
Bacterial Zinc Metalloenzyme Inhibitors: Recent Advances and Future Perspectives




Abstract
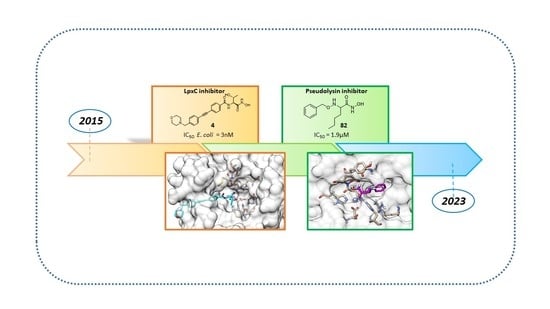
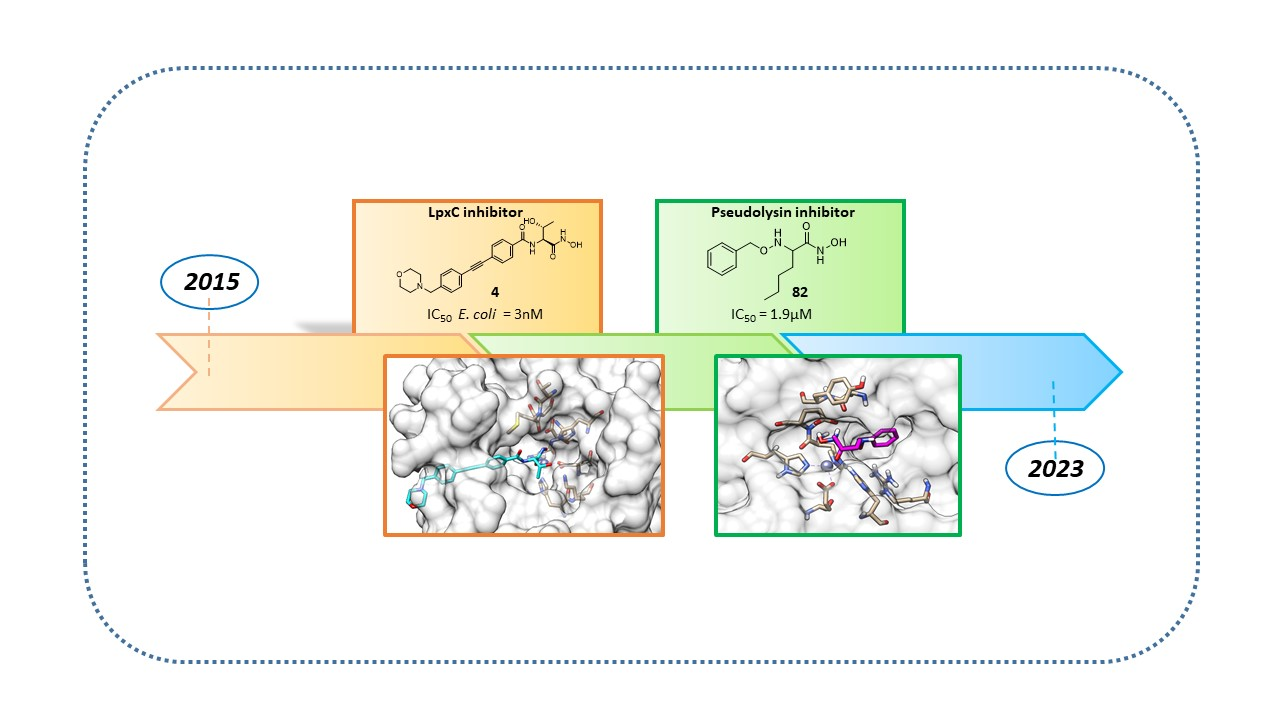
1. Introduction
1.1. Bacteremia
1.2. Multidrug Resistance
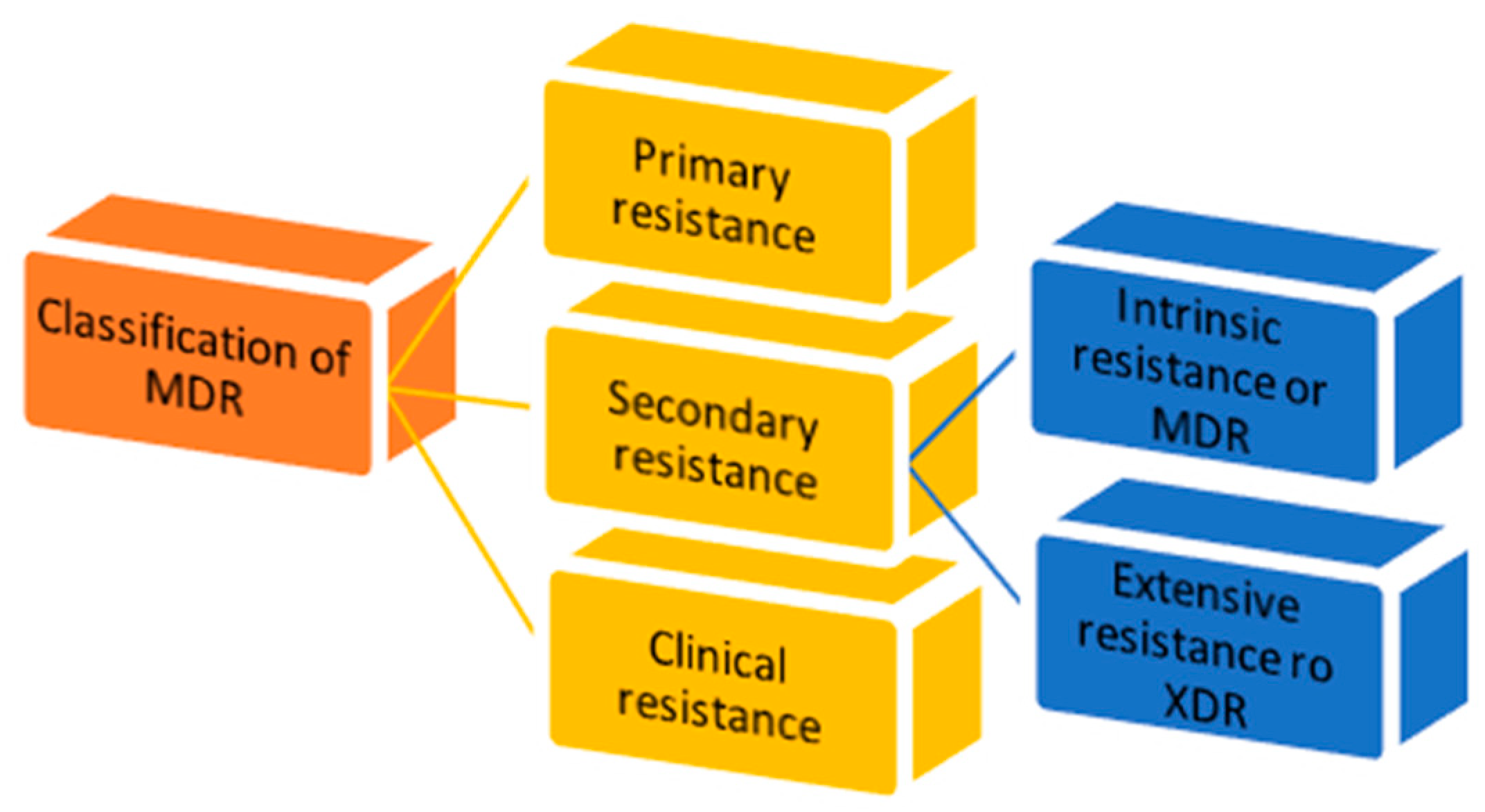
1.3. MDR Classification
- 1.
- Primary resistance occurs when the host organism interacts for the first time with the drug of interest;
- 2.
- Secondary resistance refers to the aversion which onsets after an exposure to the drug. It is also known as “acquired resistance” and can be classified into:
- intrinsic resistance: a single species of microorganisms results in insensitivity towards a first-time drug administration [11];
- extensive resistance: microorganisms are able to resist one or more of the most potent drugs. It is also named “extended detection and response” (XDR) which arises after the exposure to first-line drugs [12];
- 3.
- Clinical resistance is described when, in order to have efficacy, the drug concentration must be significantly increased, due to dysregulation of the host immune system function. This imbalance can lead to a failure of the therapy or an outbreak of different infections [11].
1.4. MDR Mechanism
1.4.1. Modifications in the Uptake of Drugs
1.4.2. Inactivation of Drugs
1.4.3. Efflux Pumps
1.5. Zinc Metalloenzymes
1.5.1. Gram-Negative Outer Membrane and Its Pathogenicity
1.5.2. M4 Enzymes
1.6. Targets of Interest
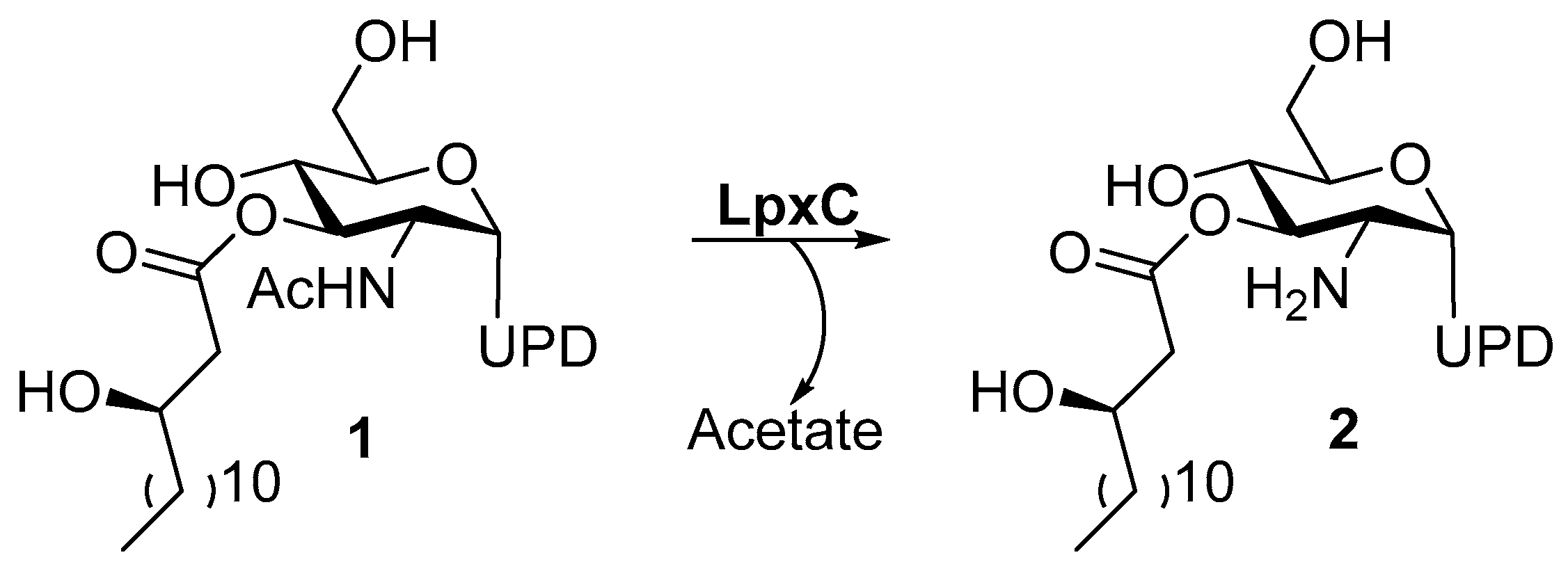
1.6.1. UDP-[3-O-(R)-3-hydroxymyristoyl]-N-acetylglucosamine Deacetylase
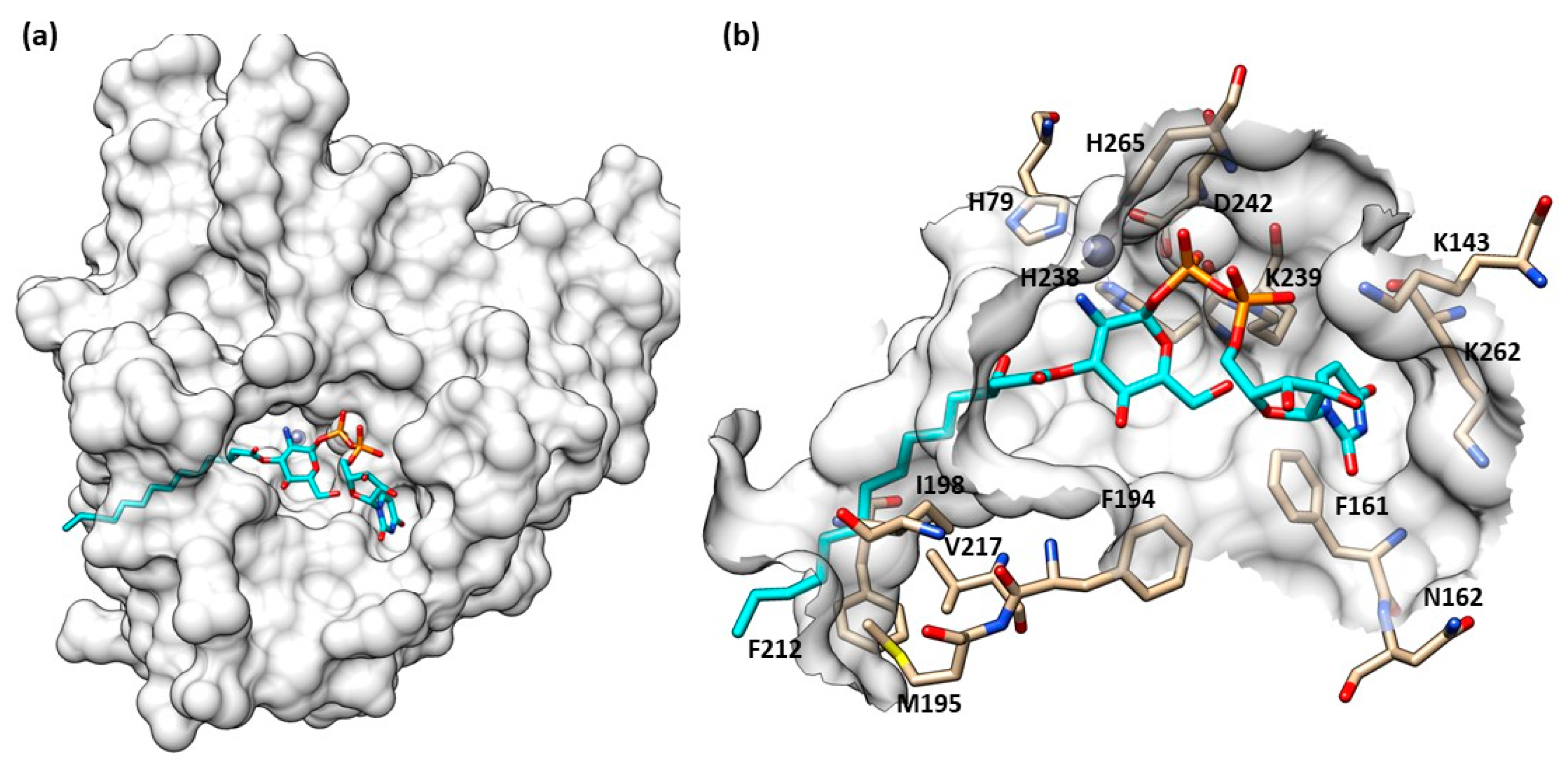
Structure of LpxC
1.6.2. Pseudolysin
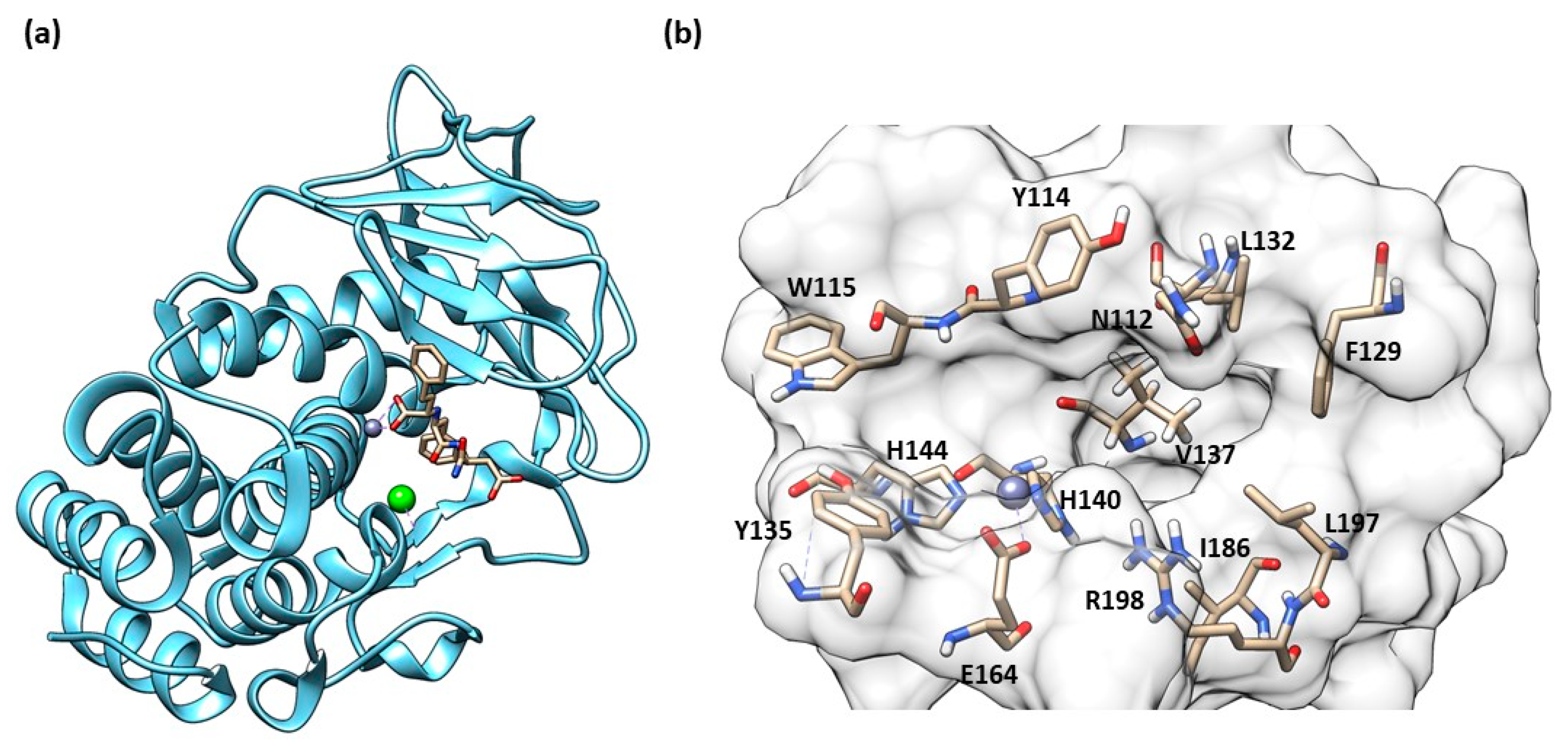
Structure of Pseudolysin
1.6.3. Thermolysin
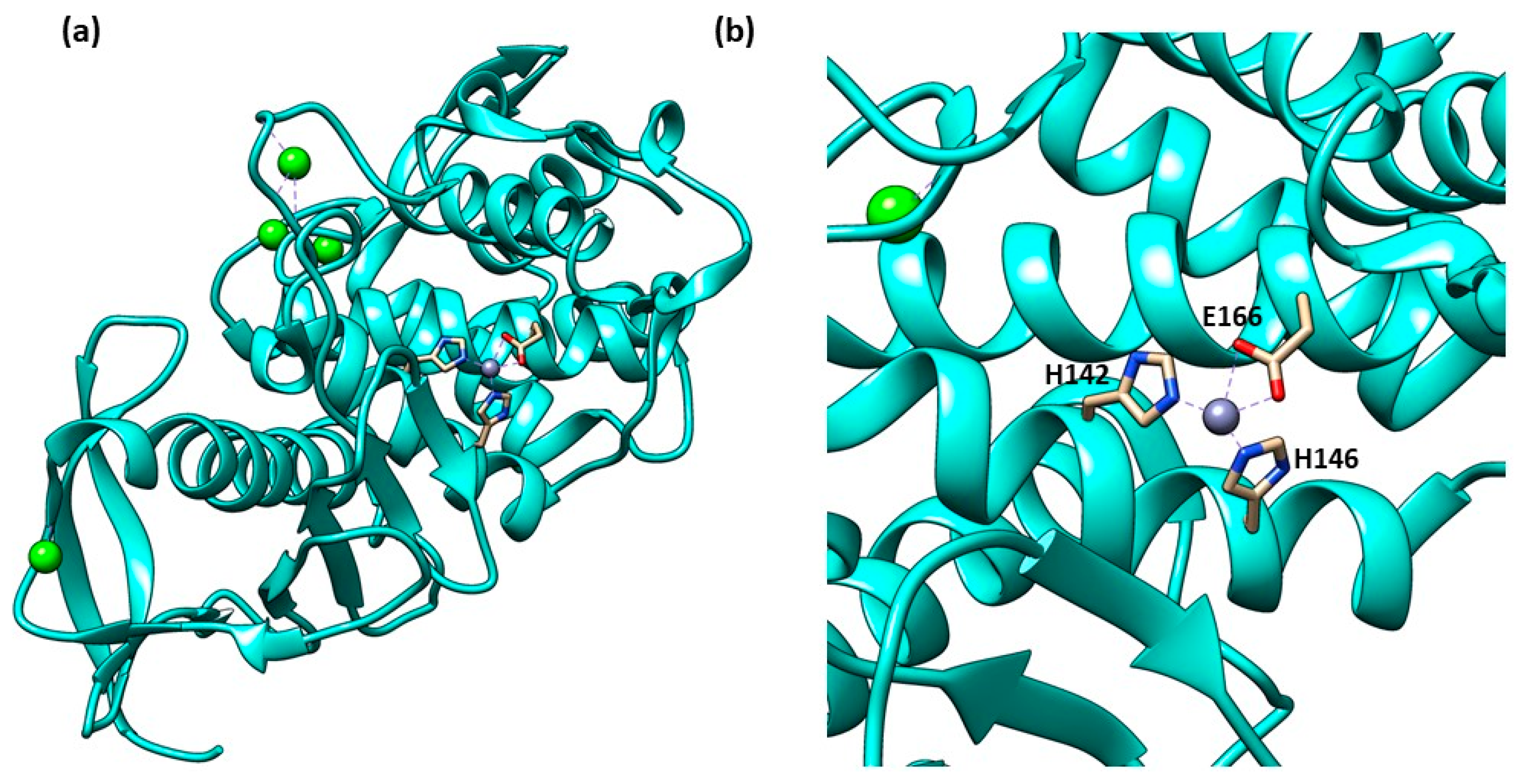
Structure of Thermolysin
2. Inhibitors
2.1. LpxC inhibitors
2.1.1. Hydroxamate-Based LpxC inhibitors
CHIR-090 and Its Analogues
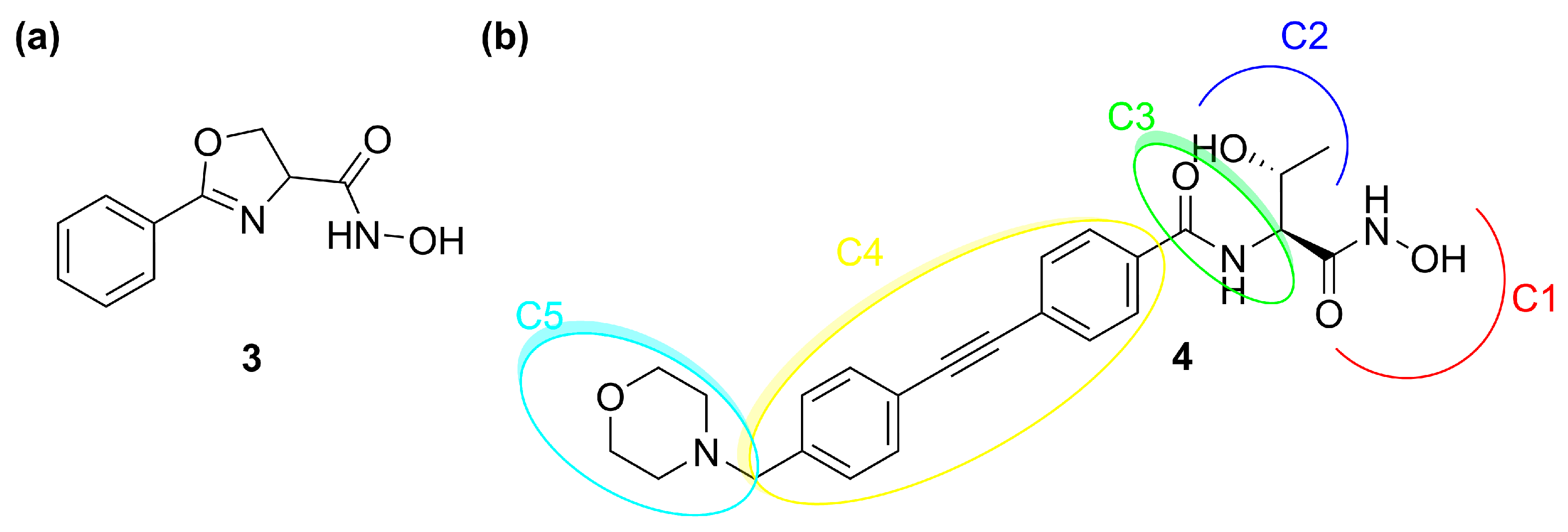
Oxazolidinones Derivatives
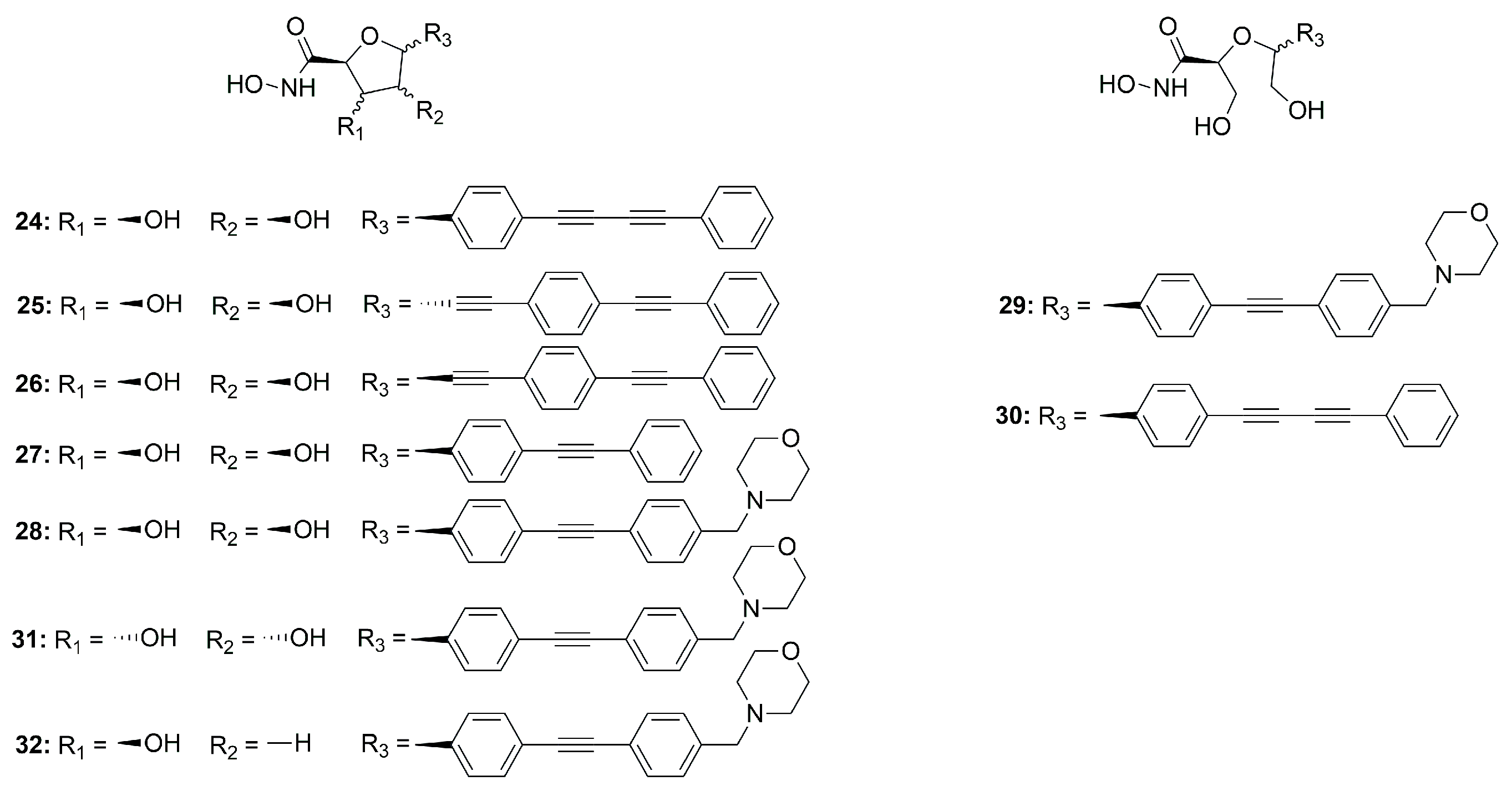
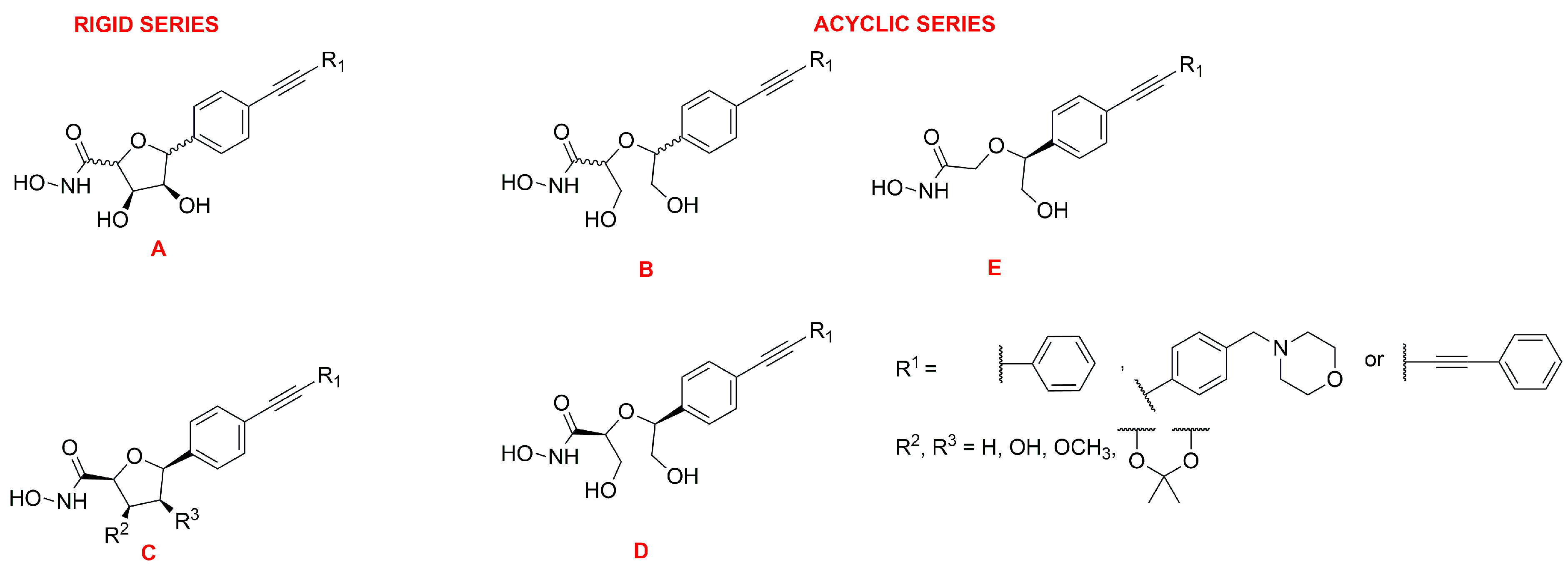
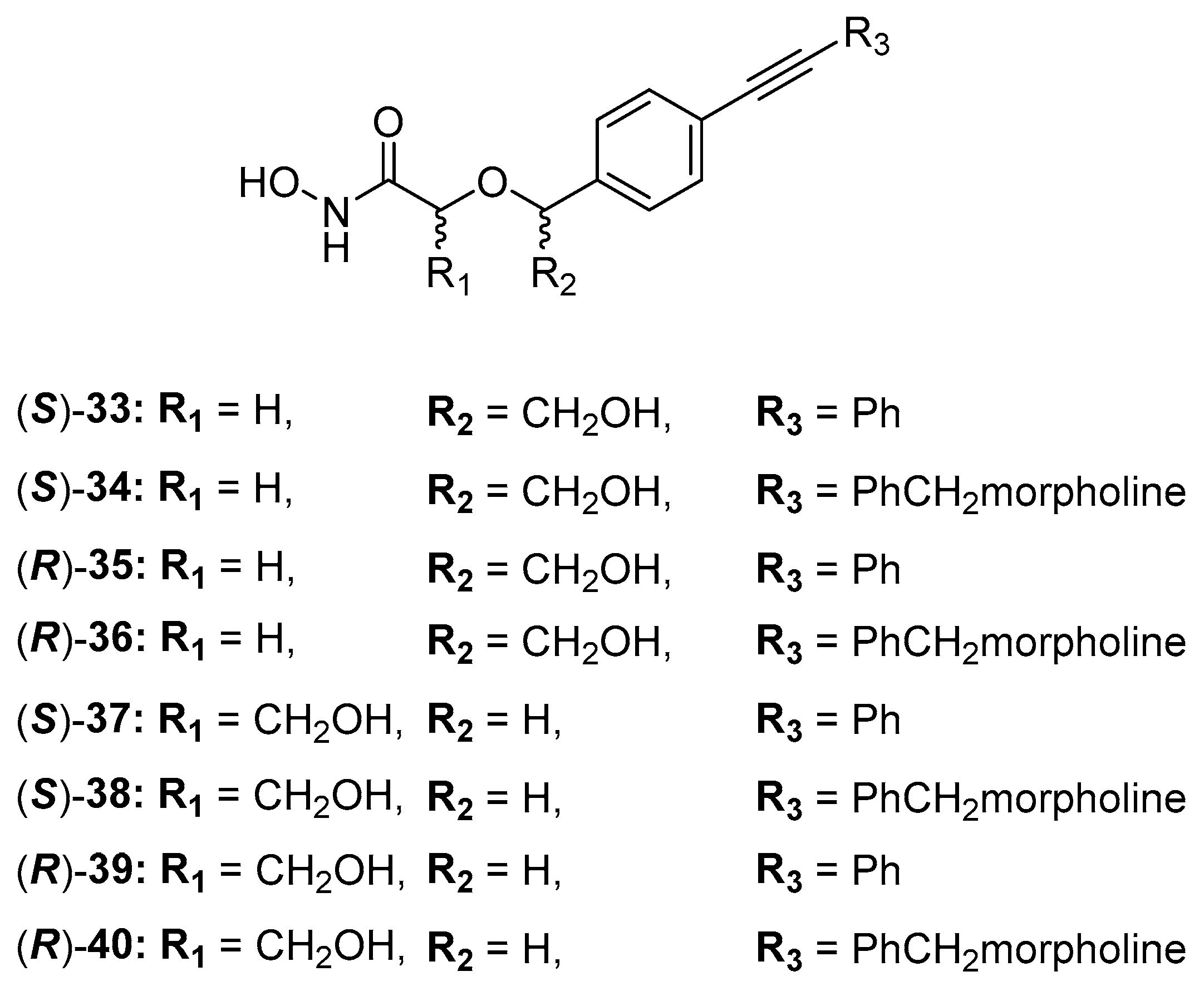
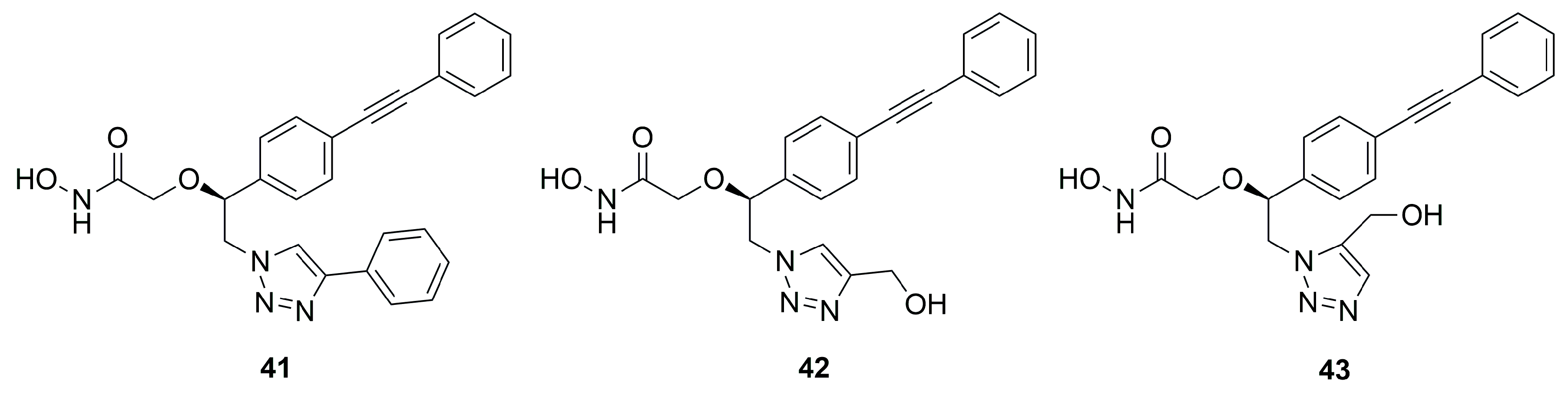
C-Furanose Derivatives as LpxC Inhibitors
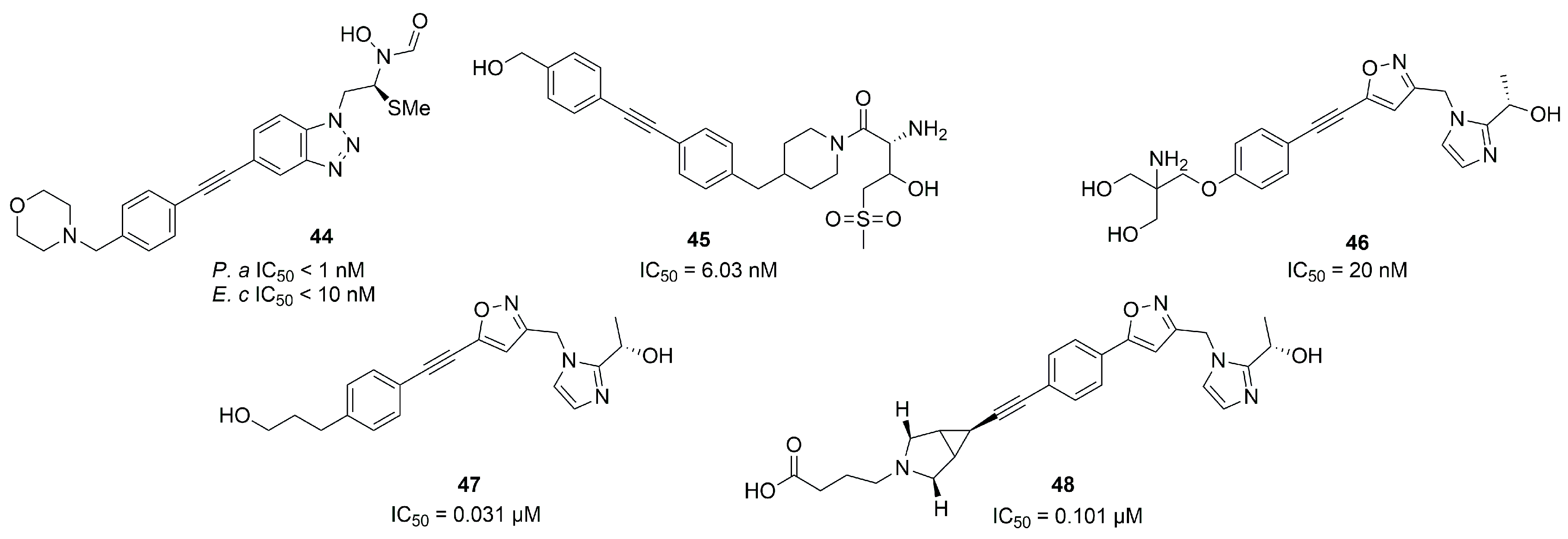
2.1.2. Non-Hydroxamate LpxC Inhibitors
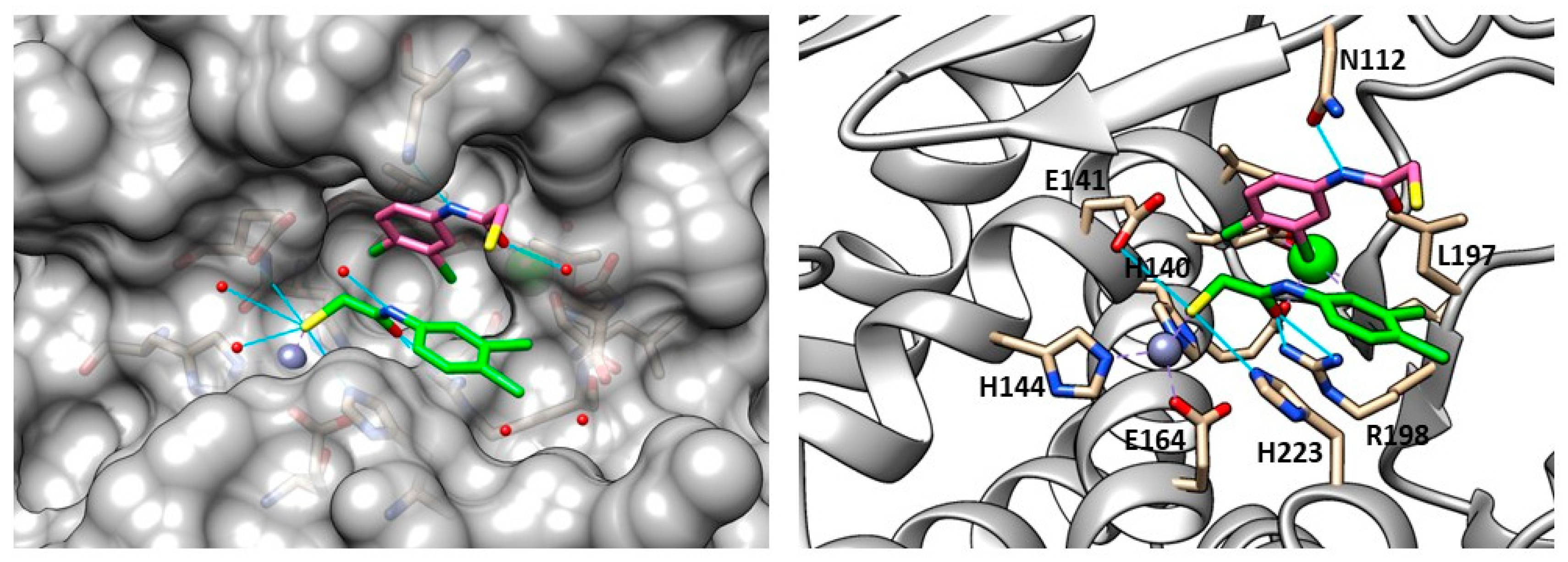
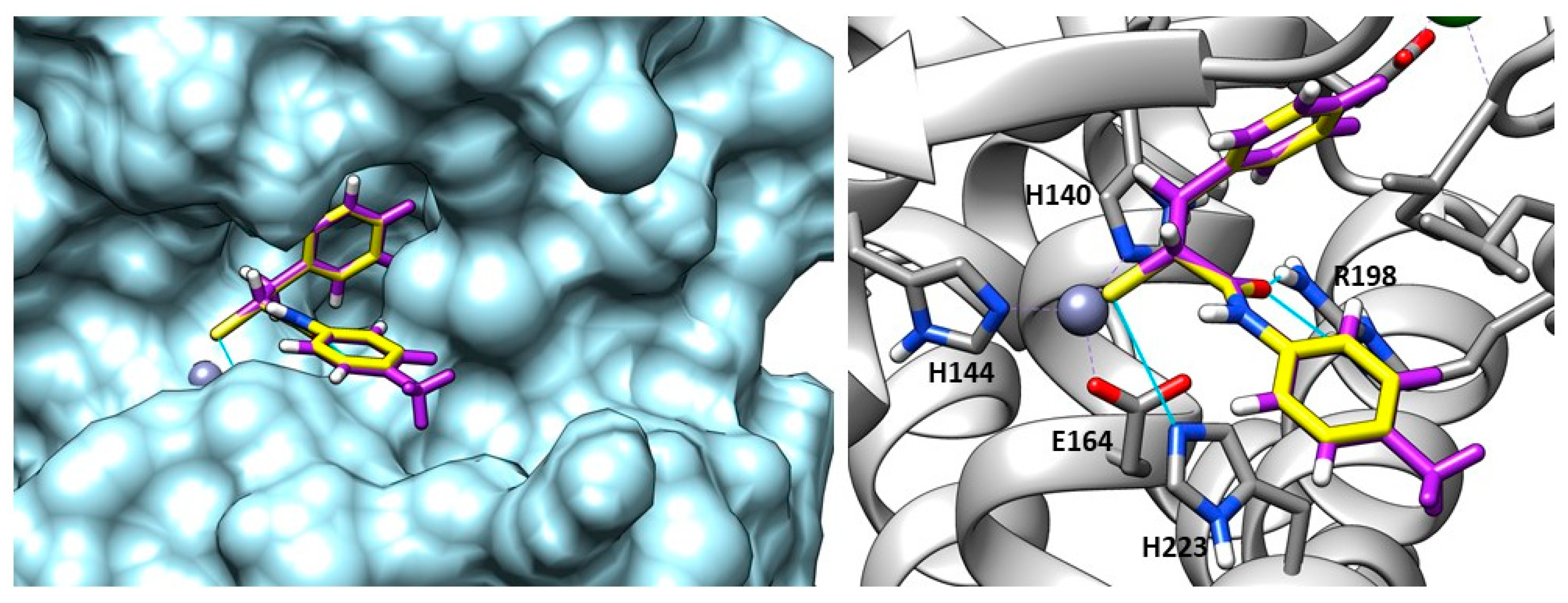
2.2. Inhibitors of Pseudolysin and Thermolysin
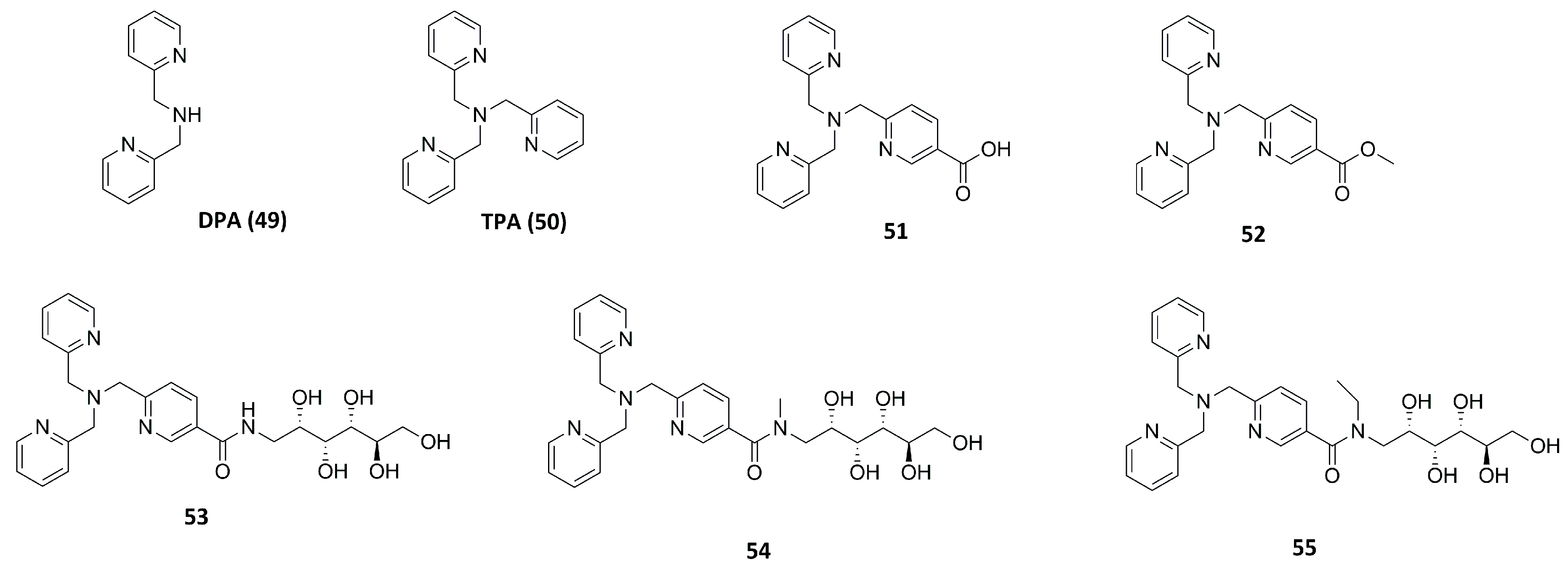
2.2.1. Dipicolylamine (DPA) and Tripicolylamine (TPA)-Based Inhibitors
2.2.2. Inhibitors with Sulfur-Based ZBGs
Thiol Derivatives
Mercaptosuccinimide Analogues
2.2.3. Hydroxamic Acid-Based Inhibitors
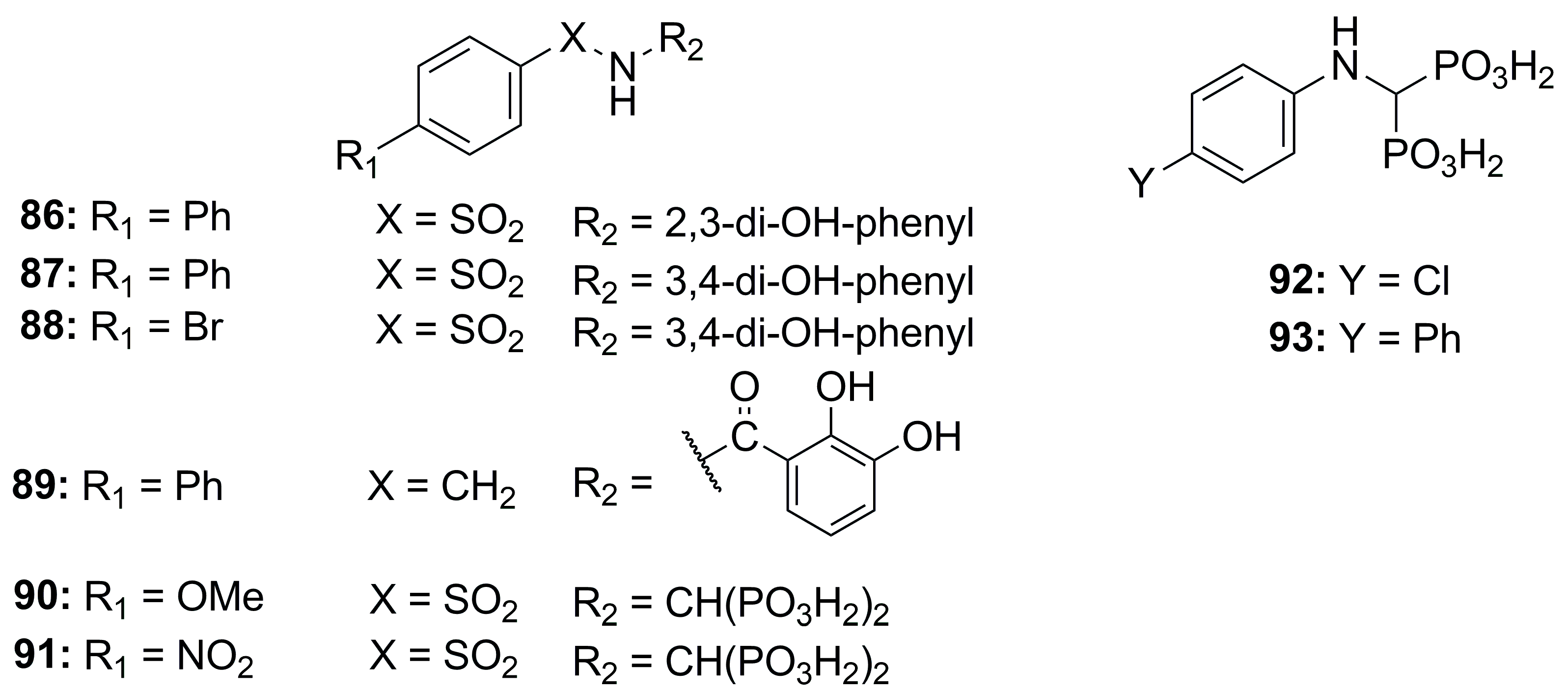
2.2.4. Bisphosphonate-, Catechol- and Carboxylate-Based Inhibitors
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Rodríguez-Alvarado, M.; Russo, R.; Connell, N.D.; Brenner-Moyer, S.E. Design, Organocatalytic Synthesis, and Bioactivity Evaluation of Enantiopure Fluorinated LpxC Inhibitors. Org. Biomol. Chem. 2020, 18, 5867–5878. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.; Sartelli, M.; McKimm, J.; Abu Bakar, M.B. Health Care-Associated Infections – an Overview. Infect. Drug Resist. 2018, 11, 2321–2333. [Google Scholar] [CrossRef]
- Khan, H.A.; Baig, F.K.; Mehboob, R. Nosocomial Infections: Epidemiology, Prevention, Control and Surveillance. Asian Pac. J. Trop. Biomed. 2017, 7, 478–482. [Google Scholar] [CrossRef]
- Holmes, C.L.; Anderson, M.T.; Mobley, H.L.T.; Bachman, M.A. Pathogenesis of Gram-Negative Bacteremia. Clin. Microbiol. Rev. 2021, 34, e00234-20. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. Lancet Infect. Dis. 2010, 10, 303–304. [Google Scholar] [CrossRef]
- Seifert, H. The Clinical Importance of Microbiological Findings in the Diagnosis and Management of Bloodstream Infections. Clin. Infect. Dis. 2009, 48, S238–S245. [Google Scholar] [CrossRef]
- Cerceo, E.; Deitelzweig, S.B.; Sherman, B.M.; Amin, A.N. Multidrug-Resistant Gram-Negative Bacterial Infections in the Hospital Setting: Overview, Implications for Clinical Practice, and Emerging Treatment Options. Microb. Drug Resist. 2016, 22, 412–431. [Google Scholar] [CrossRef]
- Iacopetta, D.; Rosano, C.; Sirignano, M.; Mariconda, A.; Ceramella, J.; Ponassi, M.; Saturnino, C.; Sinicropi, M.S.; Longo, P. Is the Way to Fight Cancer Paved with Gold? Metal-Based Carbene Complexes with Multiple and Fascinating Biological Features. Pharmaceuticals 2020, 13, 91. [Google Scholar] [CrossRef]
- Nikaido, H. Multidrug Resistance in Bacteria. Annu. Rev. Biochem. 2009, 78, 119–146. [Google Scholar] [CrossRef]
- Tanwar, J.; Das, S.; Fatima, Z.; Hameed, S. Multidrug Resistance: An Emerging Crisis. Interdiscip. Perspect. Infect. Dis. 2014, 2014, 541340. [Google Scholar] [CrossRef]
- Loeffler, J.; Stevens, D.A. Antifungal Drug Resistance. Clin. Infect. Dis. 2003, 36, S31–S41. [Google Scholar] [CrossRef]
- Lee, C.-R.; Cho, I.H.; Jeong, B.C.; Lee, S.H. Strategies to Minimize Antibiotic Resistance. Int. J. Environ. Res. Public Health 2013, 10, 4274–4305. [Google Scholar] [CrossRef]
- Yang, X.; Ye, W.; Qi, Y.; Ying, Y.; Xia, Z. Overcoming Multidrug Resistance in Bacteria Through Antibiotics Delivery in Surface-Engineered Nano-Cargos: Recent Developments for Future Nano-Antibiotics. Front. Bioeng. Biotechnol. 2021, 9, 696514. [Google Scholar] [CrossRef]
- Malouin, F.; Bryan, L.E. Modification of Penicillin-Binding Proteins as Mechanisms of Beta-Lactam Resistance. Antimicrob. Agents Chemother. 1986, 30, 1–5. [Google Scholar] [CrossRef]
- Bockstael, K.; Aerschot, A. Antimicrobial Resistance in Bacteria. Open Med. 2009, 4, 141–155. [Google Scholar] [CrossRef]
- Catalano, A.; Iacopetta, D.; Ceramella, J.; Scumaci, D.; Giuzio, F.; Saturnino, C.; Aquaro, S.; Rosano, C.; Sinicropi, M.S. Multidrug Resistance (MDR): A Widespread Phenomenon in Pharmacological Therapies. Molecules 2022, 27, 616. [Google Scholar] [CrossRef]
- Kalinin, D.V.; Holl, R. LpxC Inhibitors: A Patent Review (2010–2016). Expert Opin. Ther. Pat. 2017, 27, 1227–1250. [Google Scholar] [CrossRef]
- Adekoya, O.A.; Sjøli, S.; Wuxiuer, Y.; Bilto, I.; Marques, S.M.; Santos, M.A.; Nuti, E.; Cercignani, G.; Rossello, A.; Winberg, J.-O.; et al. Inhibition of Pseudolysin and Thermolysin by Hydroxamate-Based MMP Inhibitors. Eur. J. Med. Chem. 2015, 89, 340–348. [Google Scholar] [CrossRef]
- Supuran, C.T.; Capasso, C. An Overview of the Bacterial Carbonic Anhydrases. Metabolites 2017, 7, 56. [Google Scholar] [CrossRef]
- Nocentini, A.; Cuffaro, D.; Ciccone, L.; Orlandini, E.; Nencetti, S.; Nuti, E.; Rossello, A.; Supuran, C.T. Activation of Carbonic Anhydrases from Human Brain by Amino Alcohol Oxime Ethers: Towards Human Carbonic Anhydrase VII Selective Activators. J. Enzym. Inhib. Med. Chem. 2021, 36, 48–57. [Google Scholar] [CrossRef]
- Yedery, R.D.; Jerse, A.E. Augmentation of Cationic Antimicrobial Peptide Production with Histone Deacetylase Inhibitors as a Novel Epigenetic Therapy for Bacterial Infections. Antibiotics 2015, 4, 44–61. [Google Scholar] [CrossRef] [PubMed]
- Konovalova, A.; Kahne, D.E.; Silhavy, T.J. Outer Membrane Biogenesis. Annu. Rev. Microbiol. 2017, 71, 539–556. [Google Scholar] [CrossRef] [PubMed]
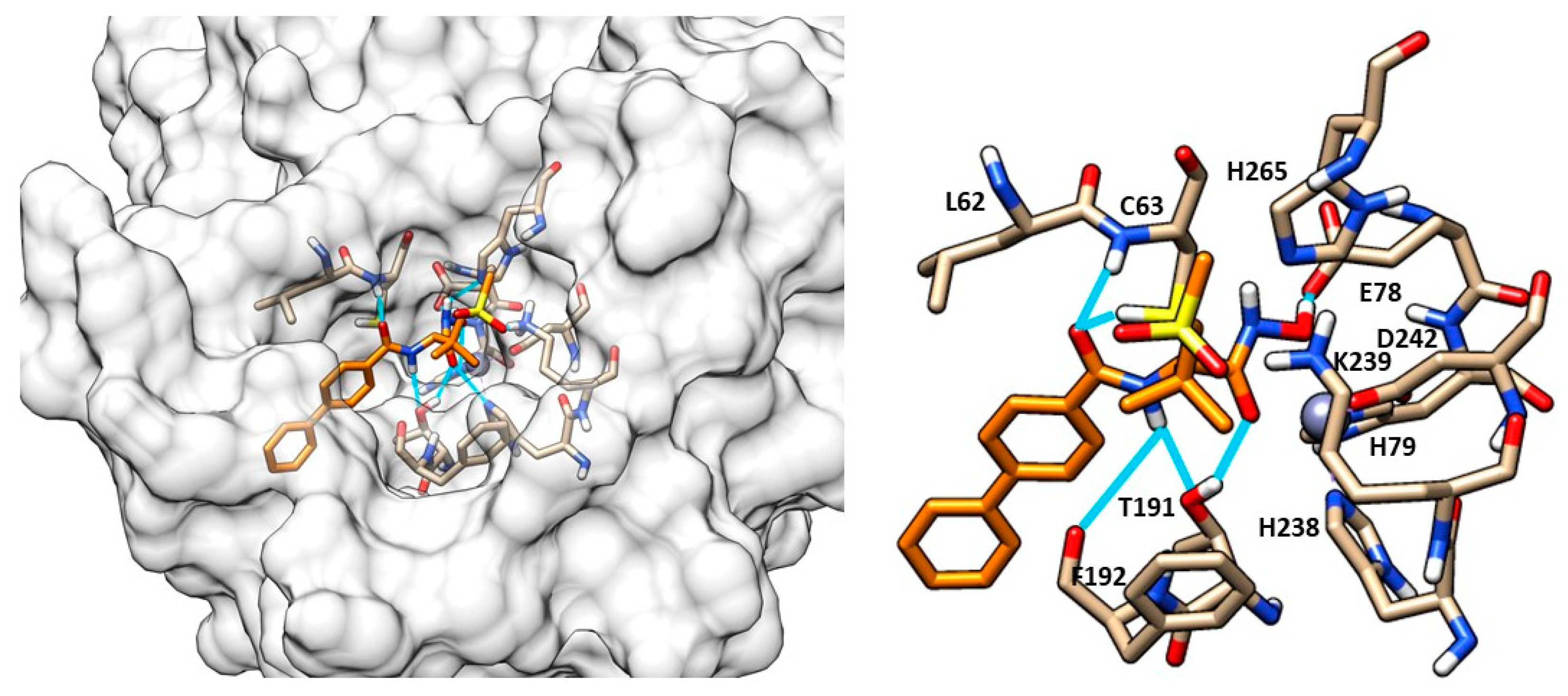
- Clayton, G.M.; Klein, D.J.; Rickert, K.W.; Patel, S.B.; Kornienko, M.; Zugay-Murphy, J.; Reid, J.C.; Tummala, S.; Sharma, S.; Singh, S.B.; et al. Structure of the Bacterial Deacetylase LpxC Bound to the Nucleotide Reaction Product Reveals Mechanisms of Oxyanion Stabilization and Proton Transfer. J. Biol. Chem. 2013, 288, 34073–34080. [Google Scholar] [CrossRef]
- Raetz, C.R.H.; Garrett, T.A.; Reynolds, C.M.; Shaw, W.A.; Moore, J.D.; Smith, D.C.; Ribeiro, A.A.; Murphy, R.C.; Ulevitch, R.J.; Fearns, C.; et al. Kdo2-Lipid A of Escherichia Coli, a Defined Endotoxin That Activates Macrophages via TLR-4. J. Lipid Res. 2006, 47, 1097–1111. [Google Scholar] [CrossRef] [PubMed]
- Bürger, M.; Chory, J. Structural and Chemical Biology of Deacetylases for Carbohydrates, Proteins, Small Molecules and Histones. Commun. Biol. 2018, 1, 217. [Google Scholar] [CrossRef]
- Miller, S.I.; Ernst, R.K.; Bader, M.W. LPS, TLR4 and Infectious Disease Diversity. Nat. Rev. Microbiol. 2005, 3, 36–46. [Google Scholar] [CrossRef]
- Zhou, P.; Barb, A.W. Mechanism and Inhibition of LpxC: An Essential Zinc-Dependent Deacetylase of Bacterial Lipid A Synthesis. Curr. Pharm. Biotechnol. 2008, 9, 9–15. [Google Scholar] [CrossRef]
- Abdel-Magid, A.F. LpxC Inhibitors as Effective Therapy Against Multidrug Resistant Bacterial Infections. ACS Med. Chem. Lett. 2015, 6, 1095–1096. [Google Scholar] [CrossRef]
- Barrett, A.J. Proteases. Curr. Protoc. Protein Sci. 2000, 21, 21.1.1–21.1.12. [Google Scholar] [CrossRef]
- Cerdà-Costa, N.; Xavier Gomis-Rüth, F. Architecture and Function of Metallopeptidase Catalytic Domains: Metallopeptidase Catalytic Domains. Protein Sci. 2014, 23, 123–144. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS Database of Proteolytic Enzymes, Their Substrates and Inhibitors in 2017 and a Comparison with Peptidases in the PANTHER Database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Adekoya, O.A.; Sylte, I. The Thermolysin Family (M4) of Enzymes: Therapeutic and Biotechnological Potential. Chem. Biol. Drug Des. 2009, 73, 7–16. [Google Scholar] [CrossRef]
- Hasan, R.; Rony, M.N.H.; Ahmed, R. In Silico Characterization and Structural Modeling of Bacterial Metalloprotease of Family M4. J. Genet. Eng. Biotechnol. 2021, 19, 25. [Google Scholar] [CrossRef]
- Sjøli, S.; Nuti, E.; Camodeca, C.; Bilto, I.; Rossello, A.; Winberg, J.-O.; Sylte, I.; Adekoya, O.A. Synthesis, Experimental Evaluation and Molecular Modelling of Hydroxamate Derivatives as Zinc Metalloproteinase Inhibitors. Eur. J. Med. Chem. 2016, 108, 141–153. [Google Scholar] [CrossRef]
- Camodeca, C.; Cuffaro, D.; Nuti, E.; Rossello, A. ADAM Metalloproteinases as Potential Drug Targets. Curr. Med. Chem. 2019, 26, 2661–2689. [Google Scholar] [CrossRef]
- Calligaris, M.; Cuffaro, D.; Bonelli, S.; Spanò, D.P.; Rossello, A.; Nuti, E.; Scilabra, S.D. Strategies to Target ADAM17 in Disease: From Its Discovery to the IRhom Revolution. Molecules 2021, 26, 944. [Google Scholar] [CrossRef]
- Rahman, F.; Wushur, I.; Malla, N.; Åstrand, O.A.H.; Rongved, P.; Winberg, J.-O.; Sylte, I. Zinc-Chelating Compounds as Inhibitors of Human and Bacterial Zinc Metalloproteases. Molecules 2021, 27, 56. [Google Scholar] [CrossRef]
- Cuffaro, D.; Nuti, E.; D’Andrea, F.; Rossello, A. Developments in Carbohydrate-Based Metzincin Inhibitors. Pharmaceuticals 2020, 13, 376. [Google Scholar] [CrossRef]
- Dreger, A.; Hoff, K.; Agoglitta, O.; Hotop, S.-K.; Brönstrup, M.; Heisig, P.; Kirchmair, J.; Holl, R. Antibacterial Activity of Xylose-Derived LpxC Inhibitors–Synthesis, Biological Evaluation and Molecular Docking Studies. Bioorg. Chem. 2021, 107, 104603. [Google Scholar] [CrossRef]
- Odunuga, O.O.; Adekoya, O.A.; Sylte, I. High-Level Expression of Pseudolysin, the Extracellular Elastase of Pseudomonas aeruginosa, in Escherichia Coli and Its Purification. Protein Expr. Purif. 2015, 113, 79–84. [Google Scholar] [CrossRef]
- Galloway, D.R. Pseudomonas aeruginosa Elastase and Elastolysis Revisited: Recent Developments. Mol. Microbiol. 1991, 5, 2315–2321. [Google Scholar] [CrossRef] [PubMed]
- Komori, Y.; Nonogaki, T.; Nikai, T. Hemorrhagic Activity and Muscle Damaging Effect of Pseudomonas aeruginosa Metalloproteinase (Elastase). Toxicon 2001, 39, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Yamamoto, T. Pathogenic Mechanisms Induced by Microbial Proteases in Microbial Infections. Biol. Chem. Hoppe-Seyler 1996, 377, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Holder, I.A.; Neely, A.N. Pseudomonas Elastase Acts as a Virulence Factor in Burned Hosts by Hageman Factor-Dependent Activation of the Host Kinin Cascade. Infect. Immun. 1989, 57, 3345–3348. [Google Scholar] [CrossRef] [PubMed]
- Azghani, A.O.; Miller, E.J.; Peterson, B.T. Virulence Factors from Pseudomonas aeruginosa Increase Lung Epithelial Permeability. Lung 2000, 178, 261–269. [Google Scholar] [CrossRef]
- Wilson, R.; Dowling, R.B. Pseudomonas aeruginosa and Other Related Species. Thorax 1998, 53, 213–219. [Google Scholar] [CrossRef]
- Everett, M.J.; Davies, D.T. Pseudomonas aeruginosa Elastase (LasB) as a Therapeutic Target. Drug Discov. Today 2021, 26, 2108–2123. [Google Scholar] [CrossRef]
- Casilag, F.; Lorenz, A.; Krueger, J.; Klawonn, F.; Weiss, S.; Häussler, S. The LasB Elastase of Pseudomonas aeruginosa Acts in Concert with Alkaline Protease AprA To Prevent Flagellin-Mediated Immune Recognition. Infect. Immun. 2016, 84, 162–171. [Google Scholar] [CrossRef]
- Ezawa, T.; Sugiyama, S.; Ara, A.; Sylte, I.; Kurita, N. Design of Galardine Analogs as Putative Psudolysin Inhibitors Based on Ab Initio Fragment Molecular Orbital Calculations. J. Biomol. Struct. Dyn. 2020, 38, 3307–3317. [Google Scholar] [CrossRef]
- Ezawa, T.; Saito, R.; Suzuki, S.; Sugiyama, S.; Sylte, I.; Kurita, N. Protonation States of Central Amino Acids in a Zinc Metalloprotease Complexed with Inhibitor: Molecular Mechanics Optimizations and Ab Initio Molecular Orbital Calculations. Biophys. Chem. 2020, 261, 106368. [Google Scholar] [CrossRef]
- Imai, K.; Saito, R.; Ezawa, T.; Sugiyama, S.; Sylte, I.; Kurita, N. Proposal of Selective Inhibitor for Bacterial Zinc Metalloprotease: Molecular Mechanics and Ab Initio Molecular Orbital Calculations. J. Mol. Graph. Model. 2022, 110, 108047. [Google Scholar] [CrossRef]
- Cuffaro, D.; Ciccone, L.; Rossello, A.; Nuti, E.; Santamaria, S. Targeting Aggrecanases for Osteoarthritis Therapy: From Zinc Chelation to Exosite Inhibition. J. Med. Chem. 2022, 65, 13505–13532. [Google Scholar] [CrossRef]
- Nuti, E.; Cuffaro, D.; Bernardini, E.; Camodeca, C.; Panelli, L.; Chaves, S.; Ciccone, L.; Tepshi, L.; Vera, L.; Orlandini, E.; et al. Development of Thioaryl-Based Matrix Metalloproteinase-12 Inhibitors with Alternative Zinc-Binding Groups: Synthesis, Potentiometric, NMR, and Crystallographic Studies. J. Med. Chem. 2018, 61, 4421–4435. [Google Scholar] [CrossRef]
- Jacobsen, J.A.; Major Jourden, J.L.; Miller, M.T.; Cohen, S.M. To Bind Zinc or Not to Bind Zinc: An Examination of Innovative Approaches to Improved Metalloproteinase Inhibition. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2010, 1803, 72–94. [Google Scholar] [CrossRef]
- Cuffaro, D.; Camodeca, C.; D’Andrea, F.; Piragine, E.; Testai, L.; Calderone, V.; Orlandini, E.; Nuti, E.; Rossello, A. Matrix Metalloproteinase-12 Inhibitors: Synthesis, Structure-Activity Relationships and Intestinal Absorption of Novel Sugar-Based Biphenylsulfonamide Carboxylates. Bioorg. Med. Chem. 2018, 26, 5804–5815. [Google Scholar] [CrossRef]
- Nuti, E.; Rossello, A.; Cuffaro, D.; Camodeca, C.; Van Bael, J.; van der Maat, D.; Martens, E.; Fiten, P.; Pereira, R.V.S.; Ugarte-Berzal, E.; et al. Bivalent Inhibitor with Selectivity for Trimeric MMP-9 Amplifies Neutrophil Chemotaxis and Enables Functional Studies on MMP-9 Proteoforms. Cells 2020, 9, 1634. [Google Scholar] [CrossRef]
- Nuti, E.; Rosalia, L.; Cuffaro, D.; Camodeca, C.; Giacomelli, C.; Da Pozzo, E.; Tuccinardi, T.; Costa, B.; Antoni, C.; Vera, L.; et al. Bifunctional Inhibitors as a New Tool To Reduce Cancer Cell Invasion by Impairing MMP-9 Homodimerization. ACS Med. Chem. Lett. 2017, 8, 293–298. [Google Scholar] [CrossRef]
- Cuffaro, D.; Camodeca, C.; Tuccinardi, T.; Ciccone, L.; Bartsch, J.W.; Kellermann, T.; Cook, L.; Nuti, E.; Rossello, A. Discovery of Dimeric Arylsulfonamides as Potent ADAM8 Inhibitors. ACS Med. Chem. Lett. 2021, 12, 1787–1793. [Google Scholar] [CrossRef]
- Pece, R.; Tavella, S.; Costa, D.; Varesano, S.; Camodeca, C.; Cuffaro, D.; Nuti, E.; Rossello, A.; Alfano, M.; D’Arrigo, C.; et al. Inhibitors of ADAM10 Reduce Hodgkin Lymphoma Cell Growth in 3D Microenvironments and Enhance Brentuximab-Vedotin Effect. Haematologica 2021, 107, 909–920. [Google Scholar] [CrossRef]
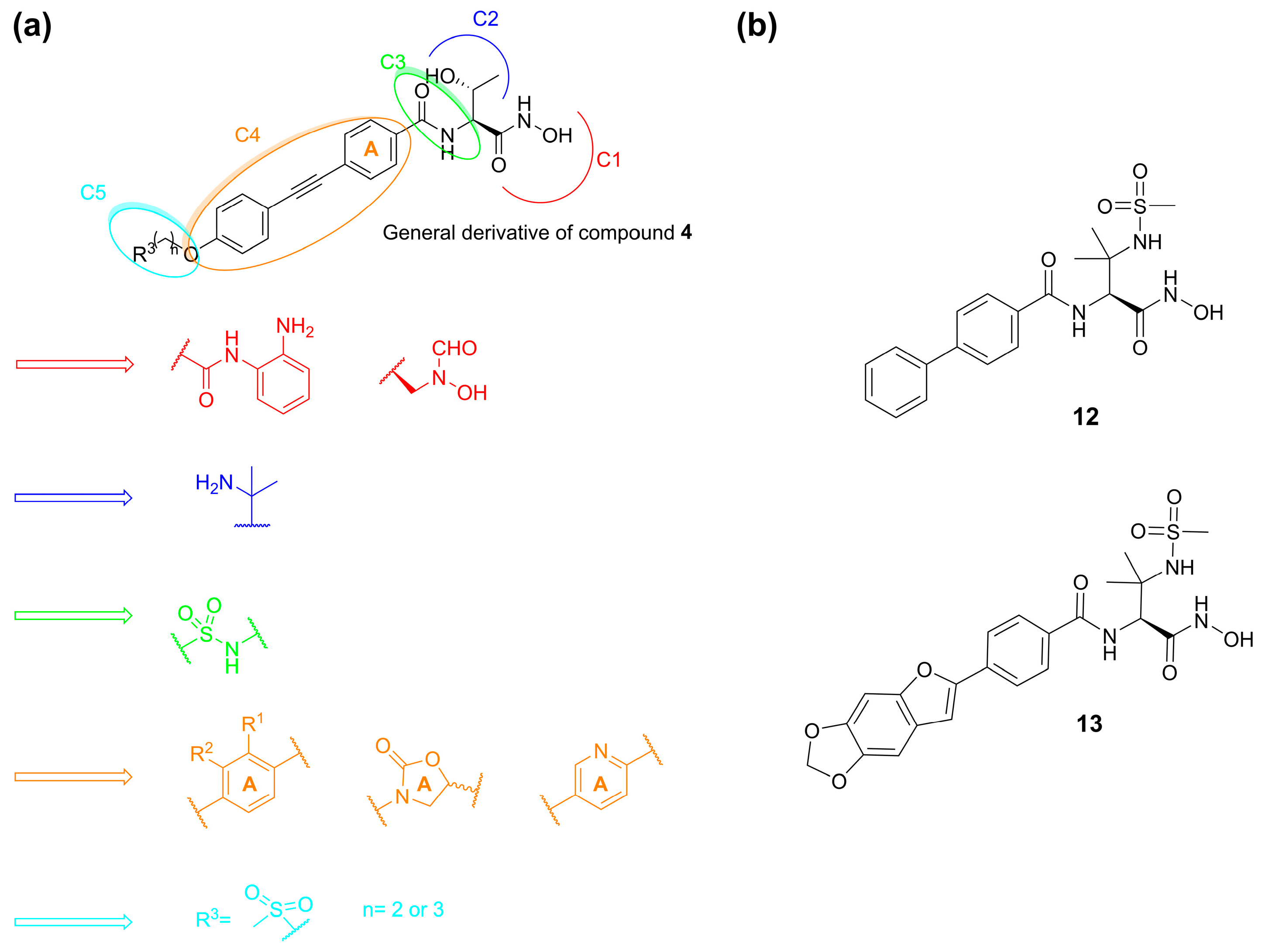
- Piizzi, G.; Parker, D.T.; Peng, Y.; Dobler, M.; Patnaik, A.; Wattanasin, S.; Liu, E.; Lenoir, F.; Nunez, J.; Kerrigan, J.; et al. Design, Synthesis, and Properties of a Potent Inhibitor of Pseudomonas aeruginosa Deacetylase LpxC. J. Med. Chem. 2017, 60, 5002–5014. [Google Scholar] [CrossRef]
- Ding, S.; Dai, R.-Y.; Wang, W.-K.; Cao, Q.; Lan, L.-F.; Zhou, X.-L.; Yang, Y.-S. Design, Synthesis and Structure-Activity Relationship Evaluation of Novel LpxC Inhibitors as Gram-Negative Antibacterial Agents. Bioorg. Med. Chem. Lett. 2018, 28, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.H.; Vidaillac, C.; Yam, J.K.H.; Chua, S.L.; Givskov, M.; Yang, L. In Vitro and In Vivo Efficacy of an LpxC Inhibitor, CHIR-090, Alone or Combined with Colistin against Pseudomonas aeruginosa Biofilm. Antimicrob. Agents Chemother. 2017, 61, e02223-16. [Google Scholar] [CrossRef] [PubMed]
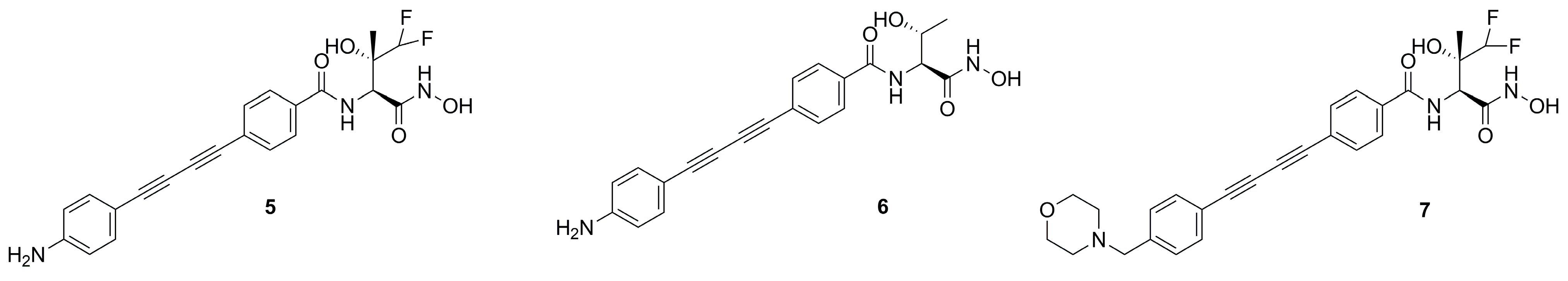
- Titecat, M.; Liang, X.; Lee, C.-J.; Charlet, A.; Hocquet, D.; Lambert, T.; Pagès, J.-M.; Courcol, R.; Sebbane, F.; Toone, E.J.; et al. High Susceptibility of MDR and XDR Gram-Negative Pathogens to Biphenyl-Diacetylene-Based Difluoromethyl-allo-Threonyl-Hydroxamate LpxC Inhibitors. J. Antimicrob. Chemother. 2016, 71, 2874–2882. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Gopalaswamy, R.; Navas, F.; Toone, E.J.; Zhou, P. A Scalable Synthesis of the Difluoromethyl-allo-Threonyl Hydroxamate-Based LpxC Inhibitor LPC-058. J. Org. Chem. 2016, 81, 4393–4398. [Google Scholar] [CrossRef] [PubMed]
- Panchaud, P.; Surivet, J.-P.; Diethelm, S.; Blumstein, A.-C.; Gauvin, J.-C.; Jacob, L.; Masse, F.; Mathieu, G.; Mirre, A.; Schmitt, C.; et al. Optimization of LpxC Inhibitor Lead Compounds Focusing on Efficacy and Formulation for High Dose Intravenous Administration. J. Med. Chem. 2020, 63, 88–102. [Google Scholar] [CrossRef]
- Surivet, J.-P.; Panchaud, P.; Specklin, J.-L.; Diethelm, S.; Blumstein, A.-C.; Gauvin, J.-C.; Jacob, L.; Masse, F.; Mathieu, G.; Mirre, A.; et al. Discovery of Novel Inhibitors of LpxC Displaying Potent in Vitro Activity against Gram-Negative Bacteria. J. Med. Chem. 2020, 63, 66–87. [Google Scholar] [CrossRef]
- Ding, S.; Wang, W.-K.; Cao, Q.; Chu, W.-J.; Lan, L.-F.; Hu, W.-H.; Yang, Y.-S. Design, Synthesis and Biological Evaluation of LpxC Inhibitors with Novel Hydrophilic Terminus. Chin. Chem. Lett. 2015, 26, 763–767. [Google Scholar] [CrossRef]
- Kawai, T.; Kazuhiko, I.; Takaya, N.; Yamaguchi, Y.; Kishii, R.; Kohno, Y.; Kurasaki, H. Sulfonamide-Based Non-Alkyne LpxC Inhibitors as Gram-Negative Antibacterial Agents. Bioorg. Med. Chem. Lett. 2017, 27, 1045–1049. [Google Scholar] [CrossRef]
- Krause, K.M.; Haglund, C.M.; Hebner, C.; Serio, A.W.; Lee, G.; Nieto, V.; Cohen, F.; Kane, T.R.; Machajewski, T.D.; Hildebrandt, D.; et al. Potent LpxC Inhibitors with In Vitro Activity against Multidrug-Resistant Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2019, 63, e00977-19. [Google Scholar] [CrossRef]
- Cohen, F.; Aggen, J.B.; Andrews, L.D.; Assar, Z.; Boggs, J.; Choi, T.; Dozzo, P.; Easterday, A.N.; Haglund, C.M.; Hildebrandt, D.J.; et al. Optimization of LpxC Inhibitors for Antibacterial Activity and Cardiovascular Safety. ChemMedChem 2019, 14, 1560–1572. [Google Scholar] [CrossRef]
- Zhang, J.; Chan, A.; Lippa, B.; Cross, J.B.; Liu, C.; Yin, N.; Romero, J.A.C.; Lawrence, J.; Heney, R.; Herradura, P.; et al. Structure-Based Discovery of LpxC Inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 1670–1680. [Google Scholar] [CrossRef]
- Ding, S.; Ji, J.; Zhang, M.; Yang, Y.; Wang, R.; Zhu, X.; Wang, L.; Zhong, Y.; Gao, L.; Lu, M.; et al. Exploration of the Structure–Activity Relationship and Druggability of Novel Oxazolidinone-based Compounds as Gram-negative Antibacterial Agents. Arch. Pharm. Chem. Life Sci. 2019, 352, 1900129. [Google Scholar] [CrossRef]
- Kurasaki, H.; Tsuda, K.; Shinoyama, M.; Takaya, N.; Yamaguchi, Y.; Kishii, R.; Iwase, K.; Ando, N.; Nomura, M.; Kohno, Y. LpxC Inhibitors: Design, Synthesis, and Biological Evaluation of Oxazolidinones as Gram-Negative Antibacterial Agents. ACS Med. Chem. Lett. 2016, 7, 623–628. [Google Scholar] [CrossRef]
- Lee, P.S.; Lapointe, G.; Madera, A.M.; Simmons, R.L.; Xu, W.; Yifru, A.; Tjandra, M.; Karur, S.; Rico, A.; Thompson, K.; et al. Application of Virtual Screening to the Identification of New LpxC Inhibitor Chemotypes, Oxazolidinone and Isoxazoline. J. Med. Chem. 2018, 61, 9360–9370. [Google Scholar] [CrossRef]
- Jana, S.K.; Löppenberg, M.; Daniliuc, C.G.; Holl, R. Synthesis and Biological Evaluation of C-Ethynyl Furanosides as LpxC Inhibitors. Tetrahedron 2015, 71, 956–966. [Google Scholar] [CrossRef]
- Müller, H.; Gabrielli, V.; Agoglitta, O.; Holl, R. Chiral Pool Synthesis and Biological Evaluation of C-Furanosidic and Acyclic LpxC Inhibitors. Eur. J. Med. Chem. 2016, 110, 340–375. [Google Scholar] [CrossRef]
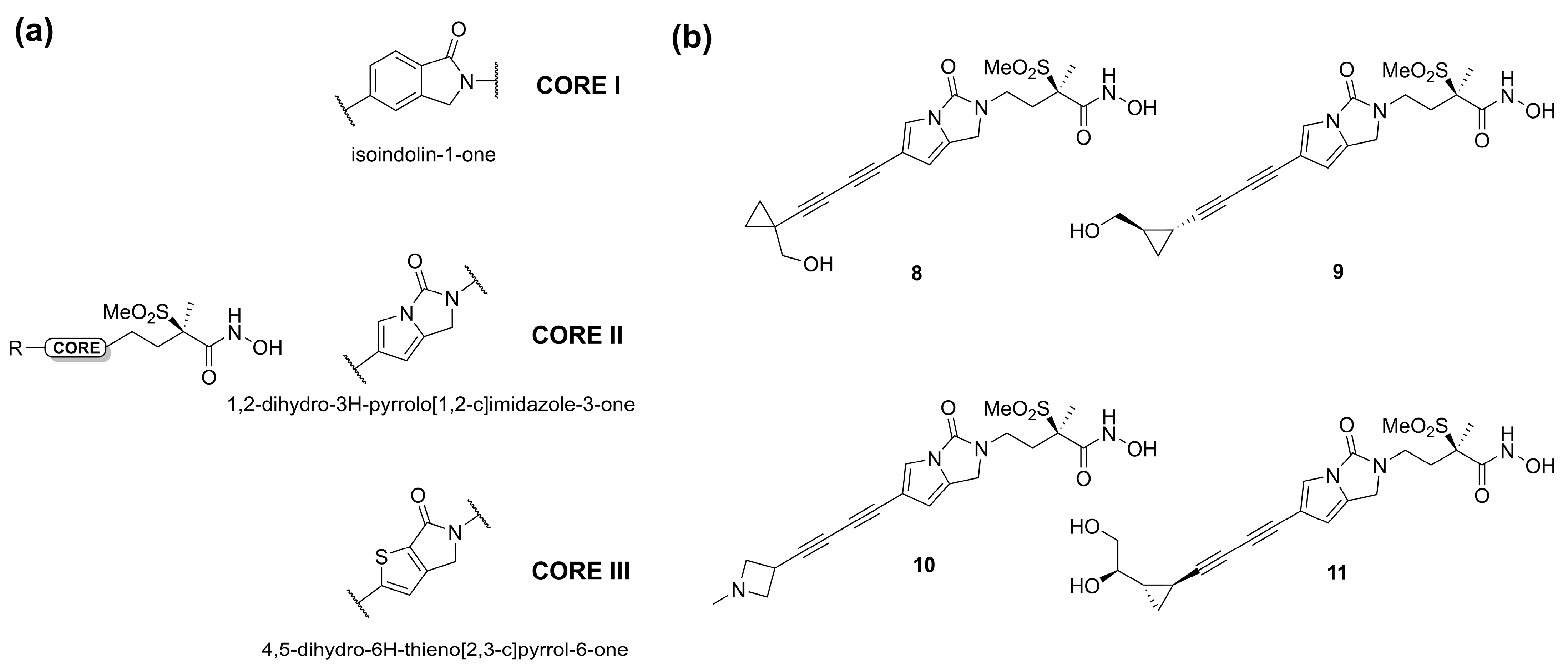
- Dreger, A.; Hoff, K.; Agoglitta, O.; Bülbül, E.F.; Melesina, J.; Sippl, W.; Holl, R. Synthesis, Biological Evaluation, and Molecular Docking Studies of Deoxygenated C-Glycosides as LpxC Inhibitors. Bioorg. Chem. 2021, 117, 105403. [Google Scholar] [CrossRef]
- Tangherlini, G.; Torregrossa, T.; Agoglitta, O.; Köhler, J.; Melesina, J.; Sippl, W.; Holl, R. Synthesis and Biological Evaluation of Enantiomerically Pure Glyceric Acid Derivatives as LpxC Inhibitors. Bioorg. Med. Chem. 2016, 24, 1032–1044. [Google Scholar] [CrossRef]
- Hoff, K.; Mielniczuk, S.; Agoglitta, O.; Iorio, M.T.; Caldara, M.; Bülbül, E.F.; Melesina, J.; Sippl, W.; Holl, R. Synthesis and Biological Evaluation of Triazolyl-Substituted Benzyloxyacetohydroxamic Acids as LpxC Inhibitors. Bioorg. Med. Chem. 2020, 28, 115529. [Google Scholar] [CrossRef]
- Galster, M.; Löppenberg, M.; Galla, F.; Börgel, F.; Agoglitta, O.; Kirchmair, J.; Holl, R. Phenylethylene Glycol-Derived LpxC Inhibitors with Diverse Zn2+-Binding Groups. Tetrahedron 2019, 75, 486–509. [Google Scholar] [CrossRef]
- Nencetti, S.; Cuffaro, D.; Nuti, E.; Ciccone, L.; Rossello, A.; Fabbi, M.; Ballante, F.; Ortore, G.; Carbotti, G.; Campelli, F.; et al. Identification of Histone Deacetylase Inhibitors with (Arylidene)Aminoxy Scaffold Active in Uveal Melanoma Cell Lines. J. Enzym. Inhib. Med. Chem. 2021, 36, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Kozikowski, A.P. Why Hydroxamates May Not Be the Best Histone Deacetylase Inhibitors-What Some May Have Forgotten or Would Rather Forget? ChemMedChem 2016, 11, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Fields, G.B. New Strategies for Targeting Matrix Metalloproteinases. Matrix Biol. 2015, 44–46, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Furuya, T.; Shapiro, A.B.; Comita-Prevoir, J.; Kuenstner, E.J.; Zhang, J.; Ribe, S.D.; Chen, A.; Hines, D.; Moussa, S.H.; Carter, N.M.; et al. N-Hydroxyformamide LpxC Inhibitors, Their in Vivo Efficacy in a Mouse Escherichia Coli Infection Model, and Their Safety in a Rat Hemodynamic Assay. Bioorg. Med. Chem. 2020, 28, 115826. [Google Scholar] [CrossRef]
- Yamada, Y.; Takashima, H.; Walmsley, D.L.; Ushiyama, F.; Matsuda, Y.; Kanazawa, H.; Yamaguchi-Sasaki, T.; Tanaka-Yamamoto, N.; Yamagishi, J.; Kurimoto-Tsuruta, R.; et al. Fragment-Based Discovery of Novel Non-Hydroxamate LpxC Inhibitors with Antibacterial Activity. J. Med. Chem. 2020, 63, 14805–14820. [Google Scholar] [CrossRef]
- Ushiyama, F.; Takashima, H.; Matsuda, Y.; Ogata, Y.; Sasamoto, N.; Kurimoto-Tsuruta, R.; Ueki, K.; Tanaka-Yamamoto, N.; Endo, M.; Mima, M.; et al. Lead Optimization of 2-Hydroxymethyl Imidazoles as Non-Hydroxamate LpxC Inhibitors: Discovery of TP0586532. Bioorg. Med. Chem. 2021, 30, 115964. [Google Scholar] [CrossRef]
- Yoshida, I.; Takata, I.; Fujita, K.; Takashima, H.; Sugiyama, H. TP0586532, a Novel Non-Hydroxamate LpxC Inhibitor: Potentiating Effect on In Vitro Activity of Meropenem against Carbapenem-Resistant Enterobacteriaceae. Microbiol. Spectr. 2022, 10, e00828-22. [Google Scholar] [CrossRef]
- Fujita, K.; Takata, I.; Yoshida, I.; Takashima, H.; Sugiyama, H. TP0586532, a Non-Hydroxamate LpxC Inhibitor, Reduces LPS Release and IL-6 Production Both in Vitro and in Vivo. J. Antibiot. 2022, 75, 136–145. [Google Scholar] [CrossRef]
- Zhu, J.; Cai, X.; Harris, T.L.; Gooyit, M.; Wood, M.; Lardy, M.; Janda, K.D. Disarming Pseudomonas aeruginosa Virulence Factor LasB by Leveraging a Caenorhabditis Elegans Infection Model. Chem. Biol. 2015, 22, 483–491. [Google Scholar] [CrossRef]
- Kany, A.M.; Sikandar, A.; Haupenthal, J.; Yahiaoui, S.; Maurer, C.K.; Proschak, E.; Köhnke, J.; Hartmann, R.W. Binding Mode Characterization and Early in Vivo Evaluation of Fragment-Like Thiols as Inhibitors of the Virulence Factor LasB from Pseudomonas aeruginosa. ACS Infect. Dis. 2018, 4, 988–997. [Google Scholar] [CrossRef]
- Kaya, C.; Walter, I.; Alhayek, A.; Shafiei, R.; Jézéquel, G.; Andreas, A.; Konstantinović, J.; Schönauer, E.; Sikandar, A.; Haupenthal, J.; et al. Structure-Based Design of α-Substituted Mercaptoacetamides as Inhibitors of the Virulence Factor LasB from Pseudomonas aeruginosa. ACS Infect. Dis. 2022, 8, 1010–1021. [Google Scholar] [CrossRef]
- Konstantinović, J.; Yahiaoui, S.; Alhayek, A.; Haupenthal, J.; Schönauer, E.; Andreas, A.; Kany, A.M.; Müller, R.; Koehnke, J.; Berger, F.K.; et al. N-Aryl-3-Mercaptosuccinimides as Antivirulence Agents Targeting Pseudomonas aeruginosa Elastase and Clostridium Collagenases. J. Med. Chem. 2020, 63, 8359–8368. [Google Scholar] [CrossRef]
- Kany, A.M.; Sikandar, A.; Yahiaoui, S.; Haupenthal, J.; Walter, I.; Empting, M.; Köhnke, J.; Hartmann, R.W. Tackling Pseudomonas aeruginosa Virulence by a Hydroxamic Acid-Based LasB Inhibitor. ACS Chem. Biol. 2018, 13, 2449–2455. [Google Scholar] [CrossRef]
- Sylte, I.; Dawadi, R.; Malla, N.; von Hofsten, S.; Nguyen, T.-M.; Solli, A.I.; Berg, E.; Adekoya, O.A.; Svineng, G.; Winberg, J.-O. The Selectivity of Galardin and an Azasugar-Based Hydroxamate Compound for Human Matrix Metalloproteases and Bacterial Metalloproteases. PLoS ONE 2018, 13, e0200237. [Google Scholar] [CrossRef]
- Rahman, F.; Nguyen, T.-M.; Adekoya, O.A.; Campestre, C.; Tortorella, P.; Sylte, I.; Winberg, J.-O. Inhibition of Bacterial and Human Zinc-Metalloproteases by Bisphosphonate- and Catechol-Containing Compounds. J. Enzym. Inhib. Med. Chem. 2021, 36, 819–830. [Google Scholar] [CrossRef]
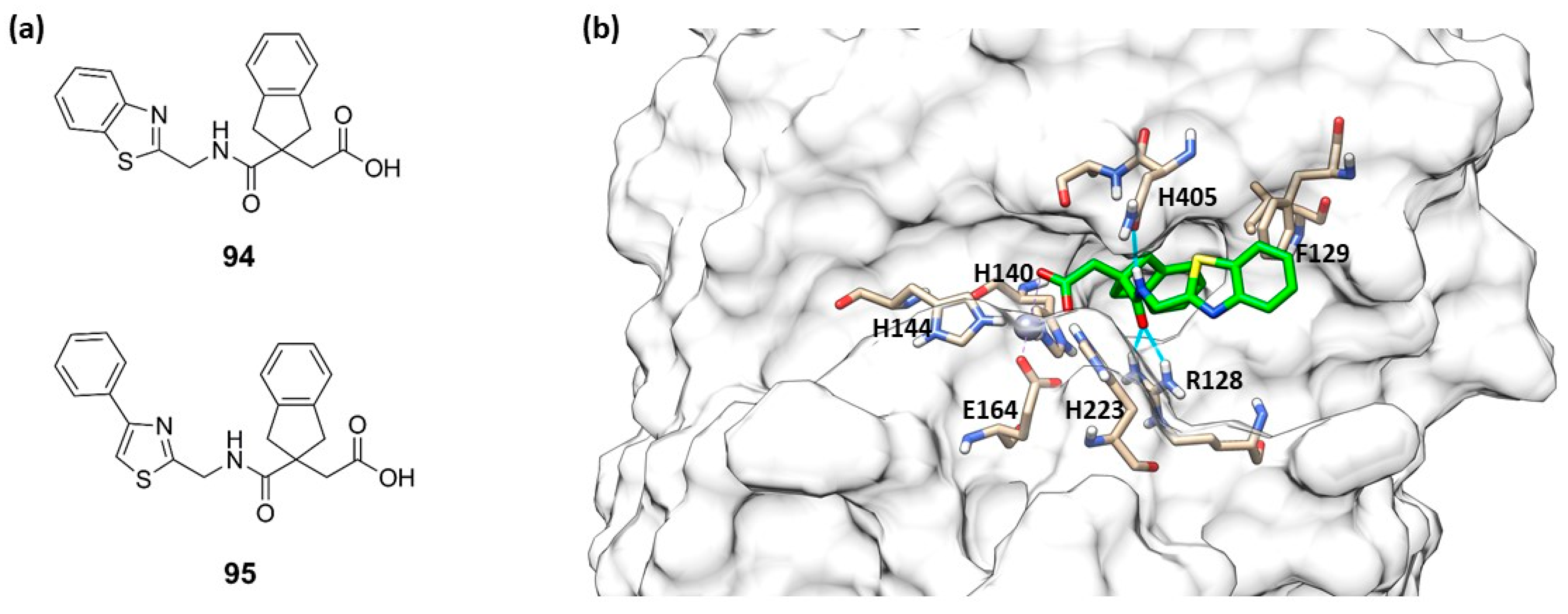
- Leiris, S.; Davies, D.T.; Sprynski, N.; Castandet, J.; Beyria, L.; Bodnarchuk, M.S.; Sutton, J.M.; Mullins, T.M.G.; Jones, M.W.; Forrest, A.K.; et al. Virtual Screening Approach to Identifying a Novel and Tractable Series of Pseudomonas aeruginosa Elastase Inhibitors. ACS Med. Chem. Lett. 2021, 12, 217–227. [Google Scholar] [CrossRef]






























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
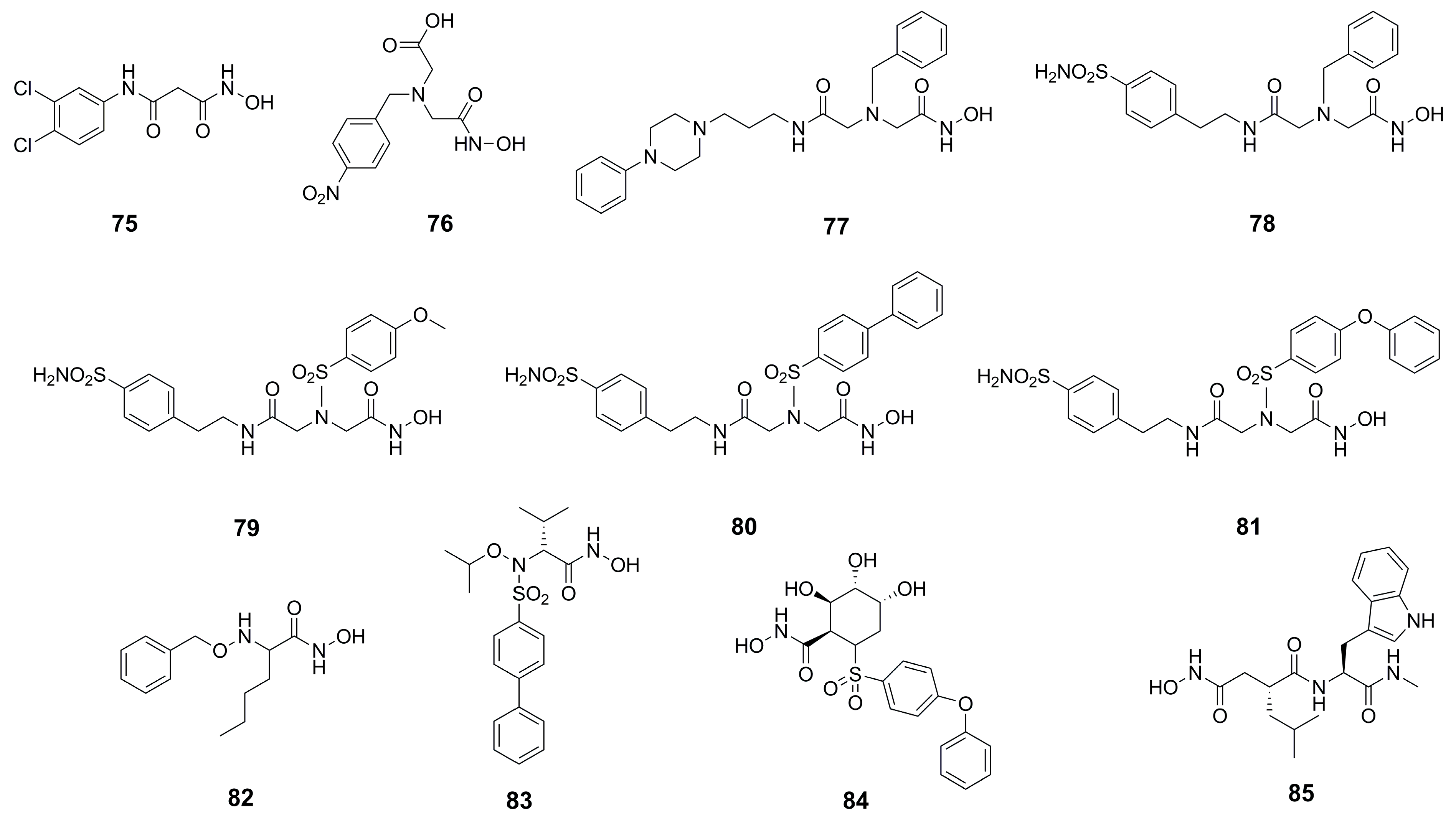{kind=link}
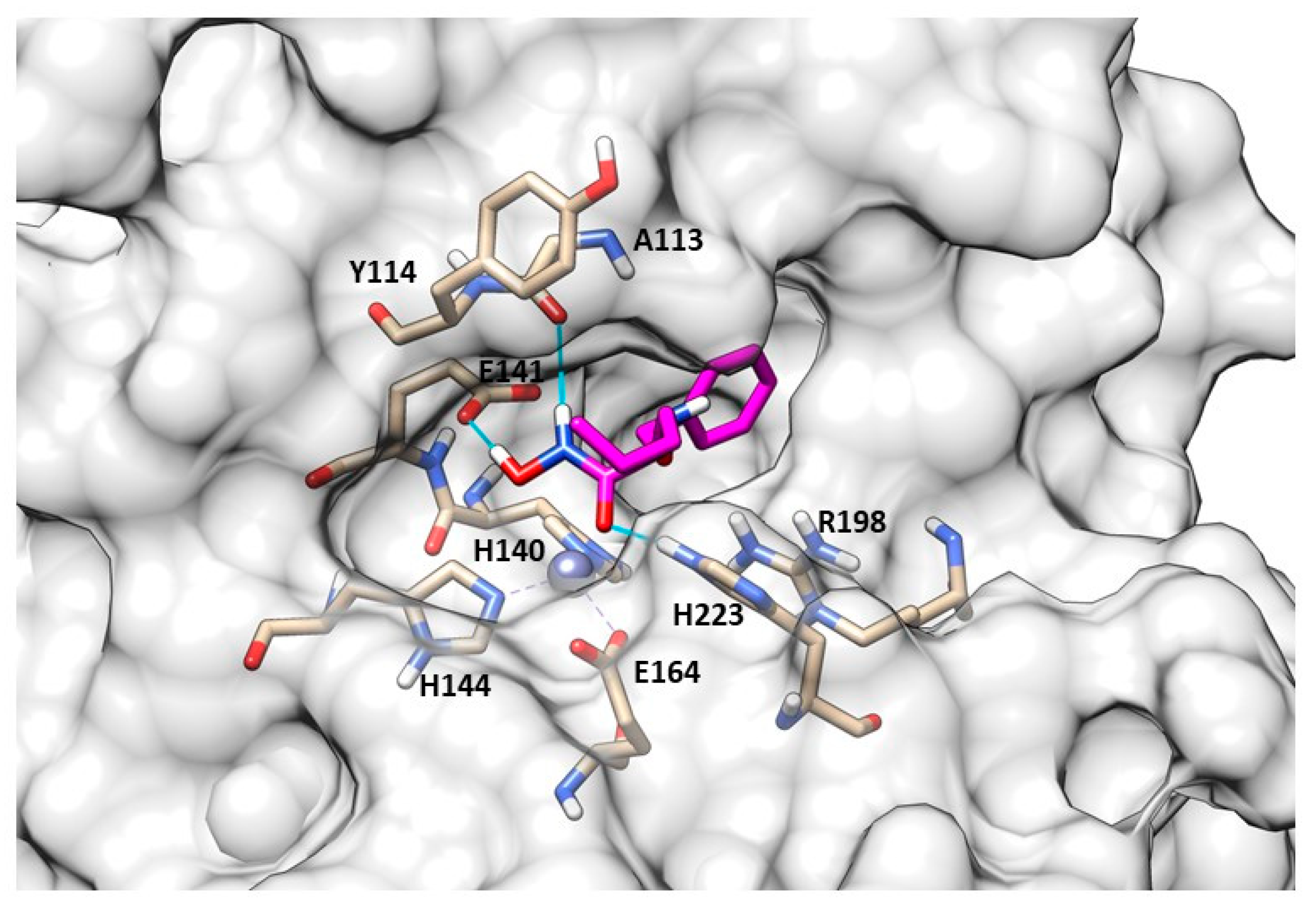{kind=link}
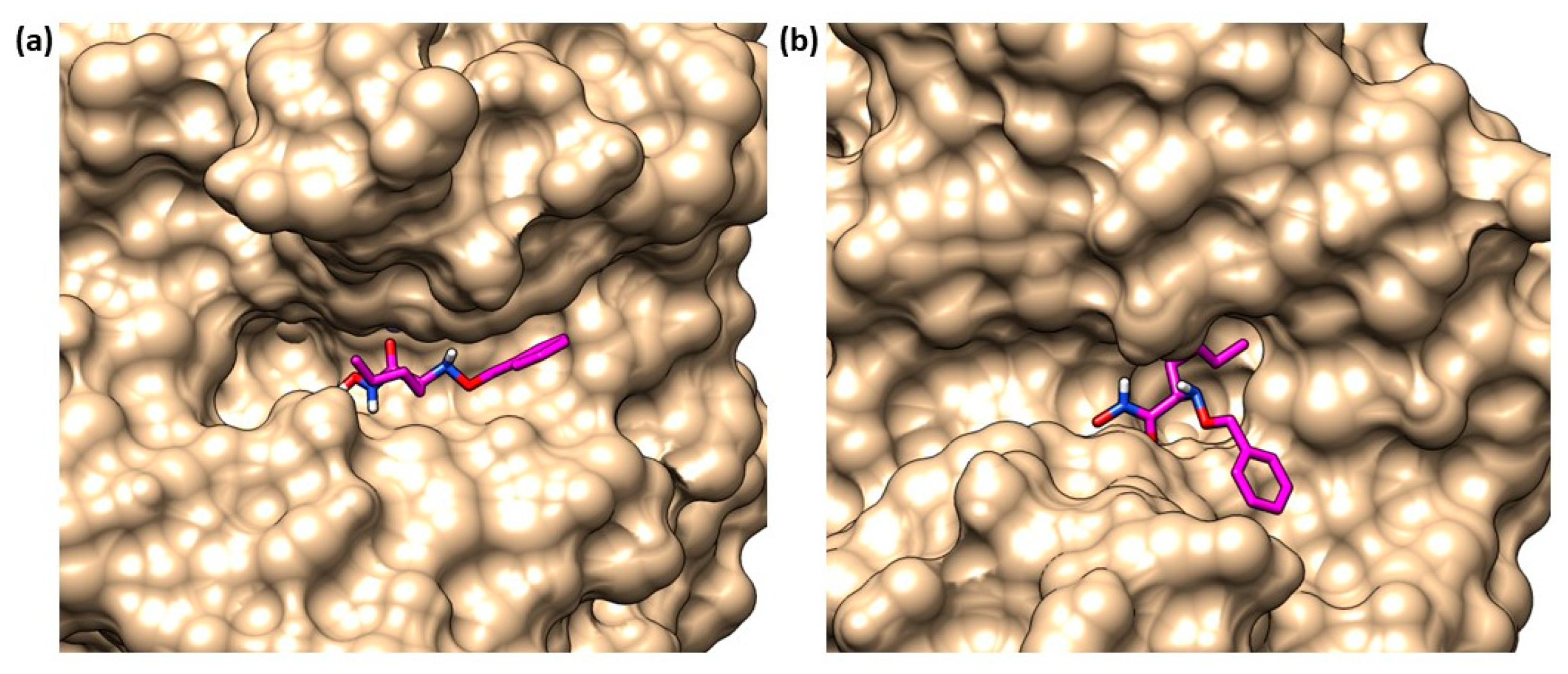{kind=link}
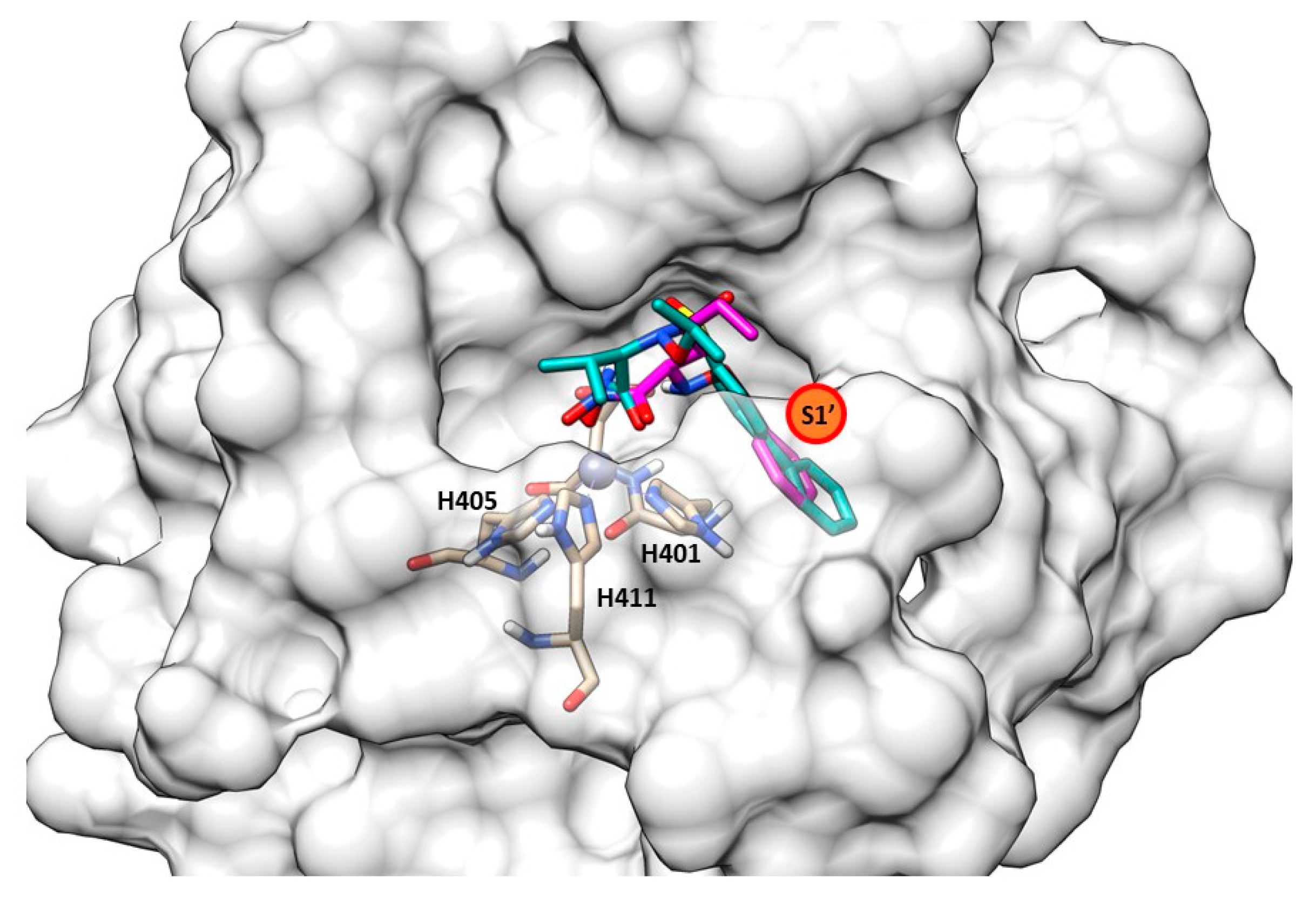{kind=link}
{kind=link}
{kind=link}
| Name of Bacterium | Drug(s) Resistant to | Typical Diseases |
|---|---|---|
| Escherichia coli | Cephalosporins and fluoroquinolones | Urinary tract infections and blood stream infections |
| Klebsiella pneumonia | Cephalosporins and carbapenems | Pneumonia, blood stream, and urinary tract infections |
| Staphylococcus aureus | Methicillin | Wound and blood stream infections |
| Streptococcus pneumoniae | Penicillin | Pneumonia, meningitis, and otitis |
| Nontyphoidal salmonella | Fluoroquinolones | Foodborne diarrhea, blood stream infections |
| Pseudomonas aeruginosa | Carbapenems | Chronic ulcers, lung infections |
| Shigella species | Fluoroquinolones | Diarrhea (bacillary dysentery) |
| Neisseria gonorrhoeae | Cephalosporins | Gonorrhea |
| Mycobacterium tuberculosis | Rifampicin, isoniazid, and fluoroquinolone | Tuberculosis |
| Compound | MIC (µg/mL) E. coli | MIC (µg/mL) K. pneumoniae | MIC (µg/mL) P. aeruginosa |
|---|---|---|---|
| 8 | 0.25 | 1 | 0.5 |
| 9 | 0.12 | 0.25 | 0.5 |
| 10 | 0.5 | 0.5 | 0.5 |
| 11 | 0.5 | 1 | 1 |
| Compound | E. coli IC50 (nM) | MIC (µg/mL) E. coli | MIC (µg/mL) K. pneumoniae | MIC (µg/mL) P. aeruginosa |
|---|---|---|---|---|
| 4 | 3 | 0.125 | 0.25 | 0.5 |
| 12 | 1200 | 2 | 64 | 1 |
| 13 | 12 | 0.063 | 0.5 | 0.5 |
| Compound | Configuration | Zone of Inhibition [mm] | Enzyme Assay | ||
|---|---|---|---|---|---|
| E. coli BLB21 (D3) | E. coli D22 | IC50 [µM] | Ki [µM] | ||
| 28 | (2S, 3R, 4S, 5S) | <6 | 13.7 ± 1.8 | >200 | >27.6 |
| 29 | (S, S) | 11.5 ± 1.5 | 24.2 ± 1.5 | 2.6 ± 0.3 | 0.36 ± 0.04 |
| 30 | (S, S) | 13.7 ± 0.6 | 21.3 ± 2.5 | 1.7 ± 0.4 | 0.23 ± 0.05 |
| 31 | (2S, 3S, 4R, 5S) | 22.3 ± 1.4 | 28.3 ± 1.4 | 3.2 ± 1.0 | 0.4 ± 0.1 |
| 32 | (2S, 3R, 5R) | 18.0 ± 1.0 | 25.3 ± 1.5 | 23.7 ± 17.6 | 3.5 ± 2.4 |
| Compound | Configuration | Zone of Inhibition [mm] | Enzyme Assay | ||
|---|---|---|---|---|---|
| E. coli BLB21 (DE3) | E. coli D22 | IC50 [µM] | Ki [µM] | ||
| 29 | (S, S) | 9.0 ± 0.5 | 20.8 ± 0.6 | 2.6 ± 0.3 | 0.36 ± 0.04 |
| 33 | (S) | 9.5 ± 0.4 | 20.5 ± 0.2 | 0.48 ± 0.23 | 0.066 ± 0.032 |
| 34 | (S) | 13.4 ± 0.5 | 21.2 ± 0.6 | 0.69 ± 0.30 | 0.095 ± 0.042 |
| 35 | (R) | 9.1 ± 0.4 | 13.0 ± 1.7 | 31.6 ± 6.0 | 4.4 ± 0.8 |
| 36 | (R) | 8.7 ± 0.7 | 12.3 ± 1.6 | 198 ± 12 | 27.3 ± 1.7 |
| 37 | (S) | 11.7 ± 0.6 | 20.7 ± 1.7 | 1.96 ± 0.36 | 0.27 ± 0.05 |
| 38 | (S) | 15.7 ± 0.6 | 25.8 ± 1.9 | 2.82 ± 0.5 | 0.39 ± 0.07 |
| 39 | (R) | 10.3 ± 2.5 | 17.0 ± 1.0 | 1.87 ± 0.85 | 0.26 ± 0.12 |
| 40 | (R) | 12.3 ± 0.6 | 22.0 ± 1.3 | 1.66 ± 0.31 | 0.23 ± 0.04 |
| Compound | LpxC | Bacterial Strain | Ref. |
|---|---|---|---|
| 4 | 3 | E. coli | [68] |
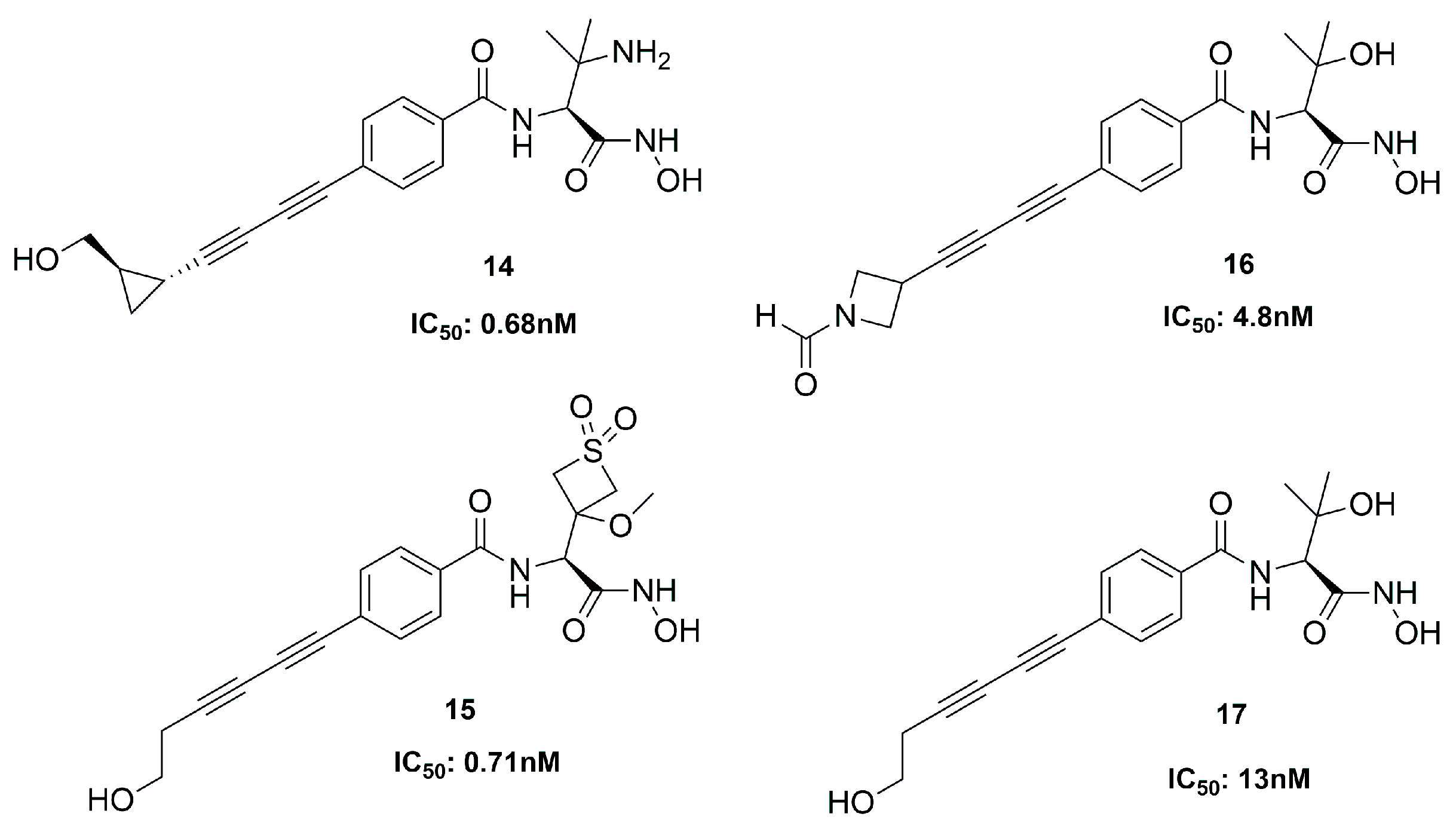
| 14 | 0.68 | P. aeruginosa | [69] |
| 15 | 0.71 | P. aeruginosa | [69] |
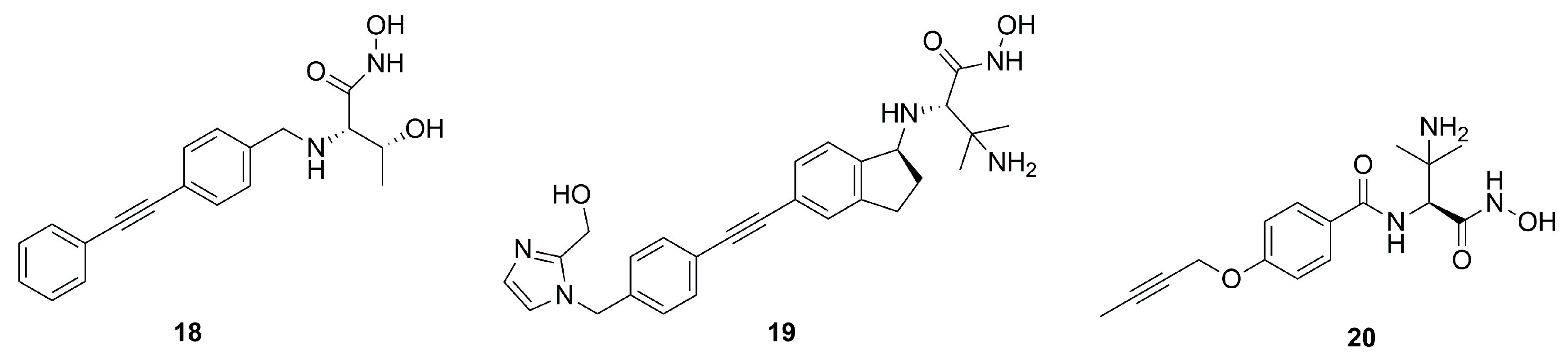
| 20 | 1.5 | P. aeruginosa | [60] |
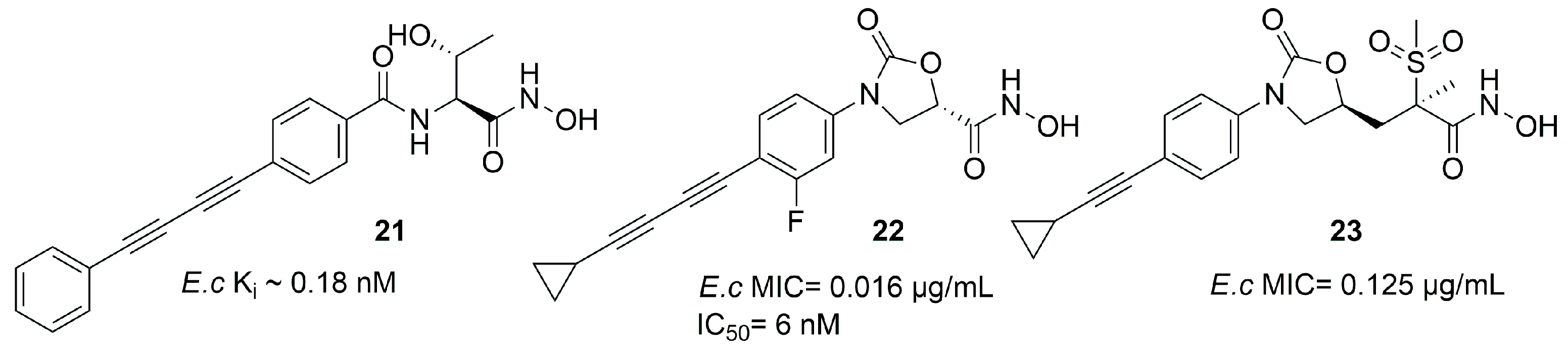
| 22 | 6 | E. coli | [73] |
| 42 | 8500 | E. coli | [79] |
| 46 | 20 | P. aeruginosa | [85] |
| Compound | MMP-14 | MMP-9 | ALN | PLN | TLN |
|---|---|---|---|---|---|
| 49 | n.d. | n.d. | n.d. | 4 ± 1 | n.d. |
| 50 | 1.2 ± 0.1 | 15 ± 4 | 16 ± 1 | 5 ± 2 | 5.5 ± 0.9 |
| 51 | 3.8 ± 0.3 | 21 ± 5 | 25 ± 2 | 4 ± 1 | n.d. * |
| 52 | 1.5 ± 0.1 | n.d. | n.d. | 12 ± 4 | n.d. |
| 53 | 3.5 ± 0.3 | 22 ± 6 | 20 ± 2 | 1.1 ± 0.3 | 12 ± 2 |
| 54 | 8.6 ± 0.7 | n.d. | n.d. | 5 ± 2 | n.d. |
| 55 | 4.5 ± 0.4 | 28 ± 7 | n.d. | 5 ± 2 | 9 ± 2 |
| Compound | Thermolysin | Pseudolysin | MMP-1 | MMP-2 | MMP-9 | MMP-14 | ADAM-17 | |
|---|---|---|---|---|---|---|---|---|
| BLS | BLS | AGLA | ||||||
| 76 | 755 ± 17 | n.d. * | 20 ± 2 | n.d. | 178 ± 6 | >300 | 313 ± 25 | - |
| 77 | 8 ± 1 | 2.8 ± 0.2 | 6.7 ± 0.5 | n.d. | >300 | >300 | >300 | - |
| 78 | 12 ± 1 | n.d. | 0.4 ± 0.3 | n.d. | >100 | >300 | >100 | - |
| 79 | 164 ± 22 | 33 ± 2 | 15 ± 2 | 0.53 ± 0.02 | 0.025 ± 0.002 | 0.054 ± 0.003 | 0.090 ± 0.004 | - |
| 80 | 67 ± 4 | n.d. | 31 ± 13 | 0.54 ± 0.02 | 0.016 ± 0.001 | 0.066 ± 0.006 | 0.26 ± 0.02 | - |
| 81 | 160 ± 31 | 276 ± 18 | 16 ± 2 | 0.14 ± 0.01 | 0.0016 ± 0.0003 | 0.00051 ± 0.00004 | 0.0021 ± 0.0002 | - |
| 82 | 9.5 | 1.9 | 1.1 | - | 34 | 81 | - | 100 |
| 83 | 800 | 798 | 57 | - | 0.0008 | 0.0067 | - | 14 |
| Ki ± sd (µM) | |||||
|---|---|---|---|---|---|
| Compound | TLN | PLN | ALN | MMP-14 | MMP-9(T) |
| 86 | n.d. * | 38 ± 8 | n.d. | 19 ± 0.8 | 51 ± 17 |
| 87 | 13 ± 2 | 9 ± 3 | 49 ± 5 | 6.6 ± 0.6 | 13 ± 2 |
| 88 | 14 ± 5 | 16 ± 3 | n.d. | 8.3 ± 0.6 | 12.6 ± 0.6 |
| 89 | n.d. | n.d. | n.d. | 12 ± 1 | 13 ± 1 |
| 90 | n.d. | 58 ± 4 | n.d. | n.d. | n.d. |
| 91 | 16.2 ± 0.4 | 22 ± 3 | n.d. | n.d. | n.d. |
| 92 | n.d. | 37 ± 6 | n.d. | 17 ± 1 | n.d. |
| 93 | 7 ± 1 | 12 ± 4 | n.d. | 7.2 ± 0.6 | 6.6 ± 0.4 |
| Compound | PLN | TLN | MMP-2 | MMP-9 | Ref. |
|---|---|---|---|---|---|
| 57 | 1.78 ± 0.01 | - a | - | - | [89] |
| 66 | 1.2 ± 0.1 | - | - | - | [91] |
| 67 | 0.48 ± 0.04 | - | - | - | [91] |
| 68 | 0.7 ± 0.03 | - | - | - | [91] |
| 69 | 0.6 ± 0.04 | - | - | - | [91] |
| 78 | 0.4 ± 0.3 | 12 ± 1 | >100 | >300 | [18] |
| 82 | 1.5 b | 9.5 | 34 | 81 | [34] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Leo, R.; Cuffaro, D.; Rossello, A.; Nuti, E. Bacterial Zinc Metalloenzyme Inhibitors: Recent Advances and Future Perspectives. Molecules 2023, 28, 4378. https://doi.org/10.3390/molecules28114378
Di Leo R, Cuffaro D, Rossello A, Nuti E. Bacterial Zinc Metalloenzyme Inhibitors: Recent Advances and Future Perspectives. Molecules. 2023; 28(11):4378. https://doi.org/10.3390/molecules28114378
Chicago/Turabian StyleDi Leo, Riccardo, Doretta Cuffaro, Armando Rossello, and Elisa Nuti. 2023. "Bacterial Zinc Metalloenzyme Inhibitors: Recent Advances and Future Perspectives" Molecules 28, no. 11: 4378. https://doi.org/10.3390/molecules28114378
APA StyleDi Leo, R., Cuffaro, D., Rossello, A., & Nuti, E. (2023). Bacterial Zinc Metalloenzyme Inhibitors: Recent Advances and Future Perspectives. Molecules, 28(11), 4378. https://doi.org/10.3390/molecules28114378