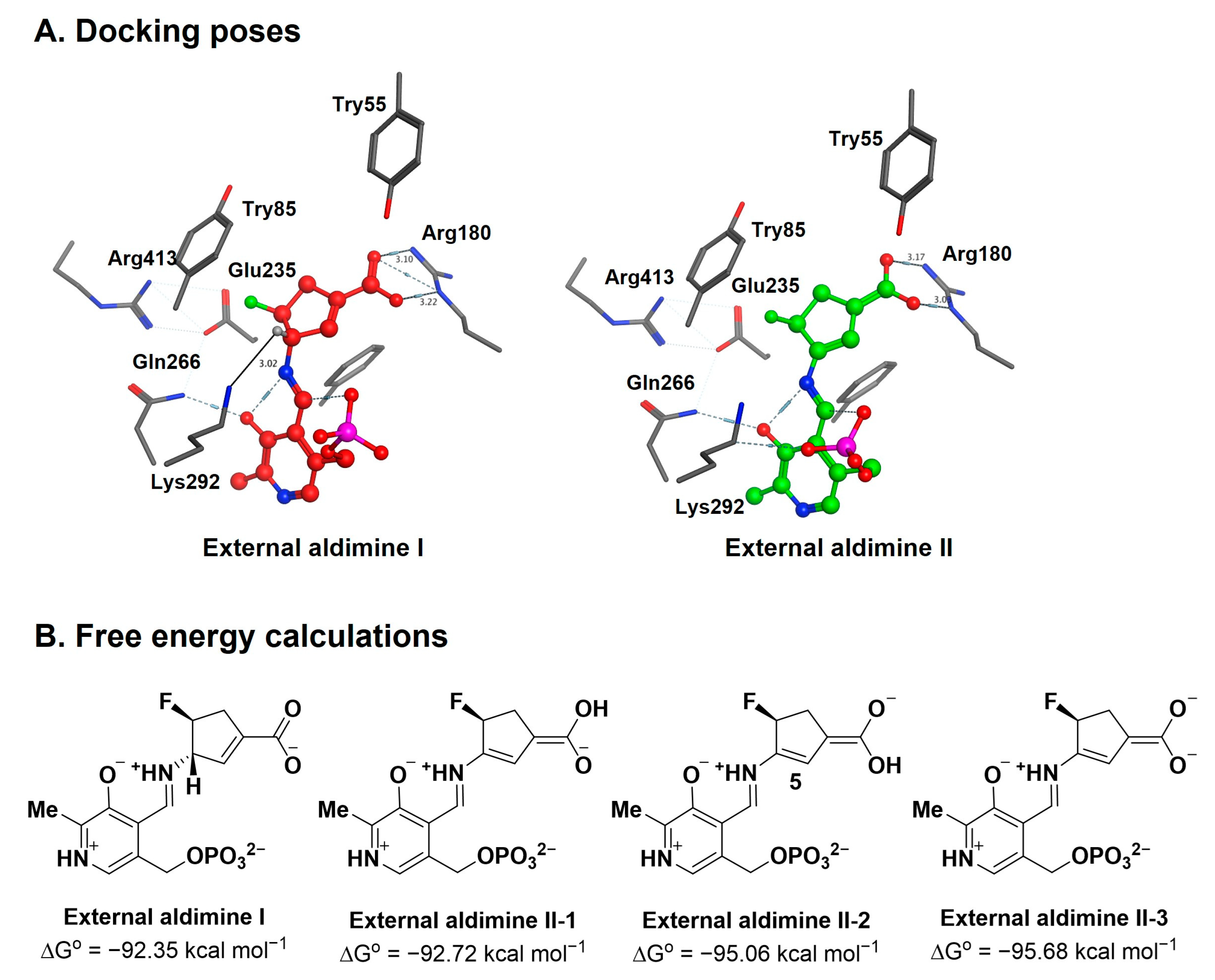
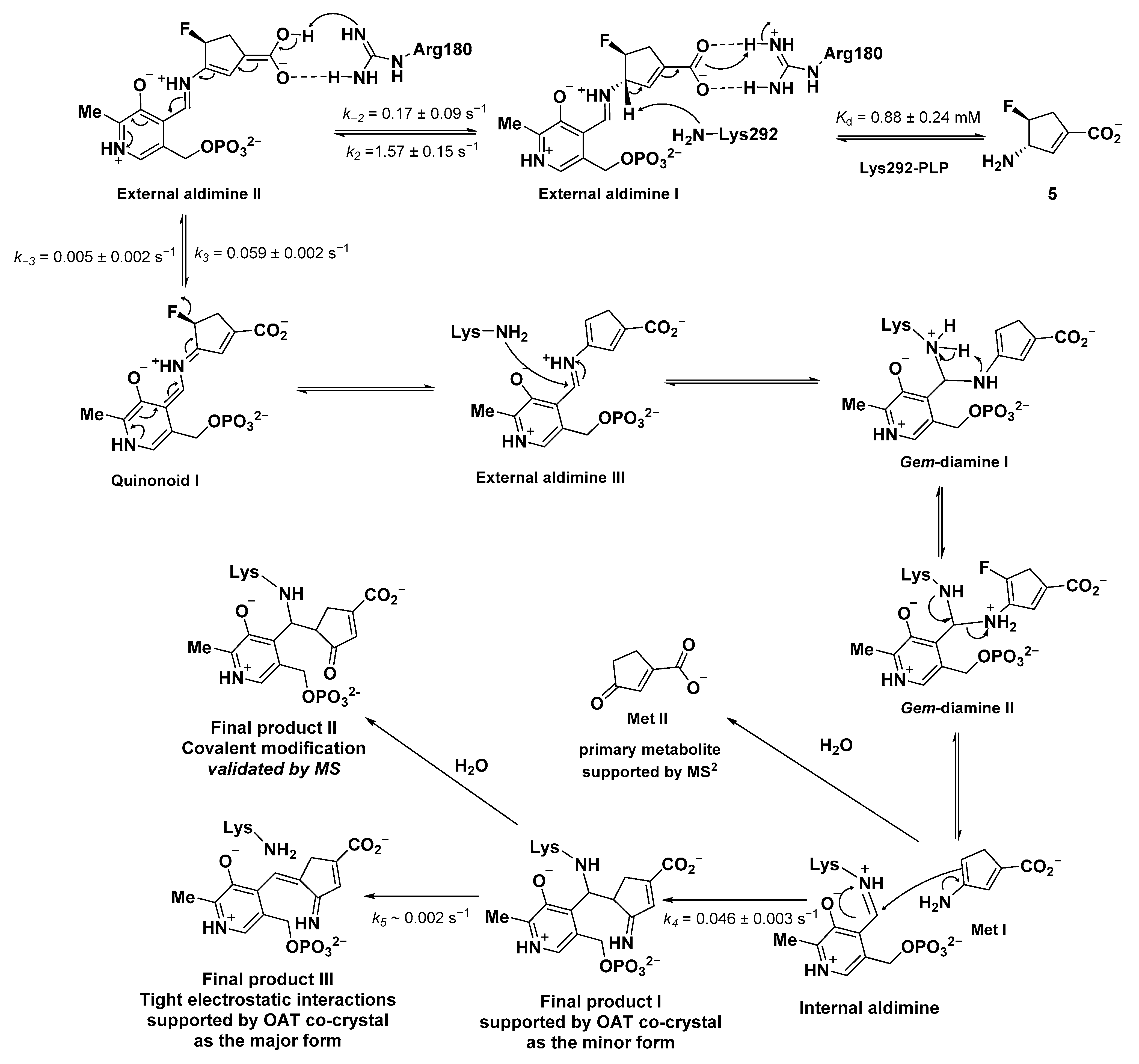
2.3. The Unusual Covalent and Noncovalent Interactions between hOAT and the Products Generated from 5
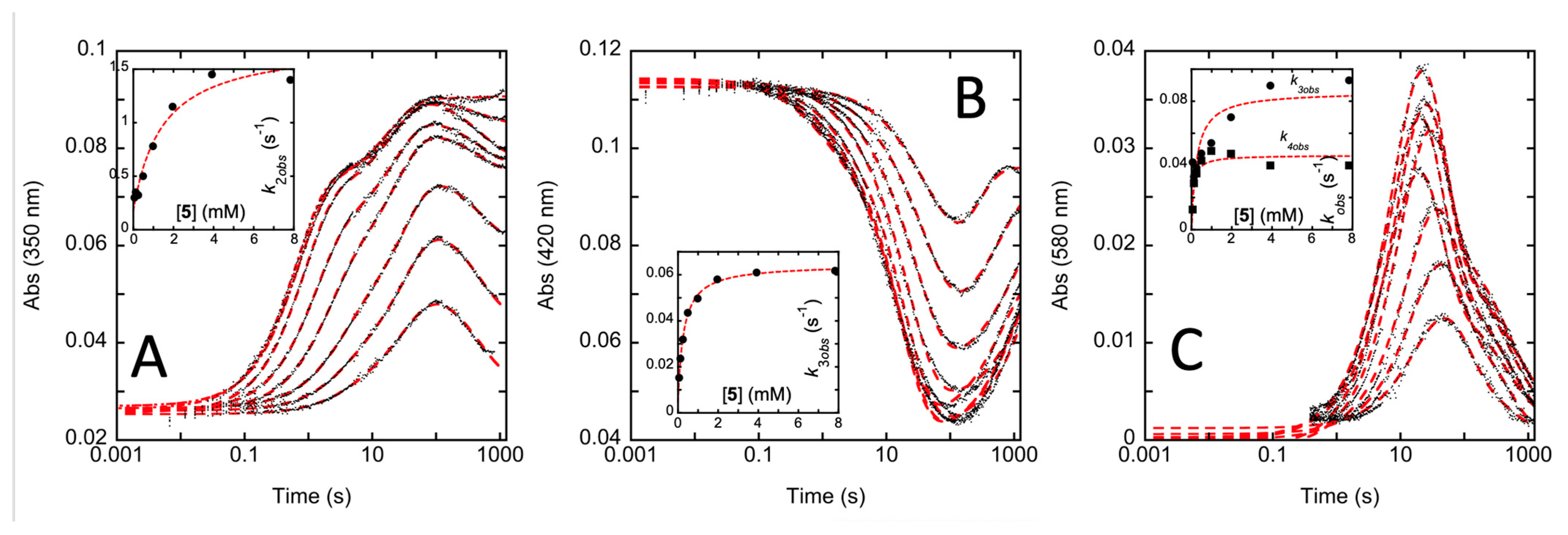
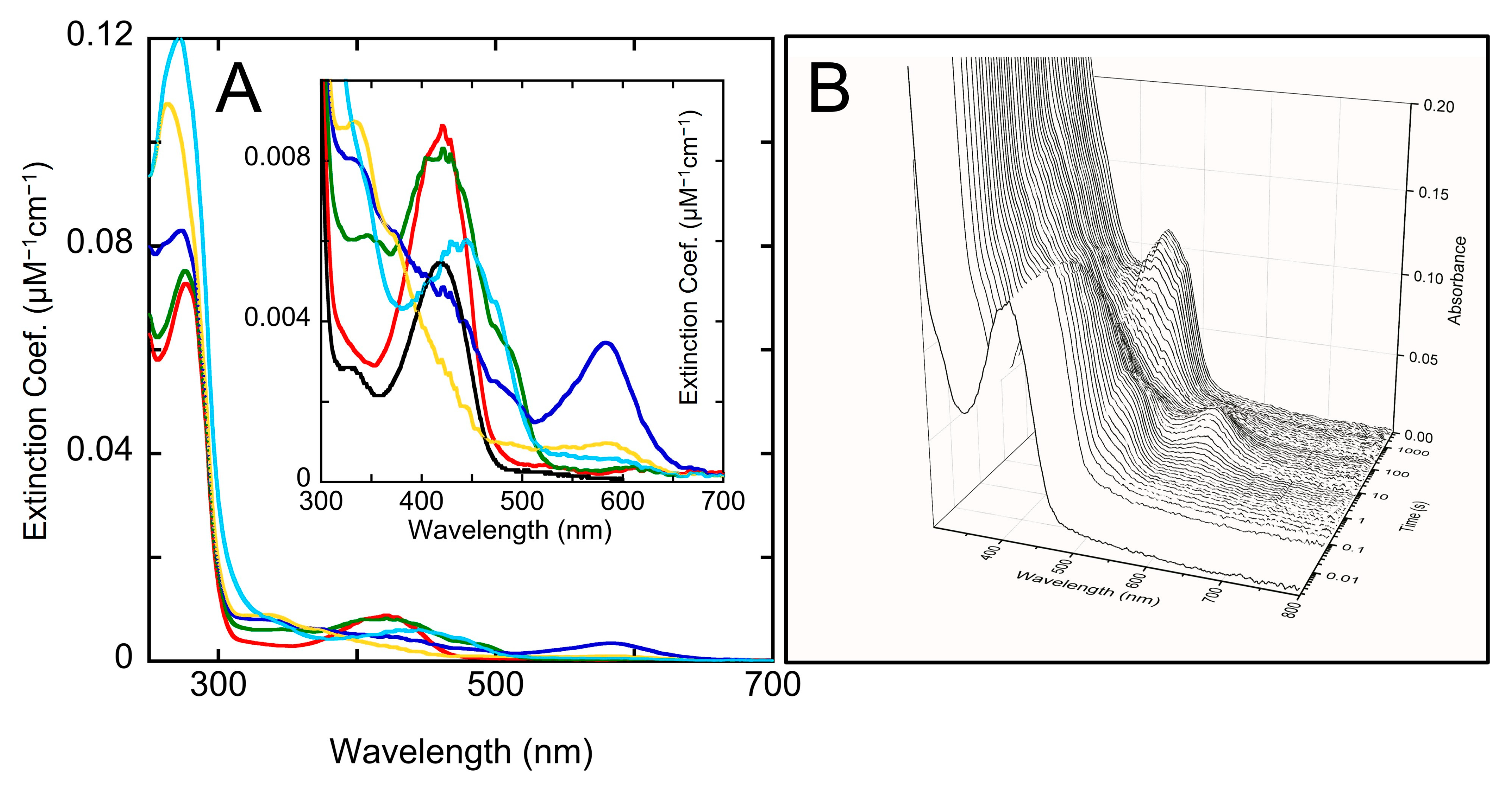
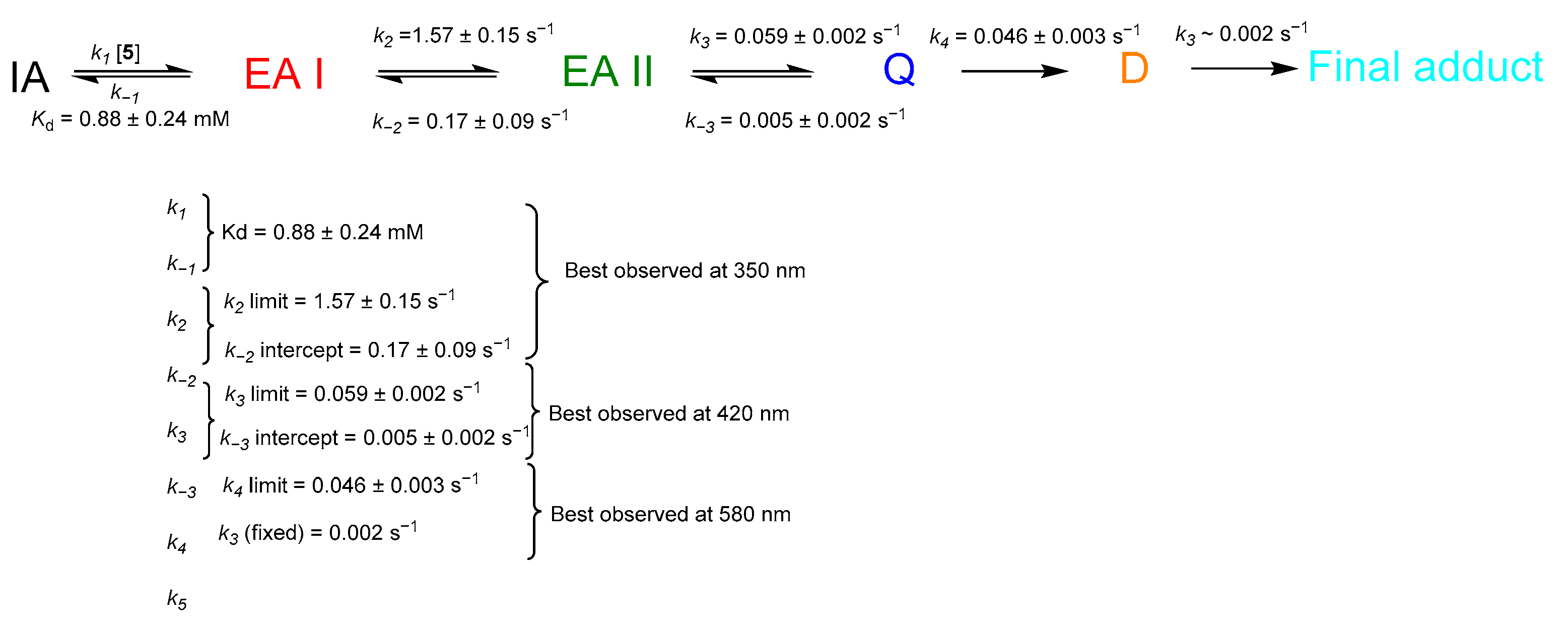
2.4. Transient-State Kinetics of hOAT Inactivation by 5
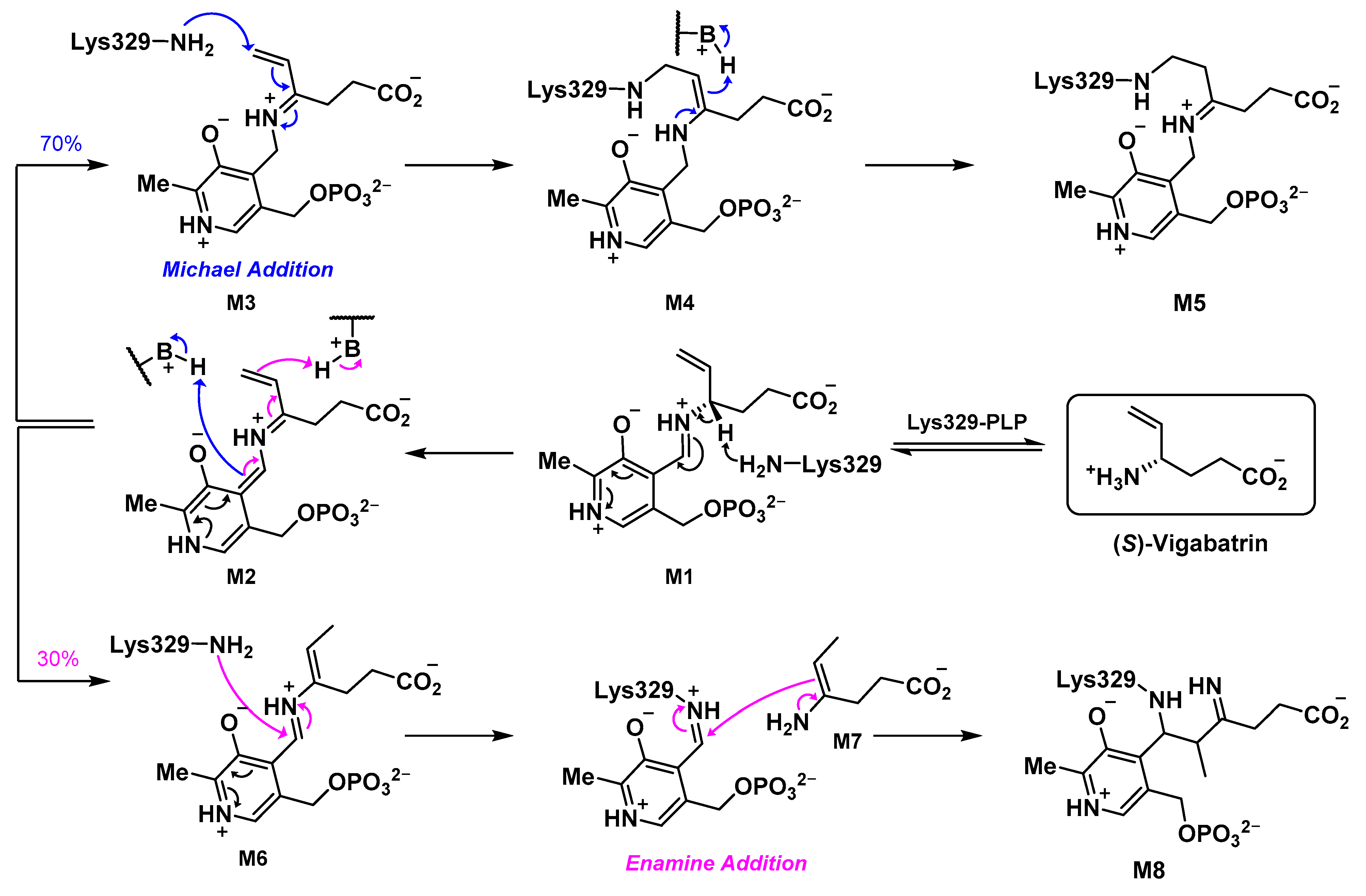
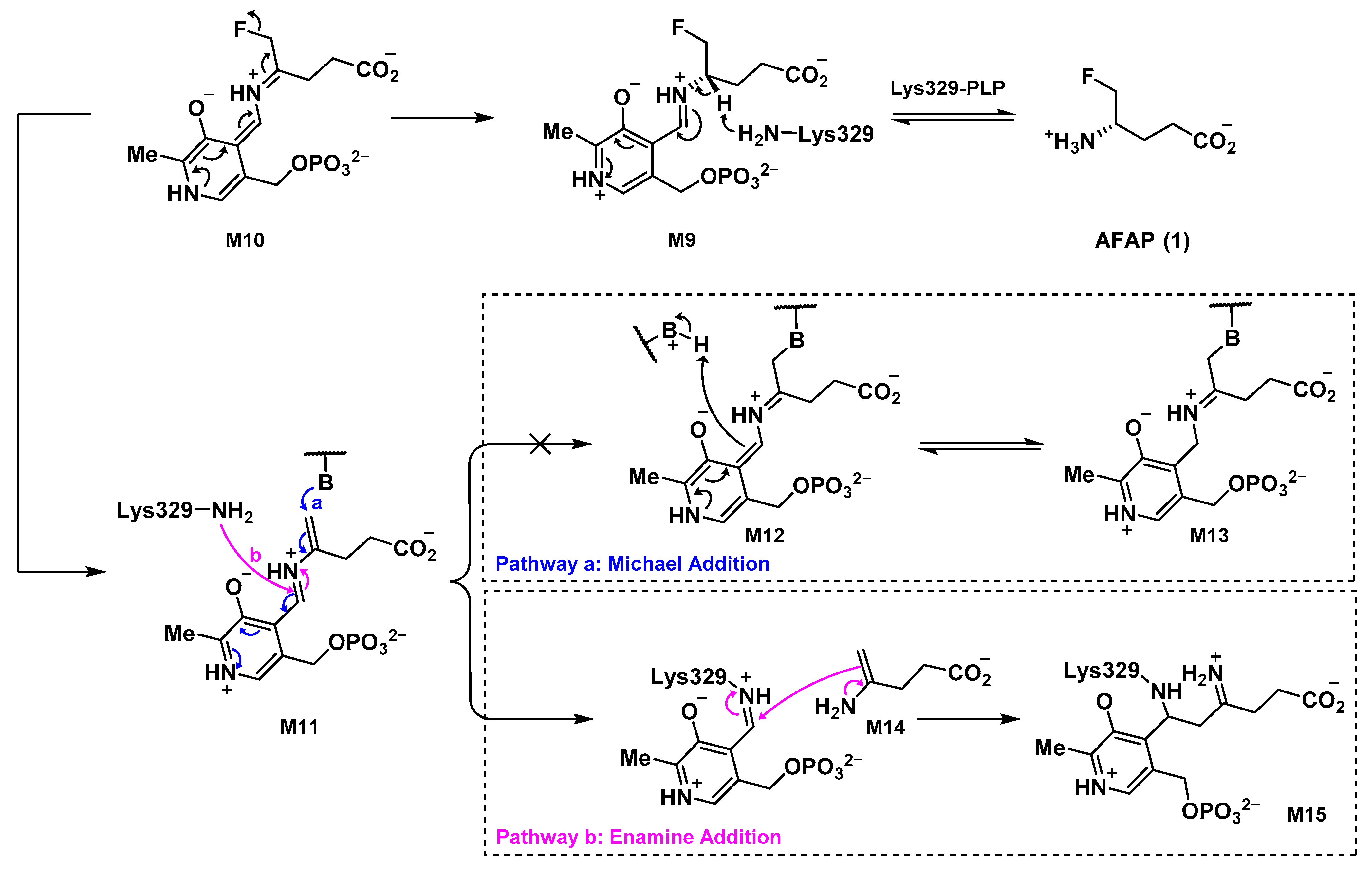
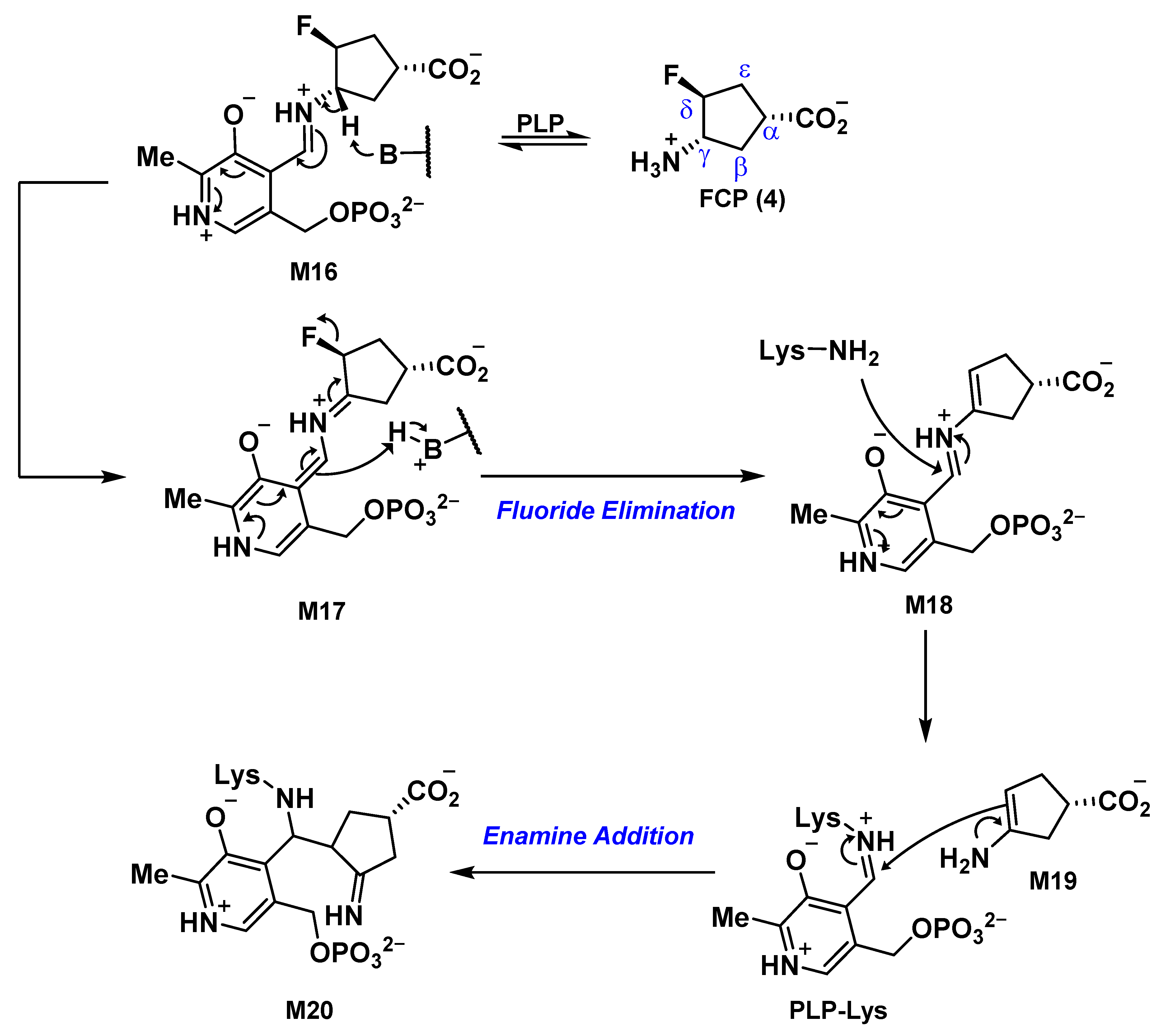
2.5. The Inactivation Pathway of Compound 5 in hOAT
3. Materials and Methods
3.1. Chemistry
3.2. Expression and Purification of hOAT
3.3. Aminotransferases and Coenzymes for Kinetic Studies
3.4. Denaturing Intact Protein and Small Molecule Mass Spectrometry
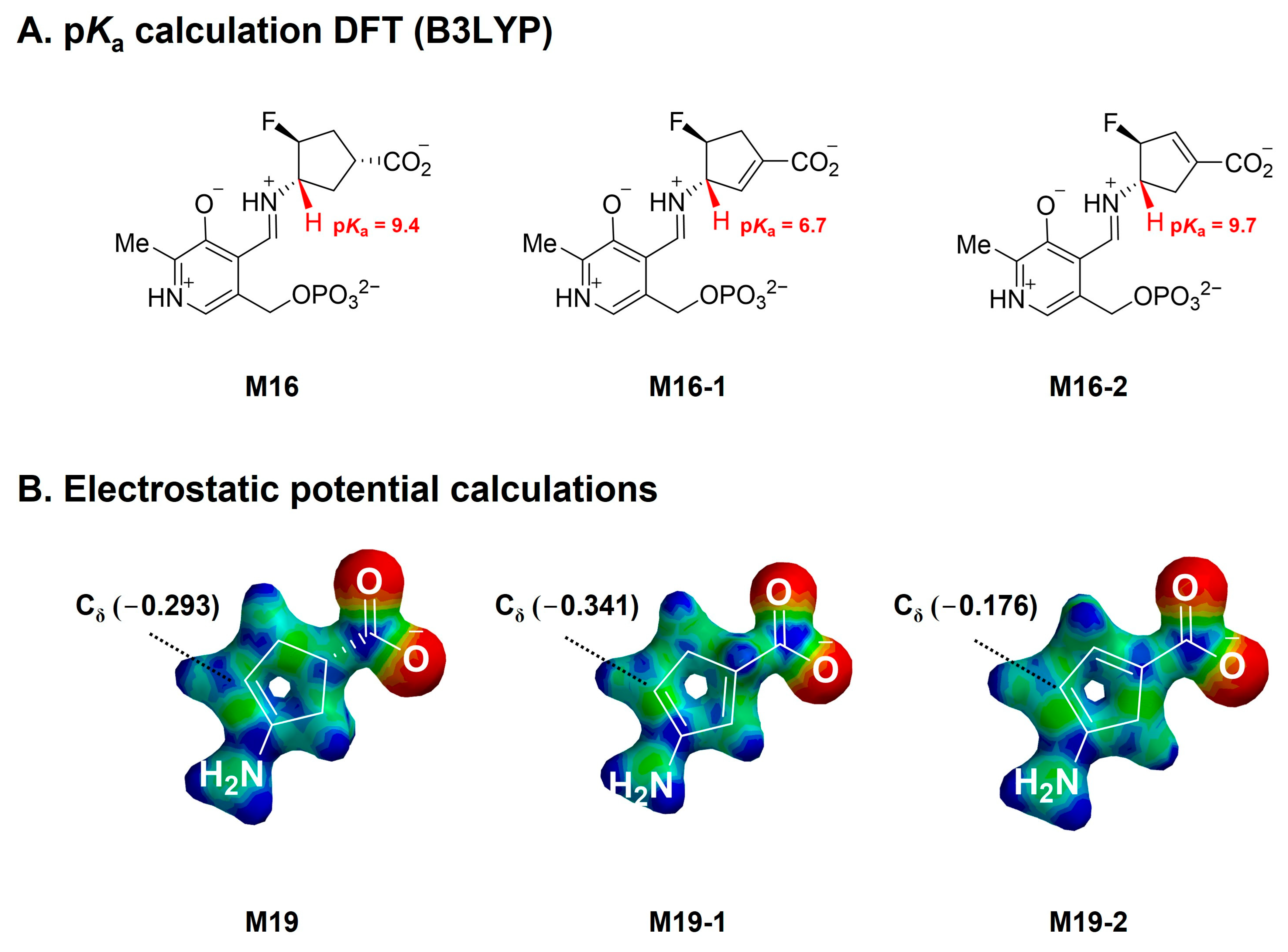
3.5. Theoretical pKa Calculations
3.6. Electrostatic Potential (ESP) Charge Calculation
3.7. Co-Crystallization of hOAT with Compound 5
3.8. Transient-State Methods
3.9. Gibb’s Free Energy Calculation
3.10. Molecular Docking Protocol
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Jansonius, J.N. Structure, evolution and action of vitamin B6-dependent enzymes. Curr. Opin. Struct. Biol. 1998, 8, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.-Y.; Cho, B.-K.; Yun, H.; Koteshwar, K.; Kim, B.-G. Revisit of aminotransferase in the genomic era and its application to biocatalysis. J. Mol. Catal. B Enzym. 2005, 37, 47–55. [Google Scholar] [CrossRef]
- Silverman, R.B. Design and Mechanism of GABA Aminotransferase Inactivators. Treatments for Epilepsies and Addictions. Chem. Rev. 2018, 118, 4037–4070. [Google Scholar] [CrossRef] [PubMed]
- Yogeeswari, P.; Sriram, D.; Vaigundaragavendran, J. The GABA shunt: An attractive and potential therapeutic target in the treatment of epileptic disorders. Curr. Drug Metab. 2005, 6, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Juncosa, J.I.; Silverman, R.B. Ornithine aminotransferase versus GABA aminotransferase: Implications for the design of new anticancer drugs. Med. Res. Rev. 2015, 35, 286–305. [Google Scholar] [CrossRef] [PubMed]
- Zigmond, E.; Ben Ya’acov, A.; Lee, H.; Lichtenstein, Y.; Shalev, Z.; Smith, Y.; Zolotarov, L.; Ziv, E.; Kalman, R.; Le, H.V.; et al. Suppression of Hepatocellular Carcinoma by Inhibition of Overexpressed Ornithine Aminotransferase. ACS Med. Chem. Lett. 2015, 6, 840–844. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, L.; Li, K.; Liu, F.; Wang, L.; Zhang, D.; Zhou, J.; Ma, X.; Wang, S.; Yang, S. Ornithine aminotransferase promoted the proliferation and metastasis of non-small cell lung cancer via upregulation of miR-21. J. Cell. Physiol. 2019, 234, 12828–12838. [Google Scholar] [CrossRef]
- Foroozan, R. Vigabatrin: Lessons Learned From the United States Experience. J. Neuroophthalmol. 2018, 38, 442–450. [Google Scholar] [CrossRef]
- Silverman, R.B. Mechanism-based enzyme inactivators. Methods Enzym. 1995, 249, 240–283. [Google Scholar] [CrossRef]
- Nanavati, S.M.; Silverman, R.B. Mechanisms of inactivation of.gamma.-aminobutyric acid aminotransferase by the antiepilepsy drug.gamma.-vinyl GABA (vigabatrin). J. Am. Chem. Soc. 1991, 113, 9341–9349. [Google Scholar] [CrossRef]
- Silverman, R.B.; Levy, M.A. Mechanism of inactivation of gamma-aminobutyric acid-alpha-ketoglutaric acid aminotransferase by 4-amino-5-halopentanoic acids. Biochemistry 1981, 20, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- Silverman, R.B.; Invergo, B.J. Mechanism of inactivation of gamma-aminobutyrate aminotransferase by 4-amino-5-fluoropentanoic acid. First example of an enamine mechanism for a gamma-amino acid with a partition ratio of 0. Biochemistry 1986, 25, 6817–6820. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Doubleday, P.F.; Weerawarna, P.M.; Zhu, W.; Kelleher, N.L.; Silverman, R.B. Mechanism-Based Design of 3-Amino-4-Halocyclopentenecarboxylic Acids as Inactivators of GABA Aminotransferase. ACS Med. Chem. Lett. 2020, 11, 1949–1955. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Doubleday, P.F.; Catlin, D.S.; Weerawarna, P.M.; Butrin, A.; Shen, S.; Wawrzak, Z.; Kelleher, N.L.; Liu, D.; Silverman, R.B. A Remarkable Difference That One Fluorine Atom Confers on the Mechanisms of Inactivation of Human Ornithine Aminotransferase by Two Cyclohexene Analogues of gamma-Aminobutyric Acid. J. Am. Chem. Soc. 2020, 142, 4892–4903. [Google Scholar] [CrossRef]
- Wang, Z.; Silverman, R.B. Syntheses and evaluation of fluorinated conformationally restricted analogues of GABA as potential inhibitors of GABA aminotransferase. Bioorg. Med. Chem. 2006, 14, 2242–2252. [Google Scholar] [CrossRef]
- Storici, P.; Capitani, G.; Muller, R.; Schirmer, T.; Jansonius, J.N. Crystal structure of human ornithine aminotransferase complexed with the highly specific and potent inhibitor 5-fluoromethylornithine. J. Mol. Biol. 1999, 285, 297–309. [Google Scholar] [CrossRef]
- Silverman, R.B.; George, C. Mechanism of inactivation of γ-aminobutyric acid aminotransferase by (S,E)-4-amino-5-fluoropent-2-enoic acid. Biochem. Biophys. Res. Commun. 1988, 150, 942–946. [Google Scholar] [CrossRef]
- Mascarenhas, R.; Le, H.V.; Clevenger, K.D.; Lehrer, H.J.; Ringe, D.; Kelleher, N.L.; Silverman, R.B.; Liu, D. Selective Targeting by a Mechanism-Based Inactivator against Pyridoxal 5’-Phosphate-Dependent Enzymes: Mechanisms of Inactivation and Alternative Turnover. Biochemistry 2017, 56, 4951–4961. [Google Scholar] [CrossRef]
- Storici, P.; Qiu, J.; Schirmer, T.; Silverman, R.B. Mechanistic crystallography. Mechanism of inactivation of gamma-aminobutyric acid aminotransferase by (1R,3S,4S)-3-amino-4-fluorocyclopentane-1-carboxylic acid as elucidated by crystallography. Biochemistry 2004, 43, 14057–14063. [Google Scholar] [CrossRef]
- Bey, P.; Gerhart, F.; Jung, M. Synthesis of (E)-4-amino-2,5-hexadienoic acid and (E)-4-amino-5-fluoro-2-pentenoic acid. Irreversible inhibitors of 4-aminobutyrate-2-oxoglutarate aminotransferase. J. Org. Chem. 1986, 51, 2835–2838. [Google Scholar] [CrossRef]
- Bolkenius, F.N.; Knodgen, B.; Seiler, N. DL-canaline and 5-fluoromethylornithine. Comparison of two inactivators of ornithine aminotransferase. Biochem. J. 1990, 268, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Silverman, R.B. A new class of conformationally rigid analogues of 4-amino-5-halopentanoic acids, potent inactivators of gamma-aminobutyric acid aminotransferase. J. Med. Chem. 2000, 43, 706–720. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.E.B.; Derrien, N.; Lloyd, M.C.; Taylor, S.J.C.; Chaplin, D.A.; McCague, R. Highly selective directed hydrogenation of enantiopure 4-(tert-butoxycarbonylamino)cyclopent-1-enecarboxylic acid methyl esters. Tetrahedron Lett. 2001, 42, 1347–1350. [Google Scholar] [CrossRef]
- Zhang, H.; Schinazi, R.F.; Chu, C.K. Synthesis of neplanocin F analogues as potential antiviral agents. Bioorganic Med. Chem. 2006, 14, 8314–8322. [Google Scholar] [CrossRef] [PubMed]
- Ghalami-Choobar, B.; Dezhampanah, H.; Nikparsa, P.; Ghiami-Shomami, A. Theoretical calculation of the pKa values of some drugs in aqueous solution. Int. J. Quantum Chem. 2012, 112, 2275–2280. [Google Scholar] [CrossRef]
- Shen, S.; Butrin, A.; Doubleday, P.F.; Melani, R.D.; Beaupre, B.A.; Tavares, M.T.; Ferreira, G.M.; Kelleher, N.L.; Moran, G.R.; Liu, D.; et al. Turnover and Inactivation Mechanisms for (S)-3-Amino-4,4-difluorocyclopent-1-enecarboxylic Acid, a Selective Mechanism-Based Inactivator of Human Ornithine Aminotransferase. J. Am. Chem. Soc. 2021, 143, 8689–8703. [Google Scholar] [CrossRef]
- Shen, B.W.; Hennig, M.; Hohenester, E.; Jansonius, J.N.; Schirmer, T. Crystal structure of human recombinant ornithine aminotransferase. J. Mol. Biol. 1998, 277, 81–102. [Google Scholar] [CrossRef]
- Moschitto, M.J.; Doubleday, P.F.; Catlin, D.S.; Kelleher, N.L.; Liu, D.; Silverman, R.B. Mechanism of Inactivation of Ornithine Aminotransferase by (1S,3S)-3-Amino-4-(hexafluoropropan-2-ylidenyl)cyclopentane-1-carboxylic Acid. J. Am. Chem. Soc. 2019, 141, 10711–10721. [Google Scholar] [CrossRef]
- Moschitto, M.J.; Silverman, R.B. Synthesis of (S)-3-Amino-4-(difluoromethylenyl)-cyclopent-1-ene-1-carboxylic Acid (OV329), a Potent Inactivator of gamma-Aminobutyric Acid Aminotransferase. Org. Lett. 2018, 20, 4589–4592. [Google Scholar] [CrossRef]
- Christensen, E.M.; Patel, S.M.; Korasick, D.A.; Campbell, A.C.; Krause, K.L.; Becker, D.F.; Tanner, J.J. Resolving the cofactor-binding site in the proline biosynthetic enzyme human pyrroline-5-carboxylate reductase 1. J. Biol. Chem. 2017, 292, 7233–7243. [Google Scholar] [CrossRef]
- Koo, Y.K.; Nandi, D.; Silverman, R.B. The multiple active enzyme species of gamma-aminobutyric acid aminotransferase are not isozymes. Arch. Biochem. Biophys. 2000, 374, 248–254. [Google Scholar] [CrossRef]
- Silverman, R.B.; Bichler, K.A.; Leon, A.J.J. Mechanisms of inactivation of γ-aminobutyric acid aminotransferase by 4-amino-5-fluoro-5-hexenoic acid. Am. Chem. Soc. 1996, 118, 1241–1252. [Google Scholar] [CrossRef]
- Lee, H.; Doud, E.H.; Wu, R.; Sanishvili, R.; Juncosa, J.I.; Liu, D.; Kelleher, N.L.; Silverman, R.B. Mechanism of inactivation of gamma-aminobutyric acid aminotransferase by (1S,3S)-3-amino-4-difluoromethylene-1-cyclopentanoic acid (CPP-115). J. Am. Chem. Soc. 2015, 137, 2628–2640. [Google Scholar] [CrossRef]
- Juncosa, J.I.; Lee, H.; Silverman, R.B. Two continuous coupled assays for ornithine-delta-aminotransferase. Anal. Biochem. 2013, 440, 145–149. [Google Scholar] [CrossRef]
- Official Gaussian 09 Literature Citation. Available online: http://sobereva.com/g09/m_citation. (accessed on 16 January 2023).
- Vonrhein, C.; Flensburg, C.; Keller, P.; Sharff, A.; Smart, O.; Paciorek, W.; Womack, T.; Bricogne, G. Data processing and analysis with the autoPROC toolbox. Acta Cryst. D Biol. Cryst. 2011, 67 Pt 4, 293–302. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Cryst. 2007, 40 Pt 4, 658–674. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Cryst. D Biol. Cryst. 2010, 66 Pt 4, 486–501. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkoczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Cryst. D Struct. Biol. 2019, 75 Pt 10, 861–877. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal Chemistry and the Molecular Operating Environment (MOE): Application of QSAR and Molecular Docking to Drug Discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Juncosa, J.I.; Takaya, K.; Le, H.V.; Moschitto, M.J.; Weerawarna, P.M.; Mascarenhas, R.; Liu, D.L.; Dewey, S.L.; Silverman, R.B. Design and Mechanism of (S)-3-Amino-4-(difluoromethylenyl)cyclopent-1-ene-1-carboxylic Acid, a Highly Potent gamma-Aminobutyric Acid Aminotransferase Inactivator for the Treatment of Addiction. J. Am. Chem. Soc. 2018, 140, 2151–2164. [Google Scholar] [CrossRef]
- Weerawarna, P.M.; Moschitto, M.J.; Silverman, R.B. Theoretical and Mechanistic Validation of Global Kinetic Parameters of the Inactivation of GABA Aminotransferase by OV329 and CPP-115. ACS Chem. Biol. 2021, 16, 615–630. [Google Scholar] [CrossRef]
- Zhu, W.; Butrin, A.; Melani, R.D.; Doubleday, P.F.; Ferreira, G.M.; Tavares, M.T.; Habeeb Mohammad, T.S.; Beaupre, B.A.; Kelleher, N.L.; Moran, G.R.; et al. Rational Design, Synthesis, and Mechanism of (3S,4R)-3-Amino-4-(difluoromethyl)cyclopent-1-ene-1-carboxylic Acid: Employing a Second-Deprotonation Strategy for Selectivity of Human Ornithine Aminotransferase over GABA Aminotransferase. J. Am. Chem. Soc. 2022, 144, 5629–5642. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmpd | Structure | hOAT | GABA-AT | ||||
|---|---|---|---|---|---|---|---|
| KI (mM) | kinact (min−1) | kinact/KI (min−1mM−1) | KI (mM) | kinact (min−1) | kinact/KI (min−1mM−1) | ||
| 1 (AFPA) b |  | 0.23 | 0.17 | 0.75 | 0.40 | 0.5 | 1.27 |
| 2 c |  | NT | 0.12 | ||||
| 3 (5FMOrn) d |  | 0.03 | 0.17 e | 5.8 | NT | ||
| 4 (FCP) f |  | 1.4 | 0.086 | 0.06 | 0.078 | 0.017 | 0.22 |
| 5 g |  | 0.25 | 0.143 | 0.57 | 0.026 | 0.132 | 5.08 |
| 6 |  | No inhibition up to 10 mM | Ki = 0.10 mM | ||||
| 7 h |  | NT | No inhibition up to 10 mM | ||||
| 8 f |  | 0.031 | 0.075 | 2.41 | 1.1 | 0.20 | 0.18 |
| 9 f |  | 4.38 | 0.075 | 0.017 | 4.28 | 0.028 | 0.0065 |
| VGA |  | Ki = 46 mM | 0.29 | 0.21 | 0.73 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, S.; Butrin, A.; Beaupre, B.A.; Ferreira, G.M.; Doubleday, P.F.; Grass, D.H.; Zhu, W.; Kelleher, N.L.; Moran, G.R.; Liu, D.; et al. Structural and Mechanistic Basis for the Inactivation of Human Ornithine Aminotransferase by (3S,4S)-3-Amino-4-fluorocyclopentenecarboxylic Acid. Molecules 2023, 28, 1133. https://doi.org/10.3390/molecules28031133
Shen S, Butrin A, Beaupre BA, Ferreira GM, Doubleday PF, Grass DH, Zhu W, Kelleher NL, Moran GR, Liu D, et al. Structural and Mechanistic Basis for the Inactivation of Human Ornithine Aminotransferase by (3S,4S)-3-Amino-4-fluorocyclopentenecarboxylic Acid. Molecules. 2023; 28(3):1133. https://doi.org/10.3390/molecules28031133
Chicago/Turabian StyleShen, Sida, Arseniy Butrin, Brett A. Beaupre, Glaucio M. Ferreira, Peter F. Doubleday, Daniel H. Grass, Wei Zhu, Neil L. Kelleher, Graham R. Moran, Dali Liu, and et al. 2023. "Structural and Mechanistic Basis for the Inactivation of Human Ornithine Aminotransferase by (3S,4S)-3-Amino-4-fluorocyclopentenecarboxylic Acid" Molecules 28, no. 3: 1133. https://doi.org/10.3390/molecules28031133
APA StyleShen, S., Butrin, A., Beaupre, B. A., Ferreira, G. M., Doubleday, P. F., Grass, D. H., Zhu, W., Kelleher, N. L., Moran, G. R., Liu, D., & Silverman, R. B. (2023). Structural and Mechanistic Basis for the Inactivation of Human Ornithine Aminotransferase by (3S,4S)-3-Amino-4-fluorocyclopentenecarboxylic Acid. Molecules, 28(3), 1133. https://doi.org/10.3390/molecules28031133