
Structure–Activity Relationship Target Prediction Studies of Clindamycin Derivatives with Broad-Spectrum Bacteriostatic Antibacterial Properties

Abstract
:1. Introduction
2. Results
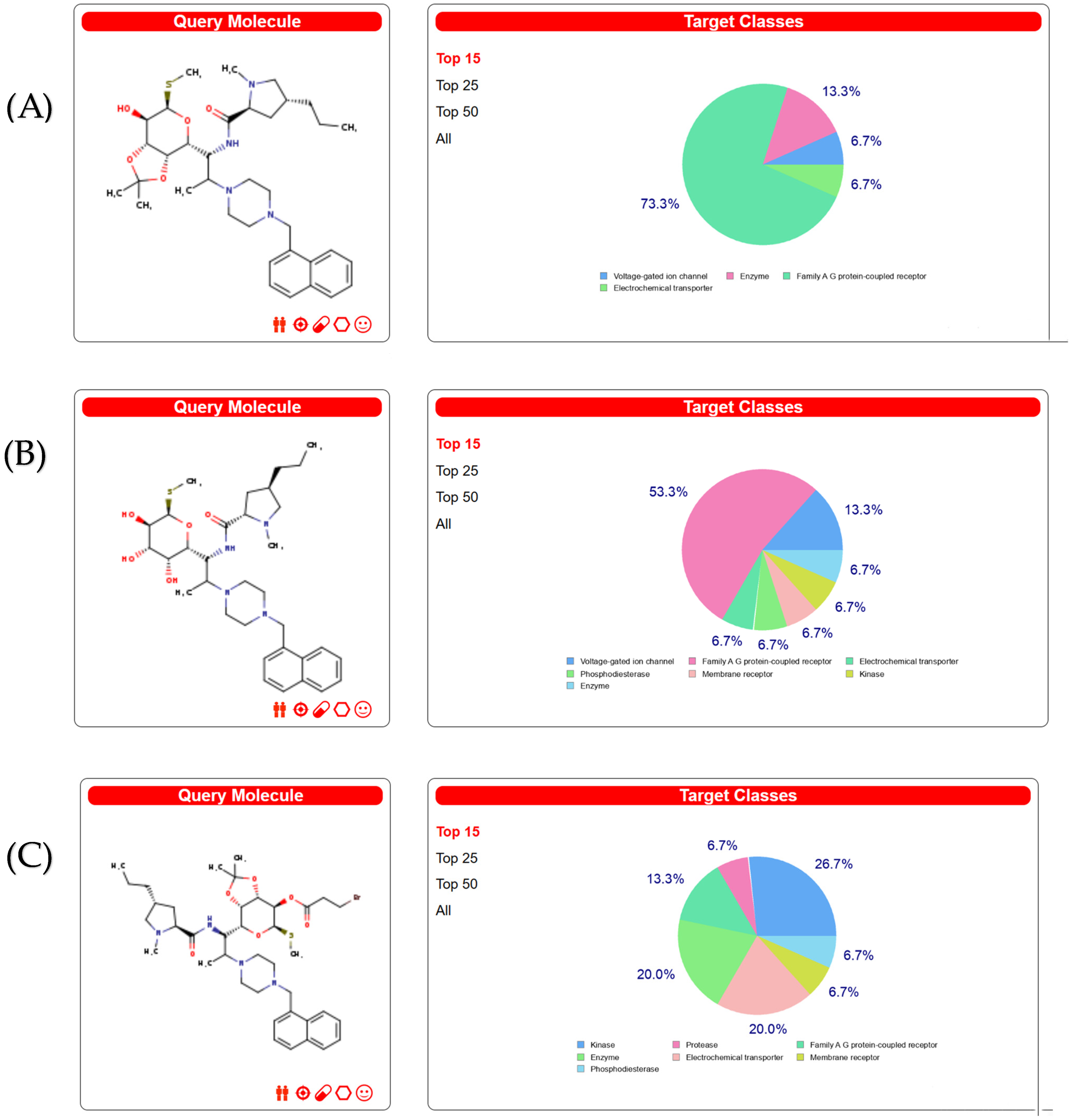
2.1. Screening of Clindamycin Targets
2.2. Intersection of Three Clindamycin Derivative Targets
2.3. Screening of the Antibacterial Targets of the Clindamycin Derivatives through the PubChem Database
2.4. Molecular Docking Simulation and Validation
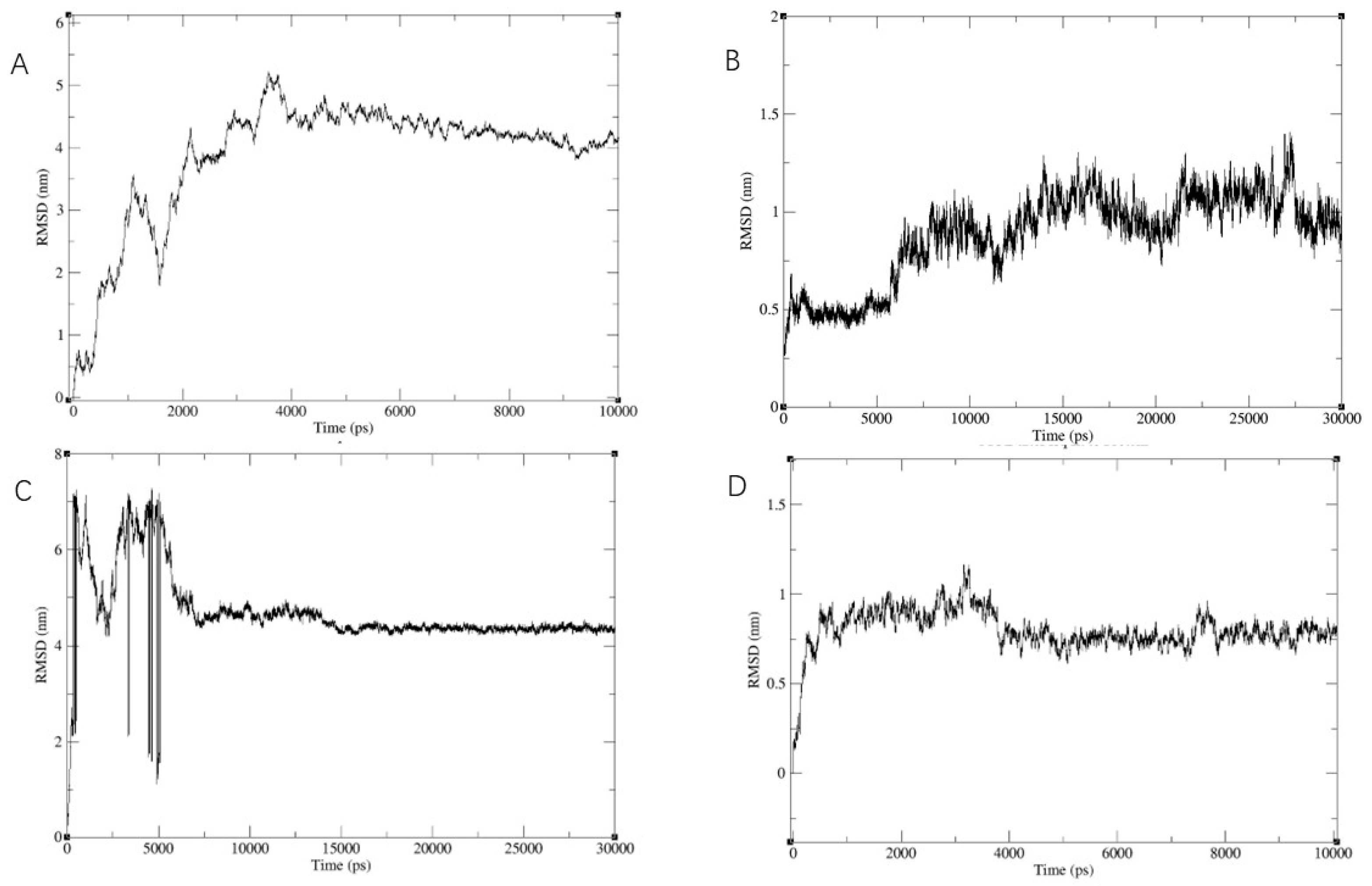
2.5. Stability of the Docked Complexes Studied via MD Simulation
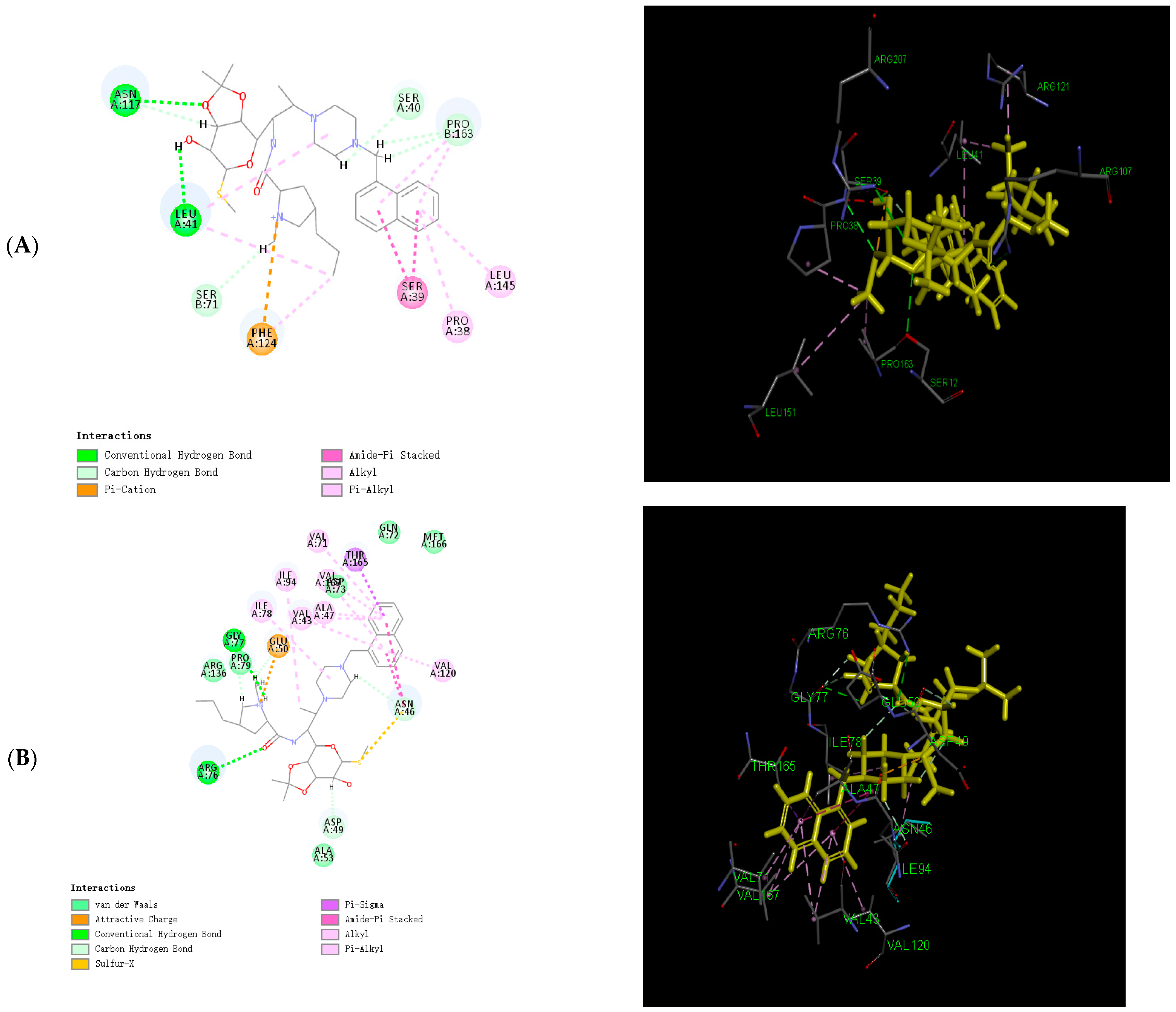
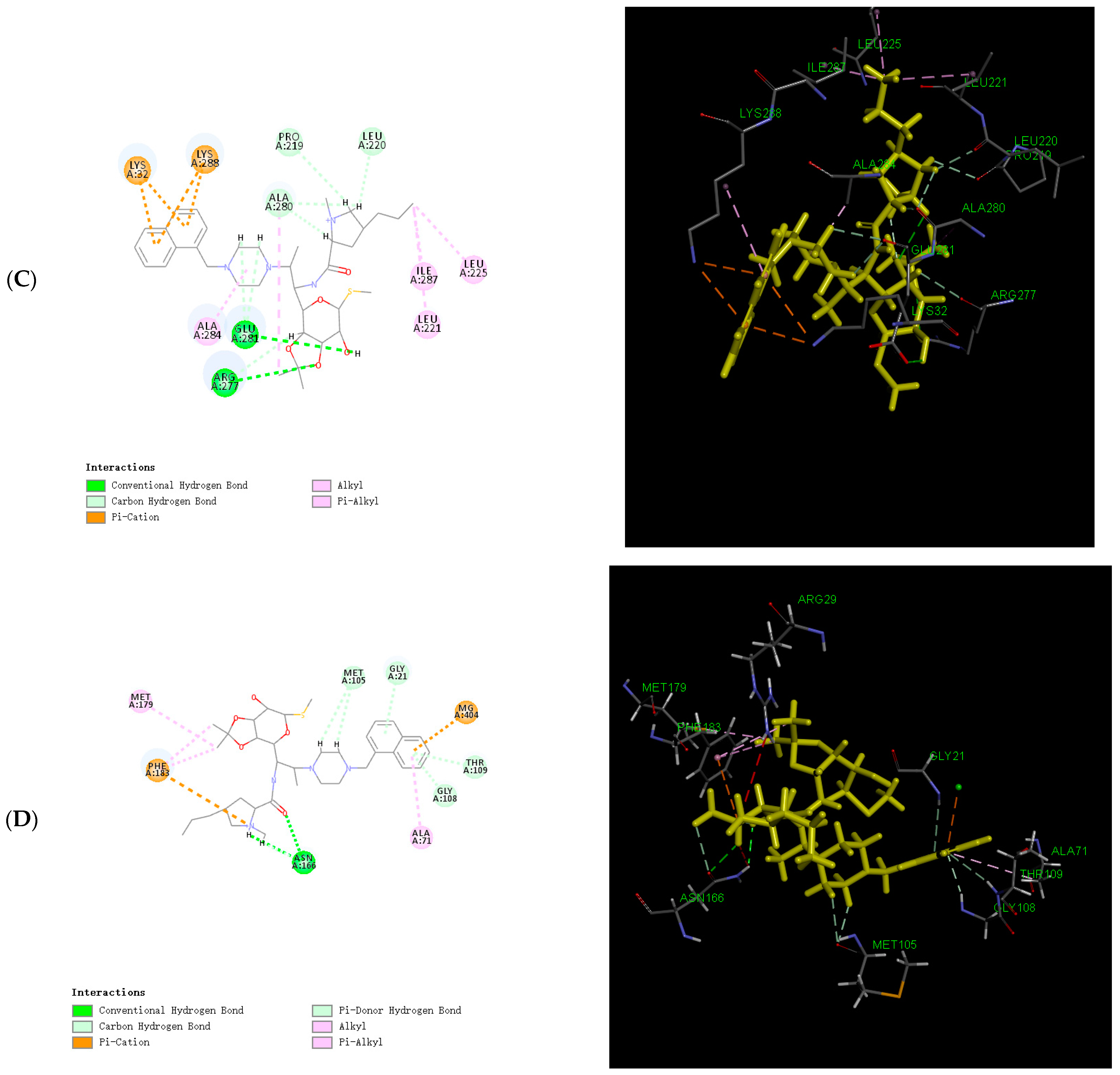
2.6. Binding Force Analysis
2.7. ADMET Prediction
2.8. Protein Subcellular Localization
3. Materials and Methods
3.1. Screening of Clindamycin Targets
3.2. Intersection of Three Clindamycin-Derivative Targets
3.3. Screening of Antibacterial Targets of Clindamycin Derivatives through PubChem Database
3.4. Molecular Docking Simulation and Validation
3.5. Stability of the Docked Complexes Studied via MD Simulation
3.6. Calculation of the Binding Energy
3.7. ADMET Prediction
3.8. Protein Subcellular Localization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Walsh, T.R.; Gales, A.C.; Laxminarayan, R.; Dodd, P.C. Antimicrobial Resistance: Addressing a Global Threat to Humanity; Public Library of Science: San Francisco, CA, USA, 2023; Volume 20, p. e1004264. [Google Scholar]
- Bakheit, A.H.; Attwa, M.W.; Kadi, A.A.; Ghabbour, H.A.; Alkahtani, H.M. Exploring the Chemical Reactivity, Molecular Docking, Molecular Dynamic Simulation and ADMET Properties of a Tetrahydrothienopyridine Derivative Using Computational Methods. Crystals 2023, 13, 1020. [Google Scholar] [CrossRef]
- Afzal, M.; Hassan, S.S.; Sohail, S.; Camps, I.; Khan, Y.; Basharat, Z.; Karim, A.; Aurongzeb, M.; Irfan, M.; Salman, M.; et al. Genomic landscape of the emerging XDR Salmonella Typhi for mining druggable targets clpP, hisH, folP and gpmI and screening of novel TCM inhibitors, molecular docking and simulation analyses. BMC Microbiol. 2023, 23, 25. [Google Scholar] [CrossRef] [PubMed]
- Rajasekhar, S.; Das, S.; Karuppasamy, R.; Musuvathi Motilal, B.; Chanda, K. Identification of novel inhibitors for Prp protein of Mycobacterium tuberculosis by structure based drug design, and molecular dynamics simulations. J. Comput. Chem. 2022, 43, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Ahmad, I.; Ahmad, I.; Amin, A.; Rather, M.A. Gene Characterization, Molecular Docking and Dynamic Simulations of Insulin-Like Growth Factor Receptor (IGF-1Ra) in Common Carp, Cyprinus carpio. In Proceedings of the Zoological Society; Springer: New Delhi, India, 2023; pp. 1–15. [Google Scholar]
- Bagewadi, Z.K.; Khan, T.Y.; Gangadharappa, B.; Kamalapurkar, A.; Shamsudeen, S.M.; Yaraguppi, D.A. Molecular dynamics and simulation analysis against superoxide dismutase (SOD) target of Micrococcus luteus with secondary metabolites from Bacillus licheniformis recognized by genome mining approach. Saudi J. Biol. Sci. 2023, 30, 103753. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, T.; Zhu, J.; Li, J. Discovery of novel targets and mechanisms of MEK inhibitor Selumetinib for LGG treatment based on molecular docking and molecular dynamics simulation. J. Mol. Model. 2022, 28, 138. [Google Scholar] [CrossRef] [PubMed]
- Omoboyede, V.; Onile, O.S.; Oyeyemi, B.F.; Aruleba, R.T.; Fadahunsi, A.I.; Oke, G.A.; Onile, T.A.; Ibrahim, O.; Adekiya, T.A. Unravelling the anti-inflammatory mechanism of Allium cepa: An integration of network pharmacology and molecular docking approaches. Mol. Divers. 2023. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, J.; Tang, J. Computational Systems Pharmacology and Molecular Docking Reveal an Anti-Apoptosis and Anti-Inflammatory Mechanism of Compound Angelica Ligusticum Wallichii Granules in the Treatment of Endometriosis. Drug Des. Dev. Ther. 2023, 17, 743–759. [Google Scholar] [CrossRef]
- Chio, H.; Guest, E.E.; Hobman, J.L.; Dottorini, T.; Hirst, J.D.; Stekel, D.J. Predicting bioactivity of antibiotic metabolites by molecular docking and dynamics. J. Mol. Graph. Model. 2023, 123, 108508. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Qiu, Y.; Baron, R.; Molinero, V. Coarse-Graining of TIP4P/2005, TIP4P-Ew, SPC/E, and TIP3P to Monatomic Anisotropic Water Models Using Relative Entropy Minimization. J. Chem. Theory Comput. 2014, 10, 4104–4120. [Google Scholar] [CrossRef] [PubMed]
- Price, D.J.; Brooks, C.L., III. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef] [PubMed]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE-AnteChamber PYthon Parser interface. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef]
- Grubmüller, H.; Heller, H.; Windemuth, A.; Schulten, K. Generalized Verlet Algorithm for Efficient Molecular Dynamics Simulations with Long-range Interactions. Mol. Simul. 1991, 6, 121–142. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2017, 27, 112–128. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Long, S.; Cao, S. Molecular docking and virtual screening of an influenza virus inhibitor that disrupts protein–protein interactions. Viruses 2021, 13, 2229. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Gupta, S.; Gupta, D.; Gulzar, Y.; Juneja, S.; Alwan, A.A.; Nauman, A. An artificial intelligence-based stacked ensemble approach for prediction of protein subcellular localization in confocal microscopy images. Sustainability 2023, 15, 1695. [Google Scholar] [CrossRef]
- Wang, L.; Cui, Z.; Li, N.; Liang, G.; Zhang, X.; Wang, Y.; Li, D.; Li, X.; Zhang, S.; Zhang, L. Comparative proteomics reveals Cryptosporidium parvum infection disrupts cellular barriers. J. Proteom. 2023, 287, 104969. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Structural Formula | Antibacterial Spectrum |
|---|---|---|
| 3 | 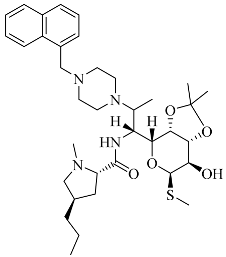 | E. coli ATCC35218 K. pneumoniae ATCC700607 S. enteritidis CICC21482 P. aeruginosa ATCC27853 MRSA clinical isolate C. albicans CMCC98001 |
| 3e | 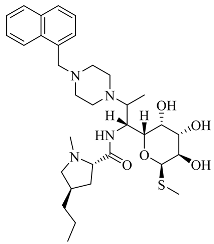 | |
| 4 | 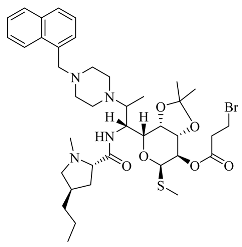 |
| Common Name | |
|---|---|
| Public Protein | Target |
| KCNH2 | HERG |
| PIK3CA | PI3-kinase p110-alpha subunit |
| ADRA1D | Alpha-1d adrenergic receptor |
| ADRA1A | Alpha-1a adrenergic receptor |
| ADRA1B | Alpha-1b adrenergic receptor |
| PIK3CG | PI3-kinase p110-gamma subunit |
| HTR1A | Serotonin 1a (5-HT1a) receptor |
| SLC6A4 | Serotonin transporter |
| CCR3 | C-C chemokine receptor type 3 |
| HTR2A | Serotonin 2a (5-HT2a) receptor |
| ADORA2A | Adenosine A2a receptor |
| F10 | Thrombin and coagulation factor X |
| PIK3CB | PI3-kinase p110-beta subunit |
| MAPK14 | MAP kinase p38 alpha |
| AURKA | Serine/threonine-protein kinase AuroraA |
| PDE5A | Phosphodiesterase |
| PDE4B | Phosphodiesterase 4B |
| PRKCB | Protein kinase C beta |
| SIGMAR1 | Sigma opioid receptor |
| ADRB2 | Adrenergic receptor beta |
| ADRB1 | Beta-1 adrenergic receptor |
| CFD | Complement factor D |
| AURKB | Serine/threonine-protein kinase Aurora-B |
| ADORA1 | Adenosine A1 receptor |
| RPS6KB1 | Ribosomal protein S6 kinase 1 |
| PIM1 | Serine/threonine-protein kinase PIM1 |
| Target | PDB ID | Uniprot ID | ChEMBL ID | Target Class |
|---|---|---|---|---|
| Adrenergic receptor beta | 4QY5, 4QY6, 4ID4, 4R4R, 4R4S | P07550 | CHEMBL210 | Family A G-protein-coupled receptor |
| Beta-1 adrenergic receptor (by homology) | 6H7J, 6H7L, 6H7M, 6H7N, 6H7O, 6IBL | P08588 | CHEMBL213 | Family A G-protein-coupled receptor |
| Adenosine A1 receptor | 6D9H, 7LD4, 7LD3 | P30542 | CHEMBL226 | Family A G-protein-coupled receptor |
| Mu opioid receptor (by homology) | 8F7R | P35372 | CHEMBL233 | Family A G-protein-coupled receptor |
| Adenosine A2a receptor | 5K2A, 5K2B,5K2D | P29274 | CHEMBL251 | Family A G-protein-coupled receptor |
| MAP kinase ERK2 | 8CJ0, 8C5P, 8C5F, 8C5E, 8BN6, 8BFT, 8BFR, 8BCJ, 8BCI, 7ZD3, 7ZD1, 7ZD0, 7Z2W, 7Z2T, 7Z18, 7Z15, 7VTG, 7VF8, 7VA3, 7V9Z, 7V6T, 7RZK, 7RM7, 7R2O, 7R1J, 7R1H, 7R0F, 7PTF, 7PJI, 7P8X, 7P8U, 7P2X, 7P2W, 7P2N, 7P2M, 7ONY, 7ON4, 7ON2, 7OJD, 7OJC, 7OJB, 7OI2, 7OHN, 7OHL, 7OHK, 7OHH, 7O5M, 7KYE, 7KRV, 7KRU, 7KPS, 7KPP, 7KDR, 7KDO | P28482 | CHEMBL4040 | Kinase |
| PDB ID | Absolute Energy | Clean Energy | Relative Energy | Lib Dock Score | Hot Spots (Average) |
|---|---|---|---|---|---|
| 7PTF | 76.0364 | 121.839 | 6.14328 | 142.136 | 16.65, −11.84, 50.63, A, 84, 29 |
| 14.65, −7.44, 48.43, A, 58, 45 | |||||
| 16.05, −6.24, 46.63, A, 35, 46 | |||||
| 7P2X | 83.8745 | 121.839 | 13.9814 | 143.521 | −22.65, −17.99, 6.92, A, 15, 21 |
| −16.45, −11.99, 8.72, A, 57, 35 | |||||
| −19.85, −10.79, 9.52, A, 67, 43 | |||||
| 7OHN | 84.5974 | 121.839 | 14.6402 | 162.369 | 0.48, −5.97, 21.08, A, 65, 26 |
| 4.68, −11.38, 25.28, A, 83, 35 | |||||
| 6.08, −10.57, 27.28, A, 98, 37 | |||||
| 7O5M | 84.6001 | 121.839 | 14.7069 | 166.496 | 17.98, 35.34, 18.83, A, 9, 25 |
| 16.98, 36.54, 19.03, A, 13, 26 | |||||
| 9.58, 34.54, 33.63, A, 85, 41 | |||||
| 8C5P | 81.269 | 121.839 | 10.961 | 181.861 | 29.18, 13.58, 20.57, P, 45, 23 |
| 35.38, 11.38, 14.77, A, 15, 35 | |||||
| 35.98, 11.78, 13.57, A, 8, 36 |
| Drug | Protein | Location (k = 23) | UniProt ID | PDB ID |
|---|---|---|---|---|
| Clindamycin derivatives | FtsZ | cyto: 51.35% cyto_nucl: 29.73% cysk: 10.81% pero: 5.40% mito: 2.70%: | Q2FZ89 | 7ohn |
| Clindamycin derivatives | β-lactamase | cyto: 38.36%, cyto_nucl: 26.03%, cysk: 19.18%, nucl: 8.22%, mito: 5.48%, pero: 2.74% | UPI00067E6531 | 4qy5 |
| Clindamycin derivatives | FleQ | cyto: 31.63% cyto_nucl: 25.17%, cyto_mito: 19.73%, nucl: 14.29%, mito: 5.10%, cysk: 4.08% | UPI0021CDE869 | 7ptf |
| Clindamycin derivatives | aspC | cyto: 59.26%, nucl: 18.52%, mito: 14.81%, cysk: 7.41% | UPI000274A8A1 | 7p2x |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, Y.; Zhang, Y.; Zhu, H. Structure–Activity Relationship Target Prediction Studies of Clindamycin Derivatives with Broad-Spectrum Bacteriostatic Antibacterial Properties. Molecules 2023, 28, 7357. https://doi.org/10.3390/molecules28217357
Jia Y, Zhang Y, Zhu H. Structure–Activity Relationship Target Prediction Studies of Clindamycin Derivatives with Broad-Spectrum Bacteriostatic Antibacterial Properties. Molecules. 2023; 28(21):7357. https://doi.org/10.3390/molecules28217357
Chicago/Turabian StyleJia, Yiduo, Yinmeng Zhang, and Hong Zhu. 2023. "Structure–Activity Relationship Target Prediction Studies of Clindamycin Derivatives with Broad-Spectrum Bacteriostatic Antibacterial Properties" Molecules 28, no. 21: 7357. https://doi.org/10.3390/molecules28217357
APA StyleJia, Y., Zhang, Y., & Zhu, H. (2023). Structure–Activity Relationship Target Prediction Studies of Clindamycin Derivatives with Broad-Spectrum Bacteriostatic Antibacterial Properties. Molecules, 28(21), 7357. https://doi.org/10.3390/molecules28217357