
Hydroxy- and Hydro-Perfluoroalkylation of Styrenes by Controlling the Quenching Cycle of Eosin Y

Abstract
: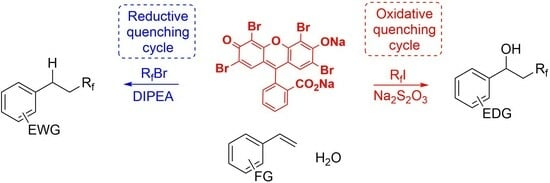
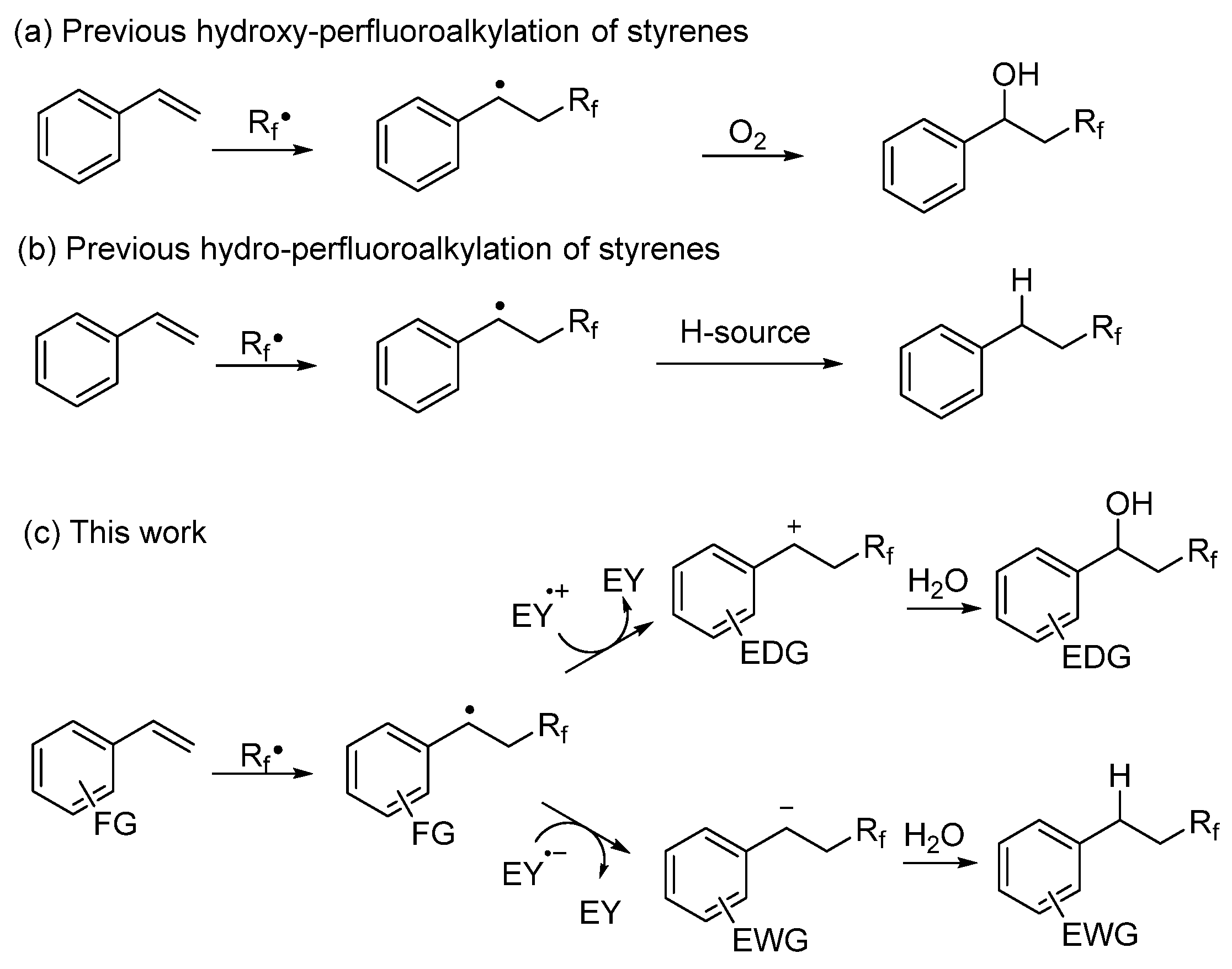
1. Introduction
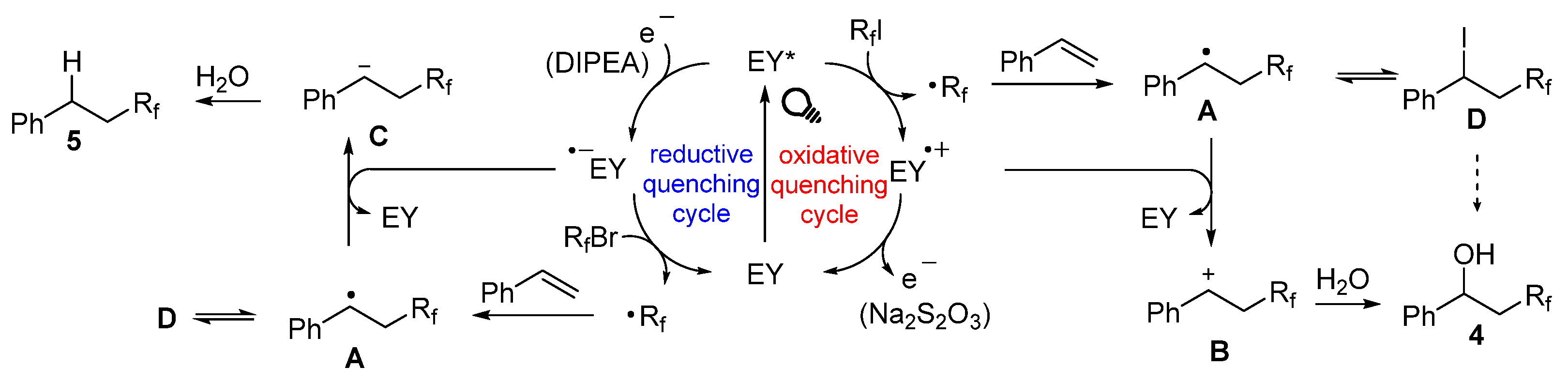
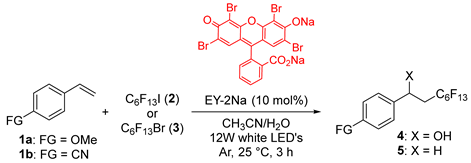
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Procedure
3.2.1. Oxidative Reaction Conditions
3.2.2. Reductive Reaction Conditions
3.3. Characterization Data of Products
3.3.1. 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluoro-1-(4-methoxyphenyl)octan-1-ol (4a)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (70.5 mg, 50%); 1H NMR (500 MHz, CDCl3) δ 7.31 (2H, d, J = 8.5 Hz), 6.91 (2H, d, J = 7.0 Hz), 5.16 (1H, dd, J = 8.5, 3.5 Hz), 3.81 (3H, s), 2.68–2.56 (1H, m), 2.45–2.33 (1H, m), 2.17 (1H, s); 13C NMR (151 MHz, CDCl3) δ 159.7, 134.2, 127.1 (2C), 119.7–110.3 (6C, m), 114.3 (2C), 67.7, 55.5, 39.8 (t, J = 21.0 Hz), 32.7, 18.3, 14.0; 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −113.0 (1F, d, J = 282.3 Hz), −114.4 (1F, d, J = 282.3 Hz), −122.3 (2F, s), −123.4 (2F, s), −124.2 (2F, s), −126.7 (2F, s); IR; 3431,3007, 2960, 2914, 1614, 1515, 1232, 1182, 1144, 1036, 812, 707; HRMS (EI+) calcd for C15H11O2F13 [M]+: 470.0551, found 470.0528.
3.3.2. 3,3,3-Trifluoro-1-(4-methoxyphenyl) propan-1-ol (4aa)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (23.3 mg, 35%); 1H NMR (400 MHz, CDCl3) δ 7.29 (2H, d, J = 8.4 Hz), 6.90 (2H, d, J = 8.8 Hz), 5.02 (1H, d, J = 8.8 Hz), 3.81 (3H, s), 2.70–2.56 (1H, m), 2.49–2.36 (1H, m), 2.16 (1H, s); 13C NMR (151 MHz, CDCl3) δ 159.7, 134.6, 130.8–112.3 (m), 127.1 (2C), 114.3 (2C), 68.5 (dd, J = 3.0, 6.0 Hz), 55.5, 42.8 (dd, J = 27.2, 54.3 Hz); 19F NMR (376 MHz, CDCl3) δ −64.2 (3F, s); IR; 3425, 3007, 2911, 2840, 1613, 1514, 1241, 1123, 1102, 1032, 824; HRMS (EI+) calcd for C10H11O2F3 [M]+: 220.0711, found 220.0718.
3.3.3. 3,3,4,4,4-Pentafluoro-1-(4-methoxyphenyl) tetran-1-ol (4ab)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow solid (25.9 mg, 32%); 1H NMR (400 MHz, CDCl3) δ 7.31 (2H, d, J = 8.8 Hz), 6.86 (2H, d, J = 8.8 Hz), 5.15 (1H, dt, J = 8.8, 3.2 Hz), 3.81 (3H, s), 2.66–2.51 (1H, m), 2.41–2.12 (1H, m), 2.11 (1H, s); 13C NMR (151 MHz, CDCl3) δ 159.7, 134.9, 127.1, 120.0–113.4 (2C, m), 114.3, 67.7, 55.5, 39.7 (t, J = 21.0 Hz); 19F NMR (376 MHz, CDCl3) δ −86.3 (3F, s), −117.1 (1F, dd, J = 263.3, 37.6 Hz), −118.3 (1F, dd, J = 263.3, 37.6 Hz); IR; 3441, 3007, 2914, 2842, 1614, 1515, 1248, 1186, 1065, 1033; HRMS (EI+) calcd for C11H11O2F5 [M]+: 270.0679, found 270.0678.
3.3.4. 3,3,4,4,5,5,5-Heptafluoro-1-(4-methoxyphenyl) pentan-1-ol (4ac)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (50.3 mg, 52%); 1H NMR (500 MHz, CDCl3) δ 7.30 (2H, d, J = 8.5 Hz), 6.91 (2H, d, J = 9.0 Hz), 5.16 (1H, dd, J = 8.5, 3.5 Hz), 3.81 (3H, s), 2.67–2.54 (1H, m), 2.44–2.32 (1H, m), 2.18 (1H, s); 13C NMR (151 MHz, CDCl3) δ 159.7, 134.9, 134.9–108.6 (3C, m), 127.1 (2C), 114.3 (2C), 67.6, 55.5, 39.5 (t, J = 21.0); 19F NMR (471 MHz, CDCl3) δ −80.9 (3F, s), −114.0 (1F, dd, J = 282.3, 47.1 Hz), −115.4 (1F, dd, J = 282.3, 47.1 Hz), −128.4 (2F, s); IR; 3455, 3007, 2842, 1613, 1515, 1304, 1155, 832, 713; HRMS (EI+) calcd for C12H11O2F7 [M]+: 320.0647, found 320.0657.
3.3.5. 3,4,4,4-Tetrafluoro-3-trifluoromethyl-1-(4-methoxyphenyl) tetran-1-ol (4ad)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (57.8 mg, 60%); 1H NMR (500 MHz, CDCl3) δ 7.30 (2H, d, J = 8.5 Hz), 6.91 (2H, d, J = 8.5 Hz), 5.12 (1H, d, J = 22.5 Hz), 3.81 (3H, s), 2.66–2.57 (1H, m), 2.41–2.33 (1H, m), 2.13 (1H, s); 13C NMR (151 MHz, CDCl3) δ 159.7, 153.73, 128.9–113.9 (2C, m), 126.9 (2C), 114.3 (2C), 92.3–90.6 (m), 68.3 (d, J = 3.0 Hz), 55.5, 38.2 (t, J = 18.0 Hz); 19F NMR (471 MHz, CDCl3) δ −76.9 (3F, s), −77.5 (3F, s), −186.0 (1F, s); IR; 3455, 3007, 2917, 2842, 1613, 1515, 1304, 1217, 1155, 1036, 832, 713; HRMS (EI+) calcd for C12H11O2F7 [M]+: 320.0647, found 320.0652.
3.3.6. 3,3,4,4,5,5,6,6,6-Nonafluoro-1-(4-methoxyphenyl) hexan-1-ol (4ae)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (46.4 mg, 42%); 1H NMR (500 MHz, CDCl3) δ 7.31 (2H, d, J = 8.5 Hz), 6.91 (2H, dd, J = 9.0 Hz), 5.16 (1H, dd, J = 8.5, 3.5 Hz), 3.81 (3H, s), 2.68–2.55 (1H, m), 2.45–2.34 (1H, m), 2.17 (1H, s); 13C NMR (151 MHz, CDCl3) δ 159.7, 134.9, 127.1 (2C), 119.5–108.7 (4C, m), 114.3 (2C), 67.7, 55.5, 39.7 (t, J = 21.0 Hz); 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −113.0 (1F, dd, J = 235.3, 47.1 Hz), −114.4 (1F, dd, J = 235.3, 47.1 Hz), −124.9 (2F, s), −126.2; IR; 3426, 2842, 2248, 1614, 1515, 1132, 1132, 1034, 881; HRMS (EI+) calcd for C13H11O2F9 [M]+: 370.0615, found 370.0617.
3.3.7. 3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-Heptadecafluoro-1-(4-methoxyphenyl) decan-1-ol (4af)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (78.1 mg, 47%); 1H NMR (500 MHz, CDCl3) δ 7.31 (2H, d, J = 8.5 Hz), 6.91 (2H, dd, J = 8.5 Hz), 5.17 (1H, dt, J = 5.5, 3.0 Hz), 3.81 (3H, s), 2.68–2.56 (1H, m), 2.45–2.34 (1H, m), 2.15 (1H, s); 13C NMR (151 MHz, CDCl3) δ 159.3, 134.9, 127.1 (2C), 119.7–108.4 (8C, m), 114.3 (2C), 67.7, 55.5, 39.9 (t, J = 21.0 Hz); 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −113.0 (1F, d, J = 282.3 Hz), −114.3 (1F, d, J = 282.3 Hz), −122.1 (2F, s), −122.4 (4F, s), −123.2 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 3326, 1517, 1199, 1145, 832; HRMS (EI+) calcd for C17H11O2F17 [M]+: 570.0487, found 570.0480.
3.3.8. 1-(4-tert-Buthoxyphenyl)3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctan-1-ol (4c)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (88.9 mg, 58%); 1H NMR (500 MHz, CDCl3) δ 7.27 (2H, d, J = 8.5 Hz), 6.98 (2H, d, J = 8.5 Hz), 5.18 (1H, d, J = 9.0 Hz), 2.68–2.56 (1H, m), 2.46–2.35 (1H, m), 2.28 (1H, s), 1.34 (3H, s); 13C NMR (151 MHz, CDCl3) δ 155.5, 137.6, 126.4 (2C), 124.6 (2C), 120.2–110.4 (6C, m), 79.0, 67.7, 39.9 (t, J = 21.5 Hz), 29.0; 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −112.9 (1F, d, J = 282.3 Hz), −114.4 (1F, d, J = 282.3 Hz), −122.3 (2F, s), −123.4 (2F, s), −124.1 (2F, s), −126.7 (2F, s); IR; 3432, 2981, 1608, 1508, 1234, 1191, 707; HRMS (ESI+) calcd for C18H17O2F13 [M+Na]+: 535.0913, found 535.0913.
3.3.9. 1-(4-(tert-butyl)phenyl)-3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctan-1-ol (4d)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (52.3 mg, 35%); 1H NMR (500 MHz, CDCl3) δ 7.41 (2H, d, J = 9.0 Hz), 7.32 (2H, d, J = 8.0 Hz), 5.20 (1H, dd, J = 9.0, 3.0 Hz), 2.69–2.56 (1H, m), 2.47–2.36 (1H, m), 2.18 (1H), 1.32 (9H, s); 13C NMR (151 MHz, CDCl3) δ 151.7, 139.8, 130.2, 128.4, 126.0, 125.5, 119.7–109.4 (6C, m), 67.9, 39.9 (J = 20.4 Hz), 34.7, 31.4 (3C); 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −112.9 (1F, d, J = 282.3 Hz), −114.5 (1F, d, J = 282.3 Hz), −123.3 (2F, s), −123.4 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 3400, 2966, 2908, 2872, 1512, 1364, 1235, 1143, 836, 731, 707; HRMS (EI+) calcd for C18H17OF13 [M]+: 496.1072, found 496.1085.
3.3.10. 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluoro-1-(4-toyl) octan-1-ol (4e)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (30.6 mg, 22%); 1H NMR (400 MHz, CDCl3) δ 7.29 (2H, d, J = 8.4 Hz), 7.20 (2H, d, J = 8.0 Hz), 5.20 (1H, dd, J = 8.8, 3.2 Hz), 2.70–2.55 (1H, m), 2.47–2.32 (1H, m), 2.36 (3H, s), 2.11 (1H, s); 13C NMR (151 MHz, CDCl3) δ 139.9, 138.5, 130.3, 129.7, 125.7, 122.8, 122.8–108.7 (6C, m), 68.0, 39.9 (d, J = 20.4 Hz), 21.3; 19F NMR (376 MHz, CDCl3) δ −81.3 (3F, s), −112.9 (1F, d, J = 263.3 Hz), −114.4 (1F, d, J = 263.3 Hz) (1F, s), −123.3 (2F, s), −123.4 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 3423, 3030, 2928, 1517, 1364, 1232, 1144, 814, 707; HRMS (EI+) calcd for C15H10OF13 [M + H]+: 454.0602, found 454.0602.
3.3.11. 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluoro-1-phenyl octan-1-ol (4f)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (47.5 mg, 36%); 1H NMR (500 MHz, CDCl3) δ 7.39 (4H, d, J = 4.5 Hz), 7.36–7.32 (1H, m), 5.22 (1H, dd, J = 9.0, 3.5 Hz), 2.69–2.57 (1H, m), 2.48–2.36 (1H, m), 2.27 (1H, br); 13C NMR (151 MHz, CDCl3) δ 142.8, 129.0 (2C), 128.6, 125.8 (2C), 120.2–108.7 (6C, m), 68.1, 40.0 (t, J = 21.0 Hz); 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −112.9 (1F, d, J = 282.3 Hz), −114.3 (1F, d, J = 282.3 Hz), −122.3 (2F, s), −123.4 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 3409, 3069, 3036, 1232, 1189, 1070, 607; HRMS (EI+) calcd for C14H9OF13 [M]+: 440.0446, found 440.0450.
3.3.12. 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluoro-1-(4-fluorophenyl) octan-1-ol (4g)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (27.4 mg, 20%); 1H NMR (500 MHz, CDCl3) δ 7.40–7.36 (2H, m), 7.10–7.06 (2H, m), 5.23 (1H, dd, J = 8.5, 3.5 Hz), 2.67–2.55 (1H, m), 2.45–2.33 (1H, m), 2.19 (1H, br); 13C NMR (151 MHz, CDCl3) δ 162.7 (d, J = 247.5 Hz), 138.5 (d, J = 3.0 Hz), 127.6 (2C), 118.5–100.3 (6C, m), 118.2 (2C), 67.5, 40.1 (t, J = 21.0 Hz); 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −112.9 (1F, d, J = 282.3 Hz), −114.0 (1F, s), −114.2 (1F, d, J = 282.3 Hz), −122.3 (2F, s), −123.3 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 3436, 1607, 1512, 1228, 1187, 1142, 838, 707; HRMS (EI+) calcd for C14H8O2F14 [M]+: 458.0352, found 458.0334.
3.3.13. 1-(4-Chlorophenyl)-3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctan-1-ol (4h)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (36.3 mg, 25%); 1H NMR (400 MHz, CDCl3) δ 7.35 (4H, dd, J = 13.2, 8.8 Hz), 5.22 (1H, d, J = 8.8 Hz), 2.63–2.57 (1H, m), 2.44–2.33 (1H, m), 2.20 (1H, s); 13C NMR (151 MHz, CDCl3) δ 141.1, 134.3, 129.2 (2C), 127.2 (2C), 119.6–108.4 (6C, m), 67.5, 40.0 (t, J = 21.0 Hz); 19F NMR (376 MHz, CDCl3) δ −81.3 (3F, s), −112.9 (1F, d, J = 263.3 Hz), −114.2 (1F, d, J = 263.3 Hz), 122.3 (2F, s), −123.4 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 3413, 1494, 1364, 1232, 1187, 1144, 831, 699; HRMS (EI+) calcd for C14H8OClF13 [M]+: 474.0056, found 474.0062.
3.3.14. 1-([1,1′-biphenyl]-4-yl)-3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctan-1-ol (4i)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). White solid (35.0 mg, 27%); 1H NMR (400 MHz, CDCl3) δ7.62–7.57 (4H, m), 7.50–7.43 (4H, m), 7.38–7.34 (1H, m), 5.29–5.25 (1H, m), 2.74–2.56 (1H, m), 2.53–2.39 (1H, m), 2.29 (1H, d, J = 1.8 Hz); 13C NMR (151 MHz, CDCl3) δ 141.7, 141.6, 140.6, 132.6, 130.2, 129.0, 128.4, 127.8, 127.7, 127.3, 126.2, 120.2–108.4 (6C, m), 67.9, 40.0 (t, J = 20.4 Hz); 19F NMR (471 MHz, CDCl3) δ −81.2 (3F, s), −112.8 (1F, d, J = 134.9 Hz), −114.2 (1F, d, J = 134.9 Hz), −122.2 (2F, s), −123.3 (2F, s), −124.0 (2F, s), −126.6 (2F, s); IR; 3299, 1234, 1189, 1144, 1007, 839, 766, 692, 651; HRMS (ESI+) calcd for C20H13OF13 [M+Na]+: 539.0651, found 539.0644.
3.3.15. 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluoro-1-(4-methoxyphenyl) octan-1-ol (4j)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (46.8 mg, 33%); 1H NMR (400 MHz, CDCl3) δ 7.40 (1H, t, J = 7.3 Hz), 6.96 (1H, d, J = 3.2 Hz), 6.95 (1H, s), 5.19 (1H, dd, J = 8.8, 3.2), 3.82 (3H, s), 2.72–2.53 (2H, m), 2.48–2.31 (1H, m); 13C NMR (151 MHz, CDCl3) δ 160.1, 144.4, 130.1, 120.3–109.2 (6C, m), 117.9, 113.9, 111.3, 68.0, 55.4, 40.0 (t, J = 21.0 Hz); 19F NMR (376 MHz, CDCl3) δ −81.2 (3F, s), −112.8 (1F, d, J = 263.3 Hz), −114.3 (1F, d, J = 263.3 Hz), −122.2 (2F, s), −123.2 (2F, s), −124.0 (2F, s), −126.5 (2F, s); IR; 3495, 2963, 2842, 1603, 1364, 1221, 1144, 1047, 786, 696; HRMS (EI+) calcd for C15H11O2F13 [M]+: 470.0551, found 470.0559.
3.3.16. 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluoro-1-(4-toyl) octan-1-ol (4k)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (56.6 mg, 42%); 1H NMR (400 MHz, CDCl3) δ 7.54 (1H, dd, J = 7.6, 1.2 Hz), 7.23 (1H, m), 5.48 (1H, dd, J = 8.8, 1.6), 2.68–2.49 (2H, m), 2.42–2.52 (1H, m), 2.34 (1H, s), 2.15 (1H, br); 13C NMR (151 MHz, CDCl3) δ 140.9, 134.0, 130.9, 128.2, 126.9, 125.2, 120.2–108.4 (6C, m), 64.3, 39.1, 18.9; 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −113.3 (1F, d, J = 263.3 Hz), −117.7 (1F, d, J = 263.3 Hz), −122.2 (2F, s), −123.2 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 3416, 3029, 2957, 1364, 1232, 1187, 1144, 812, 707; HRMS (EI+) calcd for C15H11OF13 [M]+: 454.0602, found 454.0601.
3.3.17. 1-(3,4-Dimethoxyphenyl)-3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctan-1-ol (4l)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (85.0 mg, 68%); 1H NMR (400 MHz, CDCl3) δ 6.93–6.85 (3H, m), 5.18 (1H, dd, J = 6.18, 2.52 Hz), 3.91 (3H, s), 3.89 (3H, s), 2.70–2.55 (1H, m), 2.47–2.33 (1H, m), 2.18 (1H, s); 13C NMR (151 MHz, CDCl3) δ149.4, 149.1, 135.4, 119.9–109.1 (6C, m), 118.0, 111.2, 108.6, 68.0, 56.1, 56.0, 40.0 (t, J = 21.1 Hz), 29.9; 19F NMR (376 MHz, CDCl3) δ −81.3 (3F, s), −112.9 (1F, d, J = 289.0 Hz), −114.4 (1F, d, J = 289.0 Hz), −122.3 (2F, s), −123.3 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 3498, 2841, 1518, 1231, 1188, 1141, 1116, 1026, 809, 765, 735, 707; HRMS (ESI+) calcd for C16H13O3F13 [M + Na]+: 523.0549, found 523.0557.
3.3.18. 2-(1,1,2,2,3,3,4,4,5,5,6,6,6-Tridecafluorohexyl)-1,2,3,4-tetrahydronaphthalen-1-ol (4m)
- The compound was synthesized as described in oxidative reaction conditions (62:38 diastereomer mixture, dr was measured by crude 1H NMR). The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (43.5 mg, 31%); 1H NMR (400 MHz, CDCl3) δ 7.50–7.11 (major and minor, 4H, m), 5.14 (major, 1H, t, J = 6.5 Hz), 5.10 (minor, 1H, s), 3.07–2.52 (major and minor, 3H, m), 2.33–2.09 (major and minor, 2H, m), 1.92–1.83 (major and minor, 1H, m); 13C NMR (151 MHz, CDCl3) δ 136.7 (minor), 136.4 (major), 132.6 (minor), 130.2 (major), 129.3 (minor), 129.0 (minor), 128.8 (major), 128.5 (major), 128.4 (minor), 128.2 (major), 127.0 (major), 126.8 (minor), 121.0–106.9 (major and minor, 6C, m), 66.9 (major), 66.1 (minor), 45.6 (major, t, J = 19.6 Hz), 42.4 (minor, t, J = 19.6 Hz), 28.5 (minor), 27.5 (major), 20.6 (major), 16.2 (minor); 19F NMR (471 MHz, CDCl3) δ −81.2 (major and minor, 3F, s), −113.5 (major, 1F, d, J = 288.8 Hz), −114.4 (minor, 1F, d, J = 288.8 Hz), −116.0 (minor, 1F, d, J = 288.8 Hz), −117.0 (major, 1F, d, J = 288.8 Hz), −120.5–−124.1 (major and minor, 6F, m), −125.7–−127.4 (major and minor, 2F, m); IR; 3447, 3063, 3013, 2986, 2936, 2884, 1491, 1451, 1227, 1190, 1179, 1142, 1121, 1045, 743; HRMS (EI+) calcd for C16H11OF13 [M]+: 466.0602, found 466.0565.
3.3.19. 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluoro-1-(4-(trifluoromthyl)phenyl) octan-1-ol (4n)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (25.9 mg, 17%); 1H NMR (400 MHz, CDCl3) δ 7.66 (2H, d, J = 8.0 Hz), 7.54 (2H, d, J = 8.0 Hz), 5.31 (1H, d, J = 8.6 Hz), 2.68–2.56 (1H, m), 2.48–2.36 (1H, m), 2.28 (1H, s); 13C NMR (151 MHz, CDCl3) δ: 146.3, 132.6, 130.8 (dd, J = 64.9, 33.2 Hz), 130.2, 128.3, 126.2, 126.0 (q, J = 3.0 Hz), 121.3–106.5 (6C, m), 67.5, 40.1 (t, J = 21.1 Hz); 19F NMR (376 MHz, CDCl3) δ −63.1 (3F, s), −81.2 (3F, s), −112.8 (1F, d, J = 265.8 Hz), −114.0 (1F, d, J = 265.8 Hz), −122.2 (2F, s), −123.3 (2F, s), −124.0 (2F, s), −126.6 (2F, s); IR; 3443, 1326, 1237, 1129, 1069, 1017, 844, 707; HRMS (EI+) calcd for C15H8OF16 [M]+: 508.0320, found 508.0320.
3.3.20. 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluoro-1-(2,3,4,5,6-pentafluorophenyl) octan-1-ol (4o)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (27.2 mg, 17%); 1H NMR(500 MHz, CDCl3) δ 5.60 (1H, dd, J = 12.5, 6.0 Hz), 3.00–2.89 (1H, m), 2.72–2.61 (1H, m), 2.47 (1H, d, J = 6.5 Hz); 13C NMR (151 MHz, CDCl3) δ; 145.7, 144.1, 138.6 (2C), 137.0 (2C), 119.2–106.7 (6C, m), 59.4, 37.3 (t, J = 19.5 Hz); 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −113.8 (1F, d, J = 282.3 Hz), −114.7 (1F, d, J = 282.3 Hz) −122.3 (2F, s), −123.4 (2F, s), −124.0 (2F, s), −126.6 (2F, s), −143.7 (2F, d, J = 47.1 Hz), −153.4 (1F, d, J = 47.1 Hz), 161.2 (2F, s); IR; 3486, 1656, 1523, 1504, 1235, 1192, 1142, 996, 947, 700; HRMS (EI+) calcd for C14H4OF18 [M]+: 529.9975, found 529.9976.
3.3.21. 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluoro-1,1-diphenyloctan-1-ol (4p)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (60.3 mg, 43%); 1H NMR (400 MHz, CDCl3) δ 7.45–7.42 (4H, m), 7.36–7.31 (4H, m), 7.28–7.24 (2H, m), 3.17 (2H, t, J = 18.3 Hz), 2.74 (1H, t, J = 2.1 Hz); 13C NMR (151 MHz, CDCl3) δ: 145.5 (2C), 137.7, 132.6, 130.2, 128.6 (2C), 128.4, 127.7 (2C), 125.5 (2C), 120.1–108.4 (6C, m), 76.6, 41.0 (t, J = 19.6 Hz); 19F NMR (376 MHz, CDCl3) δ −81.3 (3F, s), −109.5 (2F, s), −122.1 (2F, s), −123.3 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 3470, 3088, 3061, 2971, 3028, 1655, 1495, 1233, 1188, 1142, 1121, 812, 696; HRMS (ESI−) calcd for C20H13OF13 [M − H]−: 515.0681, found 515.0641.
3.3.22. 4-(3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluorooctyl)benzonitrile (5b)
- The compound was synthesized as described in reductive reaction conditions. The product was purified by column chromatography (Hexane). Yellow oil (96.5 mg, 72%); 1H NMR (400 MHz, CDCl3) δ 7.64–7.61 (2H, m), 7.34 (2H, d, J = 8.2 Hz), 3.01–2.98 (2H, m), 2.46–2.33 (2H, d); 13C NMR (151 MHz, CDCl3) δ: 144.7, 132.8 (2C), 129.3 (2C), 119.8–108.4 (6C, m), 118.8, 111.0, 32.4 (t, J = 22.7 Hz), 26.8 (t, J = 4.5 Hz); 19F NMR (376 MHz, CDCl3) δ −81.2 (3F, s) −115.0 (2F, s), −122.3 (2F, s), −123.3 (2F, s), −123.9 (2F, s), −126.6 (2F, s); IR; 3062, 2227, 1185, 1165, 1119, 1086, 1072, 979, 908, 862, 812, 736, 704; HRMS (ESI+) calcd for C15H8F13N [M + Na]+: 472.0347, found 472.0347.
3.3.23. 4-(3,3,4,4,5,5,5-Heptafluoropentyl)benzonitrile (5bc)
- The compound was synthesized as described in reductive reaction conditions. The product was purified by column chromatography (Hexane). Yellow oil (57.4 mg, 64%); 1H NMR (400 MHz, CDCl3) δ 7.63 (2H, dd, J = 6.6, 1.6 Hz), 7.33 (2H, d, J = 8.2 Hz), 3.02–2.97 (2H, m), 2.45–2.32 (2H, d); 13C NMR (151 MHz, CDCl3) δ: 144.6, 132.7 (2C), 129.3 (2C), 120.7–107.0 (3C, m), 118.8, 111.0, 32.1 (t, J = 22.7 Hz), 26.7 (t, J = 3.0 Hz); 19F NMR (376 MHz, CDCl3) δ −81.0 (3F, s) −116.0 (2F, s), −128.2 (2F, s); IR; 2961, 2231, 1352, 1220, 1169, 1112, 1085, 947, 910, 824, 743, 680; HRMS (ESI+) calcd for C12H8F7N [M + Na]+: 322.0442, found 322.0443.
3.3.24. 4-(3,3,4,4,5,5,6,6,6-Nonafluorohexyl)benzonitrile (5be)
- The compound was synthesized as described in reductive reaction conditions. The product was purified by column chromatography (Hexane). Yellow oil (76.0 mg, 73%); 1H NMR (400 MHz, CDCl3) δ 7.65–7.62 (2H, m), 7.34 (2H, d, J = 8.2 Hz), 3.02–2.98 (2H, m), 2.46–2.33 (2H, d); 13C NMR (151 MHz, CDCl3) δ: 144.6, 132.8 (2C), 129.3 (2C), 120.3–108.7 (4C, m), 118.8, 111.0, 32.3 (t, J = 22.7 Hz), 26.8 (t, J = 3.0 Hz); 19F NMR (376 MHz, CDCl3) δ −81.5 (3F, s) −115.2 (2F, s), −124.9 (2F, s), −126.5 (2F, s); IR; 2960, 2231, 1357, 1217, 1131, 1088, 1009, 976, 881, 824, 722, 692; HRMS (ESI+) calcd for C13H8F9N [M + Na]+: 372.0411, found 372.0411.
3.3.25. 1-(3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluorooctyl)-4-(trifluoromethyl)benzene (5n)
- The compound was synthesized as described in reductive reaction conditions. The product was purified by column chromatography (Hexane). Yellow oil (63.8 mg, 65%); 1H NMR (500 MHz, CDCl3) δ 7.59 (2H, d, J = 8.0 Hz), 7.34 (2H, d, J = 8.0 Hz), 2.99 (2H, tt, J = 8.5, 3.0 Hz), 2.39 (2H, qt, J = 19.0, 18.5 Hz); 13C NMR (151 MHz, CDCl3) δ143.3, 129.3 (q, J = 33.2 Hz), 128.8, 125.9 (q, J = 3.0 Hz), 119.9–108.4 (6C, m), 32.7 (t, J = 21.1 Hz), 26.5 (t, J = 4.5 Hz); 19F NMR (471 MHz, CDCl3) δ −62.3 (3F, s), −81.3 (3F, s) −115.0 (2F, d, J = 272.5), −122.4 (2F, s), −123.4 (2F, s), −124.0 (2F, s), −126.6 (2F, s); IR; 1612, 1422, 1325, 1235, 1166, 1069, 1020, 826, 707; HRMS (APCI+) calcd for C15H8F16 [M]+: 492.0370 found, 492.0370.
3.3.26. 1,2,3,4,5-Pentafluoro-6-(3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctyl)benzene (5o)
- The compound was synthesized as described in reductive reaction conditions. The product was purified by column chromatography (Hexane). Colorless oil (54.5 mg, 53%); 1H NMR (500 MHz, CDCl3) δ 2.99 (2H, t, J = 7.5 Hz), 2.38 (2H, qt, J = 18.0, 18.0 Hz); 13C NMR (150 MHz, CDCl3) δ 146.1–136.8 (6C, m), 120.2–108.4 (6C, m), 30.2 (t, J = 22.6 Hz), 14.0; 19F NMR (471 MHz, CDCl3) δ −81.2 (3F, s), −115.8 (2F, d, J = 272.5), −122.4 (2F, s), −123.4 (2F, s), −124.0 (2F, s), −126.6 (2F, s), −144.3 (2F,s), −156.0 (2F,s), −162.2 (2F,s); IR; 1659, 1522, 1506, 1232, 1191, 1144, 1013, 968, 697; HRMS (FD+) calcd for C14H4F18 [M − H]+: 512.9947 found 512.9938.
3.3.27. (3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluorooctane-1,1-diyl)dibenzene (5p)
- The compound was synthesized as described in reductive reaction conditions. The product was purified by column chromatography (Hexane). Yellow oil (64.0 mg, 64%); 1H NMR (500 MHz, CDCl3) δ 7.32–7.26 (8H, m), 7.24–7.19 (2H, m), 4.46 (1H, t, J = 7.2), 2.92 (2H, td, J = 28.4, 11.2); 13C NMR (151 MHz, CDCl3) δ 143.2 (2C), 128.9 (4C), 127.6 (4C), 127.0 (2C), 120.2–108.4 (6C, m), 43.8, 36.3 (t, J = 19.6 Hz); 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −113.3 (2F, s), −122.2 (2F, s), −123.3 (2F, s), 124.0 (2F, s), −126.6 (2F, s); IR; 3090, 3066, 3032, 1601, 1495, 1364, 1235, 1186, 1144, 696; HRMS (ESI+) calcd for C20H13F18 [M + Na]+: 523.0702 found 523.0702.
3.3.28. 1-Nitro-4-(3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctyl)benzene (5q)
- The compound was synthesized as described in reductive reaction conditions. The product was purified by column chromatography (Hexane). Yellow oil (19.7 mg, 21%); 1H NMR (500 MHz, CDCl3) δ 8.20 (2H, d, J = 8.4, 2.8 Hz), 7.40 (2H, d, J = 8.8 Hz), 3.05 (2H, tt, J = 8.4, 2.4 Hz), 2.42 (2H, qt, J = 17.6, 18.8 Hz); 13C NMR (151 MHz, CDCl3) δ 147.1, 146.7, 129.4 (2C), 124.2 (2C), 119.6–105.8 (6C, m), 32.4 (t, J = 21.0 Hz), 26.6; 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s) −115.0 (2F, s), −122.4 (2F, s), −123.4 (2F, s), −123.9 (2F, s), −126.7 (2F, s); IR; 2961, 1611, 1534, 1349, 1198, 1138, 1073, 977, 855, 782, 752, 693; HRMS (ESI+) calcd for C14H8NO2F13 [M + Na]+: 492.0240 found, 492.0240.
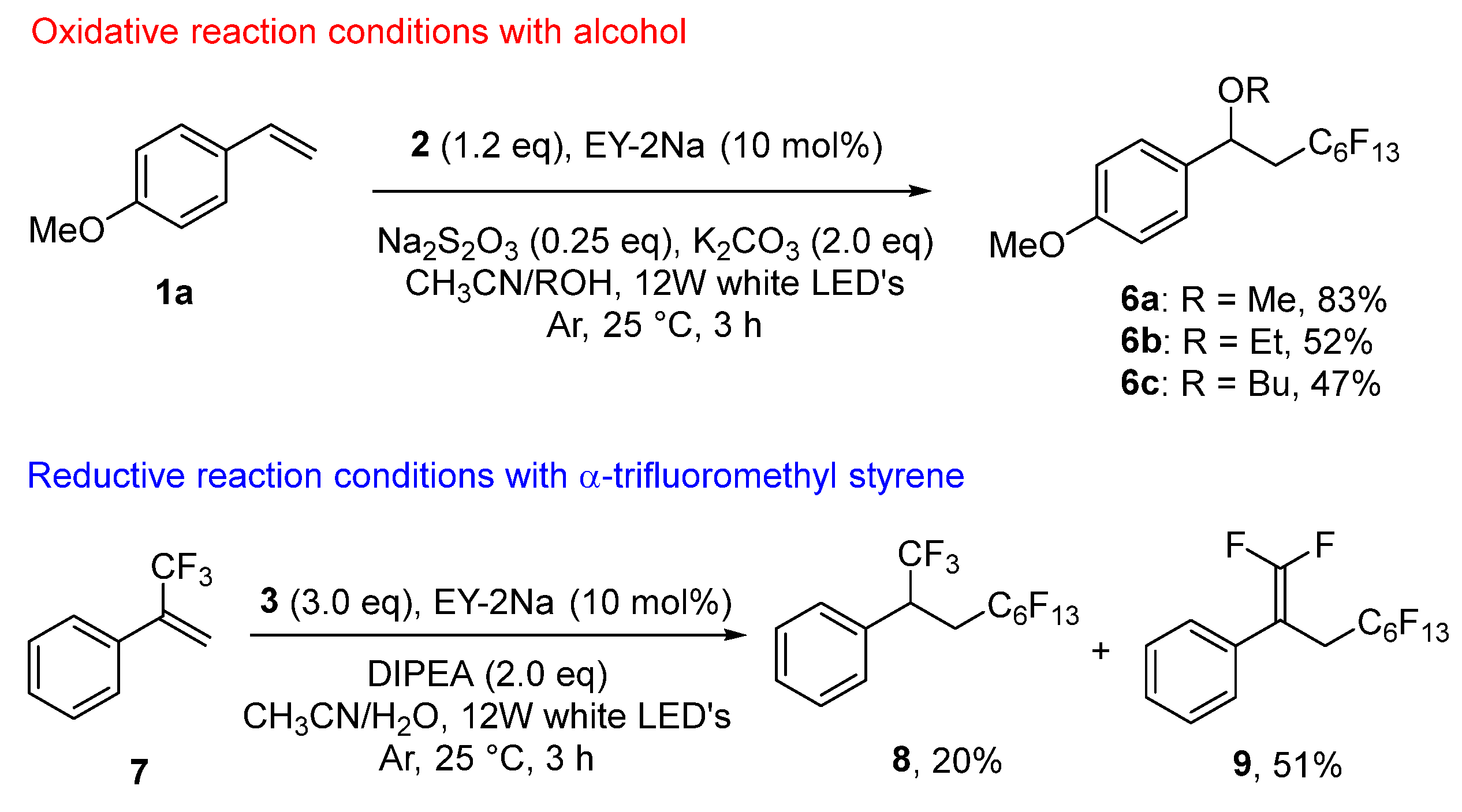
3.3.29. 1-Methoxy-4-(3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluoro-1-methoxyoctyl)benzene (6a)
- The compound was synthesized as described in oxidative reaction conditions using methanol instead of water. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Colorless oil (71.0 mg, 48%); 1H NMR (400 MHz, CDCl3) δ 7.26 (2H, d, J = 8.8 Hz), 6.92 (2H, d, J = 8.8 Hz), 4.52 (1H, dd, J = 8.4, 4.0 Hz), 3.82 (3H, s), 3.20 (3H, s), 2.71–2.55 (1H, m), 2.39–2.25 (1H, m); 13C NMR (151 MHz, CDCl3) δ 159.7, 132.4, 127.9 (2C), 119.3–111.0 (6C, m), 114.4 (2C), 76.4, 56.5, 55.3, 39.3 (t, J = 21.0); 19F NMR (376 MHz, CDCl3) δ −81.3 (3F, s), −112.7 (1F, d, J = 225.7 Hz), −114.0 (1F, d, J = 225.7 Hz), −122.3 (2F, s), −123.4 (2F, s), −124.2 (2F, s), −126.6 (2F, s); IR; 3002, 2940, 2841, 1613, 1514, 1144, 1036, 707; HRMS (EI+) calcd for C16H13O2F13 [M]+: 484.0708, found 484.0732.
3.3.30. 1-(1-Ethoxy-3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctyl)-4-methoxybenzene (6b)
- The compound was synthesized as described in oxidative reaction conditions using ethanol instead of water. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Colorless oil (77.3 mg, 52%); 1H NMR (400 MHz, CDCl3) δ 7.26 (2H, d, J = 8.4 Hz), 6.91 (2H, d, J = 8.8 Hz), 4.63 (1H, dd, J = 8.4, 4.0 Hz), 3.82 (3H, s), 3.20 (3H, s), 3.41–3.29 (2H, m), 2.71–2.56 (1H, m), 2.38–2.24 (1H, m), 1.17 (3H, t, J = 6.8 Hz); 13C NMR (151 MHz, CDCl3) δ 159.6, 133.2, 127.7, 119.3–109.2 (6C, m), 114.2, 74.5, 64.2 55.4, 39.4 (t, J = 21.0 Hz), 15.2; 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −112.7 (2F, d, J = 300.9 Hz), −113.9 (2F, d, J = 300.9 Hz), −122.3 (2F, s), −123.4 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 2981, 1613, 1514, 1235, 1102, 1037, 832, 707; HRMS (APCI+) calcd for C17H15O2F13 [M]+: 498.0864; found 498.0859.
3.3.31. 1-(1-Butoxy-3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctyl)-4-methoxybenzene (6c)
- The compound was synthesized as described in oxidative reaction conditions using n-butanol instead of water. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Colorless oil (75.8 mg, 47%); 1H NMR (500 MHz, CDCl3) δ 7.26 (2H, d, J = 8.5 Hz), 6.91 (2H, d, J = 8.5 Hz), 4.61 (1H, dd, J = 8.5, 3.5 Hz), 3.82 (3H, s), 3.32 (3H, s), 3.41–3.23 (2H, m), 2.66–2.57 (1H, m), 2.33–2.24 (1H, m), 1.54–1.48 (1H, m), 1.38–1.31 (1H, m), 0.87 (3H, t, J = 7.0 Hz); 13C NMR (151 MHz, CDCl3) δ 159.6, 133.2, 127.7 (2C), 119.3–110.4 (6C, m), 114.2 (2C), 74.8, 68.7, 55.4, 39.4 (t, J = 4.5 Hz), 31.9, 19.4, 13.0; 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −112.6 (2F, d, J = 272.5 Hz), −114.0 (2F, d, J = 272.5 Hz), −122.3 (2F, s), −123.4 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 2963, 2938, 2877, 1613, 1514, 1144, 1102, 1037, 707; HRMS (ESI+) calcd for C19H19O2F13 [M + Na]+: 549.1070 found, 549.1078.
3.3.32. (1,1,1,4,4,5,5,6,6,7,7,8,8,9,9,9-Hexadecafluorononan-2-yl)benzene (8)
- The compound was synthesized as described in reductive reaction conditions. The product was purified by column chromatography (Hexane). Colorless oil (20.0 mg, 20%); 1H NMR (500 MHz, CDCl3) 7.55–7.28 (5H, m), 3.78–3.71 (1H, m), 2.84–2.68 (2H, m); 13C NMR (151 MHz, CDCl3) δ 155.6 (t, J = 294.5 Hz), 132.5 (t, J = 4.5 Hz), 129.1 (t, J = 9.1 Hz), 128.8, 128.7, 128.3, 128.2, 120.6–108.4 (6C, m), 83.7 (t, J = 19.6 Hz), 30.7 (t, J = 22.7 Hz); 19F NMR (471 MHz, CDCl3) −71.0 (3F, s), −81.3 (3F, s), −112.4 (2F, d, J = 272.7 Hz), −114.7 (2F, d, J = 272.7 Hz), −122.4 (2F, s), −123.4 (2F, s), −123.9 (2F, s), −126.7 (2F, s); IR; 1232, 1187, 1141, 1119, 1033, 845, 812, 778, 755, 744; HRMS (FD+) calcd for C15H8F16 [M − H]+: 491.0293, found 491.0289.
3.3.33. (1,1,4,4,5,5,6,6,7,7,8,8,9,9,9-Pentadecafluoronon-1-en-2-yl)benzene (9)
- The compound was synthesized as described in reductive reaction conditions. The product was purified by column chromatography (Hexane). Colorless oil (49.0 mg, 51%); 1H NMR (500 MHz, CDCl3) δ 7.46–7.26 (5H, m), 3.21 (2H, t, J = 17.5 Hz); 13C NMR (151 MHz, CDCl3) δ 129.1 (2C), 128.3 (2C), 128.8, 128.3, 128.2, 120.6–108.2 (7C, m), 43.6 (q, J = 27.2 Hz), 31.0 (t, J = 28.7 Hz); 19F NMR (471 MHz, CDCl3) δ −81.3 (3F), −86.0 (1F, d, J = 57.9 Hz), −86.4 (1F, d, J = 27.3 Hz), −122.7 (2F, s), −122.3 (2F, s), −123.4 (2F, s),−123.7 (2F, s), −126.6 (2F, s); IR; 1736, 1232, 1197, 1139, 868, 847, 812, 799, 778, 755, 707, 696, 663; HRMS (FD+) calcd for C15H7F15 [M]+: 472.0308, found 472.0327.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Hagmann, W.K. The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Sumii, Y.; Shibata, N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5, 10633–10640. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N. Current Contributions of Organofluorine Compounds to the Agrochemical Industry. iScience 2020, 23, 101467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yan, K.; Fu, C.; Peng, H.; Hawker, C.J.; Whittaker, A.K. Biological Utility of Fluorinated Compounds: From Materials Design to Molecular Imaging, Therapeutics and Environmental Remediation. Chem. Rev. 2022, 122, 167–208. [Google Scholar] [CrossRef] [PubMed]
- Kirsh, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Application; Wiley-VCH: Weinhein, Germany, 2013. [Google Scholar]
- Ameduri, B. Fluoropolymers: The Right Material for the Right Applications. Chem. Eur. J. 2018, 24, 18830–18841. [Google Scholar] [CrossRef]
- Dolbier, W.R., Jr. Structure, Reactivity, and Chemistry of Fluoroalkyl Radicals. Chem. Rev. 1996, 96, 1557–1584. [Google Scholar] [CrossRef]
- Tsuchii, K.; Imura, M.; Kamada, N.; Hirao, T.; Ogawa, A. An Efficient Photoinduced Iodoperfluoroalkylation of Carbon−Carbon Unsaturated Compounds with Perfluoroalkyl Iodides. J. Org. Chem. 2004, 69, 6658–6665. [Google Scholar] [CrossRef]
- Murphy, O.M.; Baldwin, C.S.; Buck, R.C. Synthesis utilizing n-perfluorlakyl iodides [RFI, CnF2n+1-I] 2000–2010. J. Fluor. Chem. 2012, 138, 3–23. [Google Scholar] [CrossRef]
- Barata-Vallejo, S.; Cooke, M.V.; Postigo, A. Radical Fluoroalkylation Reactions. ACS Catal. 2018, 8, 7287–7307. [Google Scholar] [CrossRef]
- Tagami, K.; Yajima, T. Development of Electrophilic Radical Perfluoroalkylation of Electron-Deficient Olefins. Chem. Rec. 2023, 23, e202300037. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Ohkoshi, M.; Aoki, N.; Ohnuma, Y.; Iyoda, M. Photochemical Oxyfluoroalkylation of Styrenes by the Addition of Perfluoroalkyl Radicals in an Atomosphere of Oxyfem. Tetrahedron Lett. 1999, 40, 5731–5734. [Google Scholar] [CrossRef]
- Yasu, Y.; Koike, T.; Akita, M. Three-Component Oxytrifluoromethylation of Alkenes: Highly Efficient and Regioselective Difunctionalization of C=C Bonds Mediated by Photoredox Catalysts. Angew. Chem. Int. Ed. 2012, 51, 9567–9571. [Google Scholar] [CrossRef] [PubMed]
- Beatty, J.W.; Douglas, J.J.; Cole, K.P.; Stephenson, C.R.J. A Scalable and Operationally Simple Radical Trifluoromethylation. Nat. Commun. 2015, 6, 7919. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Lin, F.; Wang, X.; Jiao, N. Photoredox-Catalyzed Hydroxyfluoroalkylation of Alkene with Simple Fluoroalkyl Iodides. J. Photochem. Photobiol. A Chem. 2018, 355, 194–201. [Google Scholar] [CrossRef]
- Jud, W.; Kappe, C.O.; Cantillo, D. Catalyst-Free Oxytrifluoromethylation of Alkenes through Paired Electrolysis in Organic-Aqueous Media. Chem. Eur. J. 2018, 24, 17234–17238. [Google Scholar] [CrossRef]
- Chen, T.; Guo, Y.; Sun, K.; Wu, L.Z.; Liu, W.Q.; Liu, C.; Huang, Y.; Chen, Q.Y. Photoinduced Hydroxylperfluoroalkylation of Styrenes. Org. Chem. Front. 2018, 5, 1045–1048. [Google Scholar] [CrossRef]
- Su, Z.; Guo, Y.; Chen, Q.-Y.; Zhan, Z.-G.; Nian, B.-Y. Catalyst-Free Hydrozytrifluoromethylation of Alkenes Using Iodotrifluoromethane. Chin. J. Chem. 2019, 37, 597–604. [Google Scholar] [CrossRef]
- Shen, W.G.; Wu, Q.Y.; Gong, X.Y.; Ao, G.Z.; Liu, F. A Facile Method for Hydroxytrifluoromethylation of Alkenes with Langlois Reagent and DMSO. Green Chem. 2019, 21, 2983–2987. [Google Scholar] [CrossRef]
- Li, Q.; Fan, W.; Peng, D.; Meng, B.; Wang, S.; Huang, R.; Liu, S.; Li, S. Cobalt-Tertiary-Amine-Mediated Hydroxytrifluoromethylation of Alkenes with CF3Br and Atmospheric Oxygen. ACS Catal. 2020, 10, 4012–4018. [Google Scholar] [CrossRef]
- Quan, Y.; Shi, W.; Song, Y.; Jiang, X.; Wang, C.; Lin, W. Bifunctional Metal-Organic Layer with Organic Dyes and Iron Centers for Synergistic Photoredox Catalysis. J. Am. Chem. Soc. 2021, 143, 3075–3080. [Google Scholar] [CrossRef] [PubMed]
- Tagami, K.; Ofuji, Y.; Kanbaa, T.; Yajima, T. Metal-free visible-light-induced hydroxyperfluoroalkylation of conjugated olefins using enamine catalyst. RSC Adv. 2022, 12, 32790–32795. [Google Scholar] [CrossRef] [PubMed]
- Tagami, K.; Yajima, T. Halogen-Bond-Promoted Hydroxyperfluoroalkylation of Olefins with Molecular Oxygen under Visible-Light Irradiation. Asian J. Org. Chem. 2023, 12, e202300273. [Google Scholar] [CrossRef]
- Barata-Vallejo, S.; Postigo, A. (Me3Si)3SiH-Mediated Intermolecular Radical Perfluoroalkylation Reactions of Olefins in Water. J. Org. Chem. 2010, 75, 6141–6148. [Google Scholar] [CrossRef] [PubMed]
- Wilger, D.J.; Gesmundo, N.J.; Nicewicz, D.A. Catalytic Hydrotrifluoromethylation of Styrenes and Unactivated Aliphatic Alkenes via an Organic Photoredox System. Chem. Sci. 2013, 4, 3160–3165. [Google Scholar] [CrossRef]
- Choi, S.; Kim, Y.J.; Kim, S.M.; Yang, J.W.; Kim, S.W.; Cho, E.J. Hydrotrifluoromethylation and Iodotrifluoromethylation of Alkenes and Alkynes Using an Inorganic Electride as a Radical Generator. Nat. Commun. 2014, 5, 4881. [Google Scholar] [CrossRef]
- Straathof, N.J.W.; Cramer, S.E.; Hessel, V.; Noël, T. Practical Photocatalytic Trifluoromethylation and Hydrotrifluoromethylation of Styrenes in Batch and Flow. Angew. Chem. Int. Ed. 2016, 128, 15778–15782. [Google Scholar] [CrossRef]
- Peng, D.; Fan, W.; Zhao, X.; Chen, W.; Wen, Y.; Zhang, L.; Li, S. Zinc-Brønsted Acid Mediated Practical Hydrotrifluoromethylation of Alkenes with CF3Br. Org. Chem. Fron. 2021, 8, 6356–6363. [Google Scholar] [CrossRef]
- Jia, H.; Häring, A.P.; Berger, F.; Zhang, L.; Ritter, T. Trifluoromethyl Thianthrenium Triflate: A Readily Available Trifluoromethylating Reagent with Formal CF3+, CF3•, and CF3-Reactivity. J. Am. Chem. Soc. 2021, 143, 7623–7628. [Google Scholar] [CrossRef]
- Yang, Y.F.; Lin, J.H.; Xiao, J.C. Starting from Styrene: A Unified Protocol for Hydrotrifluoromethylation of Diversified Alkenes. Org. Lett. 2021, 23, 9277–9282. [Google Scholar] [CrossRef] [PubMed]
- Louvel, D.; Souibgui, A.; Taponard, A.; Rouillon, J.; ben Mosbah, M.; Moussaoui, Y.; Pilet, G.; Khrouz, L.; Monnereau, C.; Vantourout, J.C.; et al. Tailoring the Reactivity of the Langlois Reagent and Styrenes with Cyanoarenes Organophotocatalysts under Visible-Light. Adv. Synth. Catal. 2022, 364, 139–148. [Google Scholar] [CrossRef]
- Haria, D.P.; König, B. Synthetic Applications of Eosin Y in Photoredox Catalysis. Chem. Commun. 2014, 50, 6688–6699. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, V.; Singh, P.P. Eosin Y Catalysed Photoredox Synthesis: A Review. RSC Adv. 2017, 7, 31377–31392. [Google Scholar] [CrossRef]
- Yajima, T.; Ikegami, M. Metal-Free Visible-Light Radical Iodoperfluoroalkylation of Terminal Alkenes and Alkynes. Eur. J. Org. Chem. 2017, 15, 2126–2129. [Google Scholar] [CrossRef]
- Yajima, T.; Shigenaga, S. Metal-Free Visible Light Hydroperfluoroalkylation of Unactivated Alkenes Using Perfluoroalkyl Bromides. Org. Lett. 2019, 21, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Shigenaga, S.; Shibata, H.; Tagami, K.; Kanbara, T.; Yajima, T. Eosin Y catalyzed Visible-Light-Induced Hydroperfluoroalkylation of Electron-Deficient Alkenes. J. Org. Chem. 2022, 87, 14923–14929. [Google Scholar] [CrossRef]
- Sun, H.; Huang, C.; Bin, X.; Cui, F.; Jiang, G. Photochemical akynylperfluoroalkylation of unactive alkenes mediated by halogen-bonded charge-transfer complexes. Org. Chem. Fron. 2023, 10, 2332–2339. [Google Scholar] [CrossRef]
- Zhao, P.; Wang, L.; Guo, X.; Chen, J.; Liu, Y.; Wang, L.; Ma, Y. VisibleLight-Drivenα-Diazoketonesas Denitrogenated Synthons: Synthesis of Fluorinated N-Heterocyclesvia Multicomponent Cyclization Reactions. Org. Lett. 2023, 25, 3314–3318. [Google Scholar] [CrossRef]
- Jaffé, H.H. A Reëxamination of the Hammett Equation. Chem. Rev. 1953, 53, 191–261. [Google Scholar] [CrossRef]
- Anslyn, E.V.; Dougherty, D.A. Experimants Related to Thermodynamics and Kinetics. In Modern Physical Organic Chmistry, 1st ed.; University Science Books: Sausalito, CA, USA, 2005; p. 446. [Google Scholar]
- Bégué, J.-P.; Bonnet-Delpon, D.; Rock, M.H. A concise synthesis of functionalised gem-difluoroalkenes, via the addition of organolithium reagents to α-trifluoromethylstyrene. Tetrahedron Lett. 1995, 36, 5003–5006. [Google Scholar]
- Tian, F.; Yan, G.; Yu, J. Recent advances in the synthesis and applications of a-(trifluoromethyl)styrenes in organic synthesis. Chem. Commun. 2019, 55, 13486–13505. [Google Scholar] [CrossRef]
- Yan, G.; Qiu, K.; Guo, M. Recent advance in the C–F bond functionalization of trifluoromethyl-containing compounds. Org. Chem. Front. 2021, 8, 3915–3942. [Google Scholar] [CrossRef]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.U.; Slanina, T.; Tao, C.-J.; König, B. Metal-Free Perfluoroarylation by Visible Light Photoredox Catalysis. ACS Catal. 2016, 6, 369–375. [Google Scholar] [CrossRef]
- Tang, W.-K.; Feng, Z.-W.; Xu, Z.-W.; Cheng, Z.-F.; Xu, J.; Cai, J.-J.; Xu, H.-J. Visible-Light-Enabled Decarboxylative Mono- and Difluoromethylation of Cinnamic Acids under Metal-Free Conditions. Org. Lett. 2017, 19, 5501–5504. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.D.; Bakshi, D.; Shigh, A. Visible Light Mediated Organocatalytic Activation of EthylBromofluoroacetate: Coupling with Indoles and Anilines. J. Org. Chem. 2015, 80, 10187–10196. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | 1 | 2 or 3 | Additive | Yield of 4 (%) b | Yield of 5 (%) b |
| 1 | 1a | 2 (1.2 equiv.) | Na2S2O3 (5.0 equiv.) | 40 | n.d. c |
| 2 | 1a | 2 (1.2 equiv.) | Na2S2O3 (5.0 equiv.), K2CO3 (2.0 equiv.) | 53 | n.d. c |
| 3 | 1a | 2 (1.2 equiv.) | Na2S2O3 (0.25 equiv.), K2CO3 (2.0 equiv.) | 70 (50) | n.d. c |
| 4 | 1a | 2 (1.2 equiv.) | K2CO3 (2.0 equiv.) | 20 | n.d. c |
| 5 | 1a | 2 (3.0 equiv.) | Na2S2O3 (0.25 equiv.), K2CO3 (2.0 equiv.) | 66 | n.d. c |
| 6 | 1b | 3 (3.0 equiv.) | DIPEA (2.0 equiv.) | n.d. | 70 (72) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shibata, H.; Nakayama, M.; Tagami, K.; Kanbara, T.; Yajima, T. Hydroxy- and Hydro-Perfluoroalkylation of Styrenes by Controlling the Quenching Cycle of Eosin Y. Molecules 2023, 28, 7577. https://doi.org/10.3390/molecules28227577
Shibata H, Nakayama M, Tagami K, Kanbara T, Yajima T. Hydroxy- and Hydro-Perfluoroalkylation of Styrenes by Controlling the Quenching Cycle of Eosin Y. Molecules. 2023; 28(22):7577. https://doi.org/10.3390/molecules28227577
Chicago/Turabian StyleShibata, Haruko, Moeko Nakayama, Koto Tagami, Tadashi Kanbara, and Tomoko Yajima. 2023. "Hydroxy- and Hydro-Perfluoroalkylation of Styrenes by Controlling the Quenching Cycle of Eosin Y" Molecules 28, no. 22: 7577. https://doi.org/10.3390/molecules28227577
APA StyleShibata, H., Nakayama, M., Tagami, K., Kanbara, T., & Yajima, T. (2023). Hydroxy- and Hydro-Perfluoroalkylation of Styrenes by Controlling the Quenching Cycle of Eosin Y. Molecules, 28(22), 7577. https://doi.org/10.3390/molecules28227577