
Synthesis of 1,2-Disubstituted Adamantane Derivatives by Construction of the Adamantane Framework †


Abstract
:
{kind=link}
{kind=link}
{kind=link}
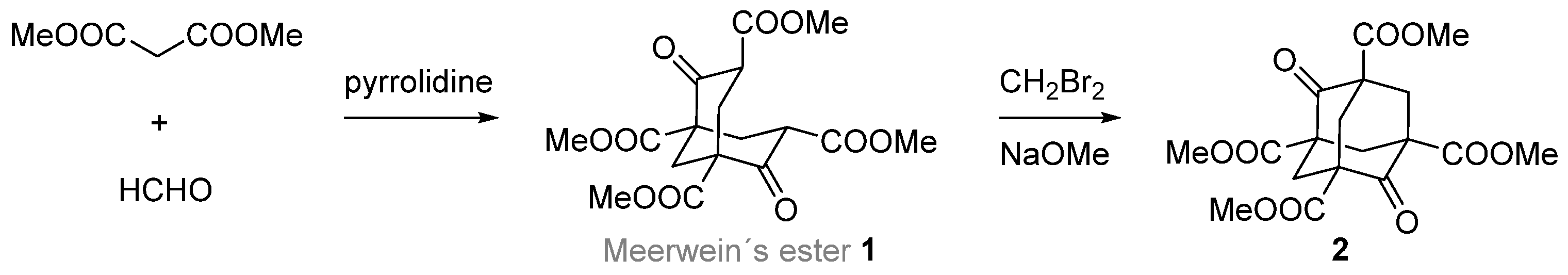{kind=link}
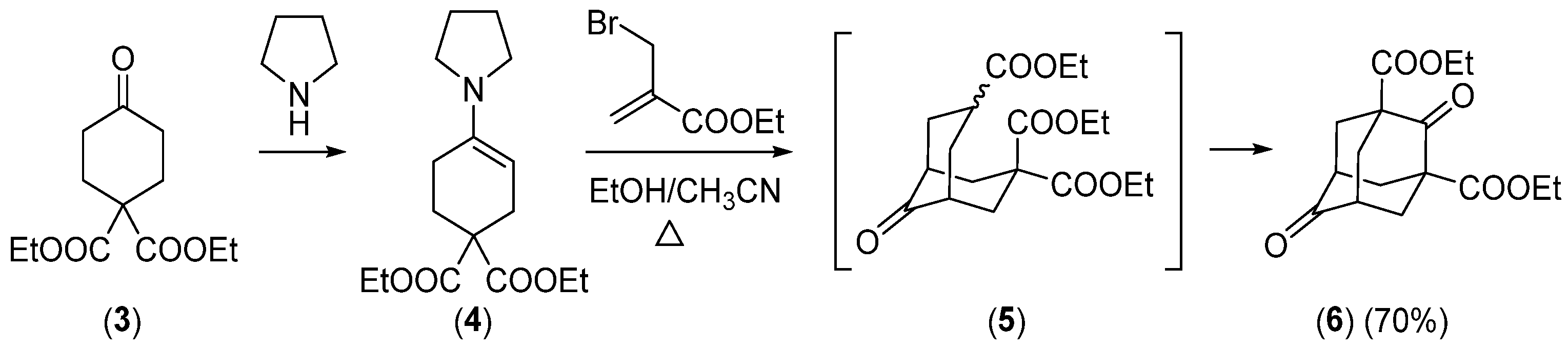{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
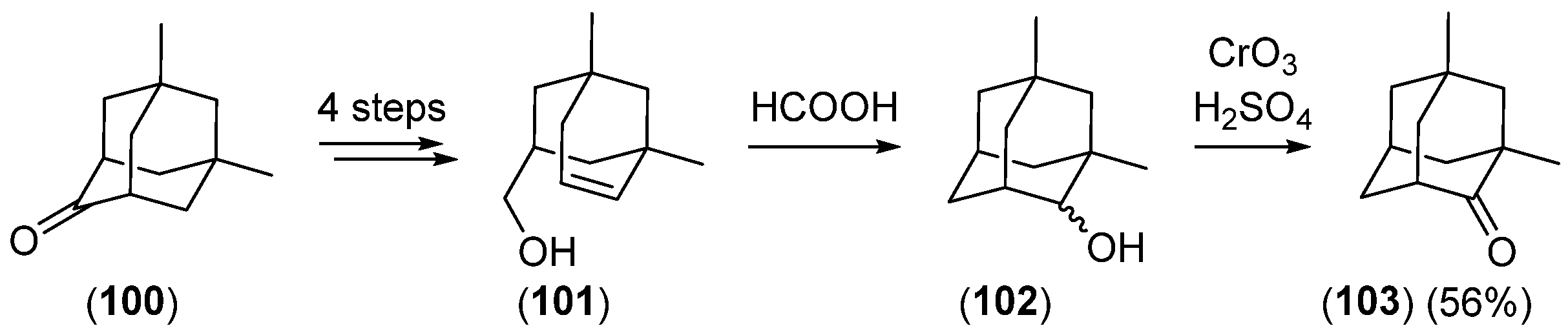{kind=link}
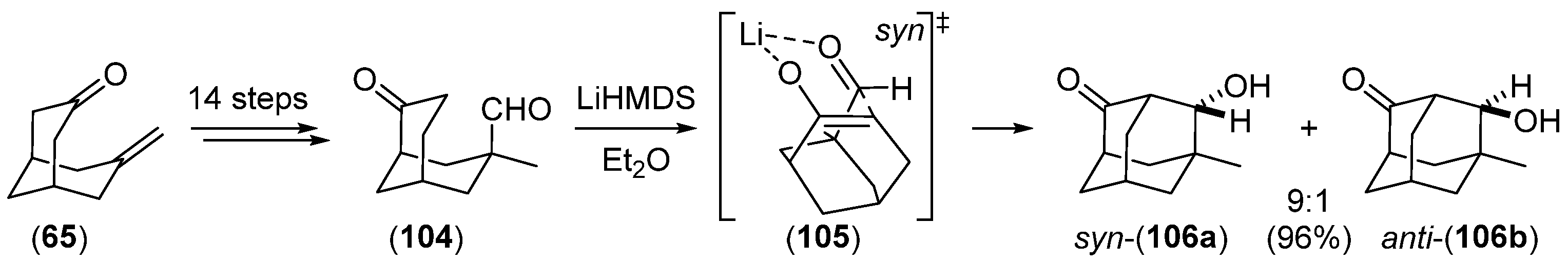{kind=link}
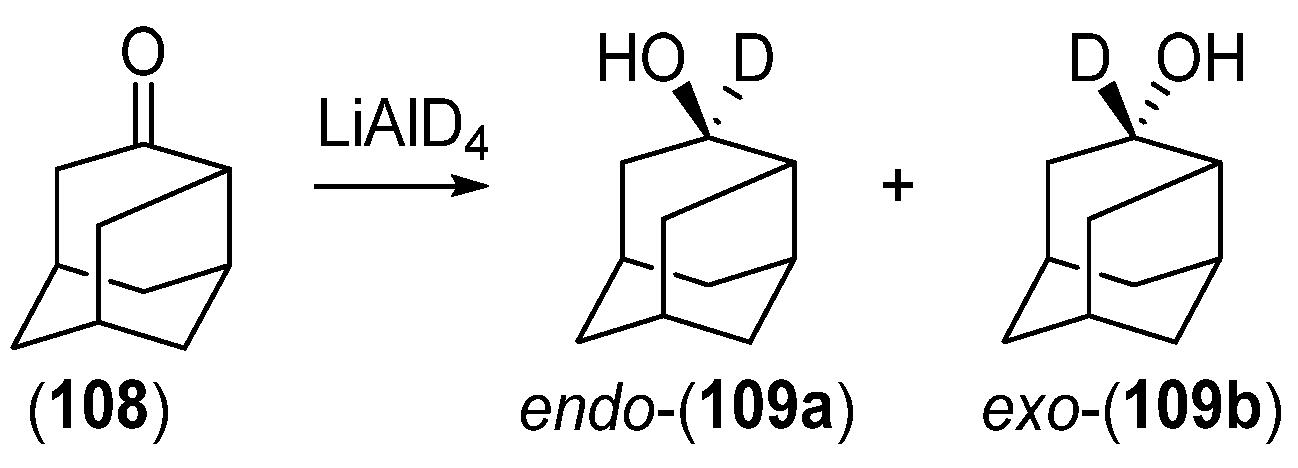{kind=link}
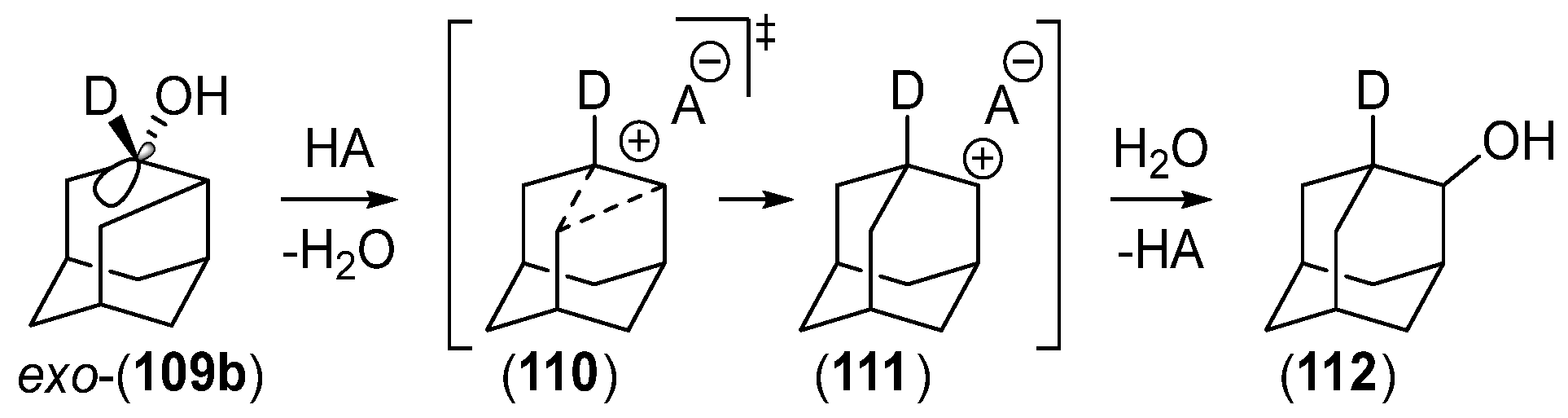{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
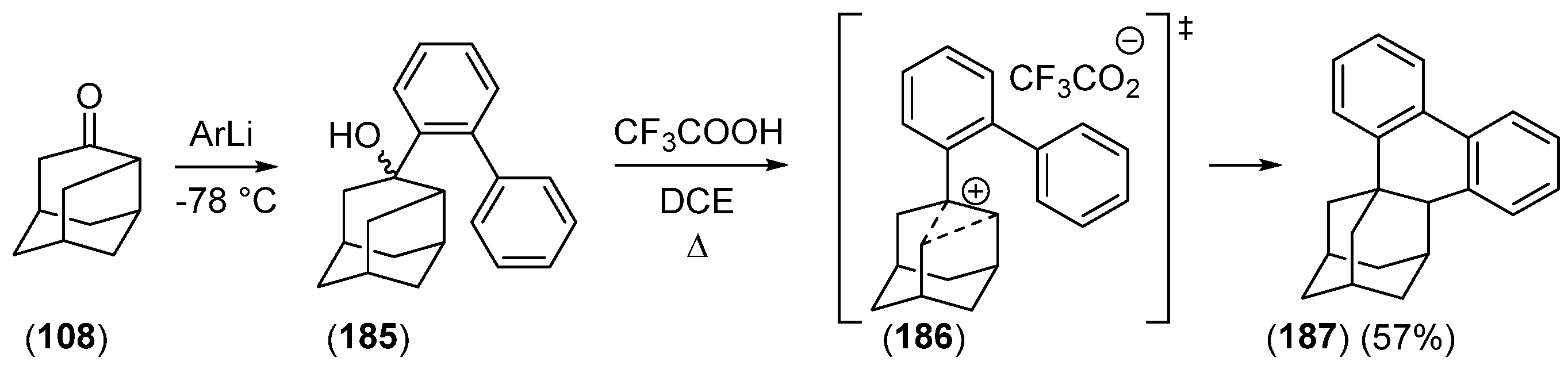{kind=link}
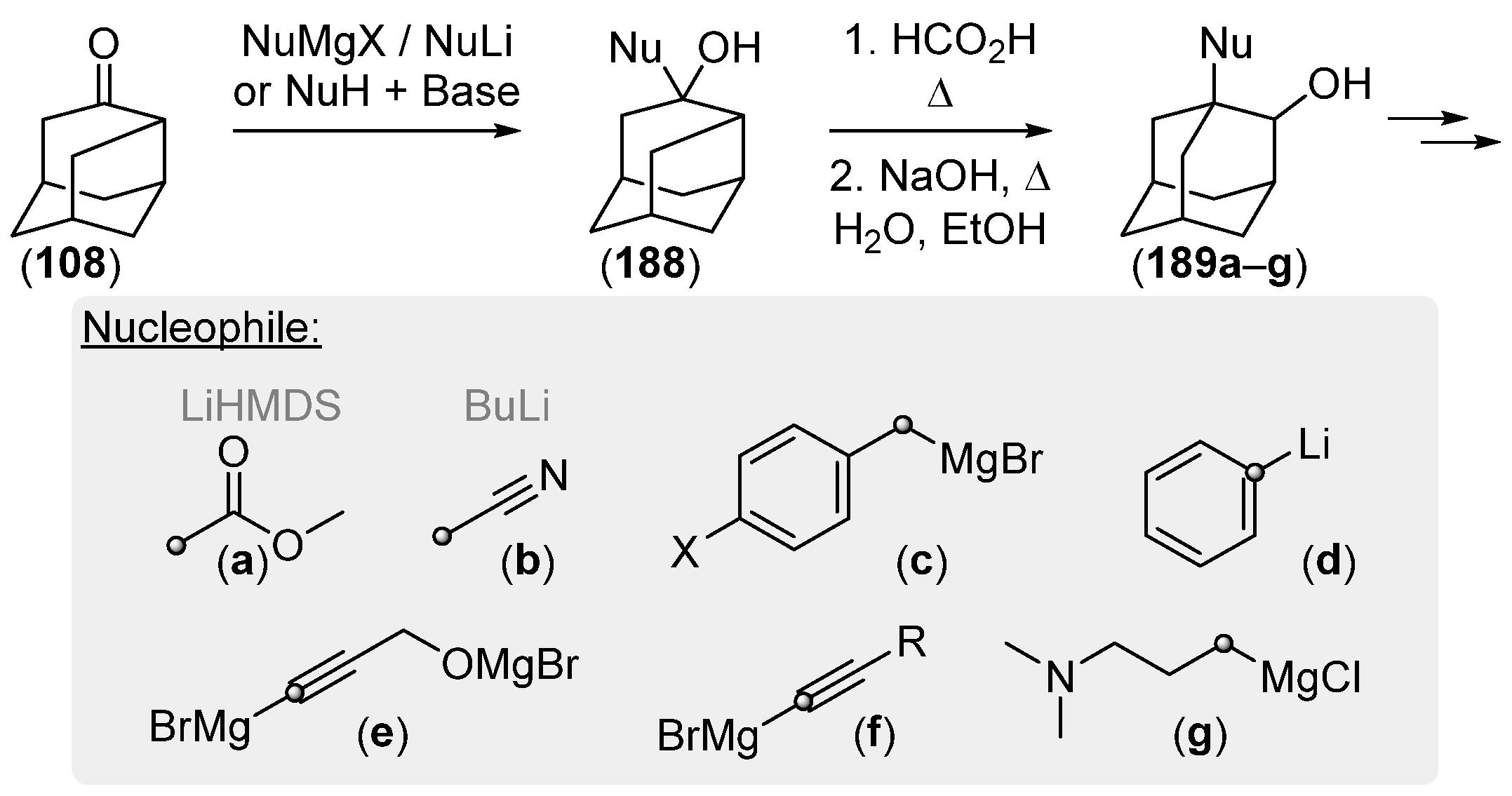{kind=link}
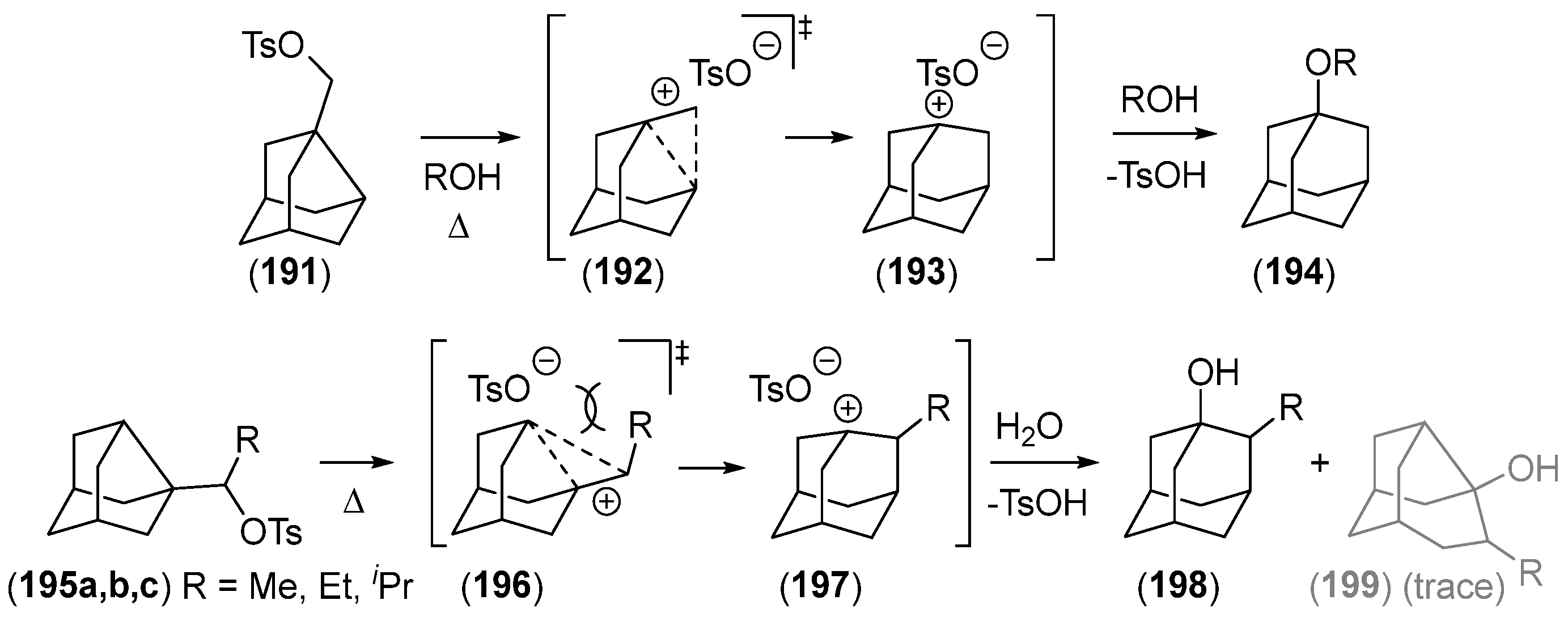{kind=link}
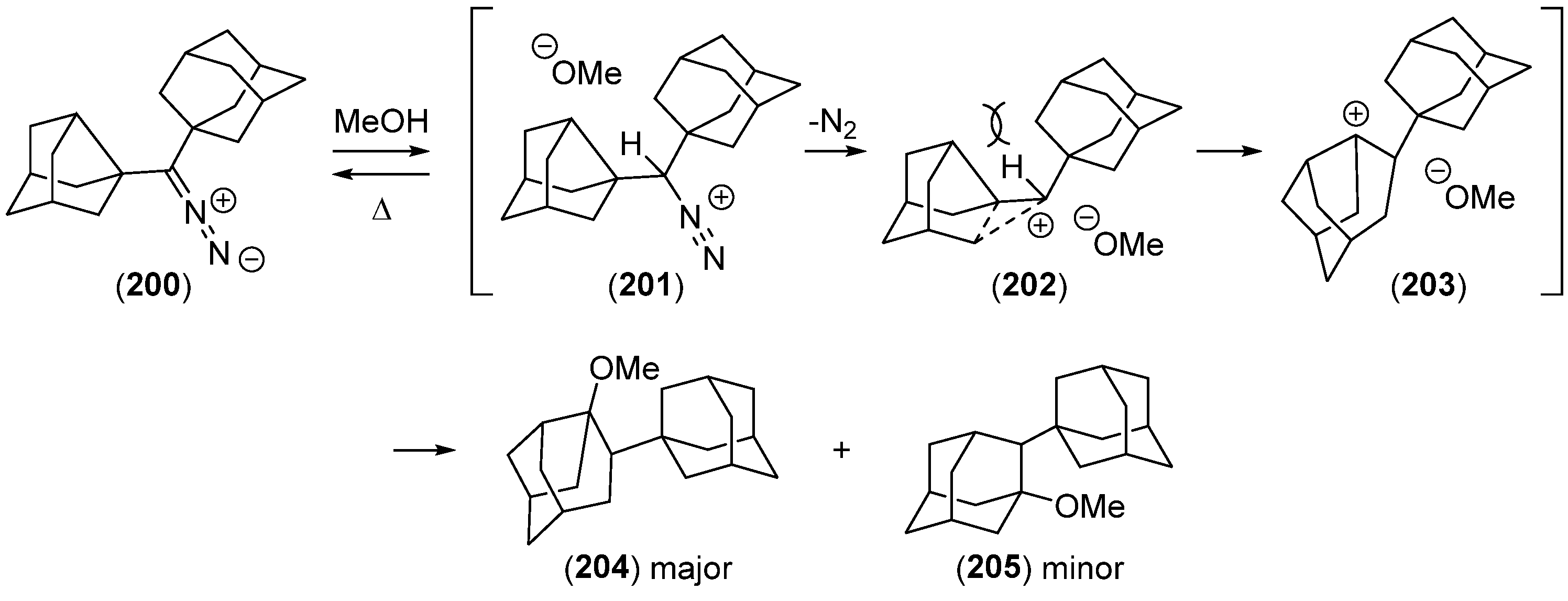{kind=link}
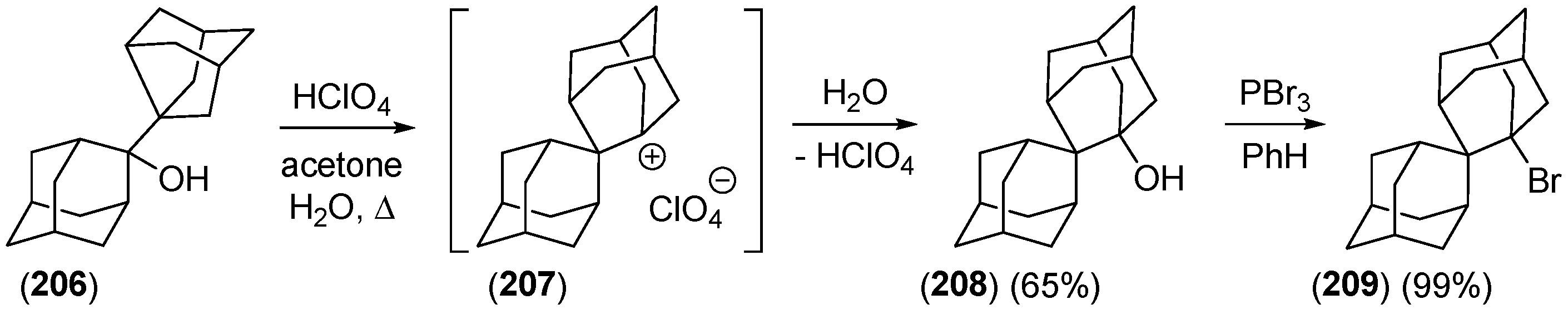{kind=link}
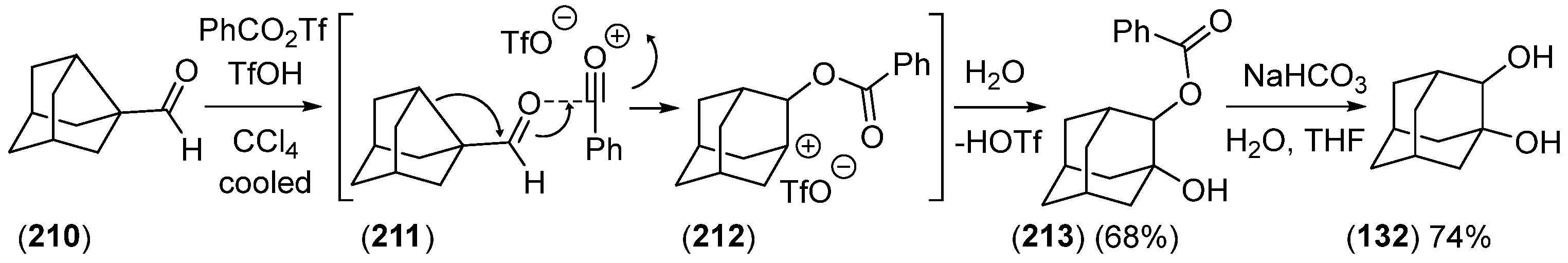{kind=link}
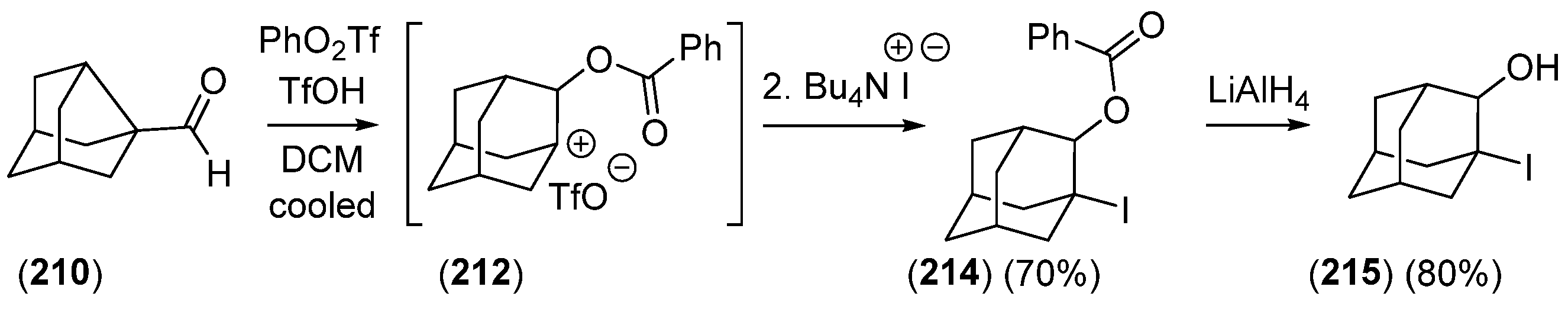{kind=link}
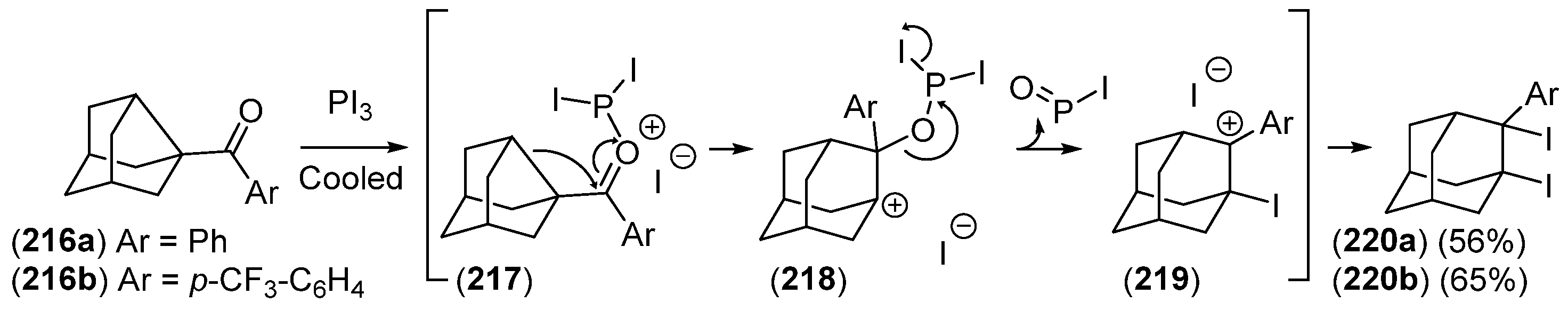{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
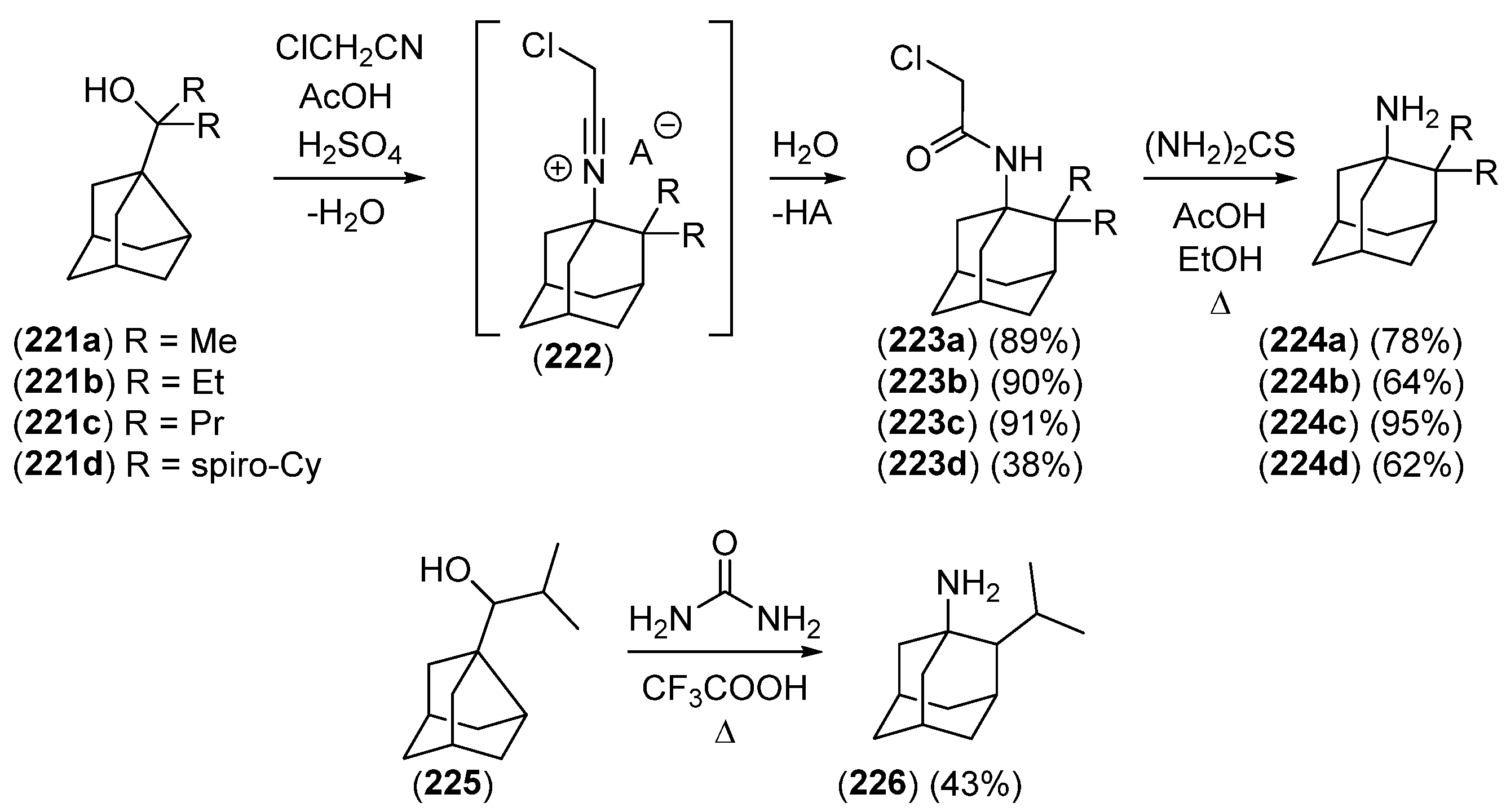
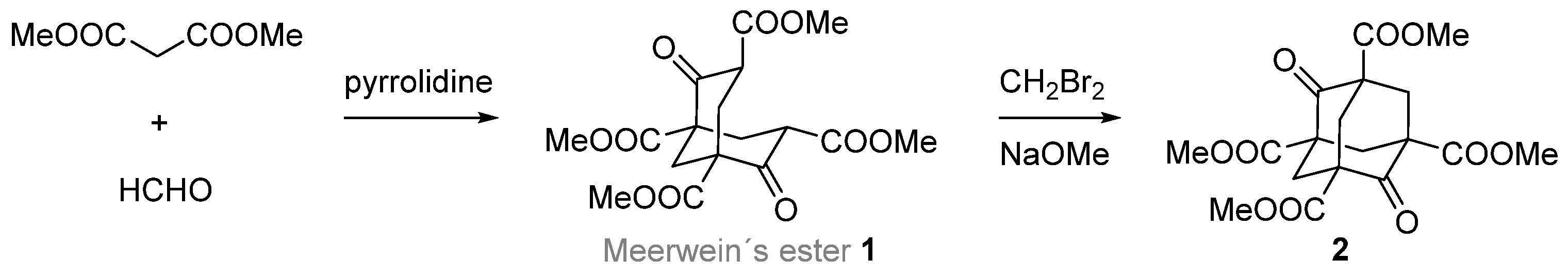
2. 1,2-Disubstituted Adamantane Derivatives
2.1. Synthesis of 1,2-Disubstituted Adamantane Derivatives by Total Synthesis from Acyclic, Monocyclic, or Bicyclic Starting Materials
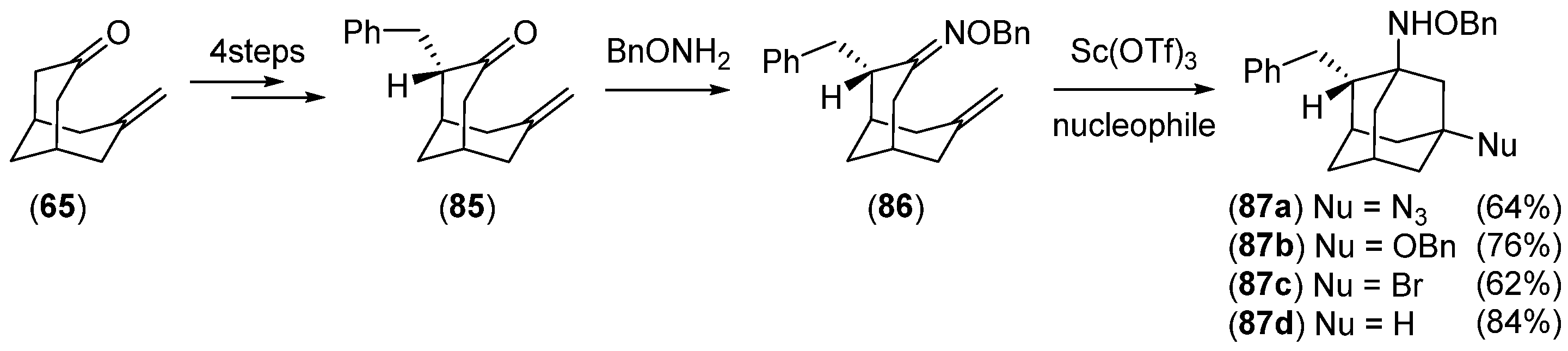
2.2. Synthesis of 1,2-Disubstituted Adamantane Derivatives from Bicyclic Starting Materials
2.2.1. Diolefines as Precursors
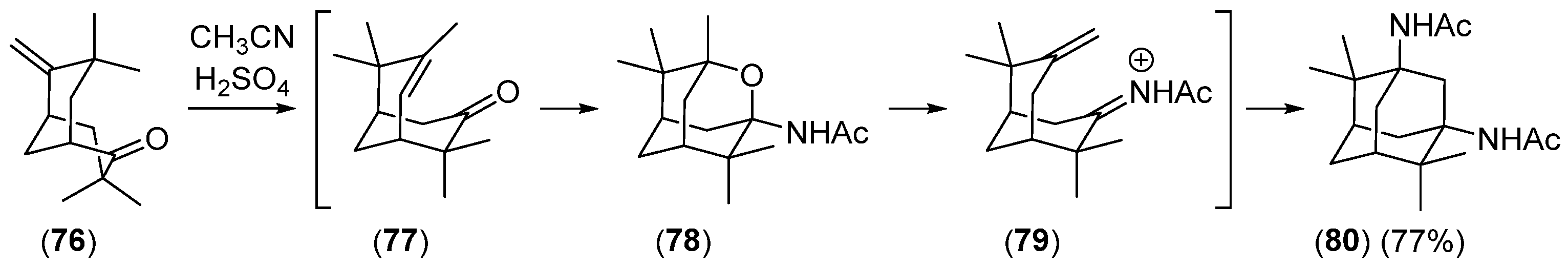
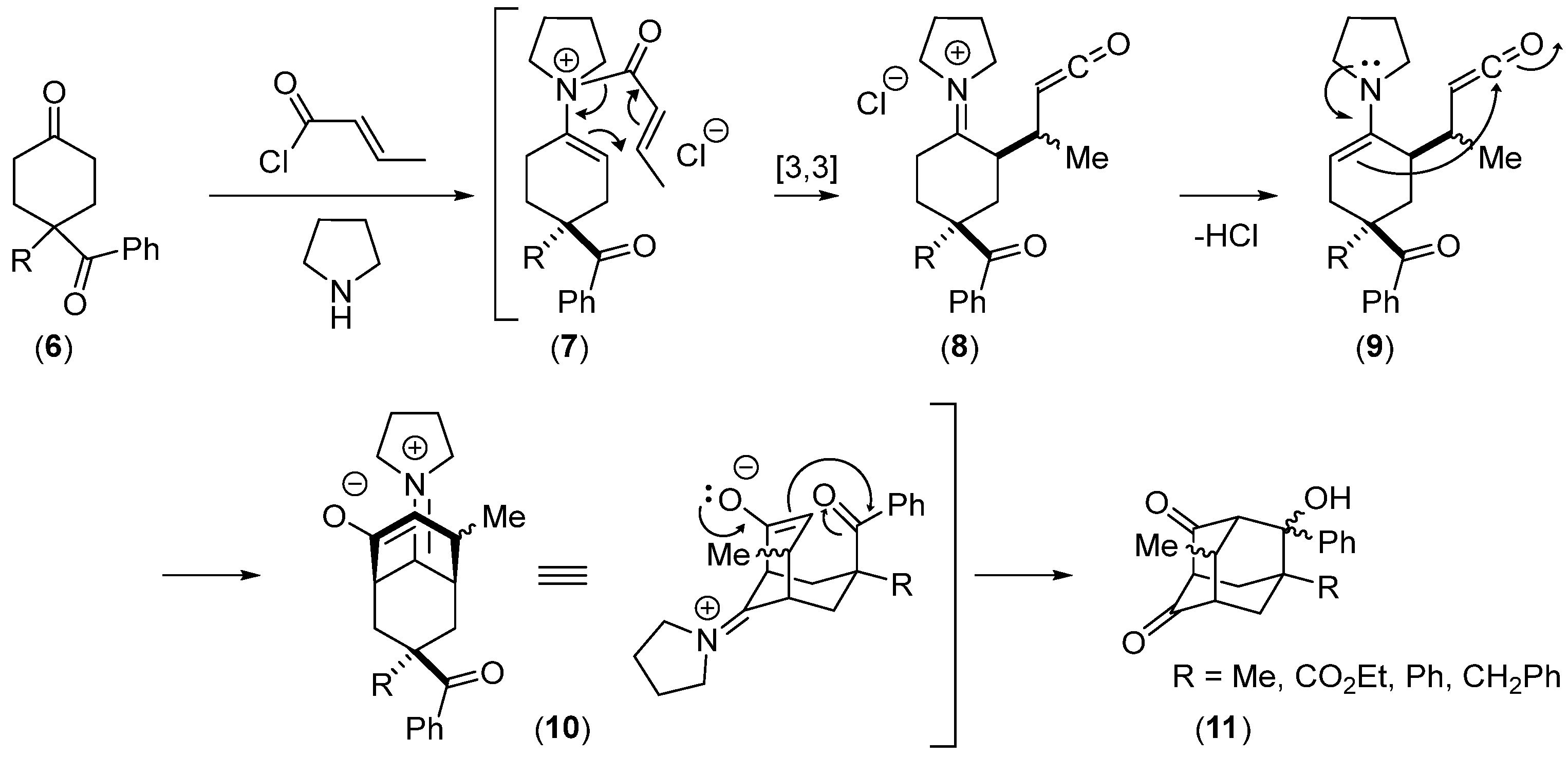
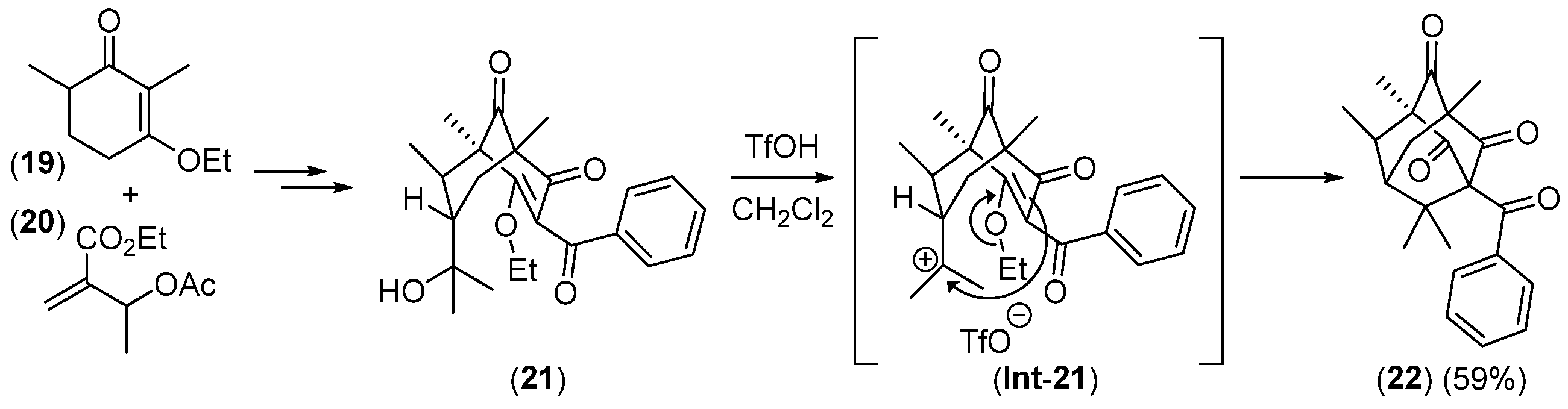
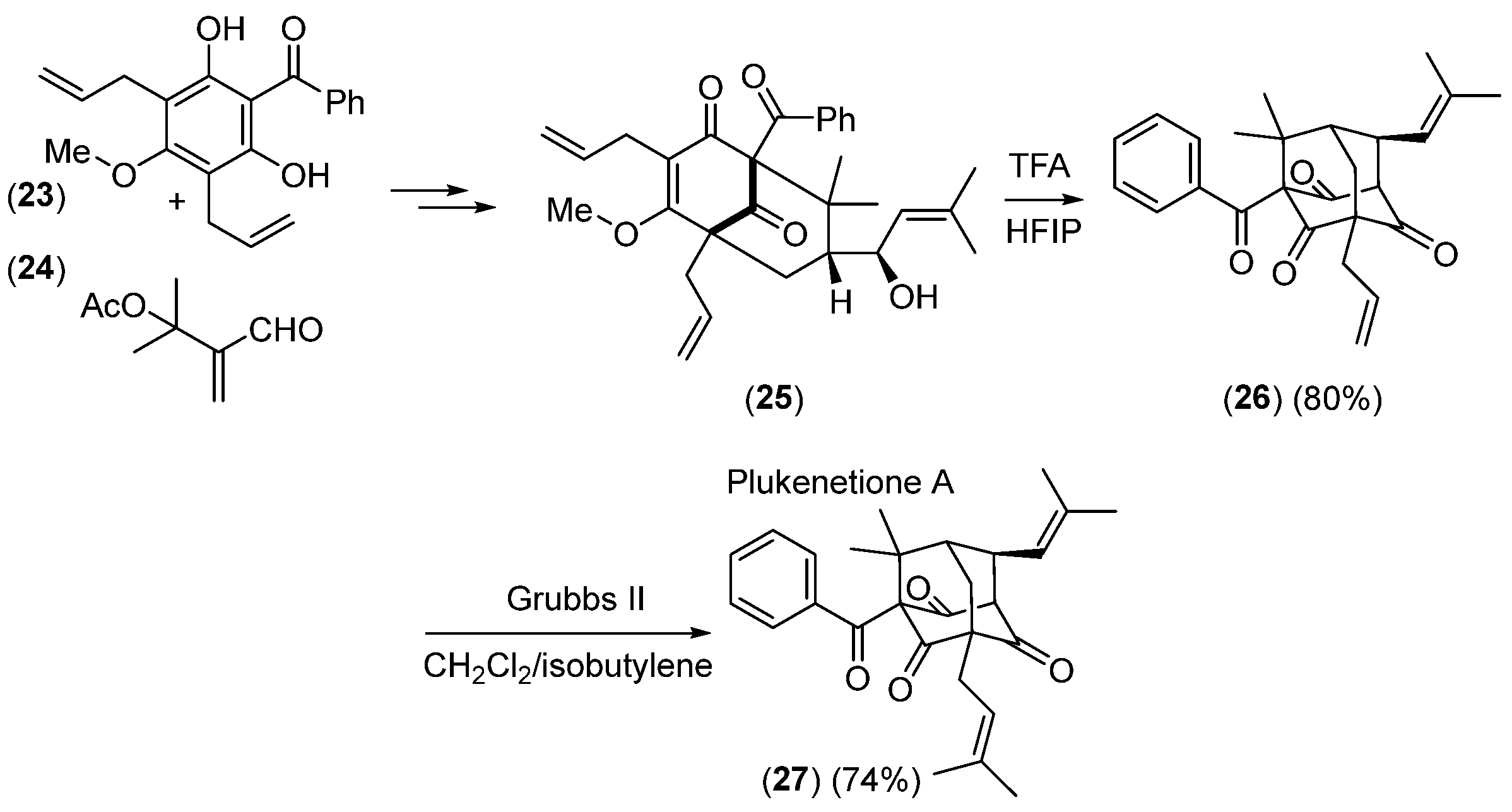
2.2.2. Ketoolefines as Precursors
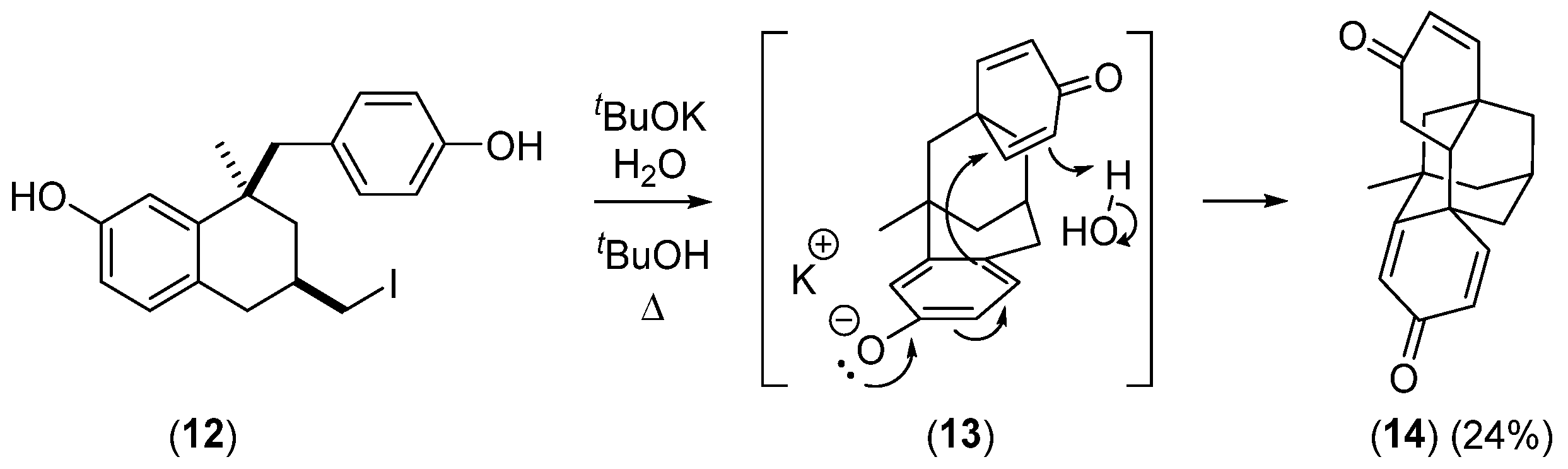
2.2.3. Other Bicyclic Precursors
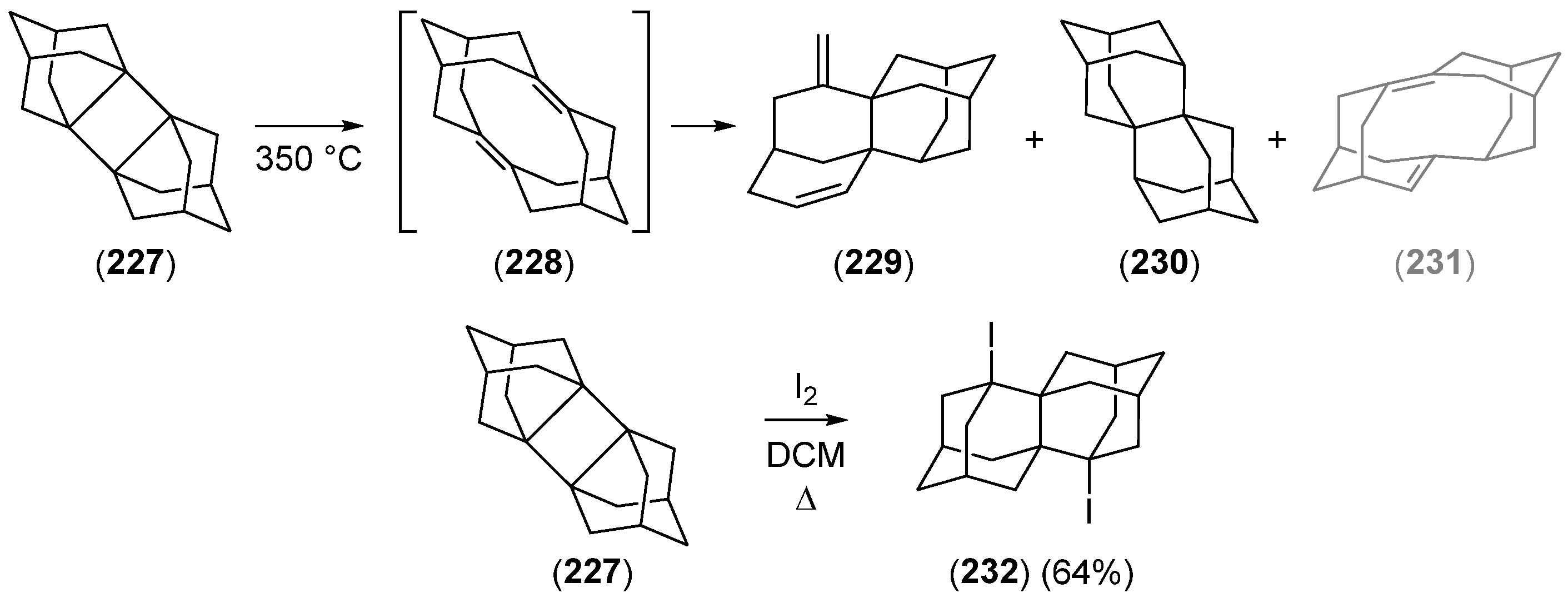
2.3. Synthesis of 1,2-Disubstituted Adamantane Derivatives by Ring Expansion Reactions
2.3.1. The Protoadamantane Route
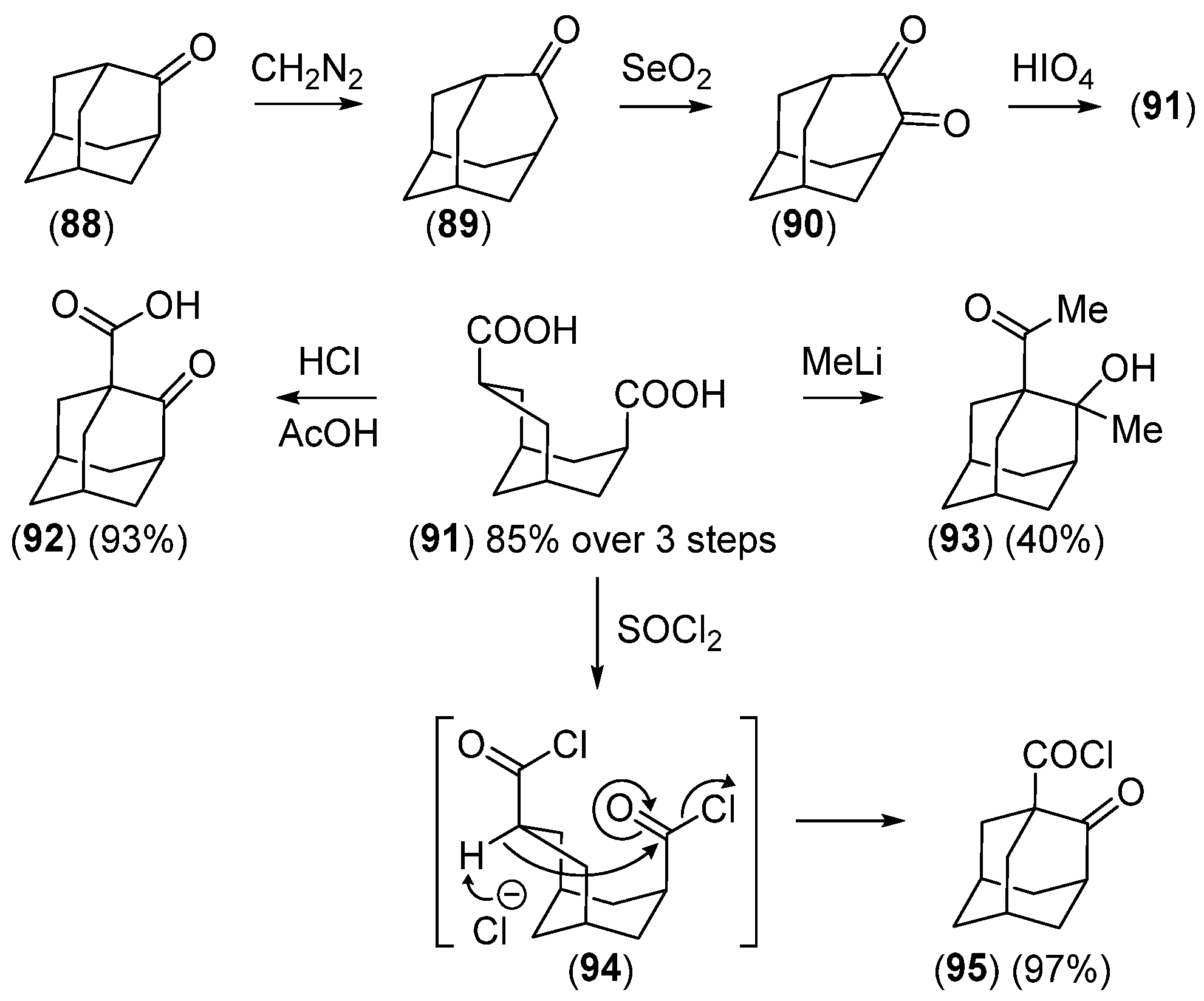
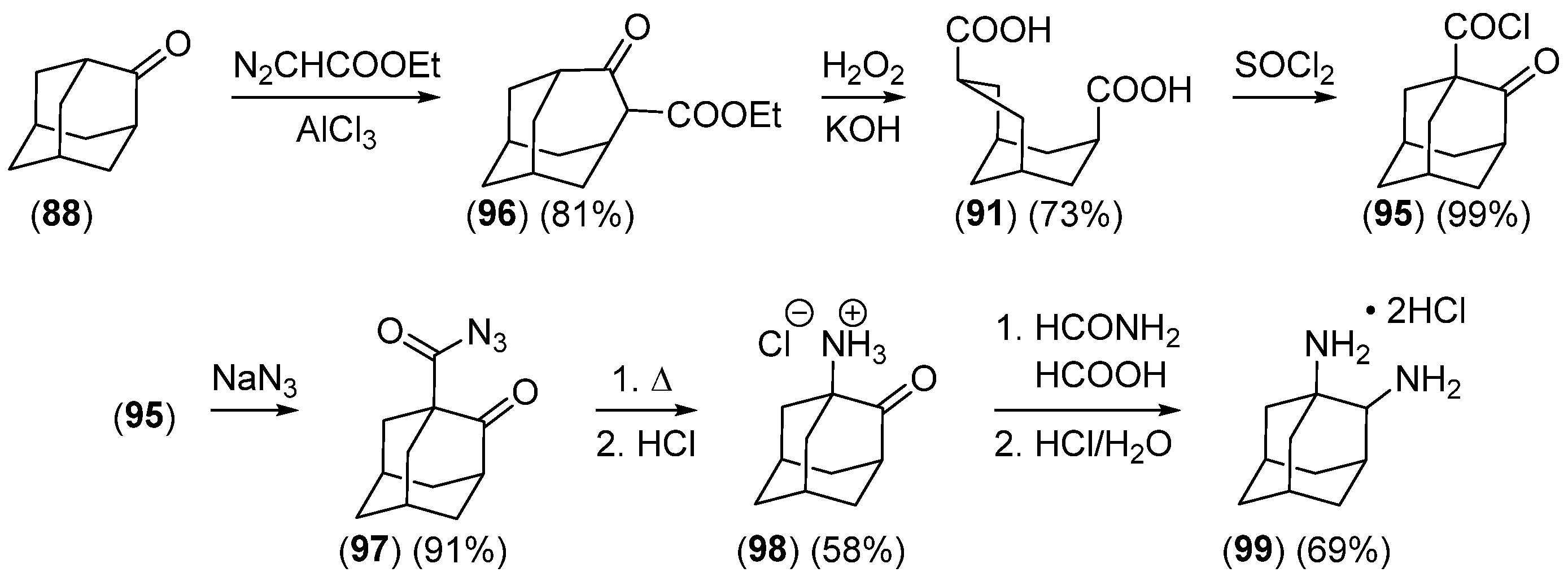
2.3.2. The Noradamantane Route

3. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Landa, S.; Macháček, V. Sur l’adamantane, Nouvel Hydrocarbure Extrait du Naphte. Collect. Czech. Chem. Commun. 1933, 5, 1–5. [Google Scholar] [CrossRef]
- Fort, R.C.; Schleyer, P.v.R. Adamantane: Consequences of the Diamondoid Structure. Chem. Rev. 1964, 64, 277–300. [Google Scholar] [CrossRef]
- Prelog, V.; Seiwerth, R. Über die Synthese des Adamantans. Ber. Dtsch. Chem. Ges. 1941, 74, 1644–1648. [Google Scholar] [CrossRef]
- Schleyer, P.v.R. A Simple Preparation of Adamantane. J. Am. Chem. Soc. 1957, 79, 3292. [Google Scholar] [CrossRef]
- Červinka, O.; Fábryová, A.; Hájíček, J. Absolute Configuration of (S)-(+)-3-Ethyl-5-methyladamantane-1-carboxylic Acid and (S)-(+)-1-Amino-3-ethyl-5-methyladamantane. Collect. Czech. Chem. Commun. 1974, 39, 1582–1588. [Google Scholar] [CrossRef]
- Fokin, A.A.; Schreiner, P.R. Chapter 12—Selective Alkane CH Bond Substitutions: Strategies for the Preparation of Functionalized Diamondoids (Nanodiamonds). In Strategies and Tactics in Organic Synthesis; Harmata, M., Ed.; Academic Press: Cambridge, MA, USA, 2012; Volume 8, pp. 317–350. [Google Scholar] [CrossRef]
- Wanka, L.; Iqbal, K.; Schreiner, P.R. The Lipophilic Bulslet Hits the Targets: Medicinal Chemistry of Adamantane Derivatives. Chem. Rev. 2013, 113, 3516–3604. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Obando, D.; Liao, V.; Lifa, T.; Codd, R. The Many Faces of the Adamantyl Group in Drug Design. Eur. J. Med. Chem. 2011, 46, 1949–1963. [Google Scholar] [CrossRef]
- De Clercq, E. Antiviral Agents Active against Influenza A Viruses. Nat. Rev. Drug Discov. 2006, 5, 1015–1025. [Google Scholar] [CrossRef]
- Stockdale, T.P.; Williams, C.M. Pharmaceuticals That Contain Polycyclic Hydrocarbon Scaffolds. Chem. Soc. Rev. 2015, 44, 7737–7763. [Google Scholar] [CrossRef]
- Spilovska, K.; Zemek, F.; Korabecny, J.; Nepovimova, E.; Soukup, O.; Windisch, M.; Kuca, K. Adamantane—A Lead Structure for Drugs in Clinical Practice. Curr. Med. Chem. 2016, 23, 3245–3266. [Google Scholar] [CrossRef]
- Štimac, A.; Šekutor, M.; Mlinarić-Majerski, K.; Frkanec, L.; Frkanec, R. Adamantane in Drug Delivery Systems and Surface Recognition. Molecules 2017, 22, 297. [Google Scholar] [CrossRef] [PubMed]
- Agnew-Francis, K.A.; Williams, C.M. Catalysts Containing the Adamantane Scaffold. Adv. Synth. Catal. 2016, 358, 675–700. [Google Scholar] [CrossRef]
- Muller, T.; Bräse, S. Tetrahedral Organic Molecules as Components in Supramolecular Architectures and in Covalent Assemblies, Networks and Polymers. RSC Adv. 2014, 4, 6886–6907. [Google Scholar] [CrossRef]
- Nasrallah, H.; Hierso, J.-C. Porous Materials Based on 3-Dimensional Td-Directing Functionalized Adamantane Scaffolds and Applied as Recyclable Catalysts. Chem. Mater. 2019, 31, 619–642. [Google Scholar] [CrossRef]
- Liangjun, L.; Deng, J.; Guo, J.; Yue, H. Synthesis and Properties of Microporous Organic Polymers Based on Adamantane. Prog. Chem. 2020, 32, 190–203. [Google Scholar] [CrossRef]
- Mohamadhoseini, M.; Mohamadnia, Z. Supramolecular Self-Healing Materials via Host-Guest Strategy between Cyclodextrin and Specific Types of Guest Molecules. Coord. Chem. Rev. 2021, 432, 213711. [Google Scholar] [CrossRef]
- Gunawan, M.A.; Hierso, J.-C.; Poinsot, D.; Fokin, A.A.; Fokina, N.A.; Tkachenko, B.A.; Schreiner, P.R. Diamondoids: Functionalization and Subsequent Applications of Perfectly Defined Molecular Cage Hydrocarbons. New J. Chem. 2013, 38, 28–41. [Google Scholar] [CrossRef]
- Yeung, K.-W.; Dong, Y.; Chen, L.; Tang, C.-Y.; Law, W.-C.; Tsui, G.C.-P. Nanotechnology of Diamondoids for the Fabrication of Nanostructured Systems. Nanotechnol. Rev. 2020, 9, 650–669. [Google Scholar] [CrossRef]
- de Araujo, P.L.B.; Mansoori, G.A.; de Araujo, E.S. Diamondoids: Occurrence in Fossil Fuels, Applications in Petroleum Exploration and Fouling in Petroleum Production. A Review Paper. Int. J. Oil Gas Coal Technol. 2012, 5, 316–367. [Google Scholar] [CrossRef]
- Gund, T.; Thielecke, W.; Schleyer, P.v.R. Diamantane: Pentacyclo [7.3. 1.14, 12. 02, 7. 06, 11] Tetradecane (3, 5, 1, 7-[1, 2, 3, 4]-Butanetetraylnaphthalene, Decahydro). Org. Synth. 1973, 53, 30–34. [Google Scholar]
- Schwertfeger, H.; Fokin, A.A.; Schreiner, P.R. Diamonds Are a Chemist’s Best Friend: Diamondoid Chemistry Beyond Adamantane. Angew. Chem. Int. Ed. 2008, 47, 1022–1036. [Google Scholar] [CrossRef] [PubMed]
- Grover, N.; Senge, M.O. Synthetic Advances in the C–H Activation of Rigid Scaffold Molecules. Synthesis 2020, 52, 3295–3325. [Google Scholar] [CrossRef]
- Fokin, A.A.; Schreiner, P.R. Metal-Free, Selective Alkane Functionalizations. Adv. Synth. Catal. 2003, 345, 1035–1052. [Google Scholar] [CrossRef]
- Weigel, W.K.; Dang, H.T.; Feceu, A.; Martin, D.B.C. Direct Radical Functionalization Methods to Access Substituted Adamantanes and Diamondoids. Org. Biomol. Chem. 2022, 20, 10–36. [Google Scholar] [CrossRef] [PubMed]
- Bagrii, E.I.; Nekhaev, A.I.; Maksimov, A.L. Oxidative Functionalization of Adamantanes (Review). Pet. Chem. 2017, 57, 183–197. [Google Scholar] [CrossRef]
- Hrdina, R. Directed C–H Functionalization of the Adamantane Framework. Synthesis 2019, 51, 629–642. [Google Scholar] [CrossRef]
- Zhou, Y.; Brittain, A.D.; Kong, D.; Xiao, M.; Meng, Y.; Sun, L. Derivatization of Diamondoids for Functional Applications. J. Mater. Chem. C 2015, 3, 6947–6961. [Google Scholar] [CrossRef]
- Man’kova, P.A.; Shiryaev, V.A.; Shmel’kova, Y.D.; Moiseev, A.V.; Reznikov, A.N.; Klimochkin, Y.N. Synthesis of 1,2-Diaminoadamantane and Chiral Ligands Derived from It. Russ. Chem. Bull. 2023, 72, 1791–1801. [Google Scholar] [CrossRef]
- Müller, C.E.; Wanka, L.; Jewell, K.; Schreiner, P.R. Enantioselective Kinetic Resolution of Trans-Cycloalkane-1,2-Diols. Angew. Chem. Int. Ed. 2008, 47, 6180–6183. [Google Scholar] [CrossRef]
- Hrdina, R.; Müller, C.E.; Schreiner, P.R. Kinetic Resolution of Trans-Cycloalkane-1,2-Diols via Steglich Esterification. Chem. Commun. 2010, 46, 2689–2690. [Google Scholar] [CrossRef]
- Berndt, J.-P.; Radchenko, Y.; Becker, J.; Logemann, C.; Bhandari, D.R.; Hrdina, R.; Schreiner, P.R. Site-Selective Nitrenoid Insertions Utilizing Postfunctionalized Bifunctional Rhodium(II) Catalysts. Chem. Sci. 2019, 10, 3324–3329. [Google Scholar] [CrossRef] [PubMed]
- Hrdina, R.; Larrosa, M.; Logemann, C. Triflic Acid Promoted Decarboxylation of Adamantane-Oxazolidine-2-One: Access to Chiral Amines and Heterocycles. J. Org. Chem. 2017, 82, 4891–4899. [Google Scholar] [CrossRef] [PubMed]
- Yasue, R.; Yoshida, K. Enantioselective Desymmetrization of 1,3-Disubstituted Adamantane Derivatives via Rhodium-Catalyzed C−H Bond Amination: Access to Optically Active Amino Acids Containing Adamantane Core. Adv. Synth. Catal. 2021, 363, 1662–1671. [Google Scholar] [CrossRef]
- Wen, Q.; di Gregorio, M.C.; Shimon, L.J.W.; Pinkas, I.; Malik, N.; Kossoy, A.; Alexandrov, E.V.; Proserpio, D.M.; Lahav, M.; van der Boom, M.E. Chiral Motifs in Highly Interpenetrated Metal–Organic Frameworks Formed from Achiral Tetrahedral Ligands. Chem. Eur. J. 2022, 28, e202201108. [Google Scholar] [CrossRef]
- Ou, G.-C.; Chen, H.-Y.; Wang, Q.; Zhou, Q.; Zeng, F. Structure and Absolute Configuration of Liquid Molecules Based on Adamantane Derivative Cocrystallization. RSC Adv. 2022, 12, 6459–6462. [Google Scholar] [CrossRef]
- Yang, C.; Chen, W.; Zhu, X.; Song, X.; Liu, M. Self-Assembly and Circularly Polarized Luminescence from Achiral Pyrene-Adamantane Conjugates by Selective Inclusion with Cyclodextrins. J. Phys. Chem. Lett. 2021, 12, 7491–7496. [Google Scholar] [CrossRef]
- Müller, J.; Kirschner, R.A.; Berndt, J.-P.; Wulsdorf, T.; Metz, A.; Hrdina, R.; Schreiner, P.R.; Geyer, A.; Klebe, G. Diamondoid Amino Acid-Based Peptide Kinase A Inhibitor Analogues. ChemMedChem 2019, 14, 663–672. [Google Scholar] [CrossRef] [PubMed]
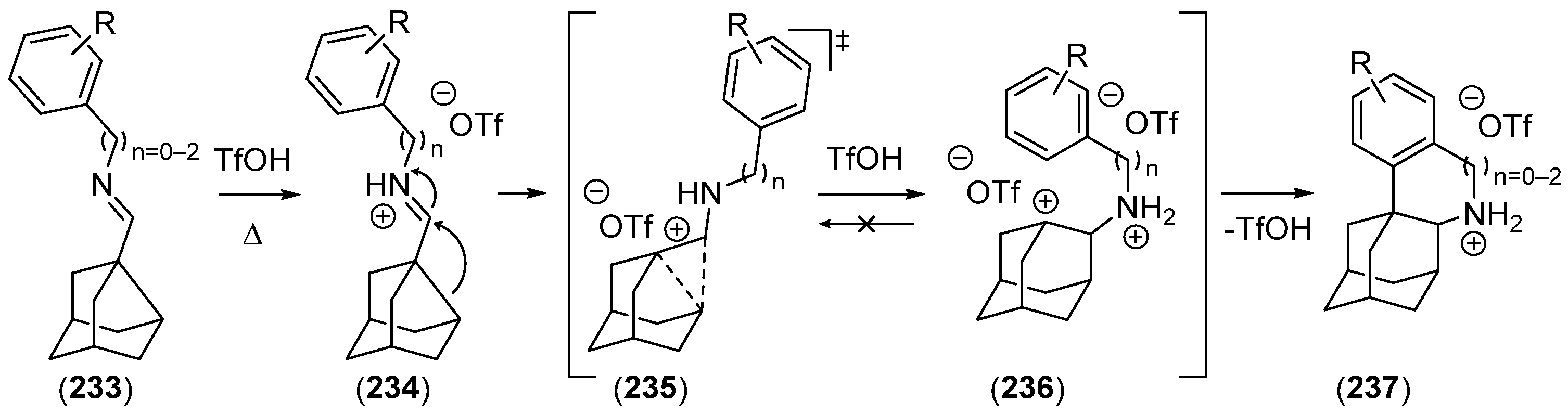
- Hrdina, R.; Metz, F.M.; Larrosa, M.; Berndt, J.-P.; Zhygadlo, Y.Y.; Becker, S.; Becker, J. Intramolecular C–H Amination Reaction Provides Direct Access to 1,2-Disubstituted Diamondoids. Eur. J. Org. Chem. 2015, 2015, 6231–6236. [Google Scholar] [CrossRef]
- Weigel, W.K.; Dang, H.T.; Yang, H.-B.; Martin, D.B.C. Synthesis of Amino-Diamondoid Pharmacophores via Photocatalytic C–H Aminoalkylation. Chem. Commun. 2020, 56, 9699–9702. [Google Scholar] [CrossRef]
- Martella, D.J.; Jones, M., Jr.; Schleyer, P.V.R. Adamantene by Ring Enlargement of 3-Noradamantylcarbene. J. Am. Chem. Soc. 1978, 100, 2896–2897. [Google Scholar] [CrossRef]
- Conlin, R.T.; Miller, R.D.; Michl, J. Adamantene. J. Am. Chem. Soc. 1979, 101, 7637–7638. [Google Scholar] [CrossRef]
- Bian, N.; Jones, M. More on Adamantene. J. Am. Chem. Soc. 1995, 117, 8957–8961. [Google Scholar] [CrossRef]
- Prelog, V.; Seiwerth, R. Über eine Neue, Ergiebigere Darstellung des Adamantans. Ber. Dtsch. Chem. Ges. 1941, 74, 1769–1772. [Google Scholar] [CrossRef]
- Stetter, H.; Thomas, H.G. A New Synthesis of the Adamantane Ring System. Angew. Chem. Int. Ed. 1967, 6, 554–555. [Google Scholar] [CrossRef]
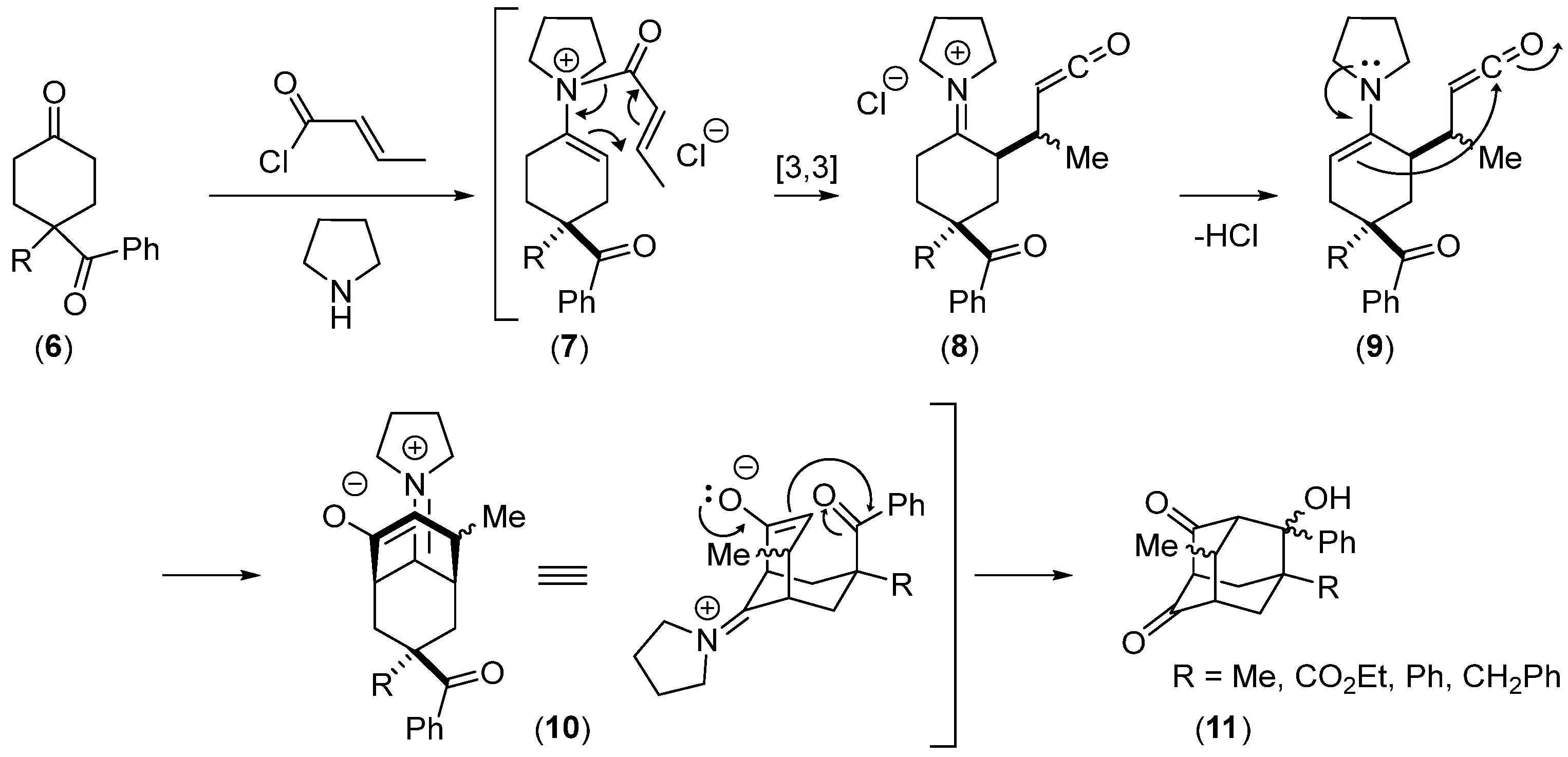
- Hickmott, P.W.; Suschitzky, H.; Urbani, R. Enamine Chemistry. Part XVIII. Reaction of Crotonoyl and Methacryloyl Chlorides with Dialkyl 4-(pyrrolidin-1-yl)cyclohex-3-ene-1,1-dicarboxylates. One-Step Synthesis of Adamantanes. J. Chem. Soc. Perkin Trans. 1 1973, 2063–2064. [Google Scholar] [CrossRef]
- Asghari Ahmed, S.; Hickmott, P.W. Enamine Chemistry. Part 26. Preparation of Substituted Adamantane-2,4-diones and Bicyclo[2.2.2]octan-2-ones. J. Chem. Soc. Perkin Trans. 1 1979, 2180–2183. [Google Scholar] [CrossRef]
- Hickmott, P.W.; Kapon, M. Enamine Chemistry. Part 29.’ Synthesis of Adamantane Derivatives from α,β-Unsaturated Acid Chlorides and 4,4-Disubstituted Cyclohexanone Enamines. Multiple [3,3]Sigmatropic Rearrangement Transition State Stereochemistry. X-ray Analysis. J. Chem. Soc. Perkin Trans. 1 1985, 1, 2559–2571. [Google Scholar] [CrossRef]
- Ahmed, M.G.; Ahmed, S.A.; Akhter, K.; Moeiz, S.M.I.; Tsuda, Y.; Kiuchi, F.; Hossain, M.M.; Forsterling, F.H. Synthesis of Some Substituted Adamantane-2,4-diones from 4, 4-Disubstituted Cyclohexanone Enamines and Methacryloyl Chloride. J. Chem. Res. 2005, 2005, 293–298. [Google Scholar] [CrossRef]
- Atkinson, R.S.; Miller, J.E. Adamantanoid Dienones from Intramolecular Ar 1 6-Participation. J. Chem. Soc. Perkin Trans. 1 1979, 3017–3021. [Google Scholar] [CrossRef]
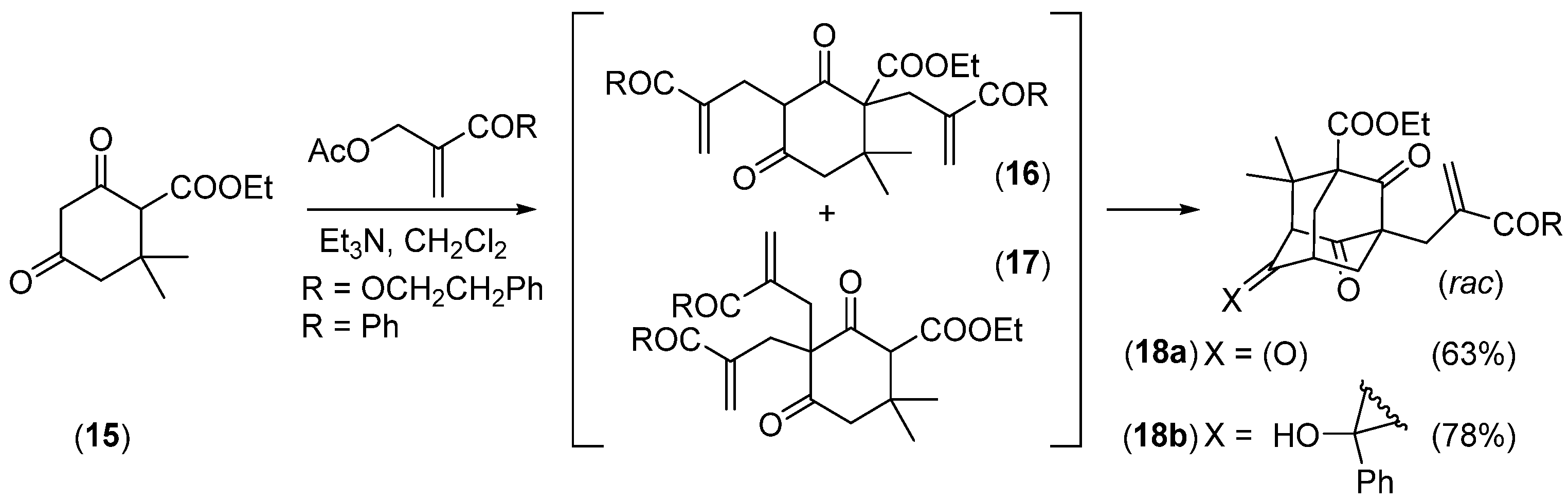
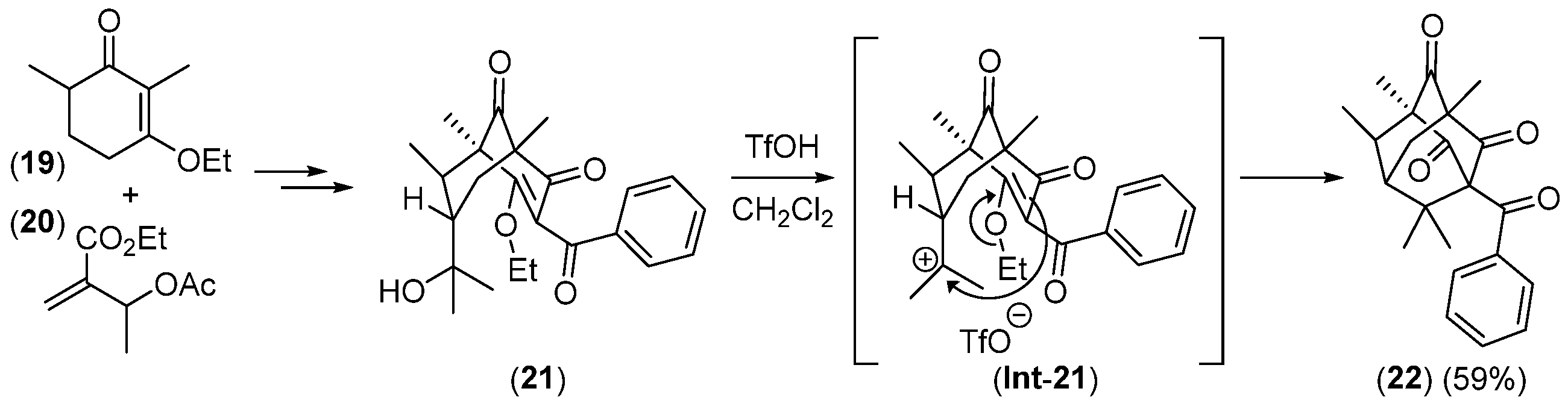
- Takagi, R.; Miwa, Y.; Matsumura, S.; Ohkata, K. One-Pot Synthesis of Adamantane Derivatives by Domino Michael Reactions from Ethyl 2,4-Dioxocyclohexanecarboxylate. J. Org. Chem. 2005, 70, 8587–8589. [Google Scholar] [CrossRef] [PubMed]
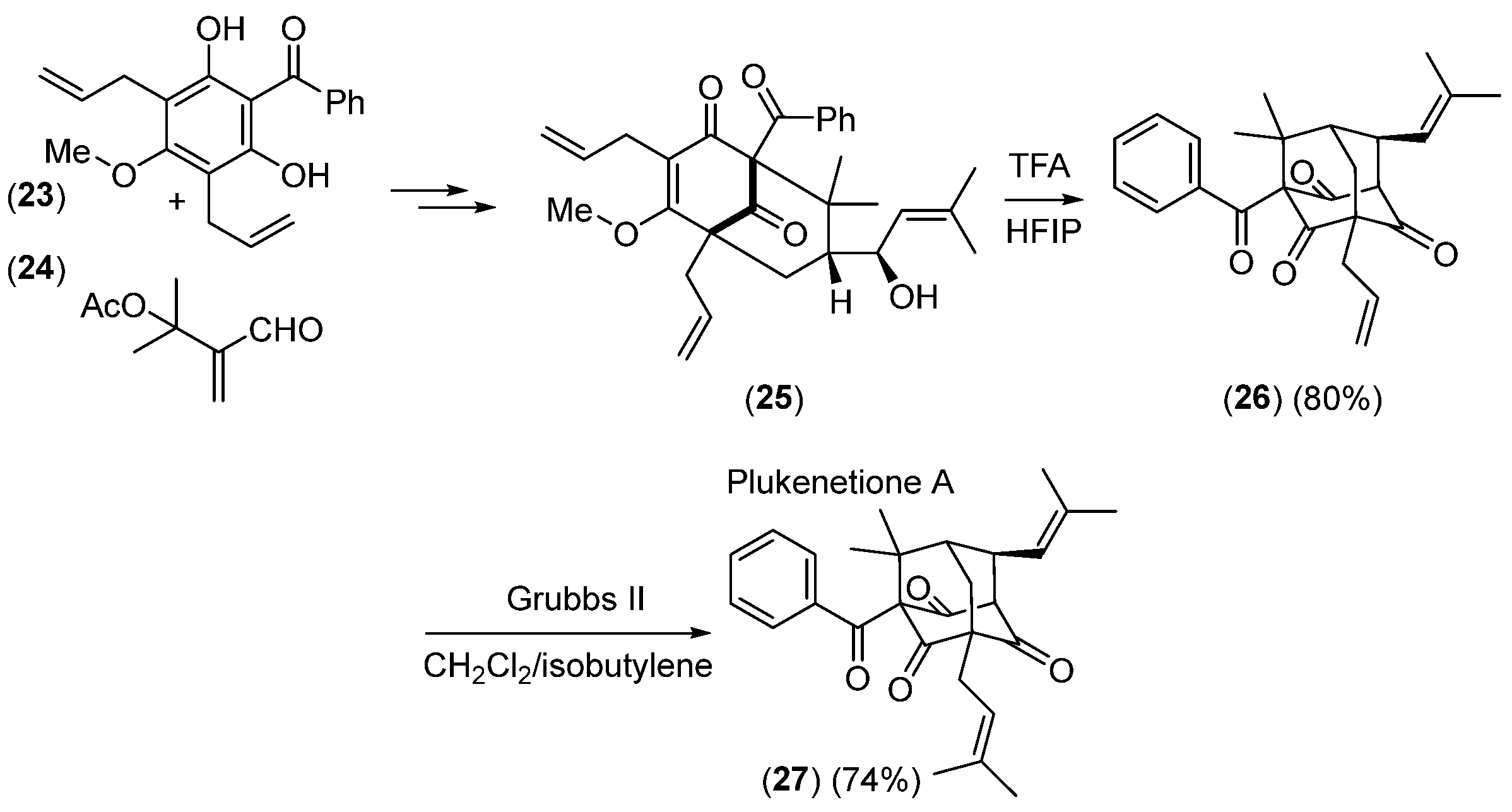
- Takagi, R.; Inoue, Y.; Ohkata, K. Construction of the Adamantane Core of Plukenetione-Type Polycyclic Polyprenylated Acylphloroglucinols. J. Org. Chem. 2008, 73, 9320–9325. [Google Scholar] [CrossRef]
- Zhang, Q.; Mitasev, B.; Qi, J.; Porco, J.A., Jr. Total Synthesis of Plukenetione A. J. Am. Chem. Soc. 2010, 132, 14212–14215. [Google Scholar] [CrossRef]
- Muraoka, O.; Wang, Y.; Okumura, M.; Nishiura, S.; Tanabe, G.; Momose, T. A Facile Synthesis of 7-Methylenebicyclo-[3.3.1]Nonan-3-One and Its Transformation Leading to the Novel Tricyclic System, Protoadamantane. Synth. Commun. 1996, 26, 1555–1562. [Google Scholar] [CrossRef]
- Gagneux, A.R.; Meier, R. 1-Substituted 2-Heteroadamantanes. Tetrahedron Lett. 1969, 10, 1365–1368. [Google Scholar] [CrossRef]
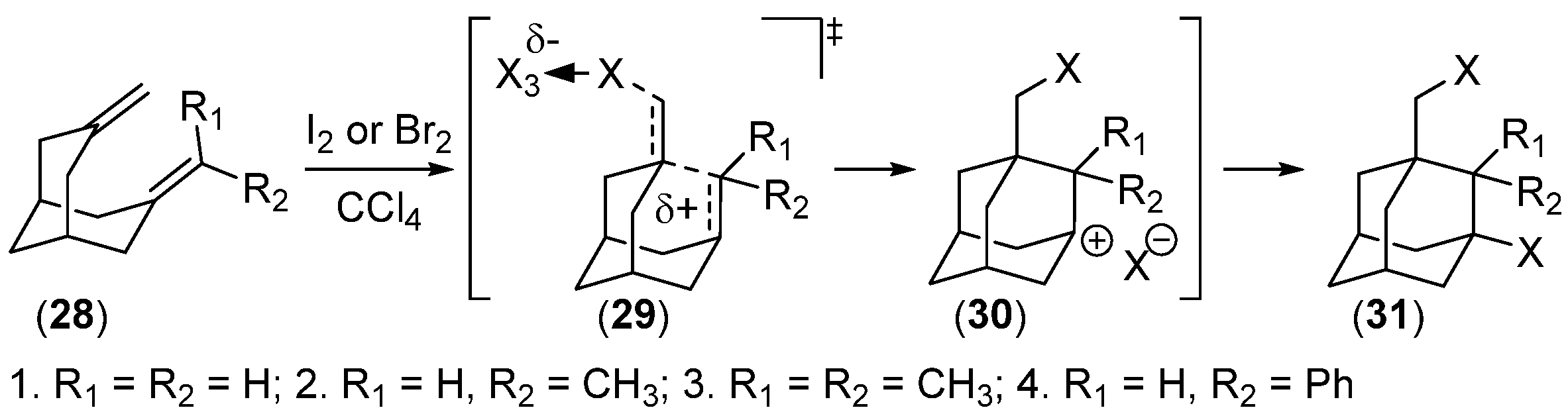
- Krasutskii, P.A.; Khotkevich, A.B.; Serguchev, Y.A.; Yurchenko, A.G. Mechanism of Transannular Bromination Reactions of Diolefins of the Bicyclo[3.3.1]nonane Series. Theor. Exp. Chem. 1985, 21, 48–52. [Google Scholar] [CrossRef]
- Serguchev, Y.A.; Krasutskii, P.A.; Khotkevich, A.B.; Yurchenko, A.G. Mechanism of Transannular Iodination of 3,7-Dimethylenebicyclo[3.3.1]nonane Derivatives. Theor. Exp. Chem. 1987, 22, 712–715. [Google Scholar] [CrossRef]
- Sohár, P.; Kuszmann, J.; Néder, Á. Synthesis and NMR Study of Adamantane Derivatives. Tetrahedron 1986, 42, 2523–2532. [Google Scholar] [CrossRef]
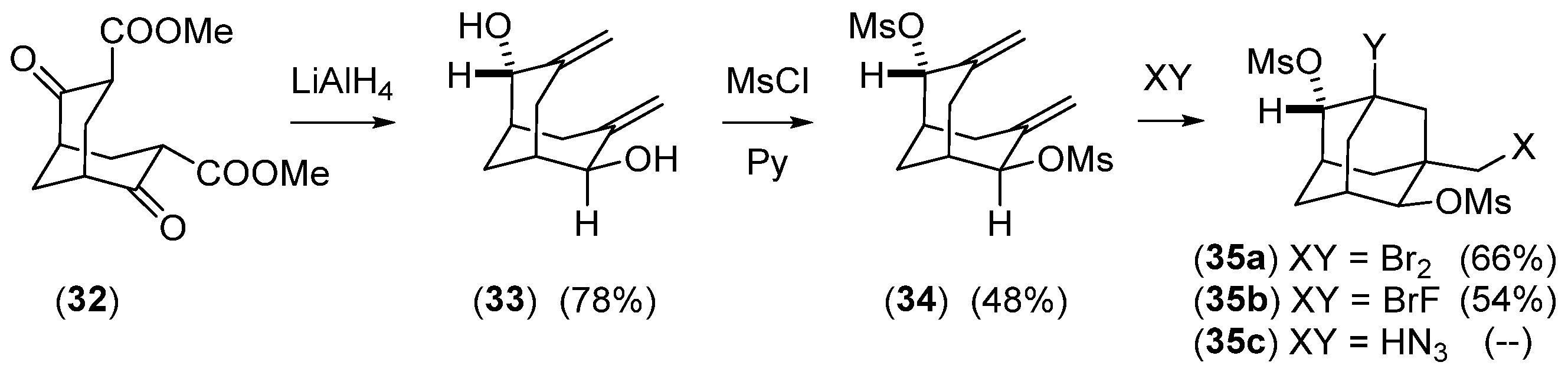
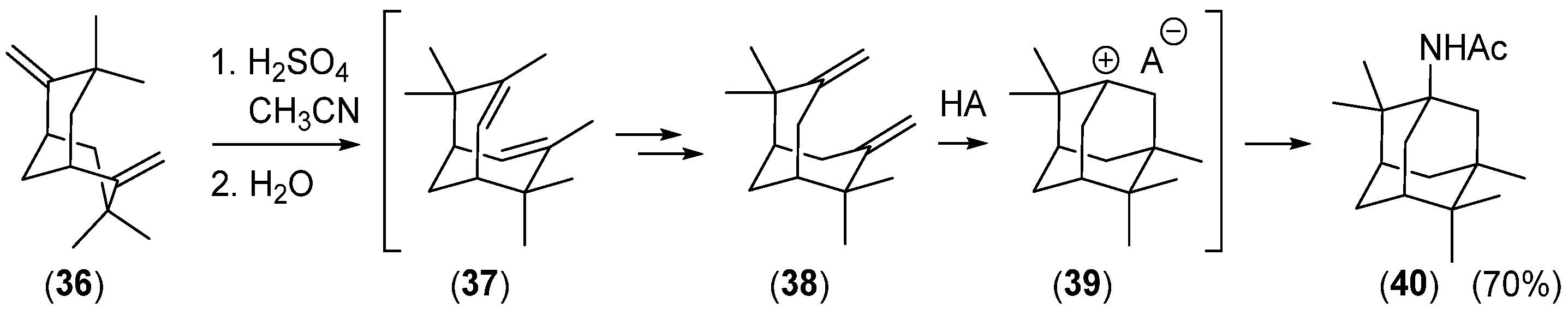
- Bishop, R.; Craig, D.C.; Rae, A.D.; Scudder, M.L. Ritter Reactions. Part 4.’Rearrangement of 3,3,7,7-Tetramethyl-2,6-Dimethylenebicyclo[3.3.1] Inonane and Crystal Structure of 1-Acetamido-2,2,5,6,6-pentamethyladamantane. J. Chem. Soc. Perkin Trans. 1 1989, 4, 733–737. [Google Scholar] [CrossRef]
- Skomorokhov, M.Y.; Klimochkin, Y.N. Halogen Addition to Esters of 7-Methylenebicyclo[3.3.1]non-2-en-3-ol. Russ. J. Org. Chem. 2001, 37, 1188–1189. [Google Scholar] [CrossRef]
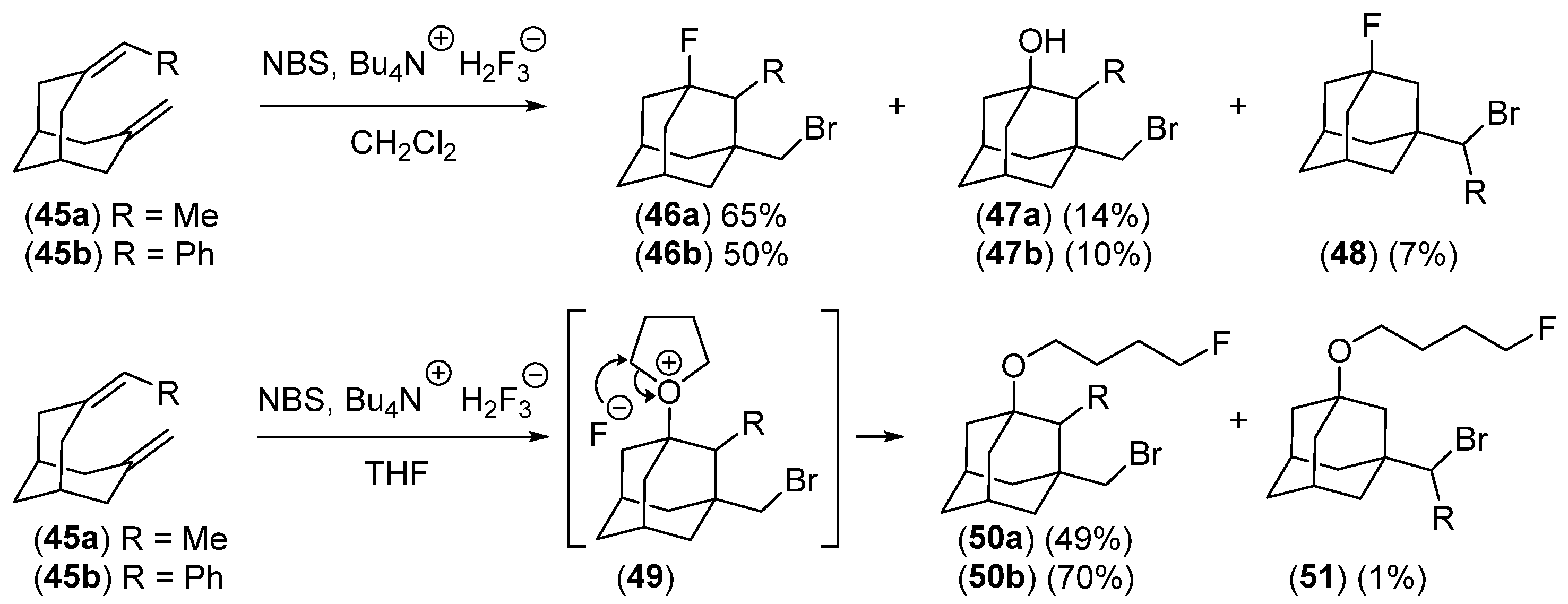
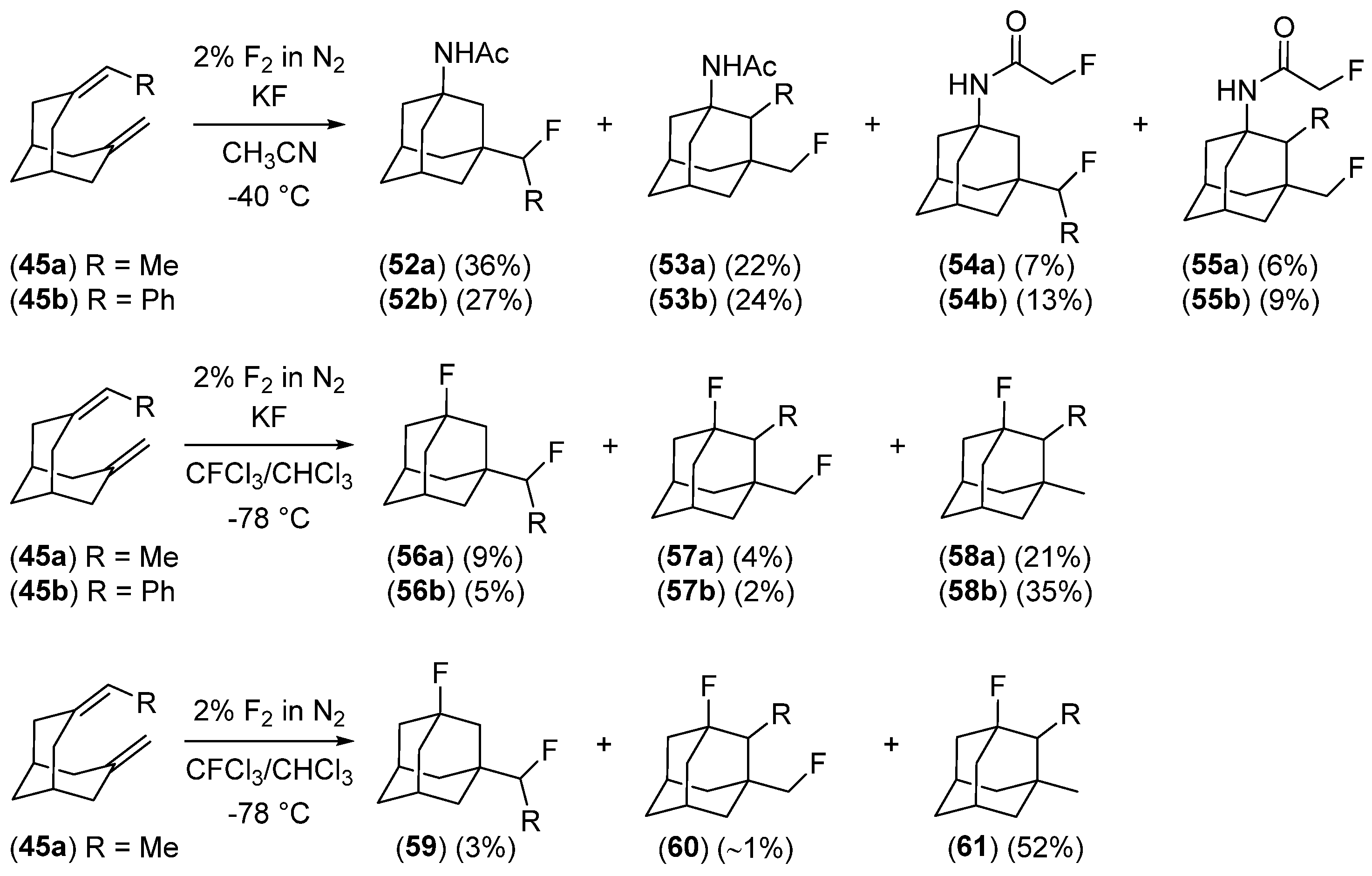
- Serguchev, Y.A.; Ponomarenko, M.V.; Lourie, L.F.; Chernega, A.N. Synthesis of Halo-Fluoro-Substituted Adamantanes by Electrophilic Transannular Cyclization of Bicyclo[3.3.1]nonane Dienes. J. Fluor. Chem. 2003, 123, 207–215. [Google Scholar] [CrossRef]
- Ponomarenko, M.V.; Serguchev, Y.A.; Hirschberg, M.E.; Röschenthaler, G.-V.; Fokin, A.A. Elemental F2 with Transannular Dienes: Regioselectivities and Mechanisms. Chem. Eur. J. 2014, 20, 10383–10391. [Google Scholar] [CrossRef]
- Serguchev, Y.A.; Ponomarenko, M.V.; Lourie, L.F.; Fokin, A.A. Transannular Additions of Selectfluor and Xenon Difluoride: Regioselectivity and Mechanism. J. Phys. Org. Chem. 2011, 24, 407–413. [Google Scholar] [CrossRef]
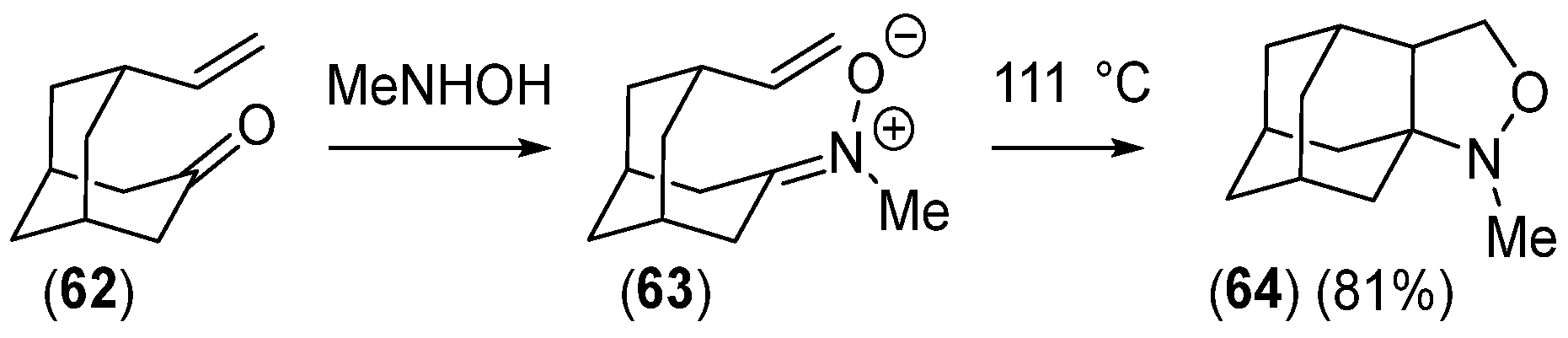
- Funk, R.L.; Bolton, G.L.; Daggett, J.U.; Hansen, M.M.; Horcher, L.H.M. Intramolecular Cycloaddition Reactions of Exocyclic Nitrones: Application in the Total Synthesis of Terpenes. Tetrahedron 1985, 41, 3479–3495. [Google Scholar] [CrossRef]
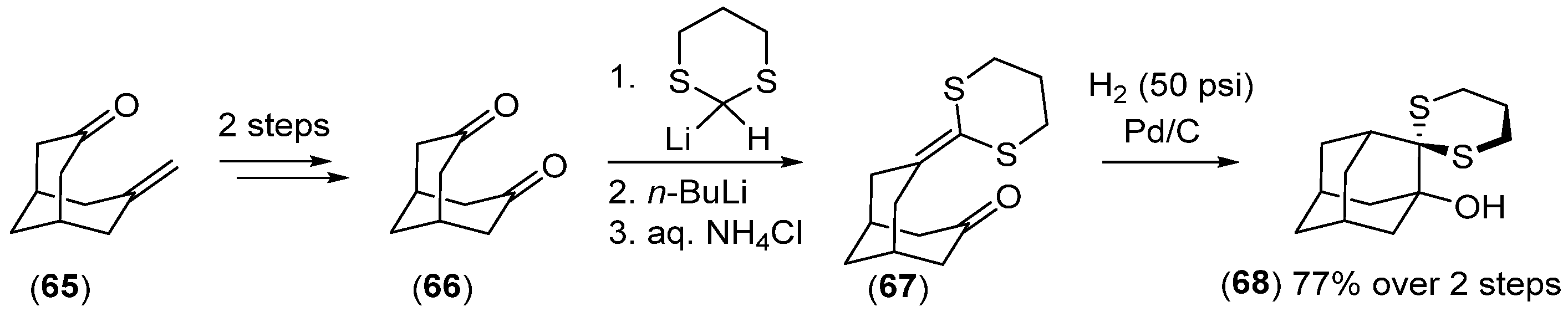
- Marchand, A.P.; Rajapaksa, D. Synthesis of Substituted Hexacyclo[5.4.1.02,6.03,10.05,9.08,11]Dodecanes. A Novel Method for Bridging Across the 8,11-Positions of Pentacyclo[5.4.0.02,6.03,10.05,9]Undecane-8,11-Dione and Related Diketones. Tetrahedron Lett. 1993, 34, 1463–1466. [Google Scholar] [CrossRef]
- Camps, P.; El Achab, R.; Görbig, D.M.; Morral, J.; Muñoz-Torrero, D.; Badia, A.; Baños, J.E.; Vivas, N.M.; Barril, X.; Orozco, M.; et al. Synthesis, in Vitro Pharmacology, and Molecular Modeling of Very Potent Tacrine−Huperzine A Hybrids as Acetylcholinesterase Inhibitors of Potential Interest for the Treatment of Alzheimer’s Disease. J. Med. Chem. 1999, 42, 3227–3242. [Google Scholar] [CrossRef] [PubMed]
- Djaidi, D.; Leung, I.S.H.; Bishop, R.; Craig, D.C.; Scudder, M.L. Ritter Reactions. Part 14. Rearrangement of 3,3,7,7-Tetramethyl-6-methylidenebicyclo[3.3.1]nonan-2-one. J. Chem. Soc. Perkin Trans. 1 2000, 13, 2037–2042. [Google Scholar] [CrossRef]
- Shibuya, M.; Taniguchi, T.; Takahashi, M.; Ogasawara, K. Chiral Modification of Adamantane. Tetrahedron Lett. 2002, 43, 4145–4147. [Google Scholar] [CrossRef]
- Kuga, T.; Sasano, Y.; Tomizawa, M.; Shibuya, M.; Iwabuchi, Y. Expedient Entry to 1-Aminoadamantane Derivatives via Aza-Prins Cyclization of 7-Methylenebicyclo[3.3.1]nonan-3-one Oximes. Synthesis 2018, 50, 1820–1826. [Google Scholar] [CrossRef]
- Peters, J.A.; Van Der Toorn, J.M.; Van Bekkum, H. 3,7-Disubstituted Bicyclo[3.3.1]Nonanes—III: Synthesis and Conformation of Bicyclo[3.3.1]nonane-3α,7α-dicarboxylic Acid, Its Dimethyl Ester and Some Other 3,7-Disubstituted Bicyclo[3.3.1]nonanes; Adamantane as an Integrated Holding System. Tetrahedron 1975, 31, 2273–2281. [Google Scholar] [CrossRef]
- Tkachenko, I.M.; Rybakov, V.B.; Klimochkin, Y.N. Convenient Synthesis of Ethyl 5-Oxohomoadamantane-4-carboxylate: A Useful Precursor of Polyfunctional Homoadamantanes. Synthesis 2019, 51, 1482–1490. [Google Scholar] [CrossRef]
- Lightner, D.A.; Van Toan, V. The Octant Rule. XX: Synthesis and Circular Dichroism of (1s,5s)-Dimethyladamantan-2-one—Predicted to Have Zero Cotton Effect. Tetrahedron 1987, 43, 4905–4916. [Google Scholar] [CrossRef]
- Denmark, S.E.; Henke, B.R. Investigations on Transition-State Geometry in the Aldol Condensation. J. Am. Chem. Soc. 1989, 111, 8032–8034. [Google Scholar] [CrossRef]
- Whitlock, H.W.; Siefken, M.W. Tricyclo[4.4.0.03,8]decane to Adamantane Rearrangement. J. Am. Chem. Soc. 1968, 90, 4929–4939. [Google Scholar] [CrossRef]
- Janjatovic, J.; Majerski, Z. Synthesis of Adamantanoid Ketones from Bridgehead Alcohols by the Hypoiodite Thermolysis-Cyclization Sequence. J. Org. Chem. 1980, 45, 4892–4898. [Google Scholar] [CrossRef]
- Kolocouris, N.; Zoidis, G.; Fytas, C. Facile Synthetic Routes to 2-Oxo-1-adamantanalkanoic Acids. Synlett 2007, 2007, 1063–1066. [Google Scholar] [CrossRef]
- Hrdina, R.; Holovko-Kamoshenkova, O.M.; Císařová, I.; Koucký, F.; Machalický, O. Annulated Carbamates Are Precursors for the Ring Contraction of the Adamantane Framework. RSC Adv. 2022, 12, 31056–31060. [Google Scholar] [CrossRef]
- Zonker, B.; Becker, J.; Hrdina, R. Synthesis of Noradamantane Derivatives by Ring-Contraction of the Adamantane Framework. Org. Biomol. Chem. 2021, 19, 4027–4031. [Google Scholar] [CrossRef]
- Lenoir, D.; Schleyer, P.V.R. Chemistry of 4-Protoadamantyl Derivatives. J. Chem. Soc. D 1970, 15, 941–942. [Google Scholar] [CrossRef]
- Lenoir, D.; Hall, R.E.; Schleyer, P.v.R. Chemistry of Protoadamantane. IV. Preparation and Solvolysis of Secondary 4-Protoadamantyl Esters. Relation to the Solvolysis of 2-Adamantyl Derivatives. J. Am. Chem. Soc. 1974, 96, 2138–2148. [Google Scholar] [CrossRef]
- Schleyer, P.v.R.; Lenoir, D.; Glaser, R.; Mison, P. Synthesis of 1,2- and 2,4-Disubstituted Adamantanes. The Protoadamantane Route. J. Org. Chem. 1971, 36, 1821–1826. [Google Scholar] [CrossRef]
- Lenoir, D.; Raber, D.J.; Schleyer, P.V.R. Chemistry of Protoadamantane. V. Solvolysis of 4-Methyl-4-protoadamantyl and Related 1-Methyl-2-adamantyl Derivatives. J. Am. Chem. Soc. 1974, 96, 2149–2156. [Google Scholar] [CrossRef]
- Herpers, E.; Kirmse, W. The 4-Protoadamantyl → 2-Adamantyl Rearrangement; Chirality of the 2-Adamantyl Cation. J. Chem. Soc. Chem. Commun. 1993, 2, 160–161. [Google Scholar] [CrossRef]
- Cuddy, B.D.; Grant, D.; McKervey, M.A. The Protoadamantane Route to 1,2- and 2,4-Disubstituted Adamantanes. J. Chem. Soc. C 1971, 3173–3179. [Google Scholar] [CrossRef]
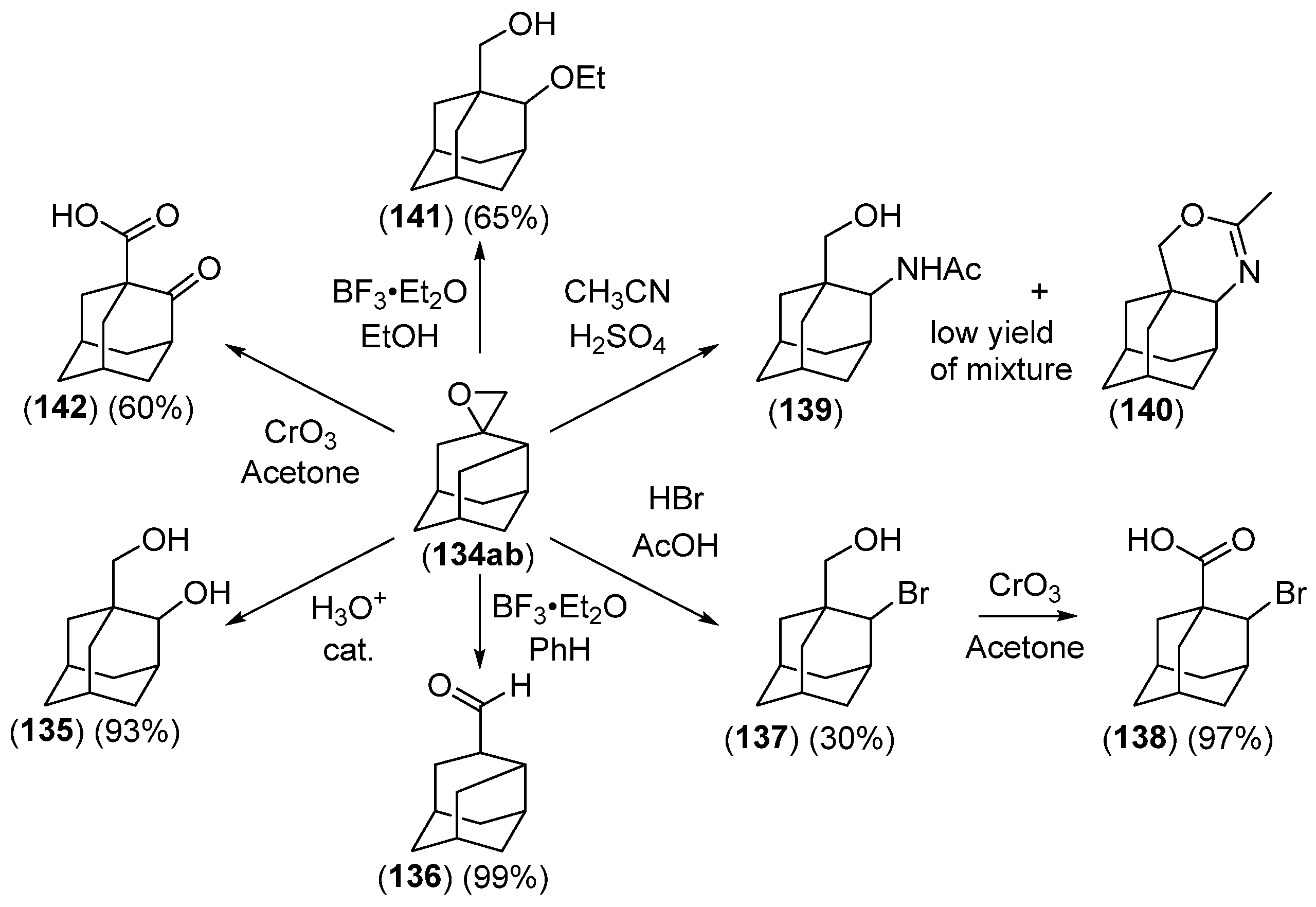
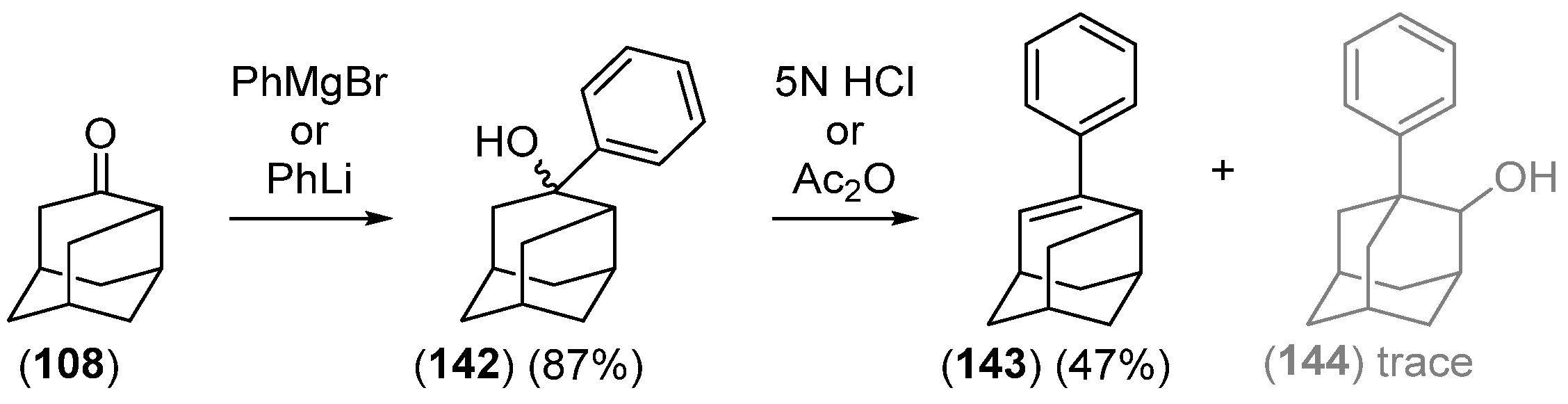
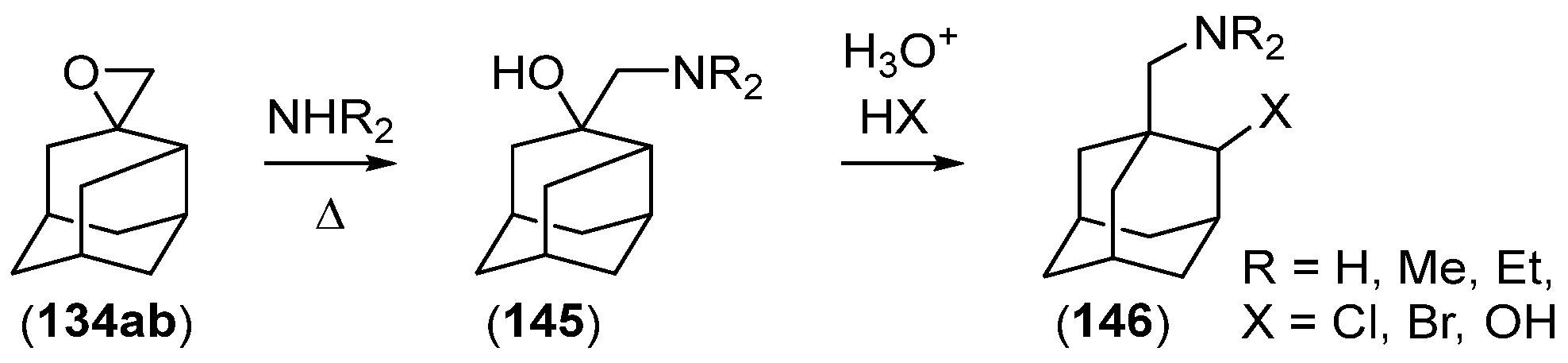
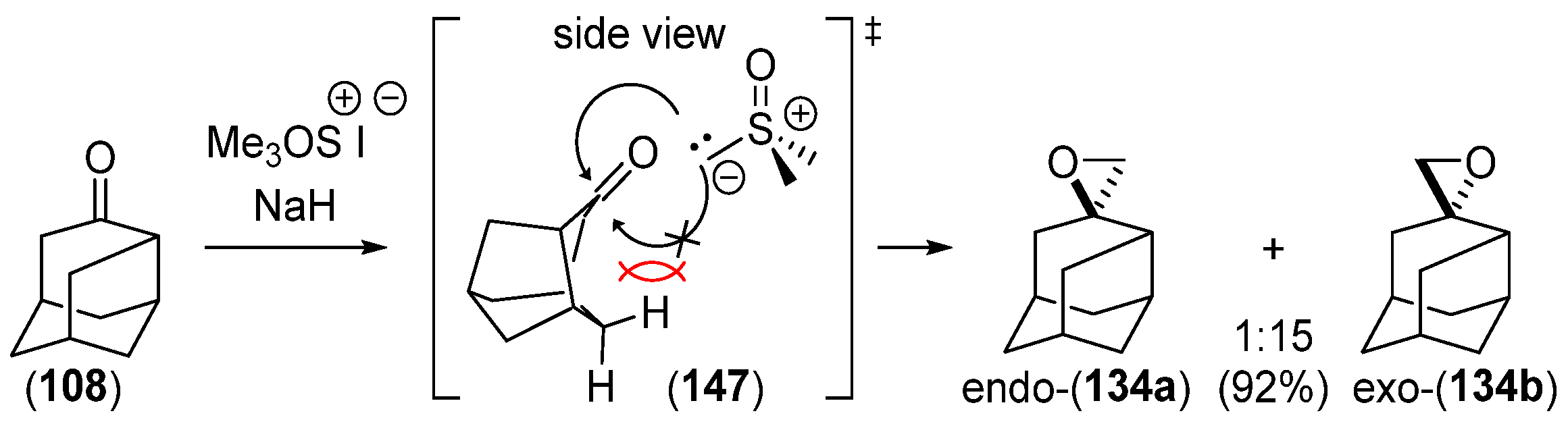
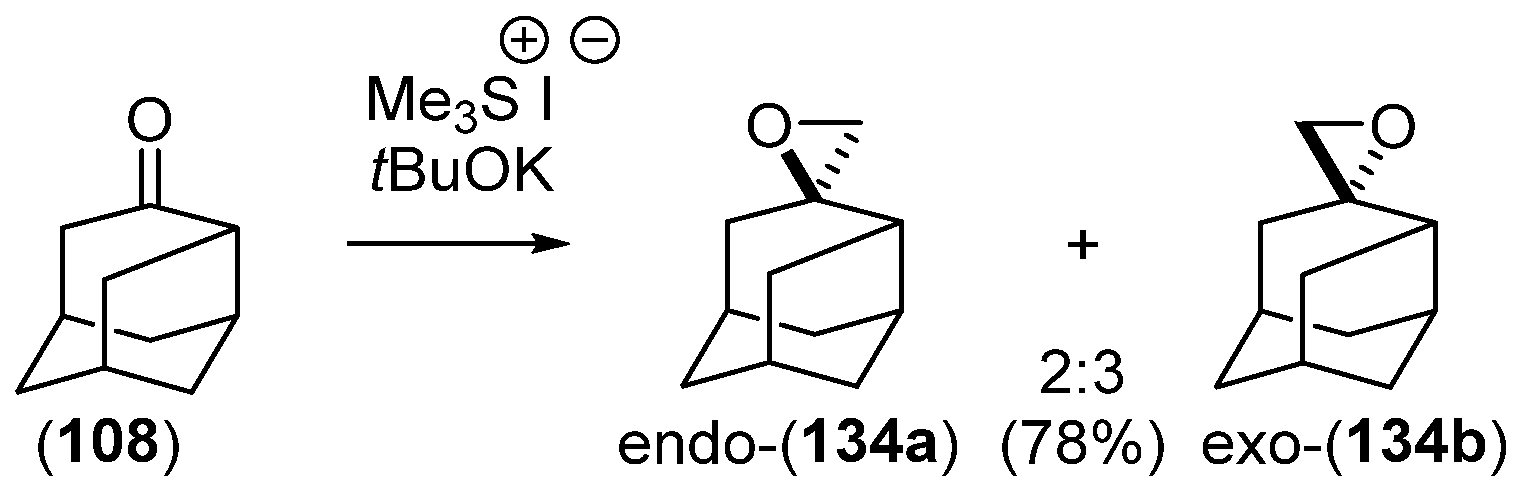
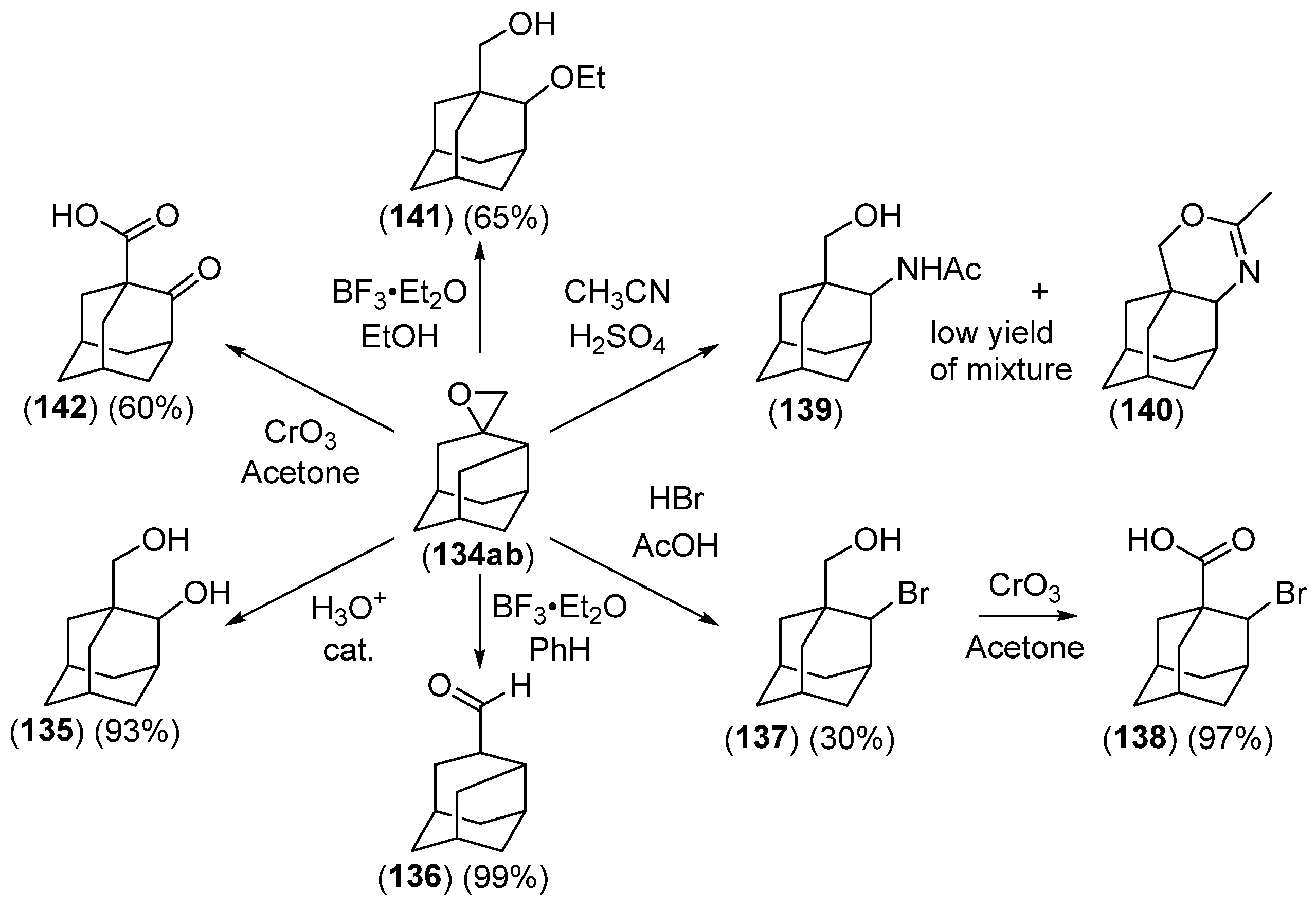
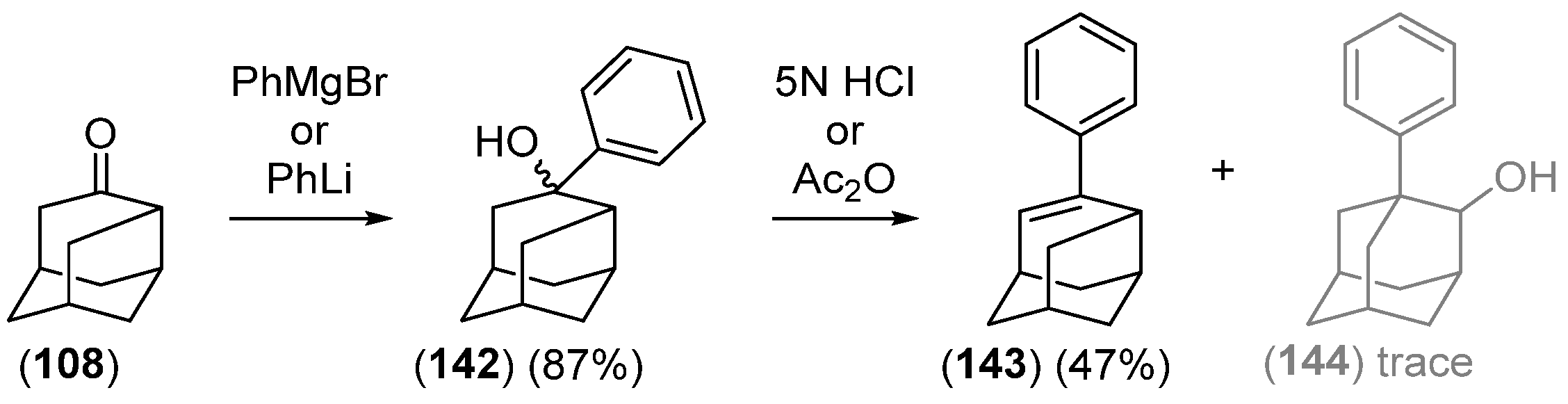
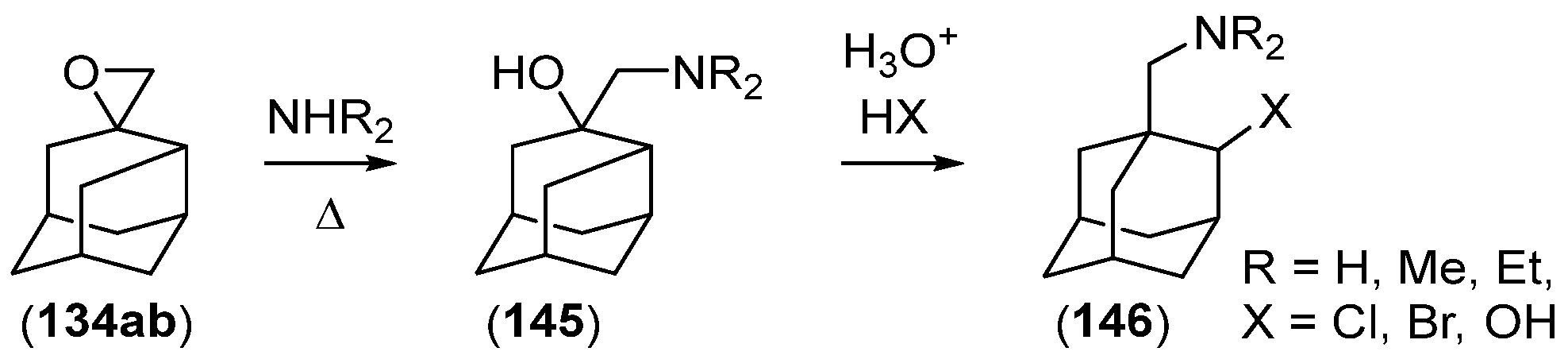
- Chakrabarti, J.K.; Hotten, T.M.; Tupper, D.E. Chemistry of Adamantane. VIII. Synthesis of 1,2-Difunctional Adamantanes Using Protoadamantane-4-Spiroöxirane as a Novel Intermediate. Tetrahedron Lett. 1975, 16, 2241–2244. [Google Scholar] [CrossRef]
- Chakrabarti, J.K.; Hotten, T.M.; Rackham, D.M.; Tupper, D.E. Chemistry of Adamantane. Part IX. 1,2-Difunctional Adamantanes; Synthesis and Reactions of Protoadamantane-4-spiro-oxiran. J. Chem. Soc. Perkin Trans. 1 1976, 17, 1893–1900. [Google Scholar] [CrossRef]
- Chakrabarti, J.K.; Hotten, T.M.; Sutton, S.; Tupper, D.E. Adamantane and Protoadamantanealkanamines as Potential Anti-Parkinson Agents. 10. J. Med. Chem. 1976, 19, 967–969. [Google Scholar] [CrossRef]
- Abdel-Sayed, A.N.; Bauer, L. Syntheses of 1,2-Distributed Adamantanes. Tetrahedron 1988, 44, 1873–1882. [Google Scholar] [CrossRef]
- Chyi Tseng, C.; Handa, I.; Abdel-Sayed, A.N.; Bauer, L. N-[(Aryl Substitute Adamantane)Alkyl] 2-Mercaptoacetamidines, Their Corresponding Disulfides and 5-Phosphorothioates. Tetrahedron 1988, 44, 1893–1904. [Google Scholar] [CrossRef]
- Mlinarić-Majerski, K.; Veljković, J.; Kaselj, M.; Marchand, A.P. Dihaloadamantanes: Ring Closure versus Rearrangement or Halogen-Displacement Reactions. Eur. J. Org. Chem. 2004, 2004, 2923–2927. [Google Scholar] [CrossRef]
- Mlinaric-Majerski, K.; Kaselj, M. 1,2-Methanoadamantane: A Molecule with a Twist Bent. Sigma. Bond. J. Org. Chem. 1994, 59, 4362–4363. [Google Scholar] [CrossRef]
- Mlinarić-Majerski, K.; Kragol, G.; Ramljak, T.Š. Transannular Cyclization with Grignard Reagents: Facile Synthetic Routes to Oxaadamantane and Protoadamantane Derivatives. Synlett 2008, 2008, 405–409. [Google Scholar] [CrossRef]
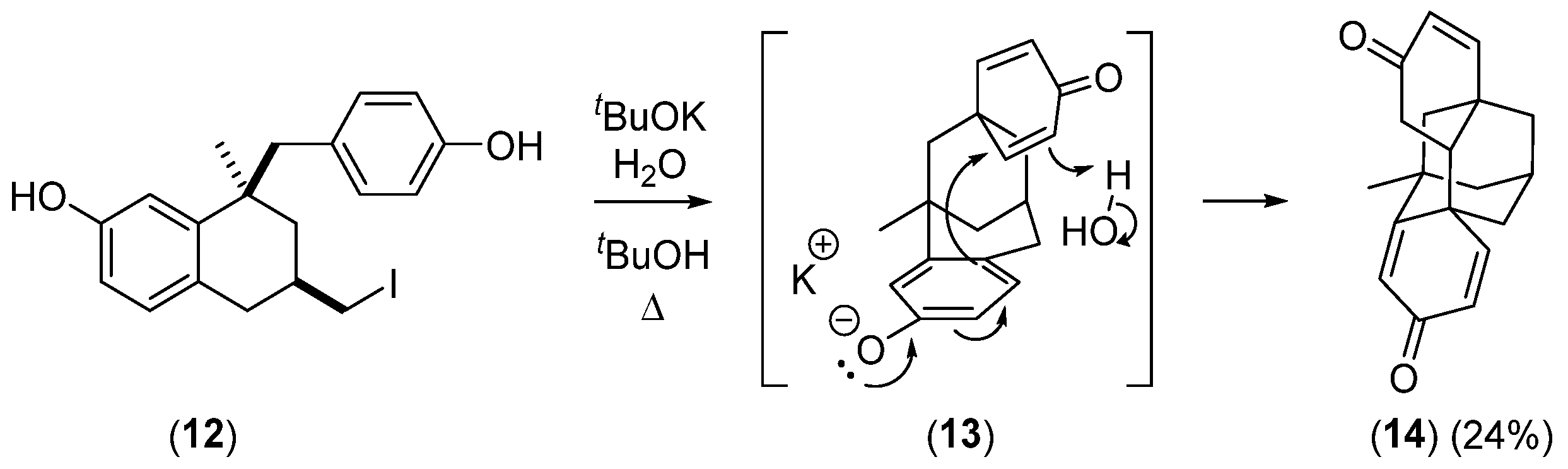
- Cindro, N.; Antol, I.; Mlinarić-Majerski, K.; Halasz, I.; Wan, P.; Basarić, N. Reactivity of Cations and Zwitterions Formed in Photochemical and Acid-Catalyzed Reactions from m-Hydroxycycloalkyl-Substituted Phenol Derivatives. J. Org. Chem. 2015, 80, 12420–12430. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dong, Y.; Ezell, E.L.; Garrison, J.C.; Wood, J.K.; Hagen, J.P.; Vennerstrom, J.L. Semipinacol and Protoadamantane-adamantane Rearrangements of 5,6-Dibromoadamantan-2-one and -2-ol. Tetrahedron 2017, 73, 2972–2976. [Google Scholar] [CrossRef]
- Yoshihara, T.; Shudo, H.; Yagi, A.; Itami, K. Adamantane Annulation to Arenes: A Strategy for Property Modulation of Aromatic π-Systems. J. Am. Chem. Soc. 2023, 145, 11754–11763. [Google Scholar] [CrossRef]
- Papanastasiou, I.; Tsotinis, A.; Kolocouris, N.; Prathalingam, S.R.; Kelly, J.M. Design, Synthesis, and Trypanocidal Activity of New Aminoadamantane Derivatives. J. Med. Chem. 2008, 51, 1496–1500. [Google Scholar] [CrossRef] [PubMed]
- Zoidis, G.; Tsotinis, A.; Kolocouris, N.; Kelly, J.M.; Radhika Prathalingam, S.; Naesens, L.; Clercq, E.D. Design and Synthesis of Bioactive 1,2-Annulated Adamantane Derivatives. Org. Biomol. Chem. 2008, 6, 3177–3185. [Google Scholar] [CrossRef] [PubMed]
- Papanastasiou, I.; Tsotinis, A.; Zoidis, G.; Kolocouris, N.; Prathalingam, S.R.; Kelly, J.M. Design and Synthesis of Trypanosoma Brucei Active 1-Alkyloxy and 1-Benzyloxyadamantano 2-Guanylhydrazones. ChemMedChem 2009, 4, 1059–1062. [Google Scholar] [CrossRef]
- Zoidis, G.; Kolocouris, N.; Kelly, J.M.; Prathalingam, S.R.; Naesens, L.; De Clercq, E. Design and Synthesis of Bioactive Adamantanaminoalcohols and Adamantanamines. Eur. J. Med. Chem. 2010, 45, 5022–5030. [Google Scholar] [CrossRef] [PubMed]
- Papanastasiou, I.; Tsotinis, A.; Kolocouris, N.; Nikas, S.P.; Vamvakides, A. New Aminoadamantane Derivatives with Antiproliferative Activity. Med. Chem. Res. 2014, 23, 1966–1975. [Google Scholar] [CrossRef]
- Zoidis, G.; Sandoval, A.; Pineda-Farias, J.B.; Granados-Soto, V.; Felix, R. Anti-Allodynic Effect of 2-(Aminomethyl)adamantane-1-carboxylic Acid in a Rat Model of Neuropathic Pain: A Mechanism Dependent on CaV2.2 Channel Inhibition. Bioorg. Med. Chem. 2014, 22, 1797–1803. [Google Scholar] [CrossRef]
- Pardali, V.; Giannakopoulou, E.; Konstantinidi, A.; Kolocouris, A.; Zoidis, G. 1,2-Annulated Adamantane Heterocyclic Derivatives as Anti-Influenza A Virus Agents. Croat. Chem. Acta 2019, 92, 211–228. [Google Scholar] [CrossRef]
- Vogt, B.R.; Hoover, J.R.E. The Synthesis of Noradamantane. Tetrahedron Lett. 1967, 8, 2841–2843. [Google Scholar] [CrossRef]
- Schleyer, P.v.R.; Wiskott, E. Noradamatane by Aluminum Halide Catalyzed Rearrangement. Tetrahedron Lett. 1967, 8, 2845–2850. [Google Scholar] [CrossRef]
- Stoelting, D.T.; Shiner, V.J., Jr. Solvolysis of 1-(3-Noradamantyl)ethyl Sulfonates. J. Am. Chem. Soc. 1993, 115, 1695–1705. [Google Scholar] [CrossRef]
- Schleyer, P.v.R.; Lam, L.K.M.; Raber, D.J.; Fry, J.L.; McKervey, M.A.; Alford, J.R.; Cuddy, B.D.; Keizer, V.G.; Geluk, H.W.; Schlatmann, J.L.M.A. Stereochemical Inhibition of Intramolecular 1,2 Shifts. Intermolecular Nature of Hydride Shifts in the Adamantane Series. J. Am. Chem. Soc. 1970, 92, 5246–5247. [Google Scholar] [CrossRef]
- Majerski, Z.; Schleyer, P.v.R.; Wolf, A.P. Stereochemical Inhibition of Intramolecular 1,2 Shifts. Mechanistic Evidence for Skeletal Rearrangement during Apparent 1,2-Methyl Shifts of Adamantane. J. Am. Chem. Soc. 1970, 92, 5731–5733. [Google Scholar] [CrossRef]
- Okazaki, T.; Isobe, H.; Kitagawa, T.; Takeuchi, K. Generation and Reactions of 2-(1-Adamantyl)adamantene. Rearrangement to 3-(1-Adamantyl)-4-protoadamantylidene. Bull. Chem. Soc. Jpn. 1996, 69, 2053–2062. [Google Scholar] [CrossRef]
- Sosnowski, J.J.; Rheingold, A.L.; Murray, R.K., Jr. Bridgehead Functionalization of [1]Diadamantane. J. Org. Chem. 1985, 50, 3788–3791. [Google Scholar] [CrossRef]
- Takeuchi, K.; Kitagawa, I.; Akiyama, F.; Shibata, T.; Kato, M.; Okamoto, K. Ring-Expansion of Bridgehead Aldehydes with 1-Adamantanecarbonyl Cation or Benzoyl Trifluoromethanesulfonate: A New Route to Bicyclic and Tricyclic 1,2-Diols. Synthesis 1987, 1987, 612–615. [Google Scholar] [CrossRef]
- Ohga, Y.; Takeuchi, K. Steric Deuterium Isotope Effect in the Solvolysis of (Z)-[Methyl-d3]-2-ethylidene-1-adamantyl Iodide Accelerated by F-Strain. J. Phys. Org. Chem. 1993, 6, 293–301. [Google Scholar] [CrossRef]
- Okazaki, T.; Tokunaga, K.; Kitagawa, T.; Takeuchi, K. Generation and Reactivity of 2-Substituted Adamantenes. Bull. Chem. Soc. Jpn. 1999, 72, 549–561. [Google Scholar] [CrossRef]
- Torres, E.; Fernández, R.; Miquet, S.; Font-Bardia, M.; Vanderlinden, E.; Naesens, L.; Vázquez, S. Synthesis and Anti-Influenza A Virus Activity of 2,2-Dialkylamantadines and Related Compounds. ACS Med. Chem. Lett. 2012, 3, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, S.; Krassos, H.; Nicolaides, A.V. Synthesis of a Novel Diene from a Cyclobutane Precursor: An Entry to 2,9-Disubstituted [2]Diadamantanes. Tetrahedron 2013, 69, 8064–8068. [Google Scholar] [CrossRef]
- Zonker, B.; Duman, E.; Hausmann, H.; Becker, J.; Hrdina, R. [1,2]-Rearrangement of Iminium Salts Provides Access to Heterocycles with Adamantane Scaffold. Org. Biomol. Chem. 2020, 18, 4941–4945. [Google Scholar] [CrossRef]


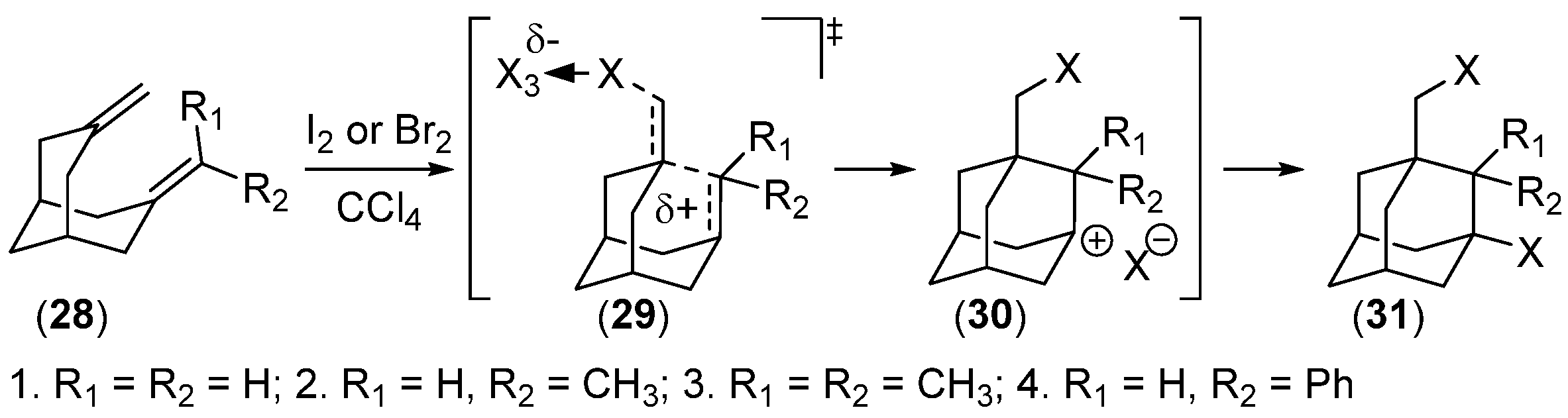
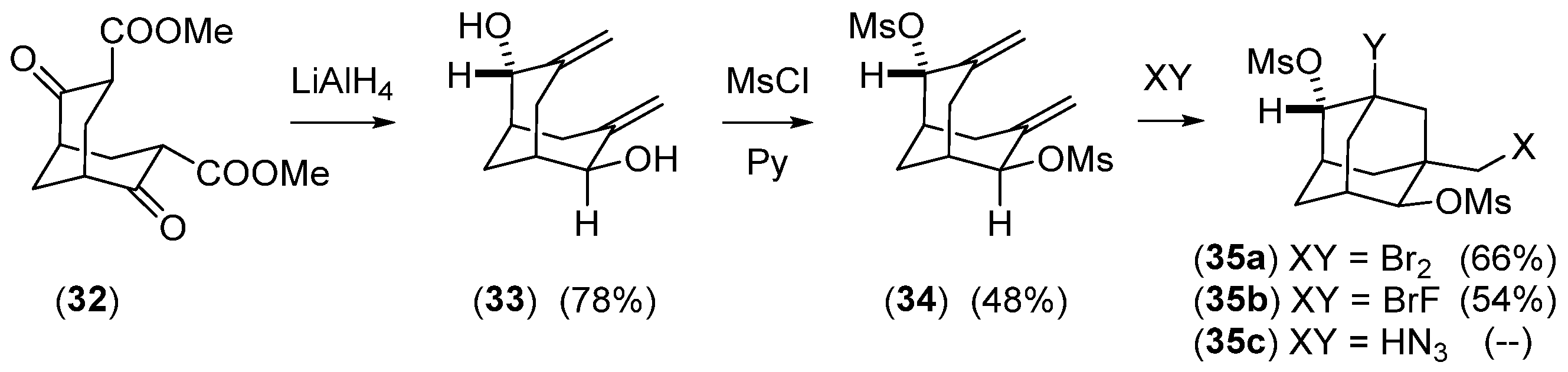
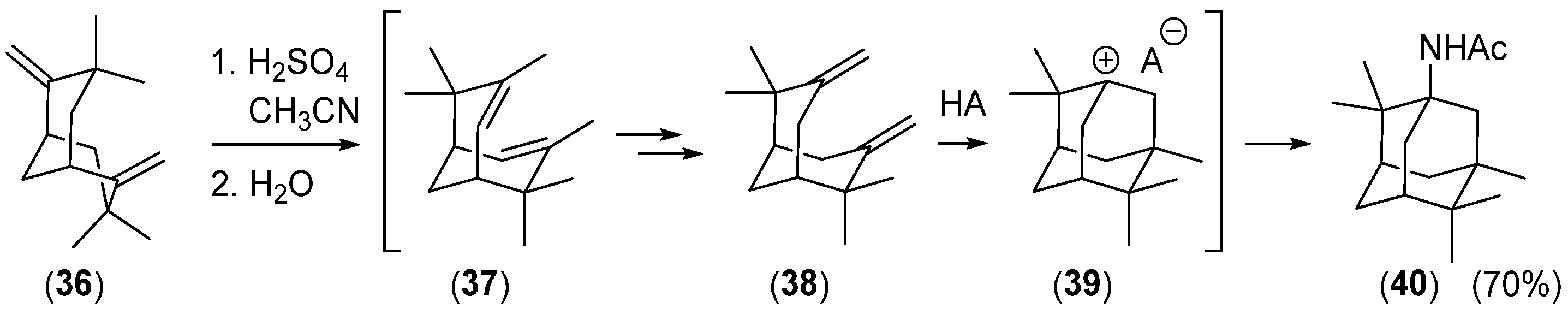

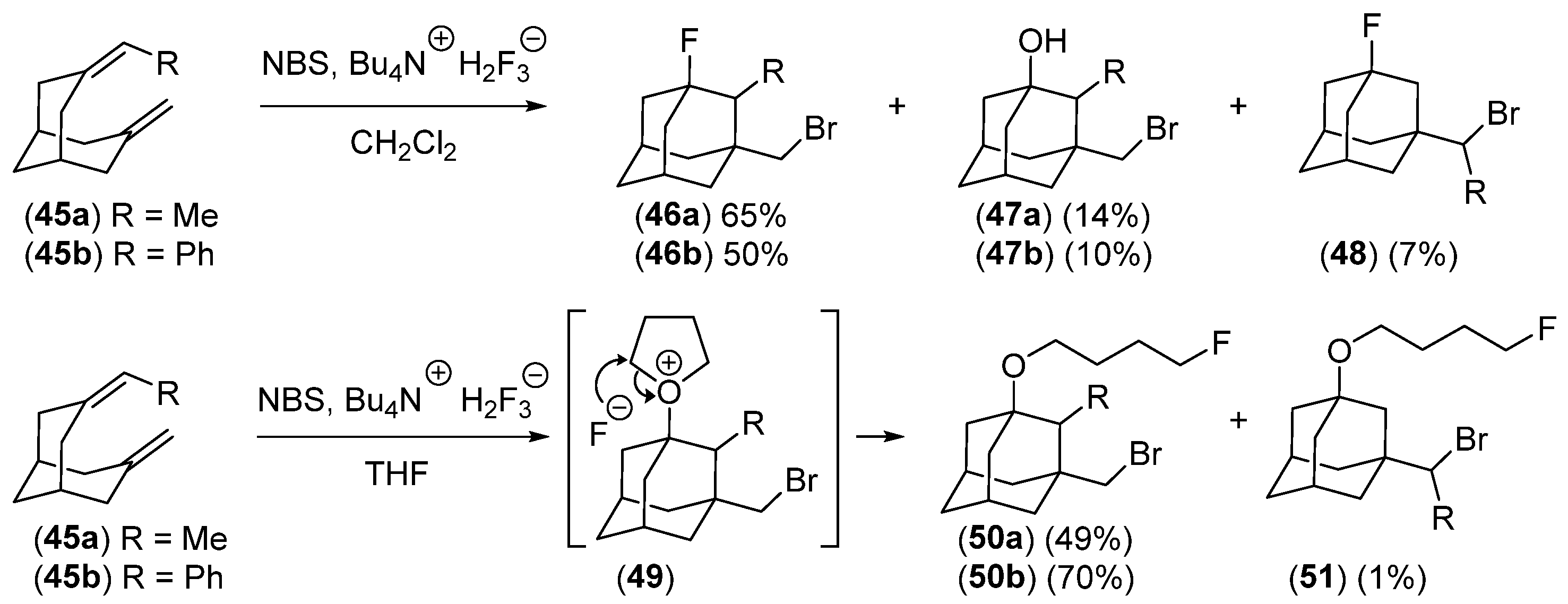

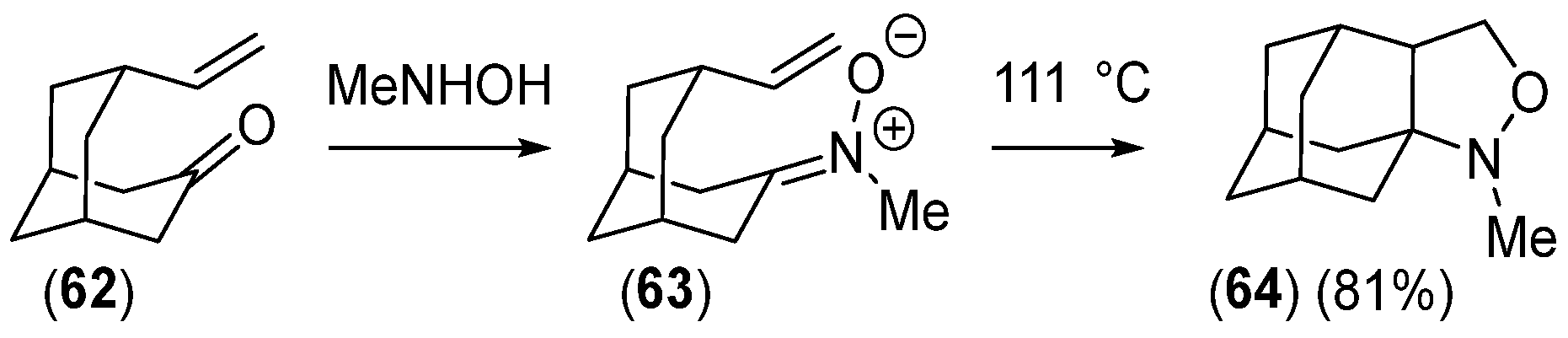
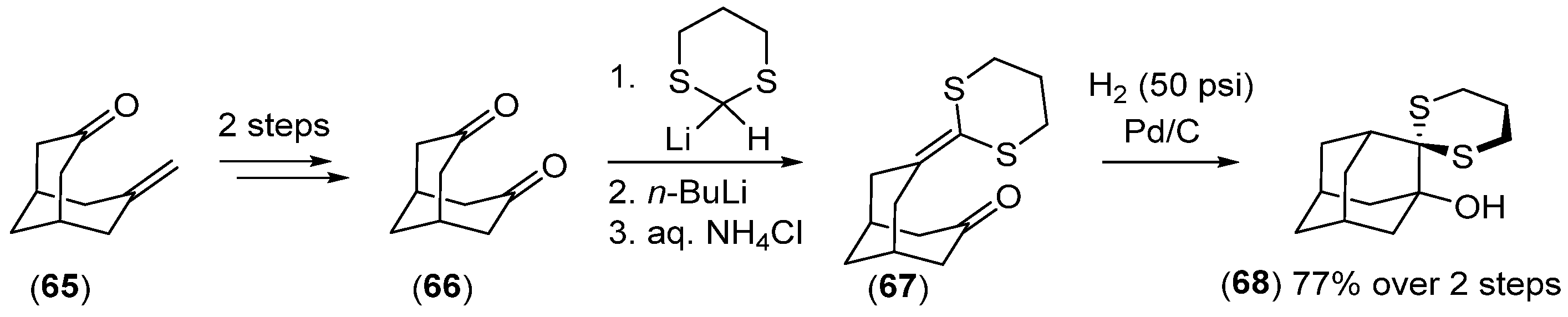
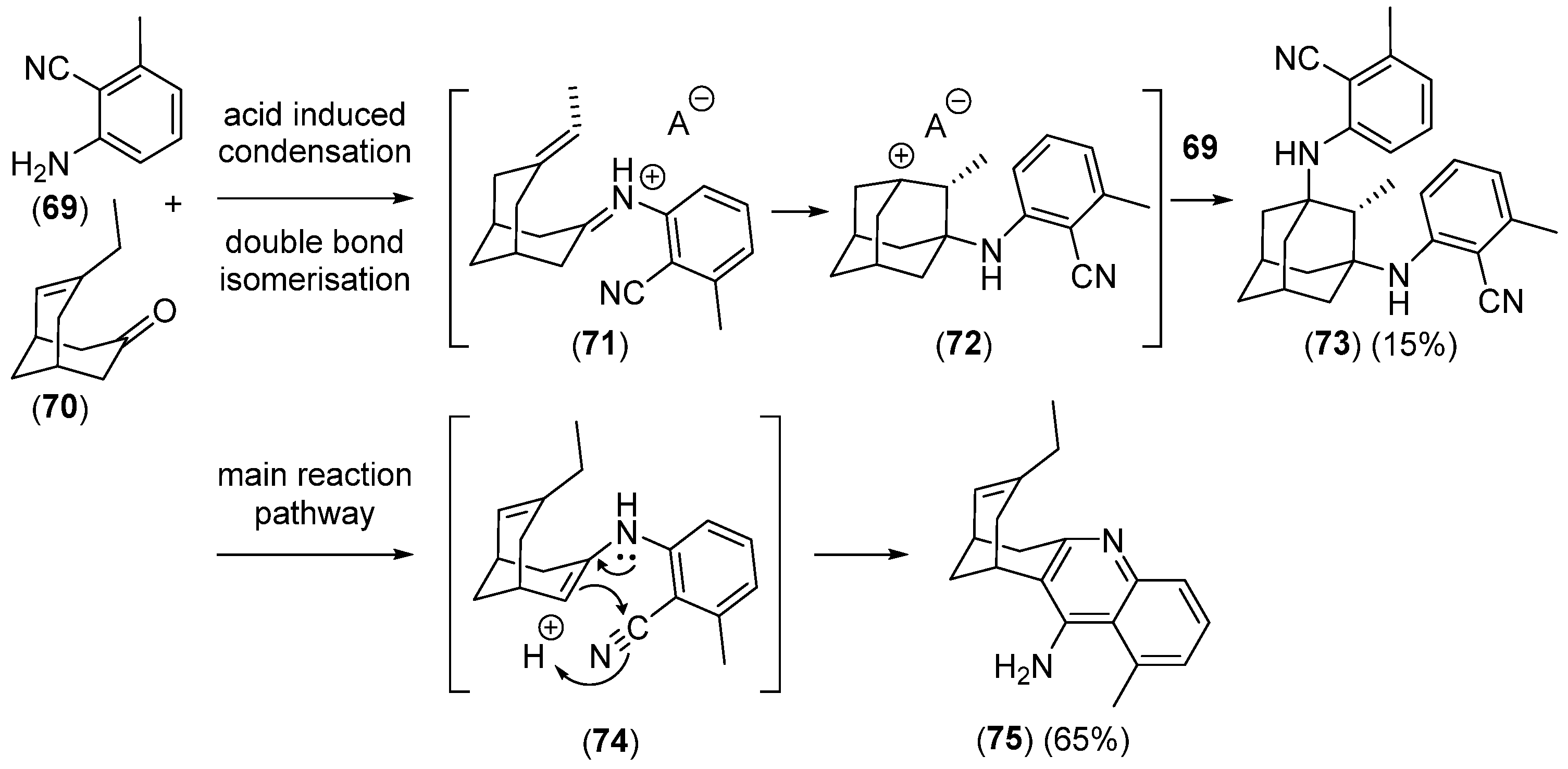
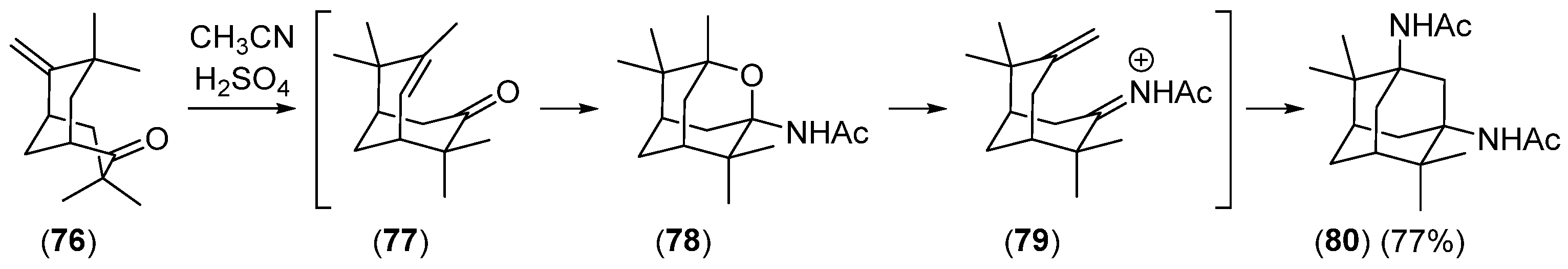
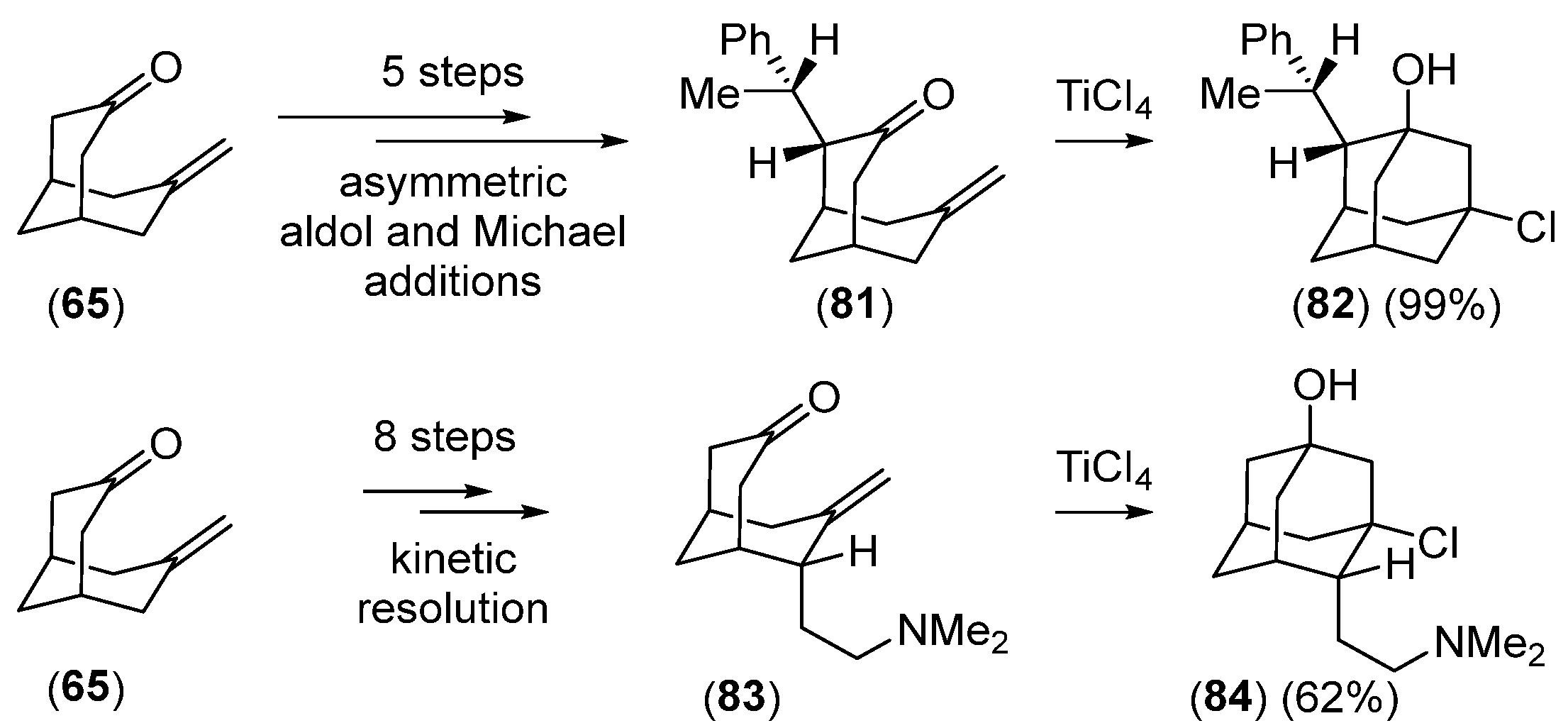
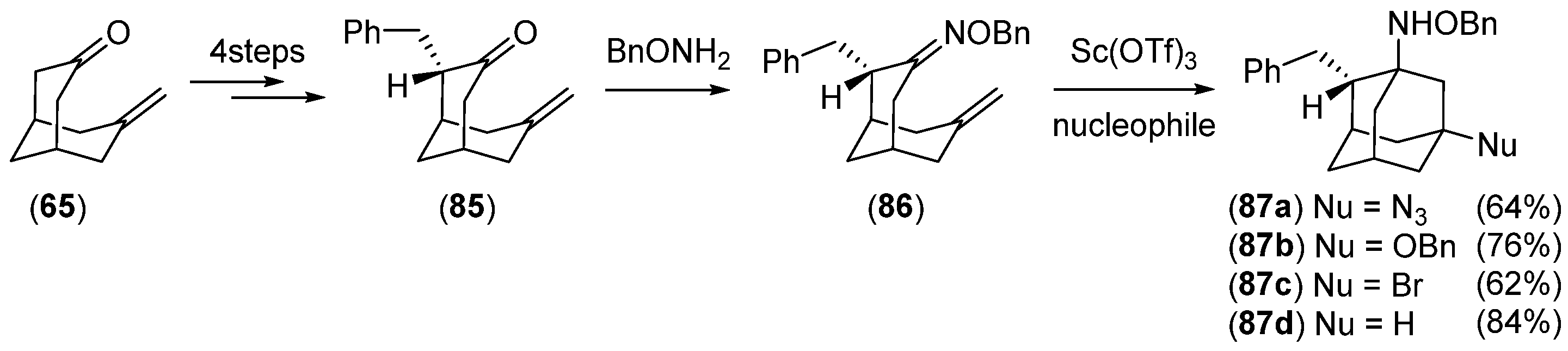
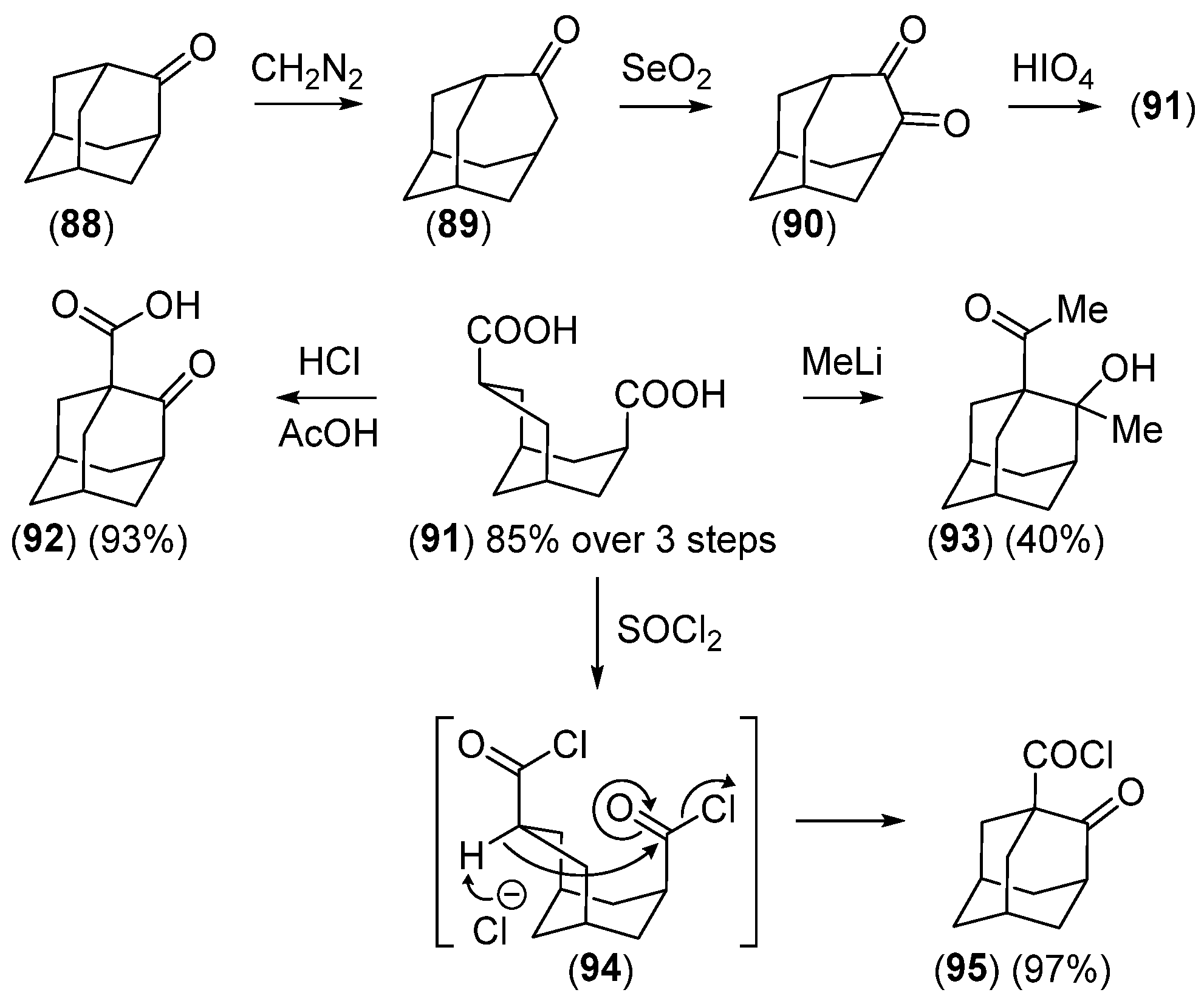

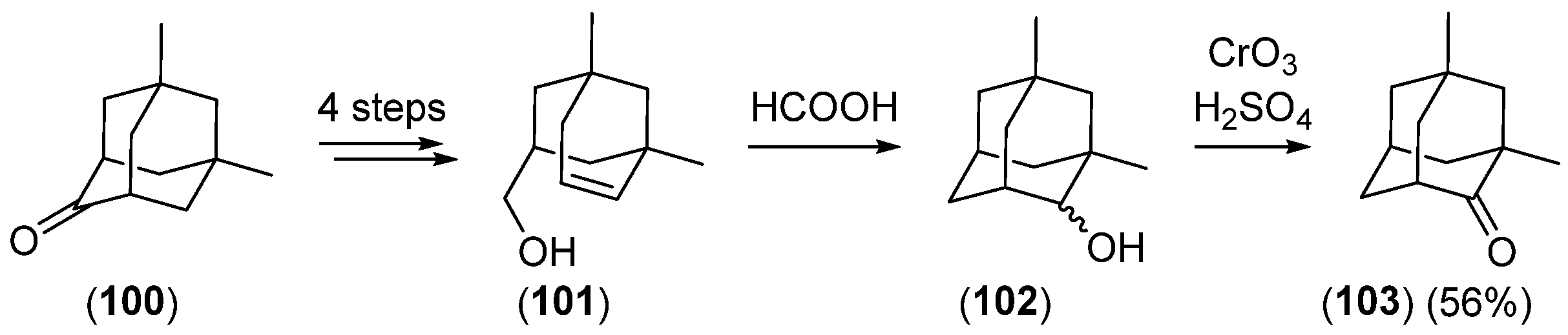
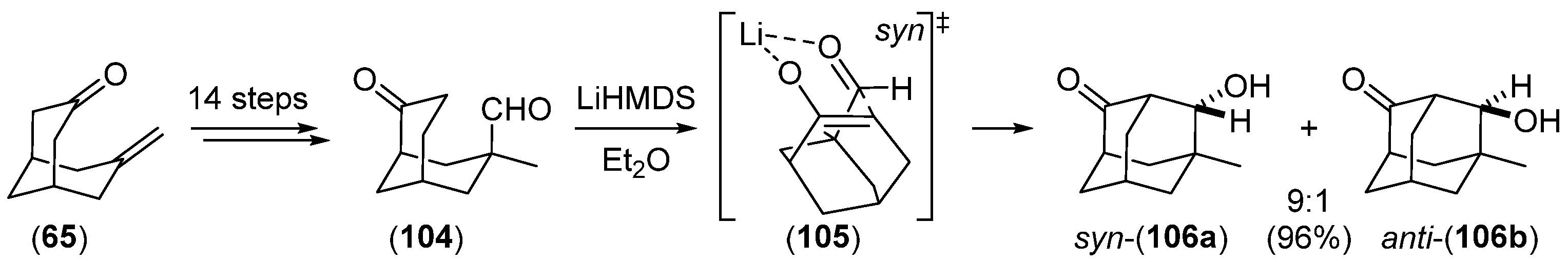


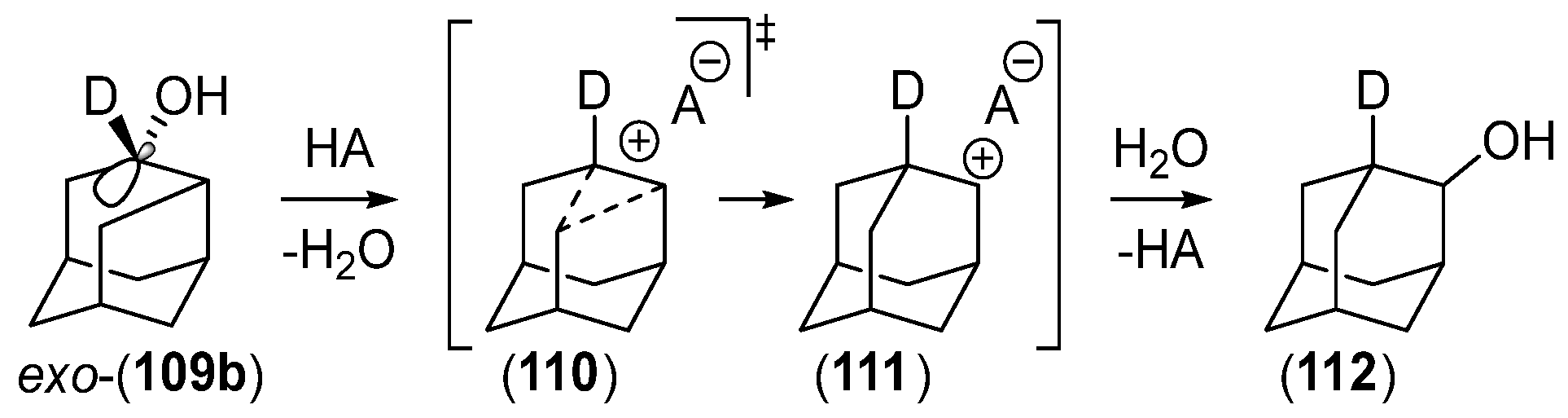
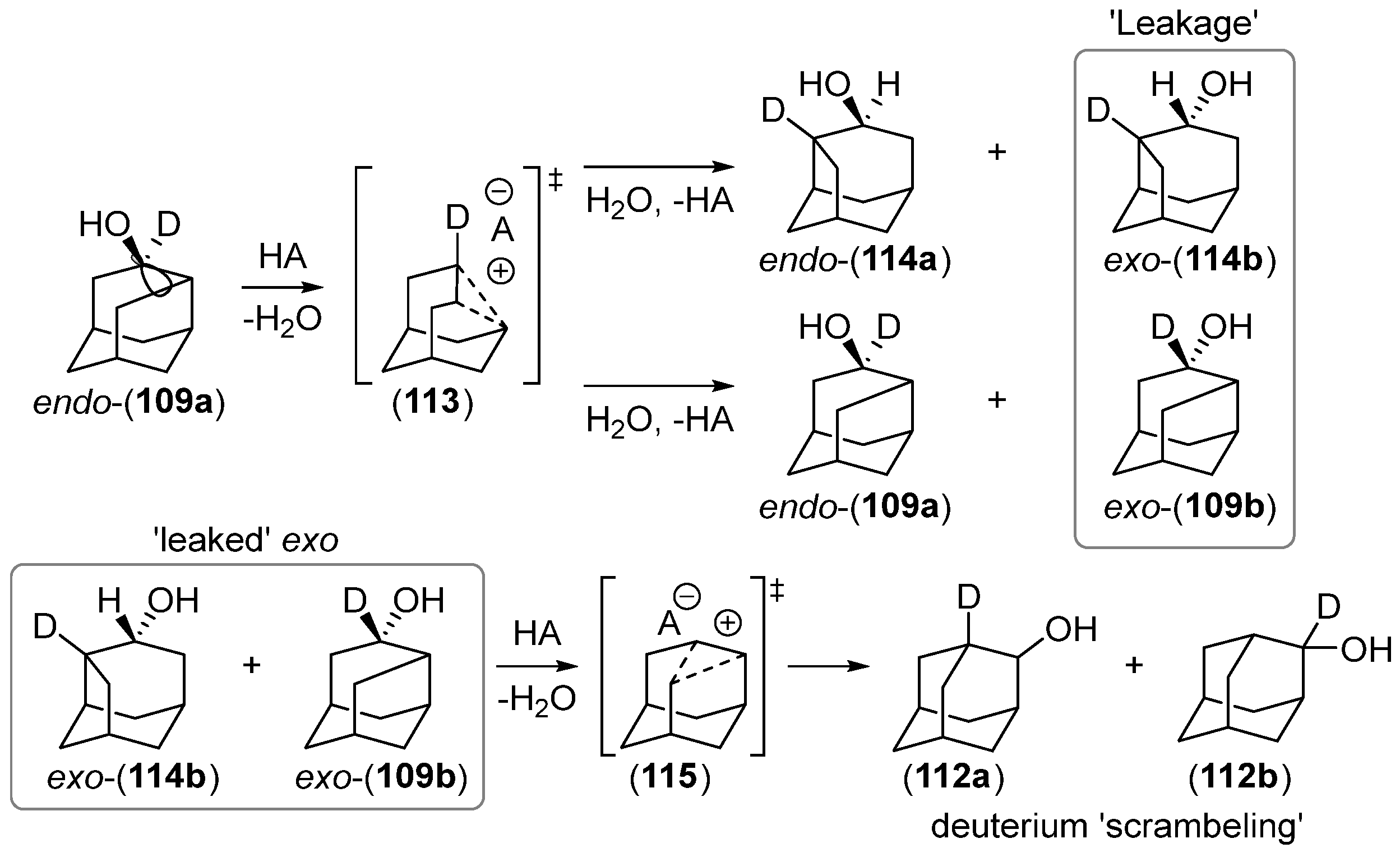
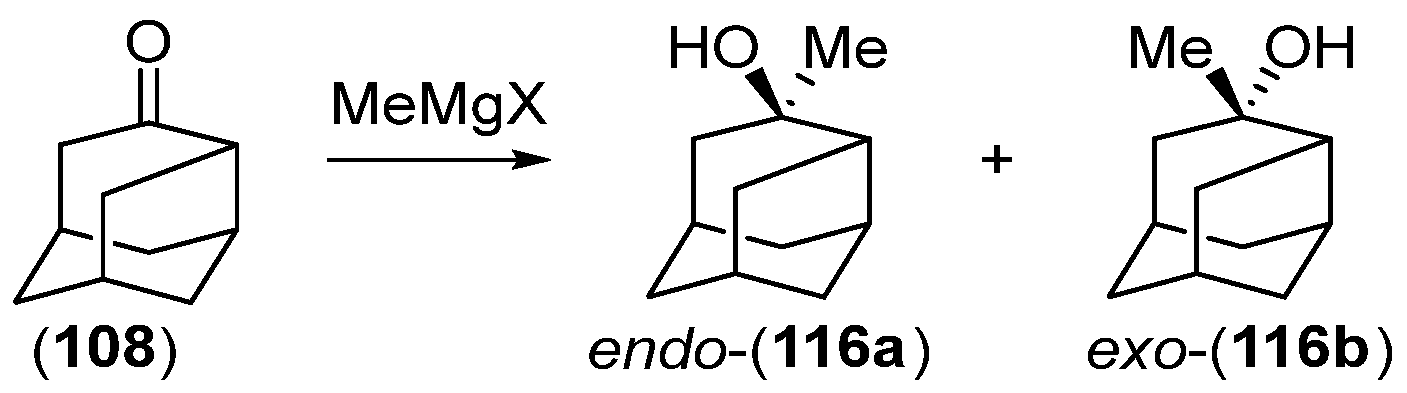
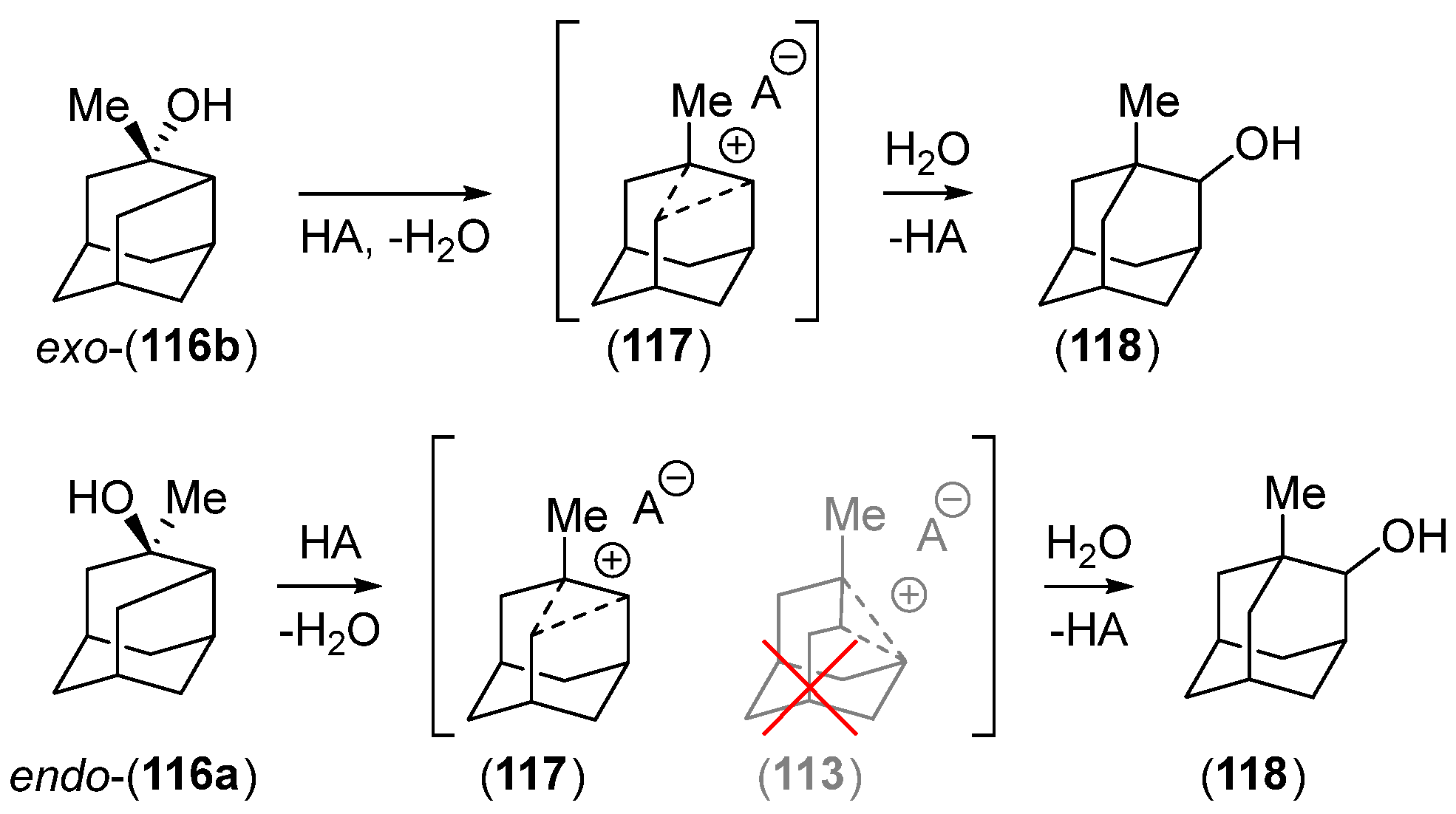
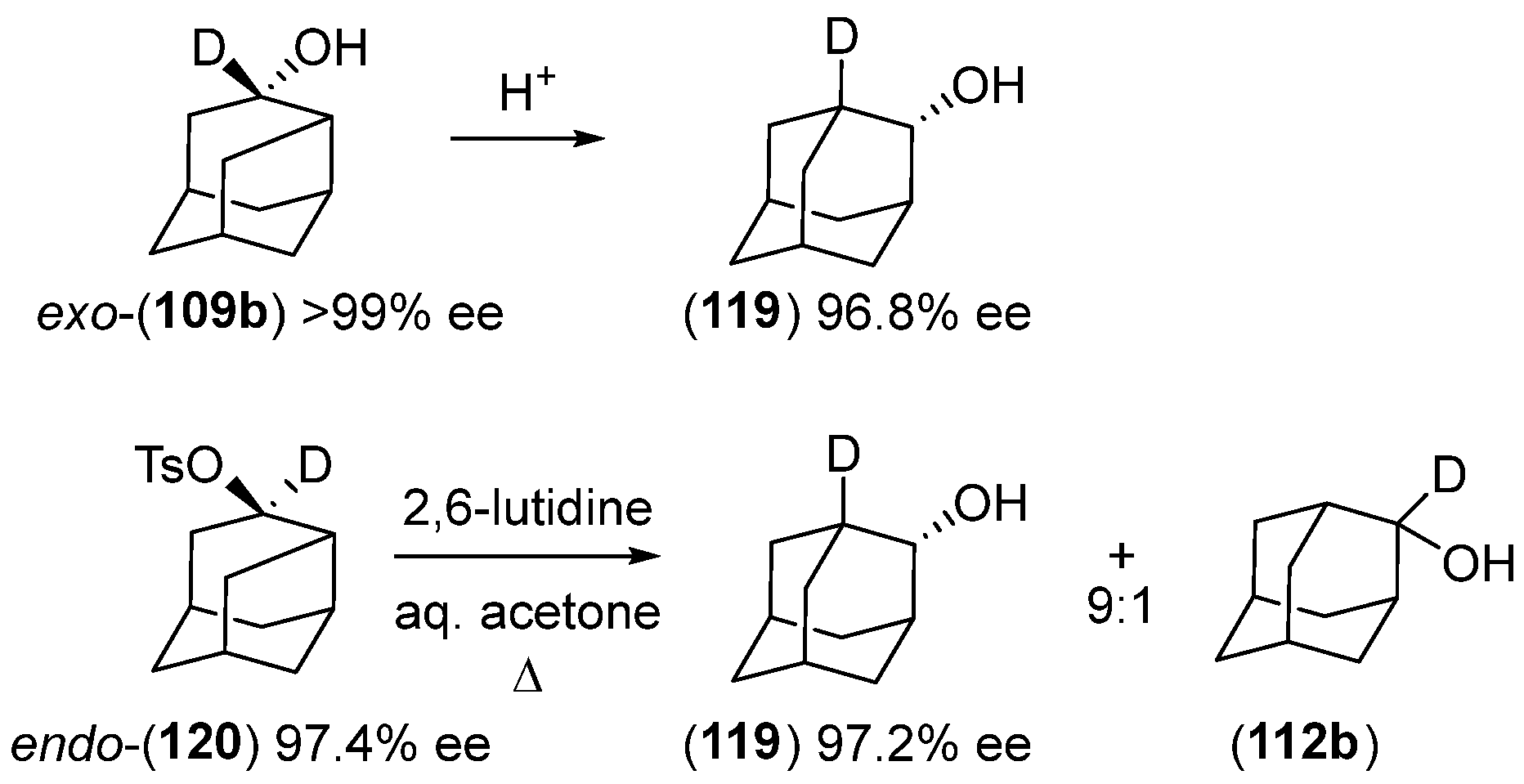
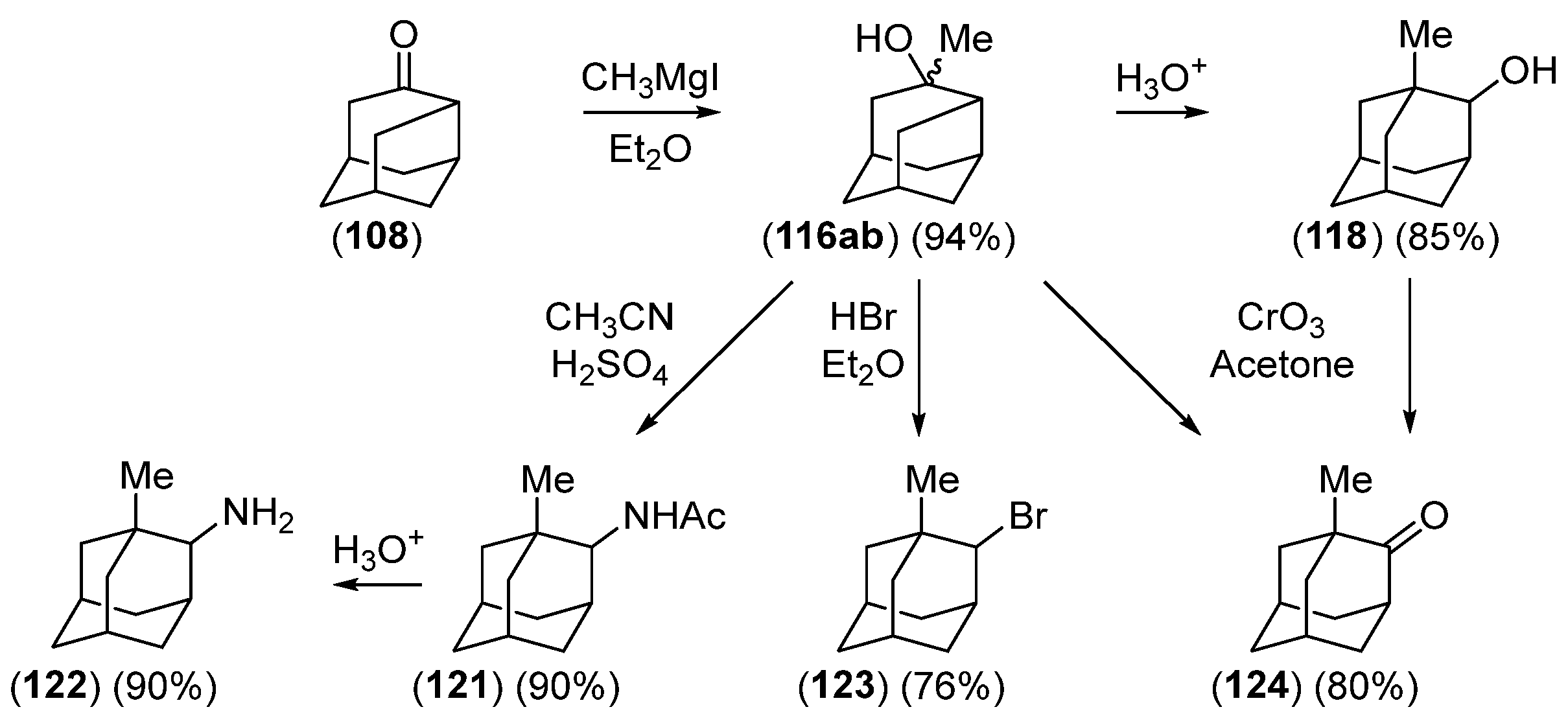
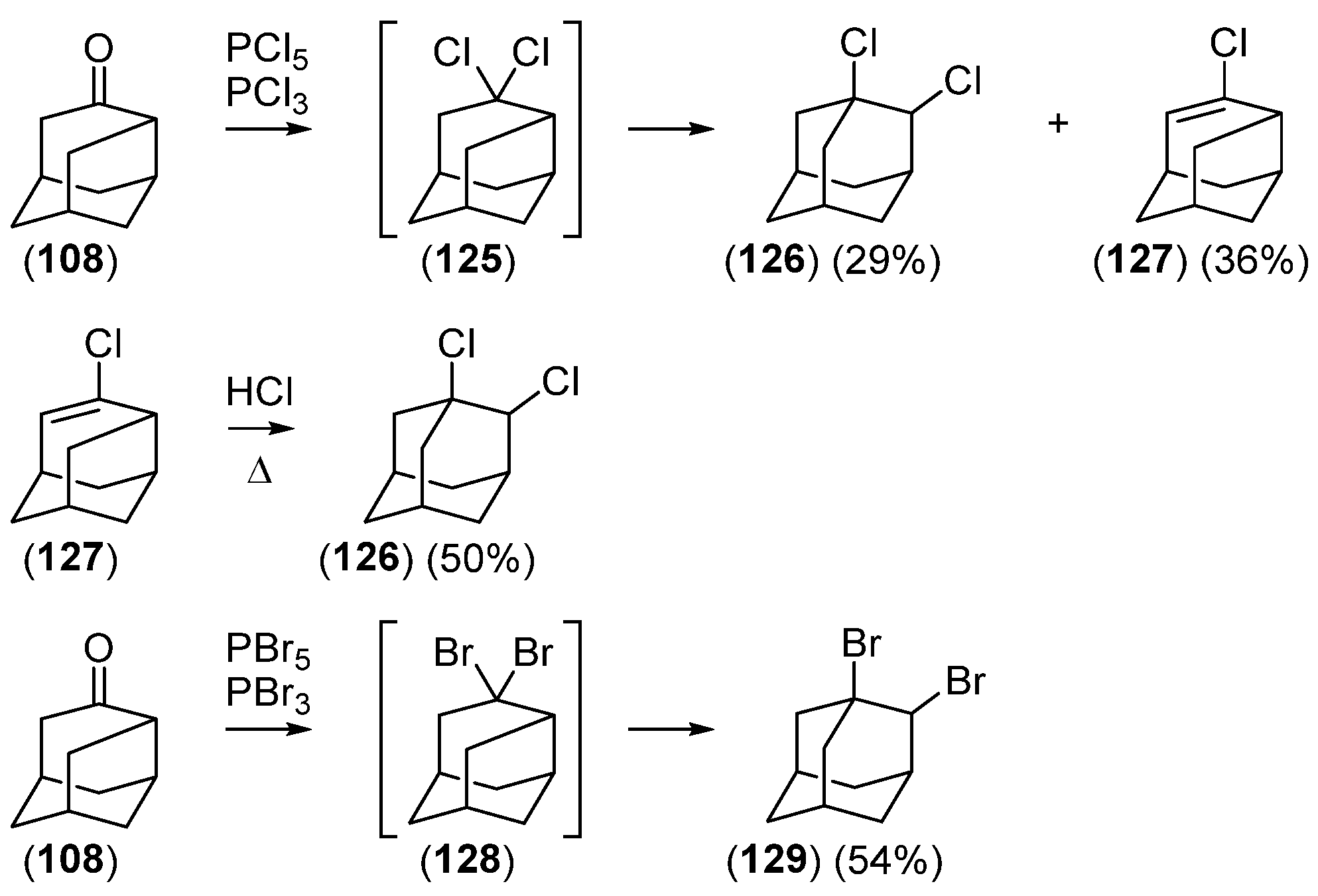









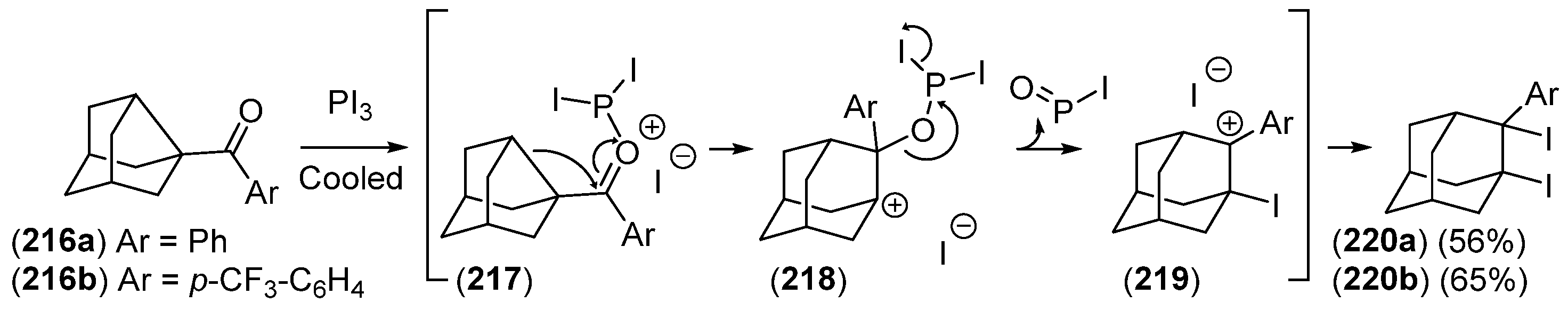
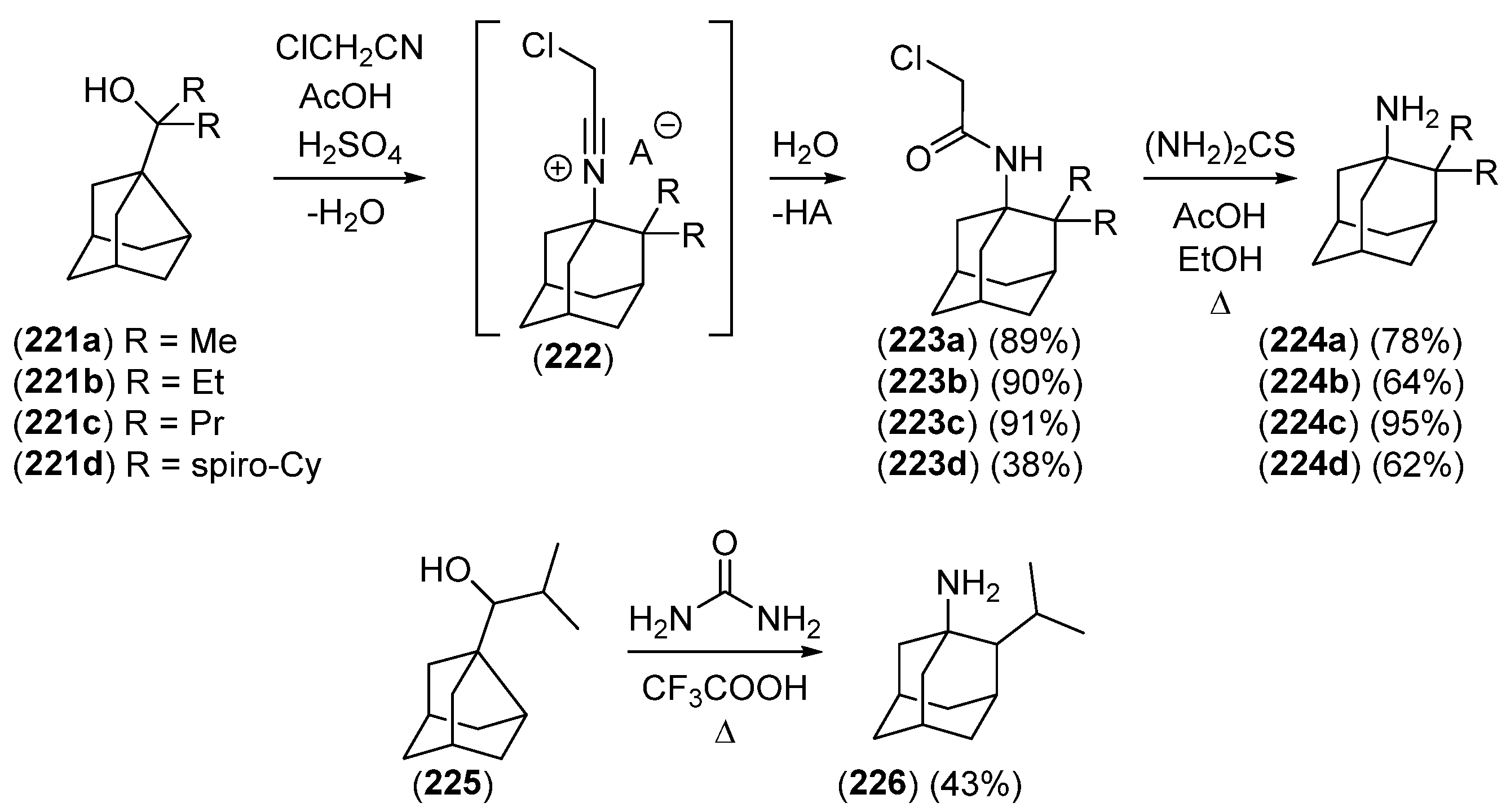


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Todd, M.; Hrdina, R. Synthesis of 1,2-Disubstituted Adamantane Derivatives by Construction of the Adamantane Framework. Molecules 2023, 28, 7636. https://doi.org/10.3390/molecules28227636
Todd M, Hrdina R. Synthesis of 1,2-Disubstituted Adamantane Derivatives by Construction of the Adamantane Framework. Molecules. 2023; 28(22):7636. https://doi.org/10.3390/molecules28227636
Chicago/Turabian StyleTodd, Matthew, and Radim Hrdina. 2023. "Synthesis of 1,2-Disubstituted Adamantane Derivatives by Construction of the Adamantane Framework" Molecules 28, no. 22: 7636. https://doi.org/10.3390/molecules28227636
APA StyleTodd, M., & Hrdina, R. (2023). Synthesis of 1,2-Disubstituted Adamantane Derivatives by Construction of the Adamantane Framework. Molecules, 28(22), 7636. https://doi.org/10.3390/molecules28227636