


Synthesis of Chalcones: An Improved High-Yield and Substituent-Independent Protocol for an Old Structure
 , , , and
, , , and
Abstract
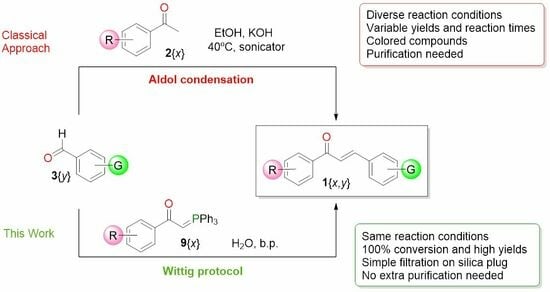
:
1. Introduction
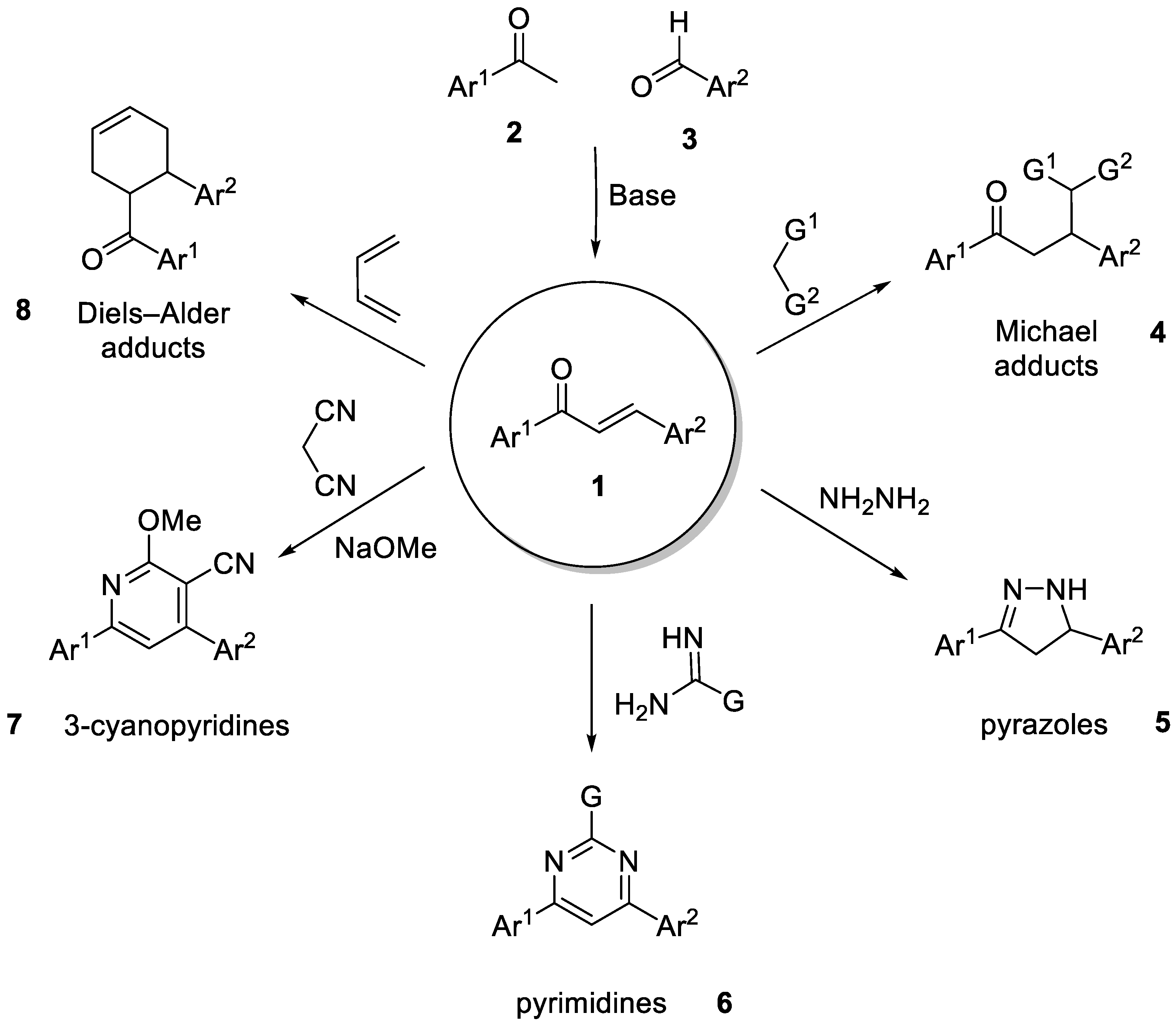
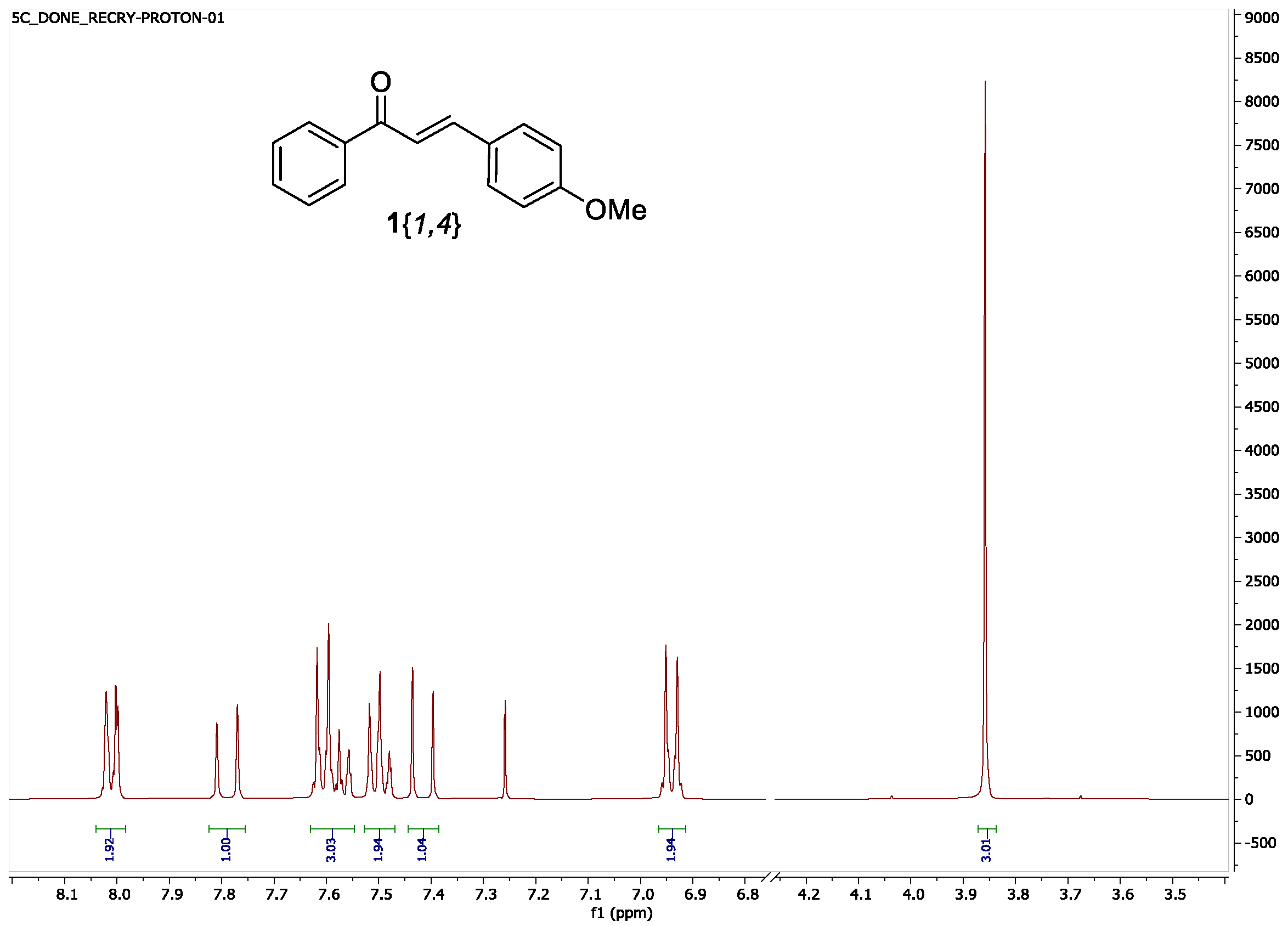
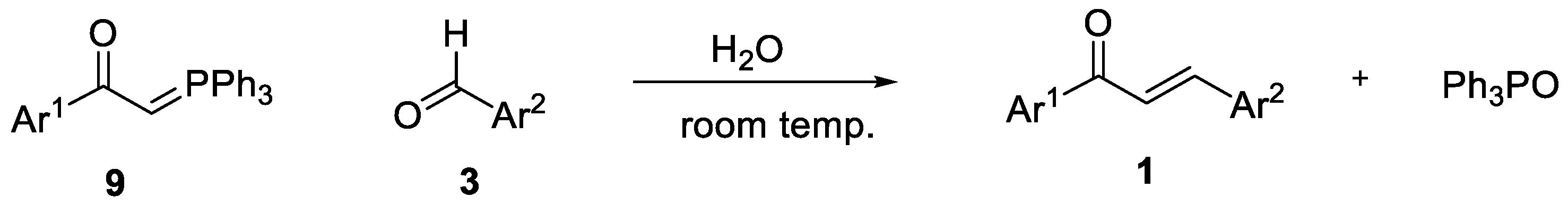
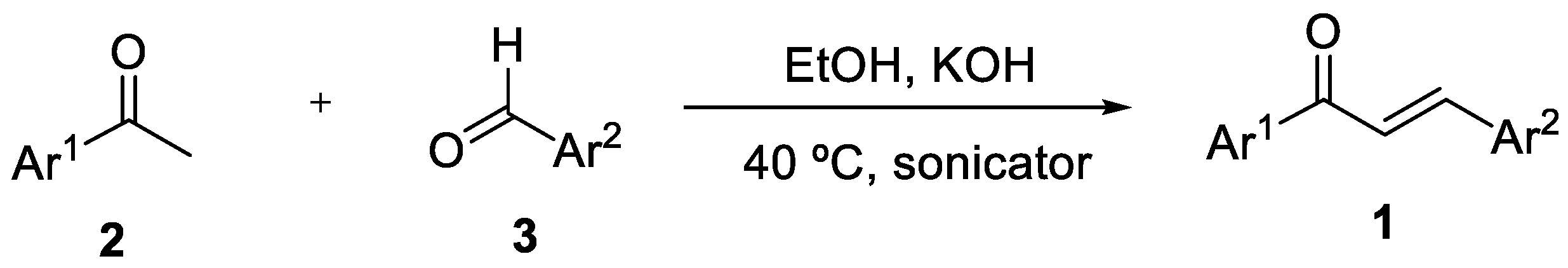
2. Results and Discussion
3. Conclusions
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Claisen, L.; Claparède, A. Condensationen von Ketonen Mit Aldehyden. Ber. Dtsch. Chem. Ges. 1881, 14, 2460–2468. [Google Scholar] [CrossRef]
- Chemical Abstracts Service. Scifinder, version 2019; Chemical Abstracts Service: Columbus, OH, USA, 2019.
- Salehi, B.; Quispe, C.; Chamkhi, I.; El Omari, N.; Balahbib, A.; Sharifi-Rad, J.; Bouyahya, A.; Akram, M.; Iqbal, M.; Docea, A.O.; et al. Pharmacological Properties of Chalcones: A Review of Preclinical Including Molecular Mechanisms and Clinical Evidence. Front. Pharmacol. 2020, 11, 592654. [Google Scholar] [CrossRef] [PubMed]
- Gaonkar, S.L.; Vignesh, U.N. Synthesis and Pharmacological Properties of Chalcones: A Review. Res. Chem. Intermed. 2017, 43, 6043–6077. [Google Scholar] [CrossRef]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef]
- Lin, N.; Wei, Q.X.; Jiang, L.H.; Deng, Y.Q.; Zhang, Z.W.; Chen, Q. Asymmetric Michael Addition of Malononitrile with Chalcones via Rosin-Derived Bifunctional Squaramide. Catalysts 2020, 10, 14. [Google Scholar] [CrossRef]
- Yang, W.; Du, D.M. Highly Enantioselective Michael Addition of Nitroalkanes to Chalcones Using Chiral Squaramides as Hydrogen Bonding Organocatalysts. Org. Lett. 2010, 12, 5450–5453. [Google Scholar] [CrossRef]
- Kim, D.Y.; Huh, S.C.; Kim, S.M. Enantioselective Michael Reaction of Malonates and Chalcones by Phase-Transfer Catalysis Using Chiral Quaternary Ammonium Salt. Tetrahedron Lett. 2001, 42, 6299–6301. [Google Scholar] [CrossRef]
- Bhat, B.A.; Puri, S.C.; Qurishi, M.A.; Dhar, K.L.; Qazi, G.N. Synthesis of 3,5-Diphenyl-1H-Pyrazoles. Synth. Commun. 2005, 35, 1135–1142. [Google Scholar] [CrossRef]
- Chimenti, F.; Fioravanti, R.; Bolasco, A.; Manna, F.; Chimenti, P.; Secci, D.; Befani, O.; Turini, P.; Ortuso, F.; Alcaro, S. Monoamine Oxidase Isoform-Dependent Tautomeric Influence in the Recognition of 3,5-Diaryl Pyrazole Inhibitors. J. Med. Chem. 2007, 50, 425–428. [Google Scholar] [CrossRef]
- Zhang, Z.; Tan, Y.-J.; Wang, C.-S.; Wu, H.-H. One-Pot Synthesis of 3,5-Diphenyl-1H-Pyrazoles from Chalcones and Hydrazine under Mechanochemical Ball Milling. Heterocycles 2014, 89, 103–112. [Google Scholar] [CrossRef]
- Von Angerer, S. Product Class 12: Pyrimidines. In Category 2, Hetarenes and Related Ring Systems; Science of Synthesis; Yamamoto, Y., Ed.; Thieme: Stuttgart, Germany, 2004; Volume 16, p. 379. [Google Scholar] [CrossRef]
- Ahmed, M.H.; El-Hashash, M.A.; Marzouk, M.I.; El-Naggar, A.M. Synthesis and Antitumor Activity of Some Nitrogen Heterocycles Bearing Pyrimidine Moiety. J. Heterocycl. Chem. 2020, 57, 3412–3427. [Google Scholar] [CrossRef]
- Siddiqui, S.M.; Azam, A. Synthesis, Characterization of 4,6-Disubstituted Aminopyrimidines and Their Sulphonamide Derivatives as Anti-Amoebic Agents. Med. Chem. Res. 2014, 23, 2976–2984. [Google Scholar] [CrossRef]
- Nie, A.; Wang, J.; Huang, Z. Microwave-Assisted Solution-Phase Parallel Synthesis of 2,4,6-Trisubstituted Pyrimidines. J. Comb. Chem. 2006, 8, 646–648. [Google Scholar] [CrossRef] [PubMed]
- Pathak, V.; Maurya, H.K.; Sharma, S.; Srivastava, K.K.; Gupta, A. Synthesis and Biological Evaluation of Substituted 4,6-Diarylpyrimidines and 3,5-Diphenyl-4,5-Dihydro-1H-Pyrazoles as Anti-Tubercular Agents. Bioorg. Med. Chem. Lett. 2014, 24, 2892–2896. [Google Scholar] [CrossRef] [PubMed]
- Victory, P.; Borrell, J.I.; Vidal-Ferran, A.; Seoane, C.; Soto, J.L. The Reaction of Malononitrile with Chalcone: A Controversial Chemical Process. Tetrahedron Lett. 1991, 32, 5375–5378. [Google Scholar] [CrossRef]
- Cong, H.; Ledbetter, D.; Rowe, G.T.; Caradonna, J.P.; Porco, J.A. Electron Transfer-Initiated Diels-Alder Cycloadditions of 2′-Hydroxychalcones. J. Am. Chem. Soc. 2008, 130, 9214–9215. [Google Scholar] [CrossRef]
- Li, X.; Han, J.; Jones, A.X.; Lei, X. Chiral Boron Complex-Promoted Asymmetric Diels-Alder Cycloaddition and Its Application in Natural Product Synthesis. J. Org. Chem. 2016, 81, 458–468. [Google Scholar] [CrossRef]
- Liu, Z.; Ganguly, R.; Vidović, D. Pursuing the Active Species in an Aluminium-Based Lewis Acid System for Catalytic Diels–Alder Cycloadditions. Dalton Trans. 2017, 46, 753–759. [Google Scholar] [CrossRef]
- Juliá, S.; Masana, J.; Vega, J.C. “Synthetic Enzymes”. Highly Stereoselective Epoxidation of Chalcone in a Triphasic Toluene-Water-Poly[(S)-Alanine] System. Angew. Chem. Int. Ed. Engl. 1980, 19, 929–931. [Google Scholar] [CrossRef]
- Rocchi, D.; González, J.F.; Menéndez, J.C. Montmorillonite Clay-Promoted, Solvent-Free Cross-Aldol Condensations under Focused Microwave Irradiation. Molecules 2014, 19, 7317–7326. [Google Scholar] [CrossRef]
- Kulkarni, P. Calcium Oxide Catalyzed Synthesis of Chalcone Under Microwave Condition. Curr. Microw. Chem. 2015, 2, 144–149. [Google Scholar] [CrossRef]
- Bou-Petit, E.; Hümmer, S.; Alarcon, H.; Slobodnyuk, K.; Cano-Galietero, M.; Fuentes, P.; Guijarro, P.J.; Muñoz, M.J.; Suarez-Cabrera, L.; Santamaria, A.; et al. Overcoming Paradoxical Kinase Priming by a Novel MNK1 Inhibitor. J. Med. Chem. 2022, 65, 6070–6087. [Google Scholar] [CrossRef] [PubMed]
- Dambacher, J.; Zhao, W.; El-Batta, A.; Anness, R.; Jiang, C.; Bergdahl, M. Water Is an Efficient Medium for Wittig Reactions Employing Stabilized Ylides and Aldehydes. Tetrahedron Lett. 2005, 46, 4473–4477. [Google Scholar] [CrossRef]
- Batesky, D.C.; Goldfogel, M.J.; Weix, D.J. Removal of Triphenylphosphine Oxide by Precipitation with Zinc Chloride in Polar Solvents. J. Org. Chem. 2017, 82, 9931–9936. [Google Scholar] [CrossRef]
- Ganesan, P.; Ranganathan, R.; Chi, Y.; Liu, X.K.; Lee, C.S.; Liu, S.H.; Lee, G.H.; Lin, T.C.; Chen, Y.T.; Chou, P.T. Functional Pyrimidine-Based Thermally Activated Delay Fluorescence Emitters: Photophysics, Mechanochromism, and Fabrication of Organic Light-Emitting Diodes. Chem. Eur. J. 2017, 23, 2858–2866. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Xiang, L.; Wen, F.; Tao, K.; Liu, Q.; Hou, T. Improved Synthesis of Chalconoid-like Compounds under Ultrasound Irradiation. Ultrason. Sonochem. 2008, 15, 681–683. [Google Scholar] [CrossRef]
- Meshram, H.M.; Goud, P.R.; Reddy, B.C.; Kumar, D.A. Triton B–Mediated Efficient and Convenient Alkoxylation of Activated Aryl and Heteroaryl Halides. Synth. Commun. 2010, 40, 2122–2129. [Google Scholar] [CrossRef]
- Ajibola, I.Y.; Ai, L.; Li, B. Synthesis of Arylfurans by Organic-Solvent-Free Method Using Phosphoric Acid as a Solvent and Catalyst. ChemistrySelect 2021, 6, 9559–9564. [Google Scholar] [CrossRef]
- Turdi, H.; Hangeland, J.J.; Lawrence, R.M.; Cheng, D.; Ahmad, S.; Meng, W.; Brigance, R.P.; Devasthale, P. Preparation of Aryl Dihydropyridinone and Piperidinone as MGAT2 Inhibitors. WO2013082345A1, 6 June 2013. [Google Scholar]
- Venkateswararao, E.; Kim, M.S.; Sharma, V.K.; Lee, K.C.; Subramanian, S.; Roh, E.; Kim, Y.; Jung, S.H. Identification of Novel Chromenone Derivatives as Interleukin-5 Inhibitors. Eur. J. Med. Chem. 2013, 59, 31–38. [Google Scholar] [CrossRef]
- Zhang, M.; Xi, J.; Ruzi, R.; Li, N.; Wu, Z.; Li, W.; Zhu, C. Domino-Fluorination-Protodefluorination Enables Decarboxylative Cross-Coupling of α-Oxocarboxylic Acids with Styrene via Photoredox Catalysis. J. Org. Chem. 2017, 82, 9305–9311. [Google Scholar] [CrossRef]
- Larionov, V.A.; Markelova, E.P.; Smol’yakov, A.F.; Savel’yeva, T.F.; Maleev, V.I.; Belokon, Y.N. Chiral Octahedral Complexes of Co(III) as Catalysts for Asymmetric Epoxidation of Chalcones under Phase Transfer Conditions. RSC Adv. 2015, 5, 72764–72771. [Google Scholar] [CrossRef]
- Kazi, I.; Guha, S.; Sekar, G. CBr4 as a Halogen Bond Donor Catalyst for the Selective Activation of Benzaldehydes to Synthesize α,β-Unsaturated Ketones. Org. Lett. 2017, 19, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Mellado, M.; Reyna-Jeldes, M.; Weinstein-Oppenheimer, C.; Coddou, C.; Jara-Gutierrez, C.; Villena, J.; Aguilar, L.F. Inhibition of Caco-2 and MCF-7 Cancer Cells Using Chalcones: Synthesis, Biological Evaluation and Computational Study. Nat. Prod. Res. 2022, 36, 4404–4410. [Google Scholar] [CrossRef] [PubMed]
- Maejima, T.; Shimoda, Y.; Nozaki, K.; Mori, S.; Sawama, Y.; Monguchi, Y.; Sajiki, H. One-Pot Aromatic Amination Based on Carbon–Nitrogen Coupling Reaction between Aryl Halides and Azido Compounds. Tetrahedron 2012, 68, 1712–1722. [Google Scholar] [CrossRef]
- Maeda, K.; Tanaka, K.; Morino, K.; Yashima, E. Synthesis of Optically Active Helical Poly(Phenylacetylene)s Bearing Oligopeptide Pendants and Their Use as Polymeric Organocatalysts for Asymmetric Epoxidation. Macromolecules 2007, 40, 6783–6785. [Google Scholar] [CrossRef]

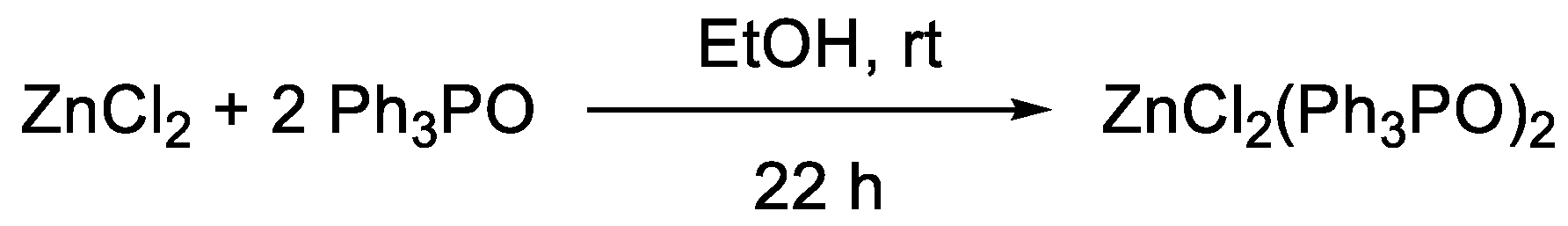




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
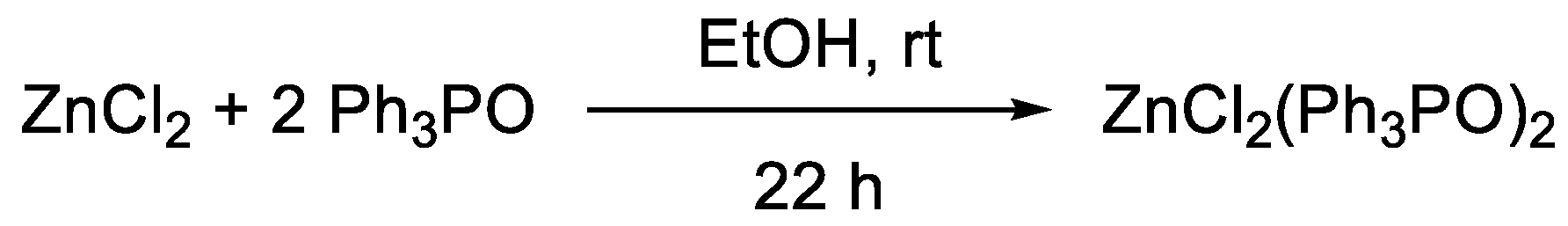{kind=link}
{kind=link}
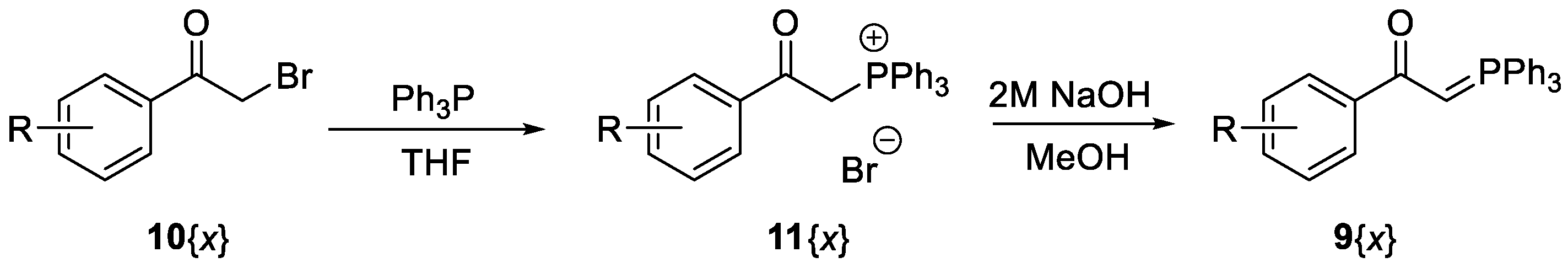{kind=link}
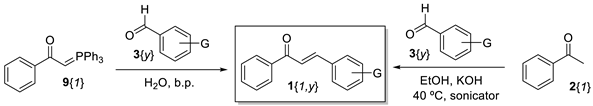 | |||||
|---|---|---|---|---|---|
| Aldehyde | Chalcone | Wittig Reaction | Aldol Condensation | ||
| Reaction Time | Yield (%) 1 | Reaction Time | Yield (%) 2 | ||
| 3{2} (G = p-NMe2) | 1{1,2} | 68 h | 93 | 2.5 h | 58 3 |
| 3{3} (G = p-Me) | 1{1,3} | 19 h | 91 | 2.5 h | 74 3 |
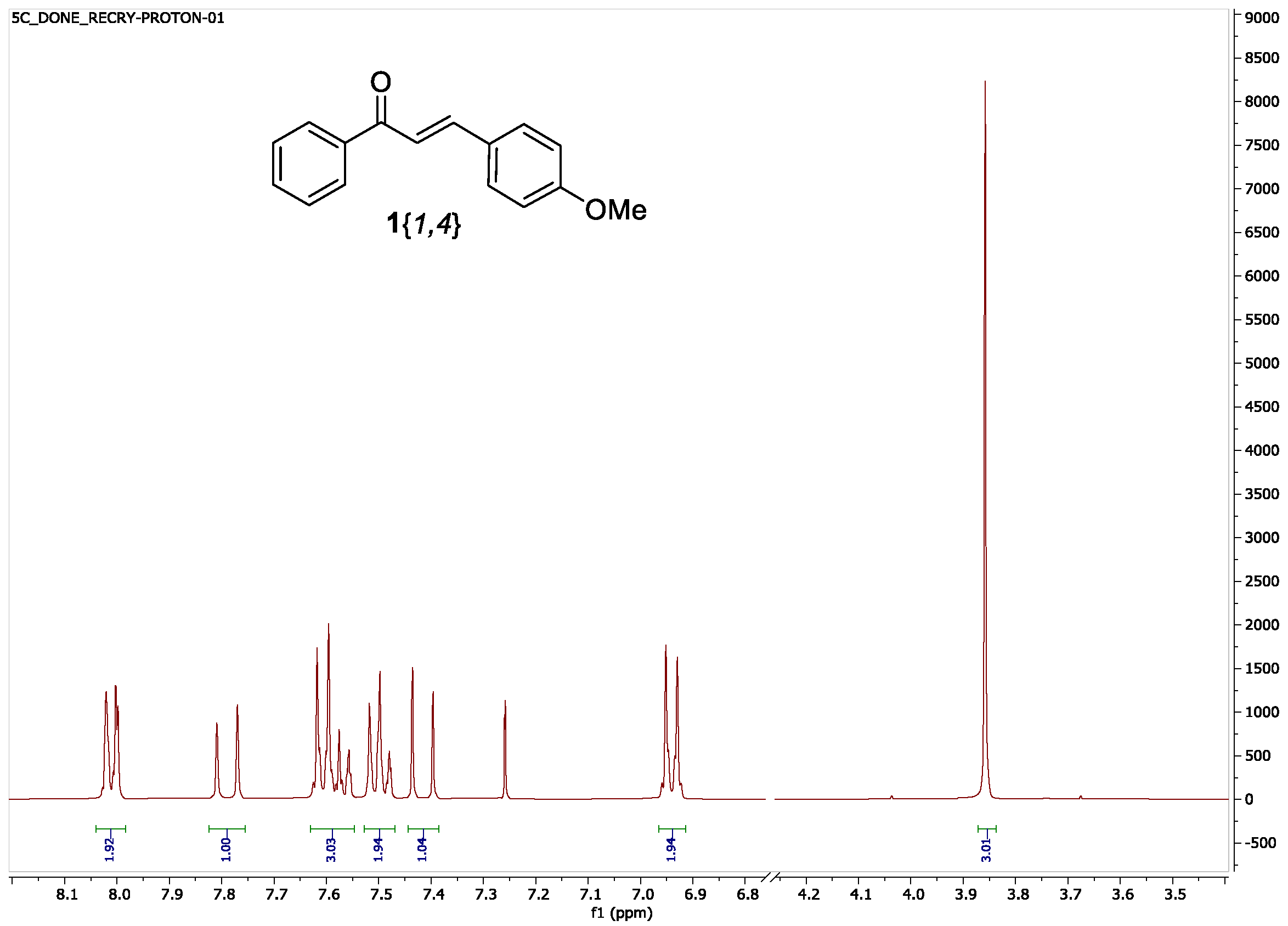
| 3{4} (G = p-OMe) | 1{1,4} | 21 h | 96 | 2.5 h | 54 3 |
| 3{5} (G = p-F) | 1{1,5} | 45 min | 95 | 2.5 h | 80 3,4 |
| 3{6} (G = p-Br) | 1{1,6} | 30 min | 97 | 40 min | 79 3 |
| 3{7} (G = p-NO2) | 1{1,7} | 30 min | 97 | 35 min | 100 3 |
| 3{8} (G = p-CN) | 1{1,8} | 45 min | 90 | 20 min | 100 3 |
| 3{9} (G = p-COOMe) | 1{1,9} | 1 h | 90 | 75 min | 100 3 |
 | |||||
|---|---|---|---|---|---|
| Aldehyde | Chalcone | Wittig Reaction | Aldol Condensation | ||
| Reaction Time | Yield (%) 1 | Reaction Time | Yield (%) 2 | ||
| 3{10} (G = o-Me) | 1{1,10} | 45 min | 81 | 1 h | 62 3 |
| 3{11} (G = o-F) | 1{1,11} | 30 min | 83 | 30 min | 72 3 |
| 3{12} (G = o-Br) | 1{1,12} | 30 min | 83 | 12 h | 65 4 |
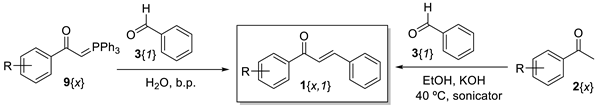 | |||||
|---|---|---|---|---|---|
| Acetophenone or Ylide | Chalcone | Wittig Reaction | Aldol Condensation | ||
| Reaction Time | Yield (%) 1 | Reaction Time | Yield (%) 2 | ||
| 2{2} or 9{2} (R = p-Me) | 1{2,1} | 3.5 h | 90 | 2 h | 70 3 |
| 2{3} or 9{3} (G = p-NO2) | 1{3,1} | 3.5 h | 73 | 2 h | 50 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donaire-Arias, A.; Poulsen, M.L.; Ramón-Costa, J.; Montagut, A.M.; Estrada-Tejedor, R.; Borrell, J.I. Synthesis of Chalcones: An Improved High-Yield and Substituent-Independent Protocol for an Old Structure. Molecules 2023, 28, 7576. https://doi.org/10.3390/molecules28227576
Donaire-Arias A, Poulsen ML, Ramón-Costa J, Montagut AM, Estrada-Tejedor R, Borrell JI. Synthesis of Chalcones: An Improved High-Yield and Substituent-Independent Protocol for an Old Structure. Molecules. 2023; 28(22):7576. https://doi.org/10.3390/molecules28227576
Chicago/Turabian StyleDonaire-Arias, Ana, Martin L. Poulsen, Jaime Ramón-Costa, Ana Maria Montagut, Roger Estrada-Tejedor, and José I. Borrell. 2023. "Synthesis of Chalcones: An Improved High-Yield and Substituent-Independent Protocol for an Old Structure" Molecules 28, no. 22: 7576. https://doi.org/10.3390/molecules28227576
APA StyleDonaire-Arias, A., Poulsen, M. L., Ramón-Costa, J., Montagut, A. M., Estrada-Tejedor, R., & Borrell, J. I. (2023). Synthesis of Chalcones: An Improved High-Yield and Substituent-Independent Protocol for an Old Structure. Molecules, 28(22), 7576. https://doi.org/10.3390/molecules28227576