
Exploration on Electronic Properties of Self-Assembled Indium Nitrogen Nanosheets and Nanowires by a Density Functional Method


Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
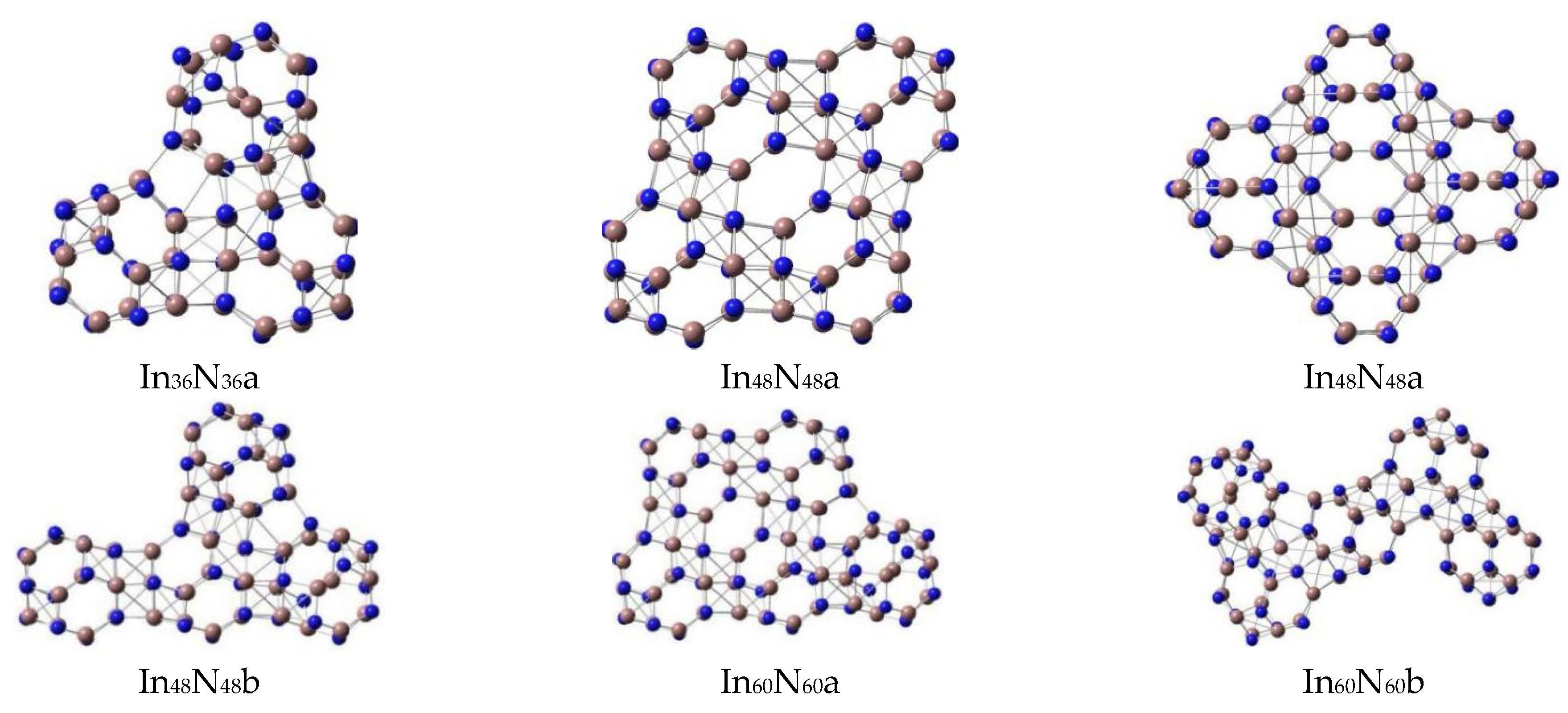
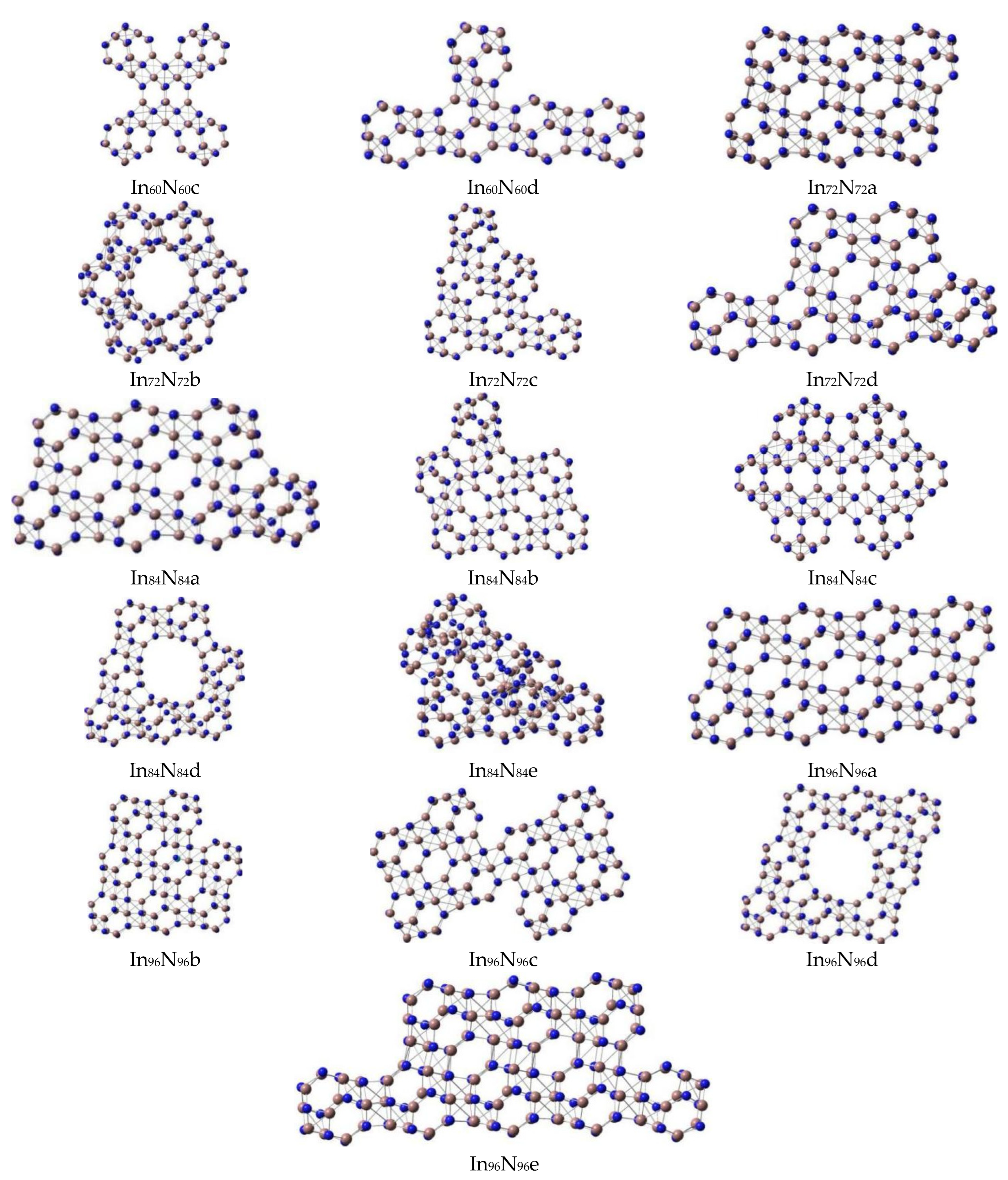
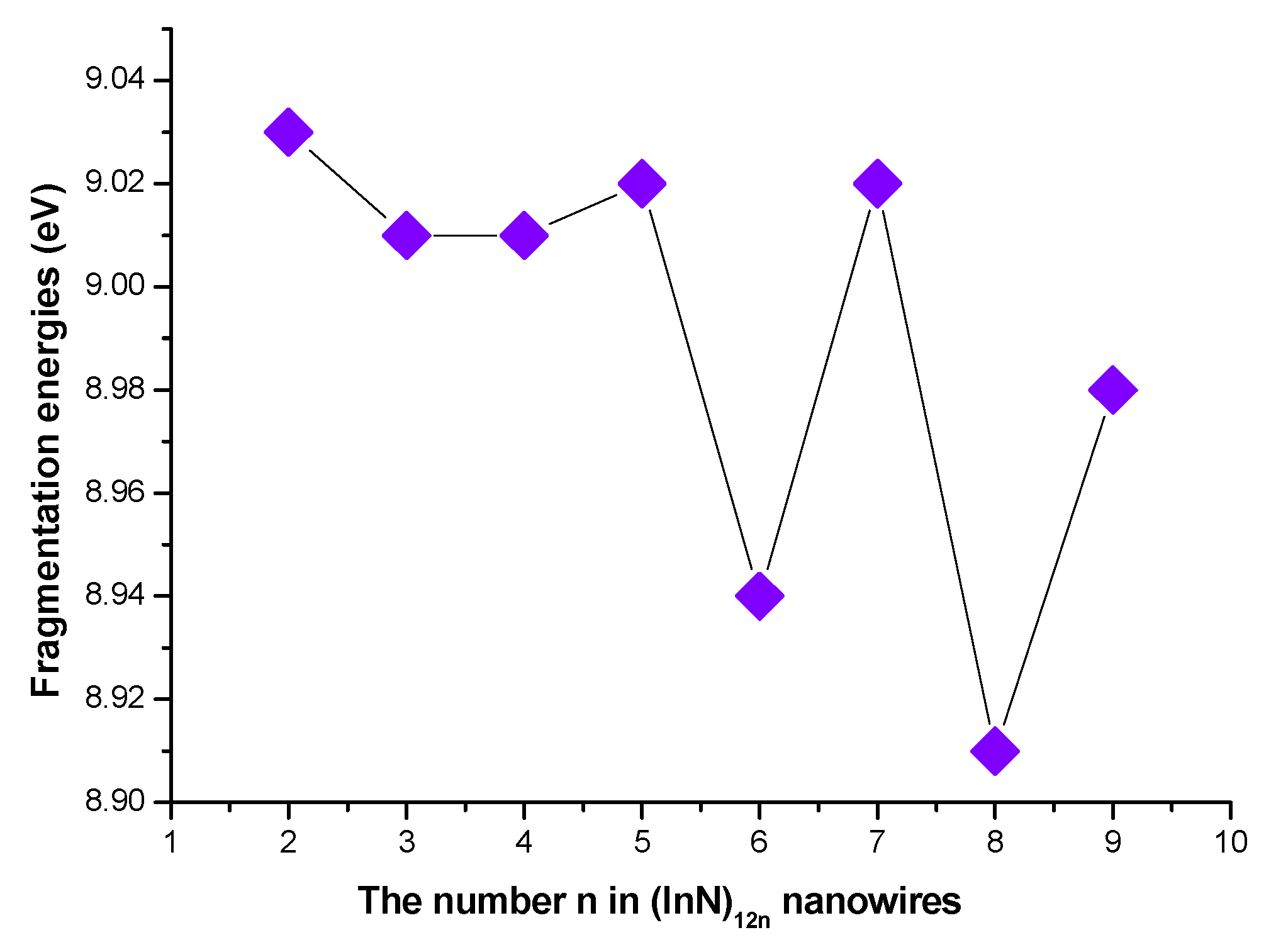
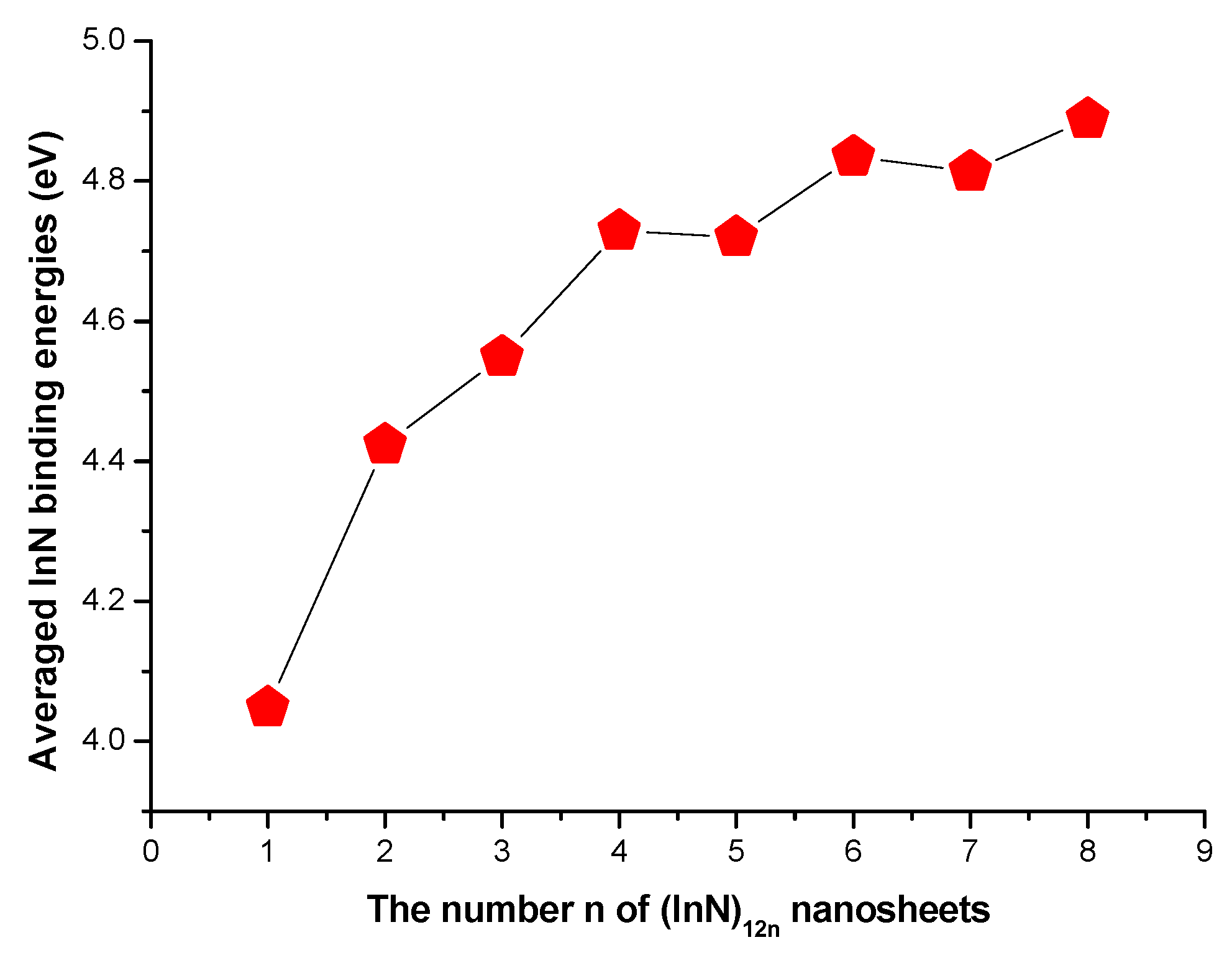
3.1. Geometries and Stabilities
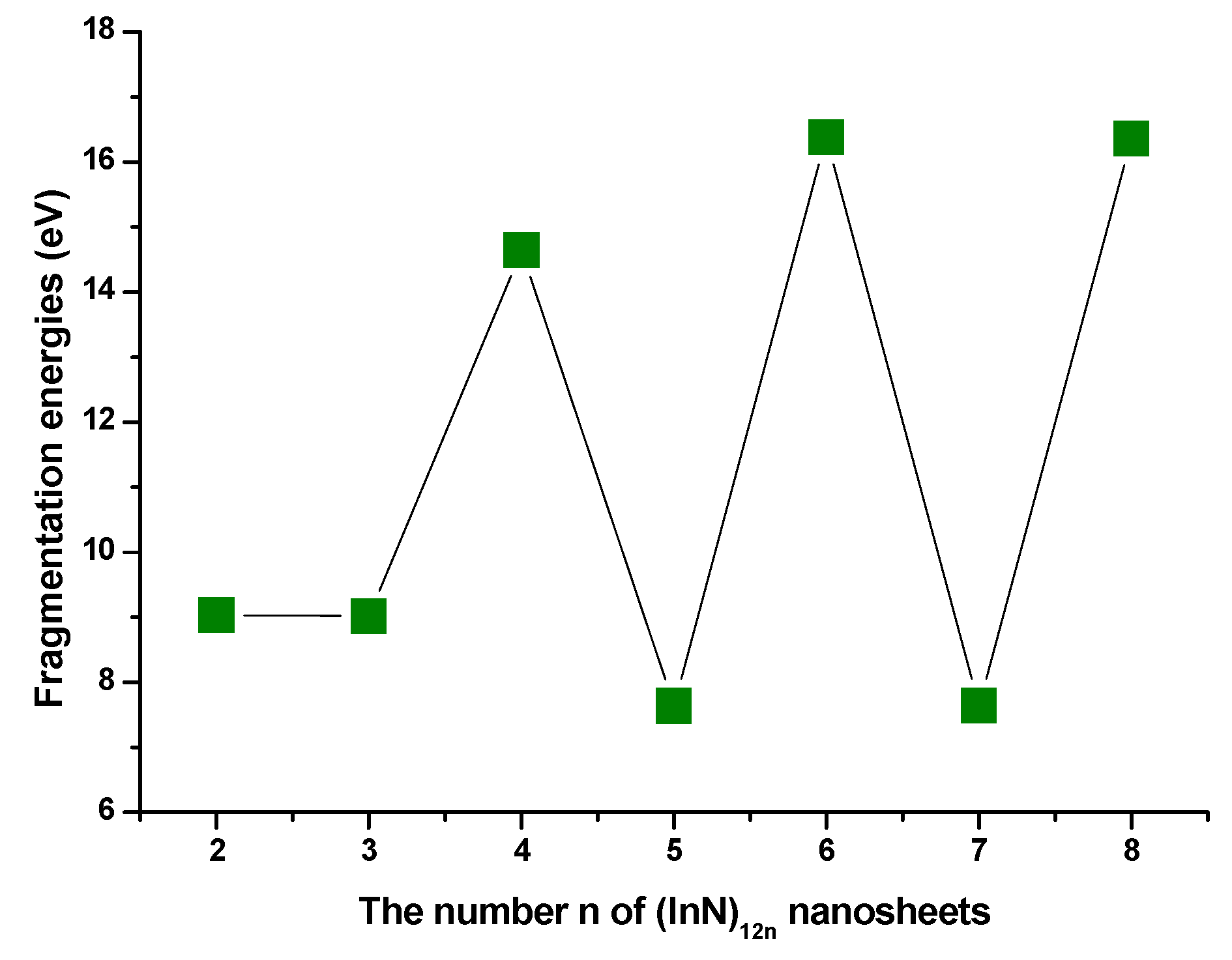
3.2. Relative Stabilities
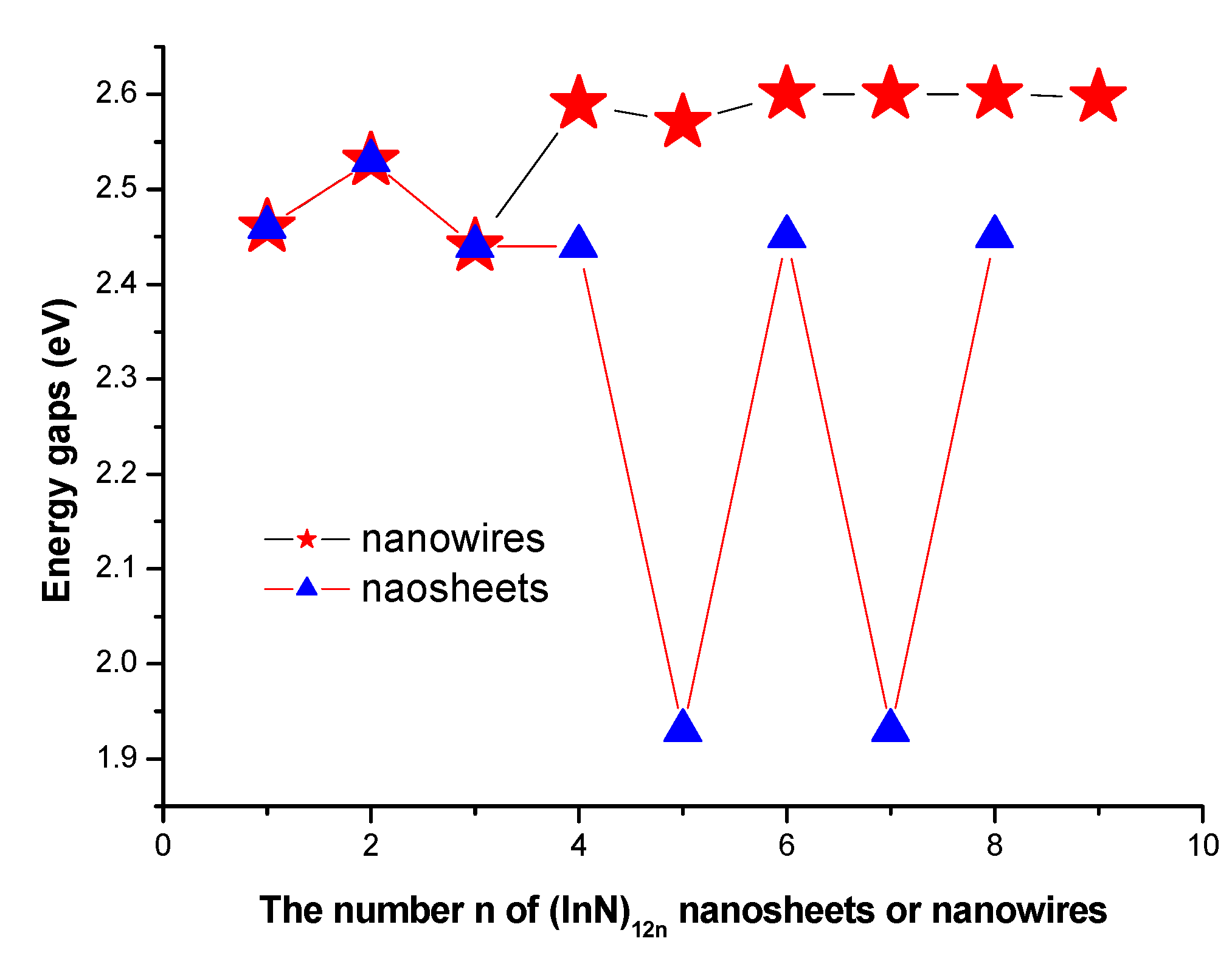
3.3. HOMO-LUMO Gaps
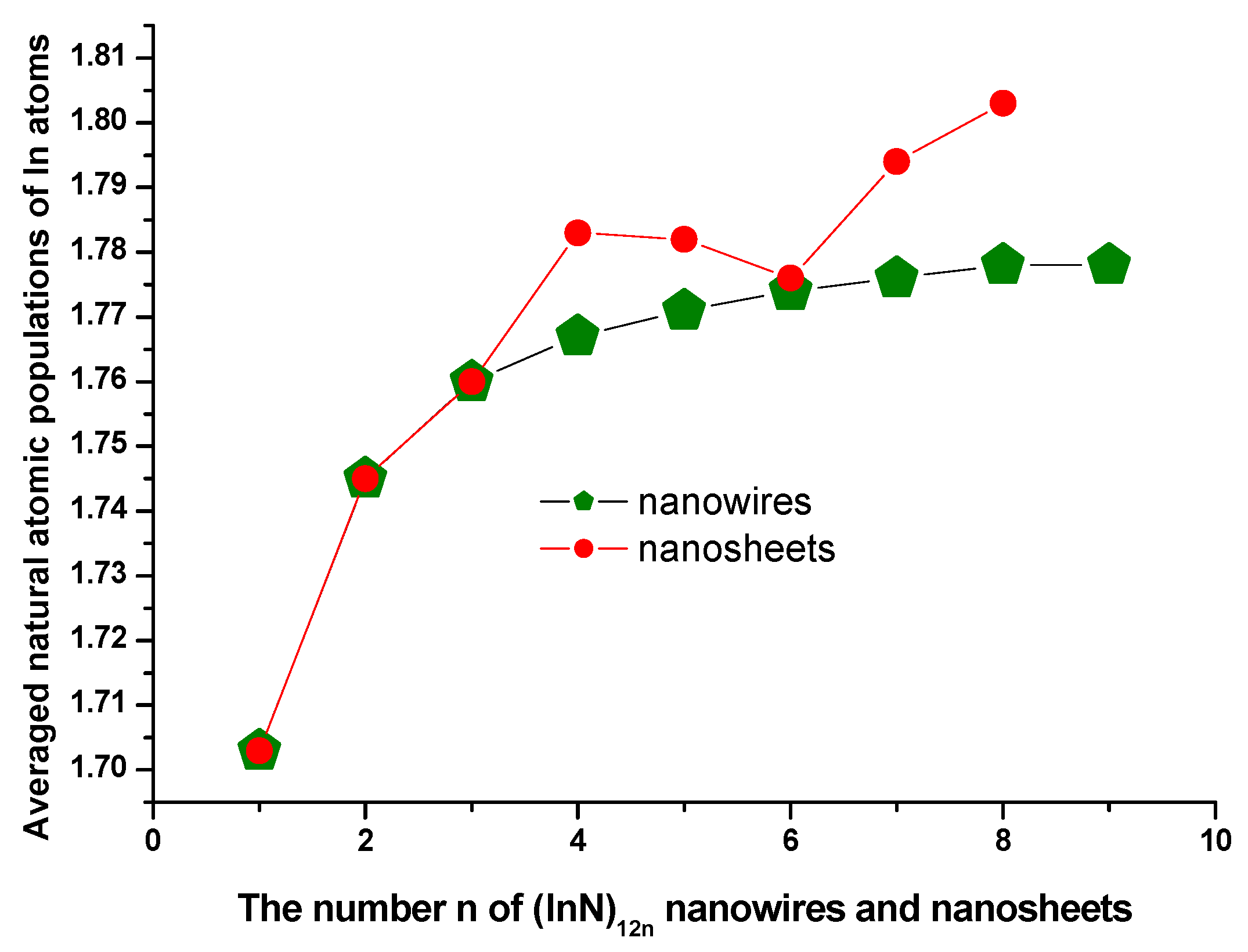
3.4. Natural Charge Population Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jimenez-Izal, E.; Ugalde, J.M.; Matxain, J.M. Nanocluster-Assembled Materials, Computational Modeling of Inorganic Nanomaterials; Taylor & Francis Group, LLC: New York, NY, USA, 2016. [Google Scholar]
- Gu, Y.B.; Xu, N.X.; Lin, M.H.; Tan, K. Structures, stabilities and properties of hollow (Al2O3)n clusters (n = 10, 12, 16, 18, 24 and 33): Studied with density functional theory. Comput. Theor. Chem. 2015, 1063, 29–34. [Google Scholar]
- Yeganeh, M.; Kafi, F.; Boochani, A. Thermoelectric properties of InN graphene-like nanosheet: A first principle study. Micro. Nanostruct. 2020, 138, 106367. [Google Scholar]
- Castillo, R.M.d.; Salcedo, E.; Martínez, A.; Ramos, E.; Sansores, L.E. Electronic Peculiarities of a Self-Assembled M12L24 Nanoball (M = Pd+2, Cr, or Mo). Molecules 2019, 24, 771. [Google Scholar] [CrossRef] [PubMed]
- Orek, C.; Keser, S.; Kaygili, O.; Zuchowski, P.; Bulut, N. Structures and optical properties of zinc oxide nanoclusters: A combined experimental and theoretical approach. J. Mol. Model. 2023, 29, 227. [Google Scholar]
- Duan, X.; Huang, Y.; Cui, Y.; Wang, J.; Lieber, C.M. Indium phosphide nanowires as building blocks for nanoscale electronic and optoelectronic devices. Nature 2001, 409, 66–69. [Google Scholar] [CrossRef]
- Zhao, R.N.; Chen, R.; Li, Q.; Duan, Y.H. Geometries, stabilities and electronic properties of photo sensitized (CdS)2n (n = 5–27) nanoclusters. J. Alloy Compd. 2018, 762, 754–762. [Google Scholar]
- Zhao, R.N.; Chen, R.; Han, J.G. Geometrical and electronic properties of the plane-like selfassembled structured (In12N12)n (n = 1–9) nanomaterials based on In12N12 cages connecting with four-membered rings. Micropor. Mesopor. Mat. 2021, 318, 111041. [Google Scholar]
- Zhao, R.N.; Han, J.G.; Duan, Y.H. A density functional prediction of geometries and stabilities and electronic properties of nanosize cagelike (InN)2n (n = 6–27, 45, 54) semiconducor material. Adv. Theor. Simul. 2018, 1, 1800001. [Google Scholar] [CrossRef]
- Inushima, T. Electronic structure of superconducting InN. Sci. Technol. Adv. Mater. 2006, 7, S112–S116. [Google Scholar]
- Zhang, Q.F.; Wang, L.S.; Zheng, H.F.; Su, A.M.; Liu, X.; Xie, X.J.; Chen, Y.Z.; Peng, D.L. Effect of in situ low-temperature annealing on anomalous Hall effect in Co nanocluster-assembled granular film. J. Alloy Compd. 2018, 748, 922–928. [Google Scholar]
- Zhao, M.; Xia, Y.; Liu, X.; Tan, Z.; Huang, B.; Song, C.; Mei, L. First-Principles Calculations of AlN Nanowires and Nanotubes: Atomic Structures, Energetics, and Surface State. J. Phys. Chem. B 2006, 110, 8764–8768. [Google Scholar] [CrossRef] [PubMed]
- Romero, C.P.; Volodin, A.; Vece, M.D.; Paddubrouskaya, H.; Wang, H.; Vantomme, A.; Haesendonck, C.V.; Lievens, P. Passivation of cobalt nanocluster assembled thin films with hydrogen. Thin Solid Film. 2012, 520, 5584–5588. [Google Scholar] [CrossRef]
- Farzan, M.; Elahi, S.M.; Salehi, H.; Abolhassani, M.R. An investigation of electronic and optical properties of InN nanosheet by first principle study. Opt. Commun. 2017, 395, 293. [Google Scholar] [CrossRef]
- Podlipskas, Z.; Jurkevicius, J.; Kadys, A.; Kolenda, M.; Kovalevskij, V.; Dobrovolskas, D.; Aleksiejunas, R.; Tamulaitis, G. Extreme radiation resistance in InN. J. Alloy Compd. 2019, 789, 48–55. [Google Scholar] [CrossRef]
- Nagai, K.; Akiyama, T.; Nakamura, K.; Ito, T. A Simple Approach to Growth Mode of InN and InGaN Thin Films on GaN(0001) Substrate. ECS Trans. 2020, 98, 155–164. [Google Scholar] [CrossRef]
- An, Y.; He, Y.; Wei, H.; Liu, S.; Li, M.; Song, Y.; Qiu, P.; Rehman, A.; Zheng, X.; Peng, M. Metallic indium segregation control of InN thin films grown on Si(100) by plasma-enhanced atomic layer deposition. Results Phys. 2019, 12, 804–809. [Google Scholar] [CrossRef]
- Ma, S.; He, C.; Sun, L.Z.; Lin, H.; Li, Y.; Zhang, K.W. Stability of two-dimensional PN monolayer sheets and their electronic properties. Phys. Chem. Chem. Phys. 2015, 17, 32009–32015. [Google Scholar] [CrossRef]
- Dong, L.H.; Liu, Y.; Zhuo, Y.J.; Chu, Y.; Wang, Y.P. Effect of alien cations on the growth mode and self-assemble fashion of ZnO nanostructures. J. Alloy Compd. 2010, 509, 2021–2030. [Google Scholar] [CrossRef]
- Wu, X.; Li, K.; Wang, H. Facile synthesis of ZnS nanostructured spheres and their photocatalytic properties. J. Alloy Compd. 2009, 487, 537–544. [Google Scholar] [CrossRef]
- Zhang, Z.R.; Chai, C.; Song, Y.; Kong, L.; Yang, Y. A DFT study on physical properties of III-V compounds (AlN, GaN, AlP, and GaP) in the P3(1)21 phase. Mater. Res. Express 2021, 8, 025908. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew—Burke—Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Lin, L.H.; Yang, J.C.; Ning, H.M.; Hao, D.S.; Fan, H.W. Silicon–sodium binary clusters SinNa (n < 10) and their anions: Structures, thermochemistry, and electron affinities. Theochem 2008, 851, 197–206. [Google Scholar]
- Foster, E.; Wong, B.M. Nonempirically Tuned Range-Separated DFT Accurately Predicts Both Fundamental and Excitation Gaps in DNA and RNA Nucleobases. J. Chem. Theory Comput. 2012, 8, 2682–2687. [Google Scholar] [CrossRef]
- Wong, B.M.; Hsieh, T.H. Optoelectronic and Excitonic Properties of Oligoacenes: Substantial Improvements from Range-Separated Time-Dependent Density Functional Theory. J. Chem. Theory Comput. 2010, 6, 3704–3712. [Google Scholar] [CrossRef]
- Chang, C.; Lin, Y.; Niu, J.; Lour, W.; Tsai, J.; Liu, W. High-Performance AlGaN/GaN Enhancement-Mode High Electron Mobility Transistor by Two-Step Gate Recess and Electroless-Plating Approaches. Sci. Adv. Mater. 2021, 13, 30–35. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Systems | Sym | Iso | NC | Dip | HOMO | LUMO | Freq | Egap | ET | Eb(n) | D(n, n−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Debye | Hartree | Hartree | cm−1 | eV | Hartree | eV | eV | ||||
| In12N12 | Th | a | 1.703 | 0.00 | −0.22082 | −0.13050 | 71.3 | 2.46 | −678.876339 | 4.048 | |
| In24N24 | C1 | a | 1.745 | 0.00 | −0.21941 | −0.12626 | 35.5 | 2.53 | −1358.084415 | 4.424 | 9.03 |
| In36N36 | S6 | a | 1.760 | 0.01 | −0.22161 | −0.13177 | 20.3 | 2.44 | −2037.291826 | 4.549 | 9.01 |
| C1 | b | 1.755 | 5.21 | −0.20426 | −0.13521 | 26.4 | −2037.234748 | ||||
| In48N48 | C1 | a | 1.783 | 0.01 | −0.21476 | −0.12515 | 27.8 | 2.44 | −2716.706404 | 4.729 | 14.645 |
| C1 | b | 1.764 | 5.61 | −0.20392 | −0.13396 | 17.3 | −2716.439412 | ||||
| In60N60 | C1 | a | 1.782 | 4.96 | −0.20447 | −0.13361 | 18.0 | 1.93 | −3395.863320 | 4.720 | 7.635 |
| C1 | b | 1.598 | 6.99 | −0.20888 | −0.13552 | 10.2 | −3395.596897 | ||||
| C1 | c | 1.767 | 0.01 | −0.21577 | −0.12998 | 10.9 | −3395.595081 | ||||
| C1 | d | 1.769 | 1.69 | −0.21508 | −0.12642 | 10.0 | −3395.652494 | ||||
| In72N72 | C1 | a | 1.776 | 0.01 | −0.21425 | −0.12415 | 18.4 | 2.45 | −4075.341547 | 4.835 | 16.377 |
| C1 | b | 1.776 | 5.02 | −0.23520 | −0.12284 | 13.0 | −4074.968946 | ||||
| C1 | c | 1.781 | 6.22 | −0.20437 | −0.13344 | 14.3 | −4075.029513 | ||||
| C1 | d | 1.783 | 4.05 | −0.20435 | −0.13351 | 12.9 | −4075.067931 | ||||
| In84N84 | C1 | a | 1.794 | 4.88 | −0.20397 | −0.13304 | 12.5 | 1.93 | −4754.498871 | 4.814 | 7.646 |
| C1 | b | 1.794 | 6.24 | −0.20149 | −0.13028 | 15.2 | −4754.495491 | ||||
| C1 | c | 1.681 | 4.54 | −0.20426 | −0.13145 | 13.2 | −4754.455641 | ||||
| C1 | d | 1.779 | 5.61 | −0.20761 | −0.12881 | 9.5 | −4754.198775 | ||||
| C1 | e | 1.776 | 7.18 | −0.20362 | −0.13557 | 9.9 | −4754.111104 | ||||
| In96N96 | C1 | a | 1.803 | 0.04 | −0.21351 | −0.12356 | 12.0 | 2.45 | −5433.976634 | 4.889 | 16.365 |
| C1 | b | 1.802 | 1.91 | −0.21697 | −0.12618 | 11.4 | −5433.936053 | ||||
| C1 | c | 1.791 | 0.20 | −0.21141 | −0.12803 | 6.8 | −5433.655668 | ||||
| C1 | d | 1.795 | 0.02 | −0.20682 | −0.12777 | 10.6 | −5433.515520 | ||||
| C1 | e | 1.793 | 1.71 | −0.21340 | −0.12581 | 9.3 | −5433.712151 |
| Systems | Sym | Iso | NC | Dip | HOMO | LUMO | Freq | Egap | ET | Eb(n) | D(n, n−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Debye | Hartree | Hartree | cm−1 | eV | Hartree | eV | eV | ||||
| In12N12 | Th | a | 1.703 | 0.00 | −0.22082 | −0.13050 | 71.3 | 2.46 | −678.876339 | 4.048 | |
| In24N24 | C1 | a | 1.745 | 0.00 | −0.21941 | −0.12626 | 35.5 | 2.53 | −1358.084415 | 4.424 | 9.03 |
| In36N36 | S6 | a | 1.760 | 0.01 | −0.22161 | −0.13177 | 20.3 | 2.44 | −2037.291826 | 4.549 | 9.01 |
| In48N48 | C1 | c | 1.767 | 0.00 | −0.21813 | −0.12329 | 12.4 | 2.59 | −2716.498627 | 4.611 | 9.01 |
| In60N60 | C1 | e | 1.771 | 0.04 | −0.21783 | −0.12264 | 8.5 | 2.57 | −3395.706352 | 4.649 | 9.02 |
| In72N72 | C1 | e | 1.774 | 0.01 | −0.21772 | −0.12218 | 5.2 | 2.60 | −4074.911066 | 4.673 | 8.94 |
| In84N84 | C1 | f | 1.776 | 0.02 | −0.21755 | −0.12193 | 3.9 | 2.60 | −4754.118833 | 4.691 | 9.02 |
| In96N96 | C1 | f | 1.778 | 0.01 | −0.21741 | −0.12181 | 2.7 | 2.60 | −5433.322507 | 4.703 | 8.91 |
| In108N108 | C1 | a | 1.778 | 0.01 | −0.21724 | −0.12178 | 1.8 | 2.597 | −6112.528888 | 4.714 | 8.98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, R.; Chen, R.; Zhao, H.; Lin, F.; Han, J.-G. Exploration on Electronic Properties of Self-Assembled Indium Nitrogen Nanosheets and Nanowires by a Density Functional Method. Molecules 2023, 28, 7358. https://doi.org/10.3390/molecules28217358
Zhao R, Chen R, Zhao H, Lin F, Han J-G. Exploration on Electronic Properties of Self-Assembled Indium Nitrogen Nanosheets and Nanowires by a Density Functional Method. Molecules. 2023; 28(21):7358. https://doi.org/10.3390/molecules28217358
Chicago/Turabian StyleZhao, Running, Rui Chen, Hua Zhao, Fan Lin, and Ju-Guang Han. 2023. "Exploration on Electronic Properties of Self-Assembled Indium Nitrogen Nanosheets and Nanowires by a Density Functional Method" Molecules 28, no. 21: 7358. https://doi.org/10.3390/molecules28217358
APA StyleZhao, R., Chen, R., Zhao, H., Lin, F., & Han, J.-G. (2023). Exploration on Electronic Properties of Self-Assembled Indium Nitrogen Nanosheets and Nanowires by a Density Functional Method. Molecules, 28(21), 7358. https://doi.org/10.3390/molecules28217358