
Phosphine Catalyzed Michael-Type Additions: The Synthesis of Glutamic Acid Derivatives from Arylidene-α-amino Esters †

 , , , , and
, , , , and
Abstract
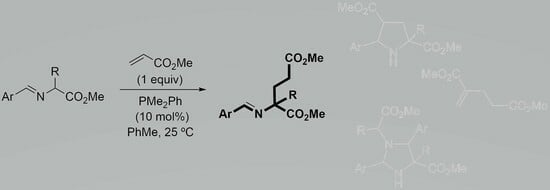
1. Introduction
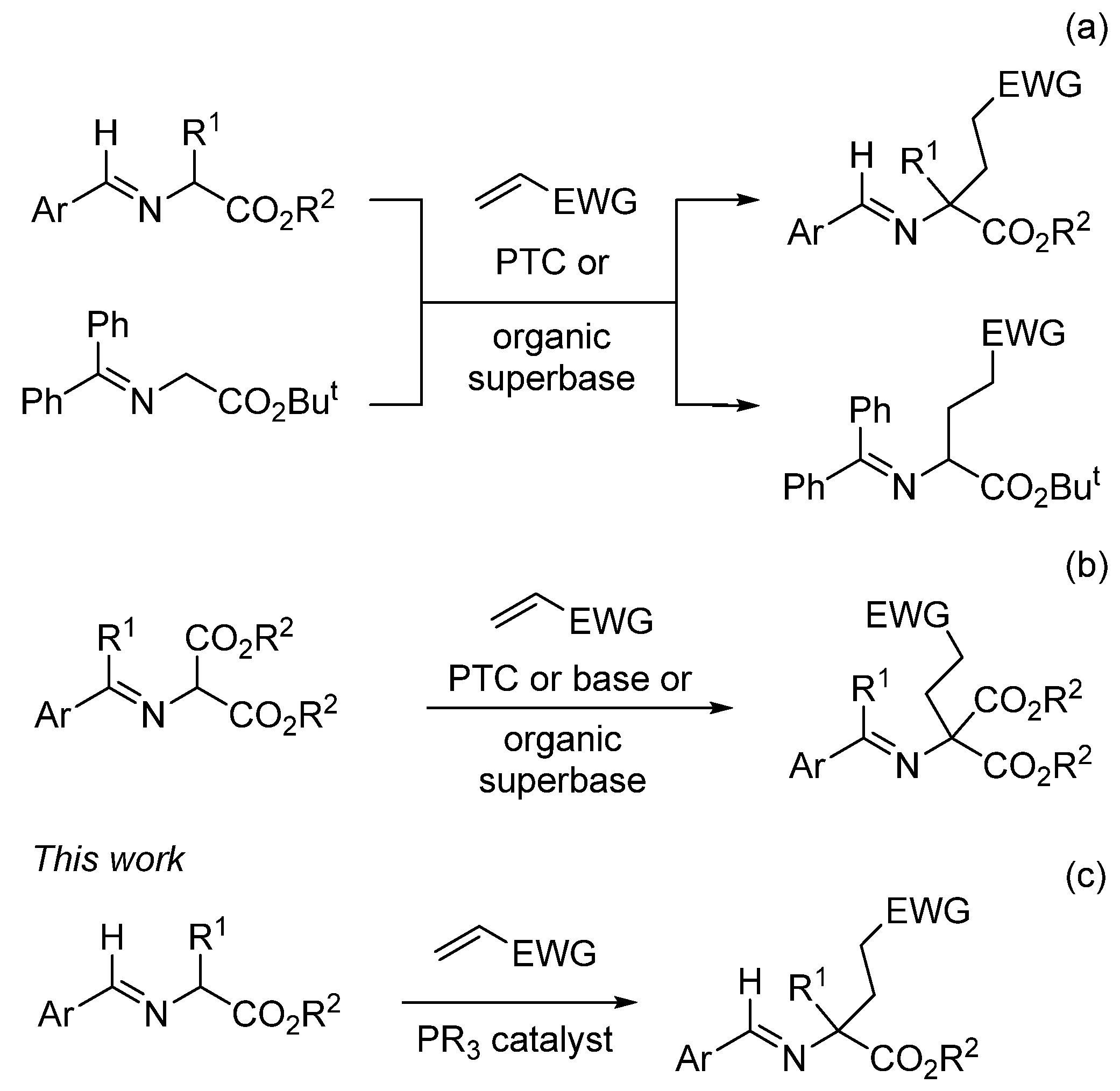
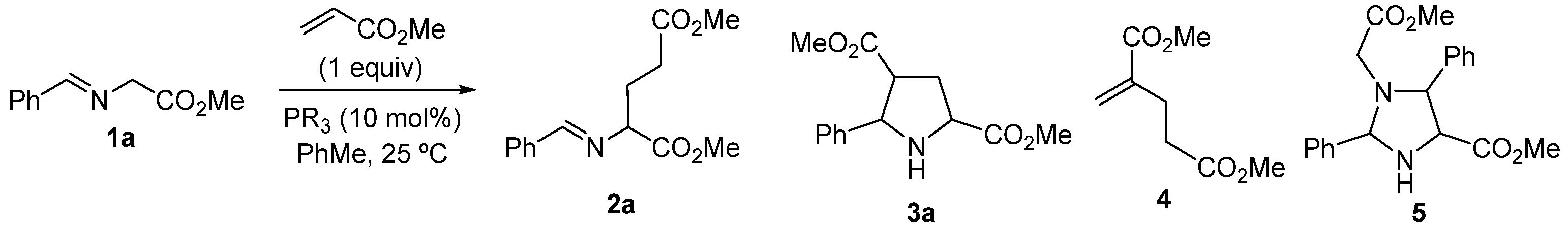
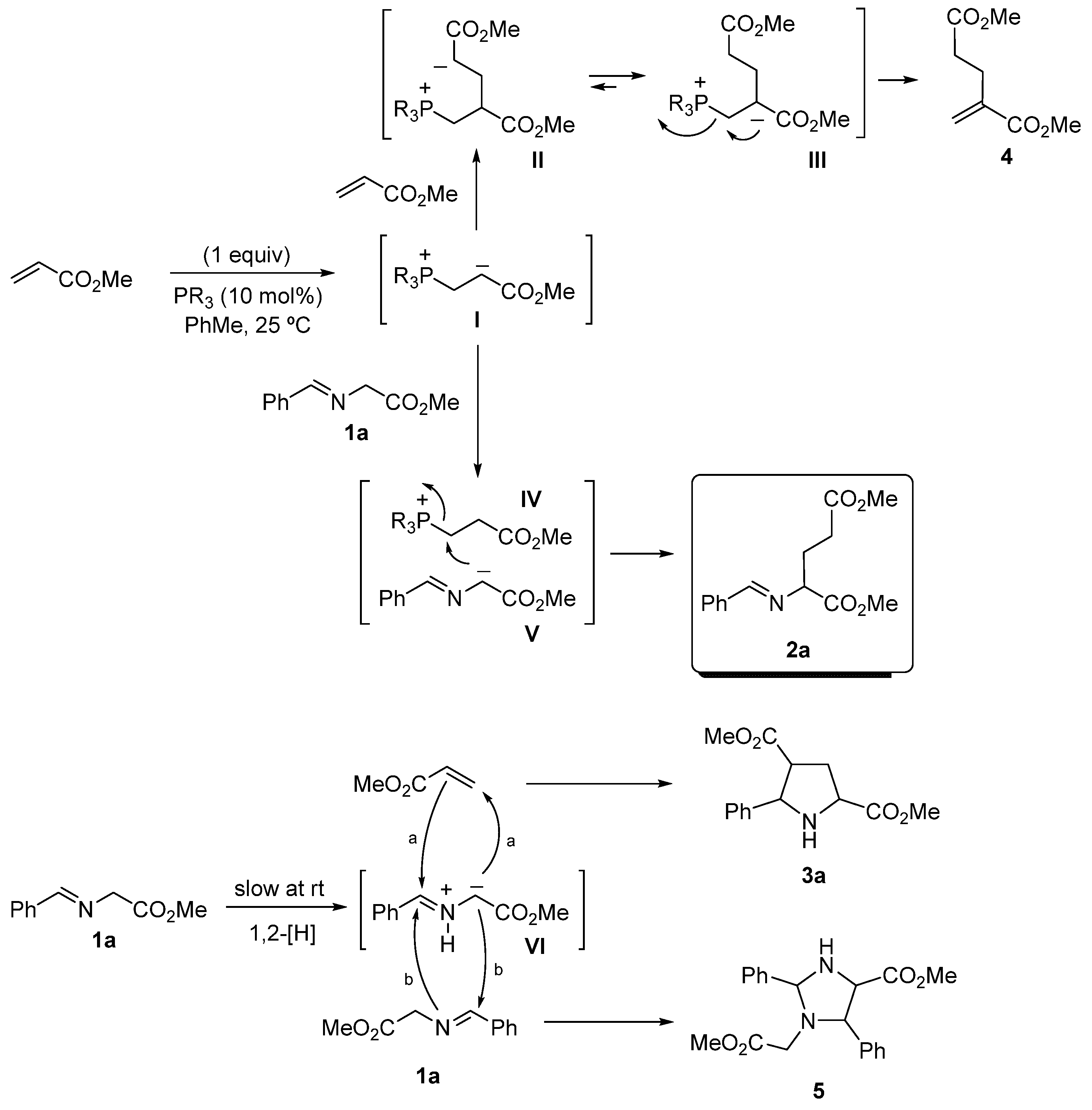
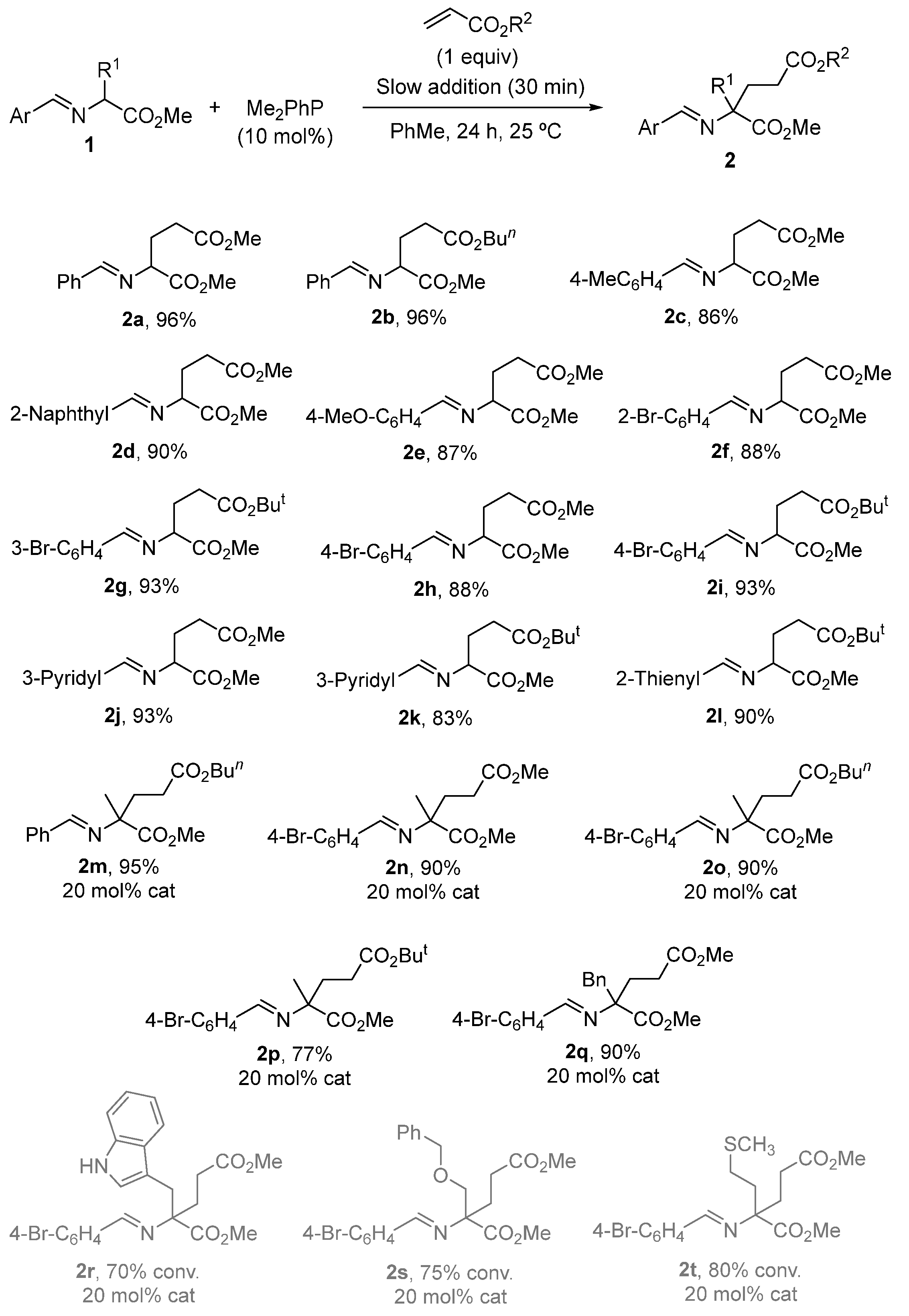
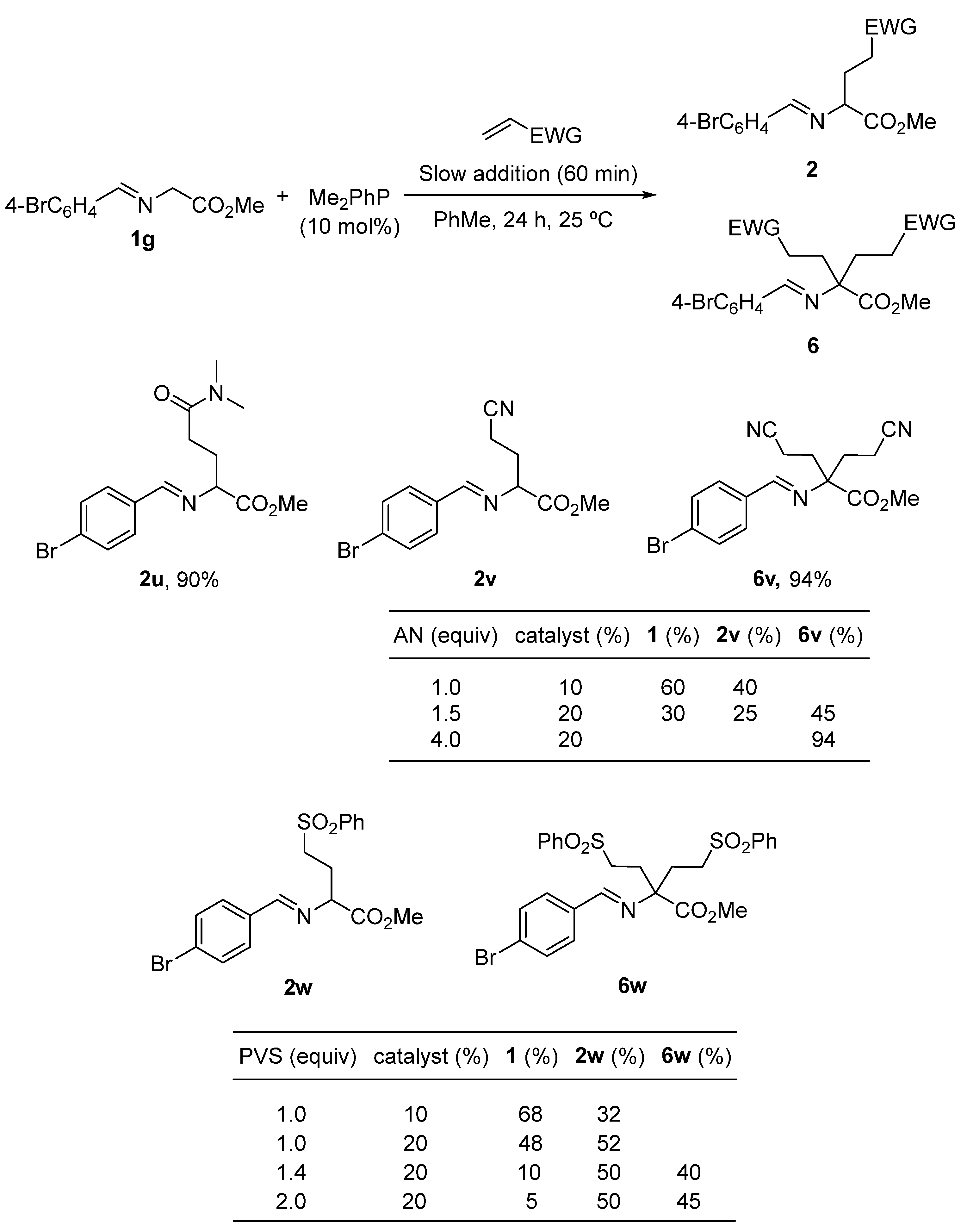
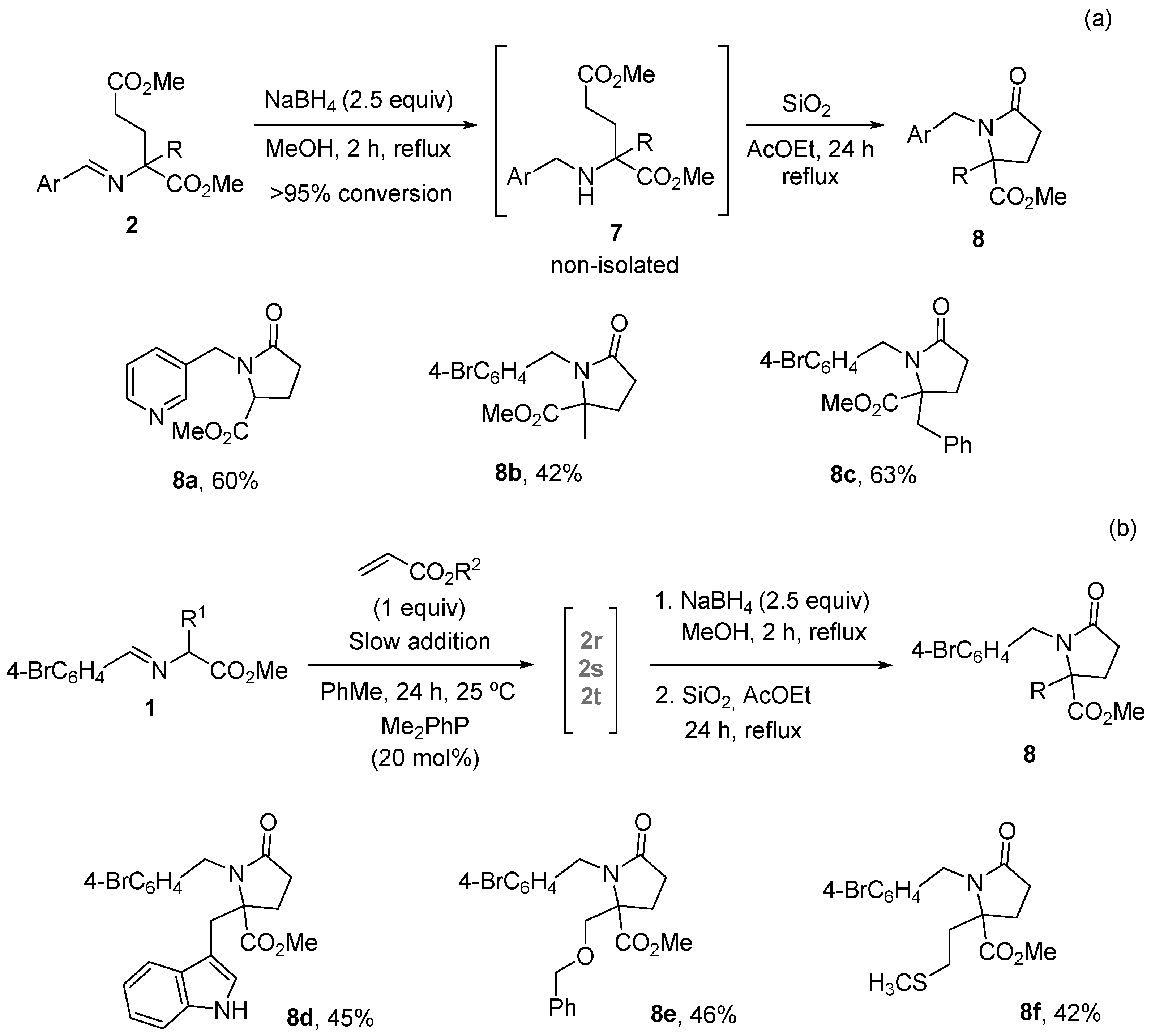
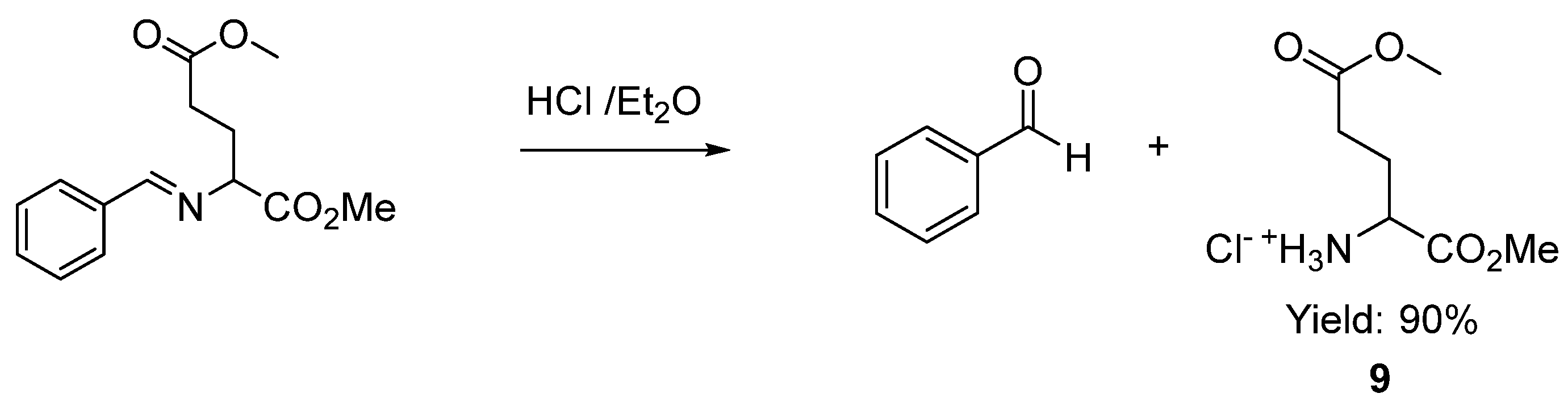
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. General Experimental Procedure for the Synthesis of Michael Type Addition Products 2
3.3. General Procedure for the Synthesis of Pyroglutamate Derivatives
3.4. Procedure to Obtain the Glutamic Acid 1,5-Dimethyl Ester Hydrochloride 9 [70]
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References and Notes
- Eder, I.; Haider, V.; Zebrowski, P.; Waser, M. Recent Progress in the Asymmetric Syntheses of α-Heterofunctionalized (Masked) α- and β-Amino Acid Derivatives. Eur. J. Org. Chem. 2021, 2021, 202–219. [Google Scholar] [CrossRef]
- Yuan, Z.; Liu, X.; Liu, C.; Zhang, Y.; Rao, Y. Recent Advances in Rapid Synthesis of Non-proteinogenic Amino Acids from Proteinogenic Amino Acids Derivatives via Direct Photo-Mediated C–H Functionalization. Molecules 2020, 25, 5270. [Google Scholar] [CrossRef] [PubMed]
- Masamba, W. Petasis vs. Strecker Amino Acid Synthesis: Convergence, Divergence and Opportunities in Organic Synthesis. Molecules 2021, 26, 1707. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Feng, Z.; Zhang, X. Recent advances in the synthesis of fluorinated amino acids and peptides. Chem. Commun. 2023, 59, 1434–1448. [Google Scholar] [CrossRef] [PubMed]
- Shatskiy, A.; Kaerkaes, M.D. Photoredox-enabled decarboxylative synthesis of unnatural α-amino acids. Synlett 2022, 33, 109–115. [Google Scholar] [CrossRef]
- Forro, E.; Fulop, F. Enzymatic Strategies for the Preparation of Pharmaceutically Important Amino Acids through Hydrolysis of Amino Carboxylic Esters and Lactams. Curr. Med. Chem. 2022, 29, 6218–6227. [Google Scholar] [CrossRef]
- Larionov, V.A.; Stoletova, N.V.; Maleev, V.I. Advances in asymmetric amino acid synthesis enabled by radical chemistry. Adv. Synth. Cat. 2020, 362, 4325–4367. [Google Scholar] [CrossRef]
- Saghyan, A.S.; Langer, P. Asymmetric Synthesis of Non-Proteinogenic Amino Acid; Wiley-VCH: Weinheim, Germany, 2016. [Google Scholar]
- O’Donnell, M.J. Benzophenone Schiff bases of glycine derivatives: Versatile starting materials for the synthesis of amino acids and their derivatives. Tetrahedron 2019, 75, 3667–3696. [Google Scholar] [CrossRef]
- Xue, Y.-P.; Cao, C.-H.; Zheng, Y.-G. Enzymatic asymmetric synthesis of chiral amino acids. Chem. Soc. Rev. 2018, 47, 1516–1561. [Google Scholar] [CrossRef]
- Wang, Y.; Song, X.; Wang, J.; Moriwaki, H.; Soloshonok, V.A.; Liu, H. Recent approaches for asymmetric synthesis of α-amino acids via homologation of Ni(II) complexes. Amino Acids 2017, 49, 1487–1520. [Google Scholar] [CrossRef]
- He, G.; Wang, B.; Nack, W.A.; Chen, G. Syntheses and Transformations of α-Amino Acids via Palladium-Catalyzed Auxiliary-Directed sp3 C-H Functionalization. Acc. Chem. Res. 2016, 49, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Nájera, C.; Sansano, J.M. Catalytic Asymmetric Synthesis of α-Amino Acids. Chem. Rev. 2007, 107, 4584–4671. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.-W.; Wu, S.-L.; Yan, J.-H.; Zhang, P.-F.; Yang, M.-M.; Li, L.; Xu, Z.; Yang, K.-F.; Xu, L.-W. Swollen-induced in-situ encapsulation of chiral silver catalysts in cross-linked polysiloxane elastomers: Homogeneous reaction and heterogeneous separation. Mol. Catal. 2021, 515, 111901. [Google Scholar] [CrossRef]
- Seibel, Z.M.; Bandar, J.S.; Lambert, T.H. Enantioenriched α-substituted glutamates/pyroglutamates via enantioselective cyclopropenimine-catalyzed Michael addition of amino ester imines. Beilst. J. Org. Chem. 2021, 17, 2077–2084. [Google Scholar] [CrossRef]
- Grigg, R.; Kemp, J.; Malone, J.P.; Rajviroongit, S.; Tangthonkum, A. X=Y-ZH Systems as potential 1,3-dipoles. Part 17. Sequential Michael addition-5-endo-trig cyclisation of arylideneimines of α-amino acid esters. Tetrahedron 1988, 44, 5361–5374. [Google Scholar] [CrossRef]
- Bai, Y.-J.; Cheng, M.L.; Zheng, X.-H.; Zhang, S.-Y.; Wang, P.A. Chiral Cyclopropenimine-catalyzed Asymmetric Michael Addition of Bulky Glycine Imine to α,β-Unsaturated Isoxazoles. chiral organosuperbase catalyst. Chem. Asian J. 2022, 17, e202200131. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, C.; Brandolese, A.; Preti, L.; Bortolini, O.; Polo, E.; Dambruoso, P.; Ragno, D.; Di Carmine, G.; Massi, A. Expanding the Toolbox of Heterogeneous Asymmetric Organocatalysts: Bifunctional Cyclopropenimine Superbases for Enantioselective Catalysis in Batch and Continuous Flow. Adv. Synth. Catal. 2021, 363, 5473–5485. [Google Scholar] [CrossRef]
- Tyszka-Gumkowska, A.; Jurczak, J. Divergent synthesis of pyrrolidine and glutamic acid derivatives using a macrocyclic phase-transfer catalyst under high-pressure conditions. Org. Chem. Front. 2021, 8, 5888–5894. [Google Scholar] [CrossRef]
- Bai, Y.-J.; Hu, X.-M.; Bai, Y.-J.; Zheng, X.-H.; Zhang, S.-Y.; Ping-An, W. DBU-catalyzed Michael addition of bulky glycine imine to α,β-unsaturated isoxazoles and pyrazolamides. Tetrahedron 2021, 101, 132511. [Google Scholar] [CrossRef]
- Meninno, S.; Carratu, M.; Overgaard, J.; Lattanzi, A. Diastereoselective Synthesis of Functionalized 5-Amino-3,4-Dihydro-2H-Pyrrole-2-Carboxylic Acid Esters: One-Pot Approach Using Commercially Available Compounds and Benign Solvents. Chem. Eur. J. 2021, 27, 4573–4577. [Google Scholar] [CrossRef]
- O’Donnell, M.J.; Boniece, J.M.; Earp, S.E. The synthesis of amino acids by phase-transfer reaction. Tetrahedron Lett. 1978, 19, 2641–2644. [Google Scholar] [CrossRef]
- Duan, X.-Y.; Tian, Z.; Liu, B.; He, T.; Zhao, L.-L.; Dong, M.; Zhang, P.; Qi, J. Highly Enantioselective Synthesis of Pyrroloindolones and Pyrroloquinolinones via an N-Heterocyclic Carbene-Catalyzed Cascade Reaction. Org. Lett. 2021, 23, 3777–3781. [Google Scholar] [CrossRef] [PubMed]
- Ren, N.; Zhang, L.; Hu, Y.; Wang, X.; Deng, Z.; Chen, J.; Deng, H.; Zhang, H.; Tang, X.-J.; Cao, W. Perfluoroalkyl-Promoted Synthesis of Perfluoroalkylated Pyrrolidine-Fused Coumarins with Methyl β-Perfluoroalkylpropionates. J. Org. Chem. 2021, 86, 15717–15725. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Xu, S.M.; Dong, X.-Q.; Wang, C.-J. Allylations Ir-Catalyzed Asymmetric Tandem Allylation/Iso-Pictet−Spengler Cyclization Reaction for the Enantioselective Construction of Tetrahydro-γ-carbolines. Org. Lett. 2021, 23, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Barlow, S.R.; Callaghan, L.J.; Franckevicius, V. Investigation of the palladium-catalysed cyclisation of α-amido malonates with propargylic compounds. Tetrahedron 2021, 80, 131866. [Google Scholar] [CrossRef]
- Cui, Z.; Zhang, K.; Gu, L.; Bu, Z.; Zhao, J.; Wang, Q. Diastereoselective trifunctionalization of pyridinium salts to access structurally crowded azaheteropolycycles. Chem. Commun. 2021, 57, 9402–9405. [Google Scholar] [CrossRef]
- Ha, M.W.; Lee, M.; Choi, S.; Kim, S.; Hong, S.; Park, Y.; Kim, M.; Kim, T.-S.; Lee, J.; Lee, J.K.; et al. Construction of Chiral α-Amino Quaternary Stereogenic Centers via Phase-Transfer Catalyzed Enantioselective α-Alkylation of α-Amidomalonates. J. Org. Chem. 2015, 80, 3270–3279. [Google Scholar] [CrossRef]
- Li, J.-T.; Luo, J.-N.; Wang, J.-L.; Wang, D.-K.; Yu, Y.-Z.; Zhuo, C.-X. Stereoselective intermolecular radical cascade reactions of tryptophans or ɤ-alkenyl-α-amino acids with acrylamides via photoredox catalysis. Nat. Commun. 2022, 13, 1778–1785. [Google Scholar] [CrossRef]
- Banerjee, I.; Ghosh, K.C.; Oheix, E.; Jean, M.; Naubron, J.V.; Réglier, M.; Iranzo, O.; Sinha, S. Synthesis of Protected 3,4- and 2,3-Dimercaptophenylalanines as Building Blocks for Fmoc-Peptide Synthesis and Incorporation of the 3,4-Analogue in a Decapeptide Using Solid-Phase Synthesis. J. Org. Chem. 2021, 86, 2210–2223. [Google Scholar] [CrossRef]
- Liao, H.-S.; Chung, Y.-H.; Hsieh, M.-H. Glutamate: A multifunctional amino acid in plants. Plant Sci. 2022, 318, 111238. [Google Scholar] [CrossRef]
- Bayer, T.A. Pyroglutamate Aβ cascade as drug target in Alzheimer’s disease. Mol. Psychiatry 2022, 27, 1880–1885. [Google Scholar] [CrossRef]
- Lin, T.-Y.; Lu, C.-W.; Hsieh, P.-W.; Chiu, K.-M.; Lee, M.-Y.; Wang, S.-J. Natural Product Isoliquiritigenin Activates GABAB Receptors to Decrease Voltage-Gate Ca2+ Channels and Glutamate Release in Rat Cerebrocortical Nerve Terminals. Biomolecules 2021, 11, 1537. [Google Scholar] [CrossRef]
- Gulder, T.A.M.; Moore, B.S. Salinosporamide Natural Products: Potent 20 S Proteasome Inhibitors as Promising Cancer Chemotherapeutics. Angew. Chem. Int. Ed. 2010, 49, 9346–9367. [Google Scholar] [CrossRef] [PubMed]
- Panday, S.K.; Prasad, J.; Dikshit, D.K. Pyroglutamic acid: A unique chiral synthon. Tetrahedron Asymmetry 2009, 20, 1581–1632. [Google Scholar] [CrossRef]
- Zhang, J.; Flippen-Anderson, J.L.; Kozikowski, A.P. A Tandem Michael Addition Ring-Closure Route to the Metabotropic Receptor Ligand α-(Hydroxymethyl)glutamic Acid and Its γ-Alkylated Derivatives. J. Org. Chem. 2001, 66, 7555–7559. [Google Scholar] [CrossRef] [PubMed]
- Tekkam, S.; Alam, M.A.; Just, M.J.; Berry, S.M.; Johnson, J.L.; Jonnalagadda, S.C.; Mereddy, V.R. Stereoselective Synthesis of Pyroglutamate Natural Product Analogs from α-Aminoacids and their Anti-Cancer Evaluation. Anti-Cancer Agents Med. Chem. 2013, 13, 1514–1530. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Hisa, C.; Honda, T. Enantioselective synthesis of (R)-deoxydysibetaine and (−)-4-epi-dysibetaine. Tetrahedron Lett. 2007, 48, 4691–4694. [Google Scholar] [CrossRef]
- Isaacson, J.; Loo, M.; Kobayashi, Y. Total Synthesis of (±)-Dysibetaine. Org. Lett. 2008, 10, 1461–1463. [Google Scholar] [CrossRef]
- Ma, G.; Nguyen, H.; Romo, D. Concise Total Synthesis of (±)-Salinosporamide A, (±)-Cinnabaramide A, and Derivatives via a Bis-cyclization Process: Implications for a Biosynthetic Pathway? Org. Lett. 2007, 9, 2143–2146. [Google Scholar] [CrossRef]
- Ling, T.; Macherla, V.R.; Manam, R.R.; McArthur, K.A.; Potts, B.C.M. Enantioselective Total Synthesis of (−)-Salinosporamide a (NPI-0052). Org. Lett. 2007, 9, 2289–2292. [Google Scholar] [CrossRef]
- Shibasaki, M.; Kanai, M.; Fukuda, N. Total Synthesis of Lactacystin and Salinosporamide A. Chem. Asian J. 2007, 2, 20–38. [Google Scholar] [CrossRef]
- Masse, C.E.; Morgan, A.J.; Adams, J.; Panek, J.S. Syntheses and Biological Evaluation of (+)-Lactacystin and Analogs. Eur. J. Org. Chem. 2000, 2000, 2513–2528. [Google Scholar] [CrossRef]
- Bai, X.-F.; Li, L.; Xu, Z.; Zheng, Z.-J.; Xia, C.G.; Cui, Y.M.; Xu, L.-W. Asymmetric Michael-addition of aldimino esters with chalcones catalyzed by Silver/Xing-Phos: Mechanism-oriented divergent synthesis of chiral pyrrolines. Chem. Eur. J. 2016, 22, 10399–10404. [Google Scholar] [CrossRef]
- Guo, H.; Fan, Y.C.; Sun, Z.; Wu, Y.; Kwon, O. Phosphine Organocatalysis. Chem. Rev. 2018, 118, 10049–10293. [Google Scholar] [CrossRef] [PubMed]
- Molteni, G.; Silvani, A. Spiro-2-oxindoles via 1,3-dipolar cycloadditions. A decade update. Eur. J. Org. Chem. 2021, 2021, 1653–1675. [Google Scholar] [CrossRef]
- Nájera, C.; Sansano, J.M. Synthesis of pyrrolizidines and indolizidines by multicomponent 1,3-dipolar cycloaddition of azomethine ylides. Pure Appl. Chem. 2019, 91, 575–596. [Google Scholar] [CrossRef]
- Dondas, H.A.; Retamosa, M.G.; Sansano, J.M. Current trends towards the synthesis of bioactive heterocycles and natural products using 1, 3-dipolar cycloadditions (1, 3-DC) with azomethine ylides. Synthesis 2017, 49, 2819–2851. [Google Scholar] [CrossRef]
- Bdiri, B.; Zhao, B.-J.; Zhou, Z.-M. Recent advances in the enantioselective 1,3-dipolar cycloaddition of azomethine ylides and dipolarophiles. Tetrahedron Asymmetry 2017, 28, 876–899. [Google Scholar] [CrossRef]
- Wei, L.; Chang, X.; Wang, C.-J. Catalytic Asymmetric Reactions with N-Metallated Azomethine Ylides. Acc. Chem. Res. 2020, 53, 1084–1100. [Google Scholar] [CrossRef]
- Adrio, J.; Carretero, J.C. Stereochemical diversity in pyrrolidine synthesis by catalytic asymmetric 1,3-dipolar cycloaddition of azomethine ylides. Chem. Commun. 2019, 55, 11979–11991. [Google Scholar] [CrossRef]
- Fang, X.; Wang, C.-J. Catalytic asymmetric construction of spiropyrrolidines via 1,3-dipolar cycloaddition of azomethine ylides. Org. Biomol. Chem. 2018, 16, 2591–2601. [Google Scholar] [CrossRef]
- De, N.; Yoo, E.J. Recent Advances in the Catalytic Cycloaddition of 1,n-Dipoles. ACS Catal. 2018, 8, 48–58. [Google Scholar] [CrossRef]
- Stewart, I.C.; Bergman, R.G.; Toste, F.D. Phosphine-Catalyzed Hydration and Hydroalkoxylation of Activated Olefins: Use of a Strong Nucleophile to Generate a Strong Base. J. Am. Chem. Soc. 2003, 125, 8696–8697. [Google Scholar] [CrossRef] [PubMed]
- Rundlöf, T.; Mathiasson, M.; Bekiroglu, S.; Hakkarainen, B.; Bowden, T.; Arvidsson, T. Survey and qualification of internal standards for quantification by 1 H NMR Spectroscopy. J. Pharm. Biomed. Anal. 2010, 52, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Purities of compounds were determined accordingly. See experimental section of this article.
- The addition of a substituent at the β-position in the acrylate did not give the expected reaction.
- Zhang, X.; Zhang, J.; Jia, M.; Peng, L.; Zhang, N.; Qi, S.; Zhang, L. Bifunctional additive phenyl vinyl sulfone for boosting cyclability of lithium metal batteries. Green. Chem. Engin. 2023, 4, 49–56. [Google Scholar] [CrossRef]
- Fahim, A.M.; Ghabbour, H.A.; Kabil, M.M.; Al-Rashood, S.T.; Abdel-Aziz, H.A. Synthesis, X-ray crystal structure, Hirshfeld analysis and computational investigation of bis(methylthio)acrylonitrile with antimicrobial and docking evaluation. J. Mol. Struct. 2022, 1260, 132793. [Google Scholar] [CrossRef]
- Ishihara, M.; Fujisawa, S. A structure-activity relationship study on the mechanisms of methacrylate-induced toxicity using NMR chemical shift of β-carbon, RP-HPLC log P and semiempirical molecular descriptor. Dental Mat. J. 2009, 28, 113–120. [Google Scholar] [CrossRef]
- Pérez-Garmendia, R.; Gevorkian, G. Pyroglutamate-Modified Amyloid Beta Peptides: Emerging Targets for Alzheimer’s Disease Immunotherapy. Curr. Neuropharm. 2013, 11, 491–498. [Google Scholar] [CrossRef]
- Cynis, H.; Frost, J.L.; Crehan, H.; Lemere, C.A. Immunotherapy targeting pyroglutamate-3 Aβ: Prospects and challenges. Mol. Neurodegener. 2016, 11, 48/1–48/11. [Google Scholar] [CrossRef]
- Piechotta, A.; Parthier, C.; Kleinschmidt, M.; Gnoth, K.; Pillot, T.; Lues, I.; Demuth, H.-U.; Schilling, S.; Rahfeld, J.-U.; Stubbs, M.T. Structural and functional analyses of pyroglutamate-amyloid-β-specific antibodies as a basis for Alzheimer immunotherapy. J. Biol. Chem. 2017, 292, 12713–12724. [Google Scholar] [CrossRef]
- Xu, C.; Wang, Y.-n.; Wu, H. Glutaminyl cyclase, diseases, and development of glutaminyl cyclase inhibitors. J. Med. Chem. 2021, 64, 6549–6565. [Google Scholar] [CrossRef]
- Casas, J.; Grigg, R.; Nájera, C.; Sansano, J.M. The Effect of Phase-Transfer Catalysis in the 1,3-Dipolar Cycloaddition Reactions of Azomethine Ylides− Synthesis of Substituted Prolines Using AgOAc and Inorganic Base in Substoichiometric Amounts. Eur. J. Org. Chem. 2001, 2001, 1971–1982. [Google Scholar] [CrossRef]
- Caleffi, G.; Larrañaga, O.; Ferrándiz-Saperas, M.; Costa, P.; Nájera, C.; de Cózar, A.; Cossío, F.; Sansano, J.M. Switching Diastereoselectivity in Catalytic Enantioselective (3+2) Cycloadditions of Azomethine Ylides Promoted by Metal Salts and Privileged Segphos-Derived Ligands. J. Org. Chem. 2019, 84, 10593–10605. [Google Scholar] [CrossRef]
- Beksultanova, N.; Doğan, Ö. Asymmetric synthesis of aryl-substituted pyrrolidines by using CFAM ligand–AgOAc chiral system via 1,3-dipolar cycloaddition reaction. Chirality 2023, 35, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Moins, S.; Coulembier, O. Dimerization of Methyl Acrylate through CO2-pressurized DBU Mediated Process. Asian J. Org. Chem. 2022, 11, e202100734. [Google Scholar] [CrossRef]
- Yu, B.; Bai, X.-F.; Ly, J.-Y.; Yuan, Y.; Cao, J.; Zheng, Z.-J.; Xu, Z.; Cui, Y.-M.; Yang, K.F.; Xu, L.-W. Enantioselective Synthesis of Chiral Imidazolidine Derivatives by Asymmetric Silver/Xing-Phos-Catalyzed Homo-1,3-Dipolar [3+2] Cycloaddition of Azomethine Ylides. Adv. Synth. Cat. 2017, 359, 3577–3584. [Google Scholar] [CrossRef]
- Weigl, M.; Wunsch, B. Synthesis of chiral non-racemic 3-(dioxopiperazin-2-yl)propionic acid derivatives. Tetrahedron 2002, 58, 1173–1183. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Slow Addition | Products’ Ratio a | |||||||
|---|---|---|---|---|---|---|---|---|
| Entry | PR3 (10 mol%) | (min) | t (h) | 2a | 3a | 4 | 5 | 1a |
| 1 | PPh3 | --- | 15 | 0 | 9 | 0 | 0 | 91 |
| 2 | dppe | --- | 15 | 37 | 22 | 0 | 4 | 37 |
| 3 | dppe | --- | 48 | 22 | 32 | 0 | 30 | 16 |
| 4 | dppe | --- | 72 | 56 | 39 | 0 | 5 | 0 |
| 5 | dppe | 1a (60) | 72 | 80 | 0 | 20 | 0 | 0 |
| 6 | dppe | MA (60) | 72 | 88 | 0 | 0 | 12 | 0 |
| 7 | dppe | MA (60) | 12 | 71 | 0 | 0 | 9 | 20 |
| 8 | Bu3P | --- | 72 | 0 | 0 | 0 | 100 | 0 |
| 9 | Bu3P | MA (60) | 72 | 0 | 0 | 100 | 0 | 0 |
| 10 | But3P | --- | 72 | 0 | 0 | 100 | 0 | 0 |
| 11 | Me2PhP | --- | 12 | 23 | 0 | 38 | 38 | 0 |
| 12 | Me2PhP | --- | 2 | 70 | 0 | 15 | 15 | 0 |
| 13 | Me2PhP | MA (60) | 2 | 70 | 5 | 0 | 5 | 20 |
| 14 | Me2PhP | MA (60) | 24 | 80 | 10 | 0 | 5 | 5 |
| 15 | Me2PhP | MA (30) | 24 | 97 | 0 | 0 | 0 | 3 |
| 16 | Me2PhP | MA (5) | 24 | 64 | 0 | 29 | 0 | 7 |
| 17 | Me2PhP b | MA (5) | 24 | 50 | 0 | 0 | 0 | 50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Flórez, L.V.; González-Marcos, M.; García-Mingüens, E.; Retamosa, M.d.G.; Kawase, M.; Selva, E.; Sansano, J.M. Phosphine Catalyzed Michael-Type Additions: The Synthesis of Glutamic Acid Derivatives from Arylidene-α-amino Esters. Molecules 2024, 29, 342. https://doi.org/10.3390/molecules29020342
Rodríguez-Flórez LV, González-Marcos M, García-Mingüens E, Retamosa MdG, Kawase M, Selva E, Sansano JM. Phosphine Catalyzed Michael-Type Additions: The Synthesis of Glutamic Acid Derivatives from Arylidene-α-amino Esters. Molecules. 2024; 29(2):342. https://doi.org/10.3390/molecules29020342
Chicago/Turabian StyleRodríguez-Flórez, Lesly V., María González-Marcos, Eduardo García-Mingüens, María de Gracia Retamosa, Misa Kawase, Elisabet Selva, and José M. Sansano. 2024. "Phosphine Catalyzed Michael-Type Additions: The Synthesis of Glutamic Acid Derivatives from Arylidene-α-amino Esters" Molecules 29, no. 2: 342. https://doi.org/10.3390/molecules29020342
APA StyleRodríguez-Flórez, L. V., González-Marcos, M., García-Mingüens, E., Retamosa, M. d. G., Kawase, M., Selva, E., & Sansano, J. M. (2024). Phosphine Catalyzed Michael-Type Additions: The Synthesis of Glutamic Acid Derivatives from Arylidene-α-amino Esters. Molecules, 29(2), 342. https://doi.org/10.3390/molecules29020342