Abstract
A simple and rapid access to fluorinated dithioesters was developed by a one-pot sequence corresponding to a Grignard reaction—Mitsunobu type substitution. These activated dithioesters have shown excellent reactivity in an aminolysis reaction from simple or more complex primary amines such as cinchona alkaloids. A stoichiometric amount of amine was sufficient to prepare various thioamides, including a 4-styrenylthioamide cinchonidine monomer, under environmentally friendly conditions, at room temperature, and in a very short time.
1. Introduction
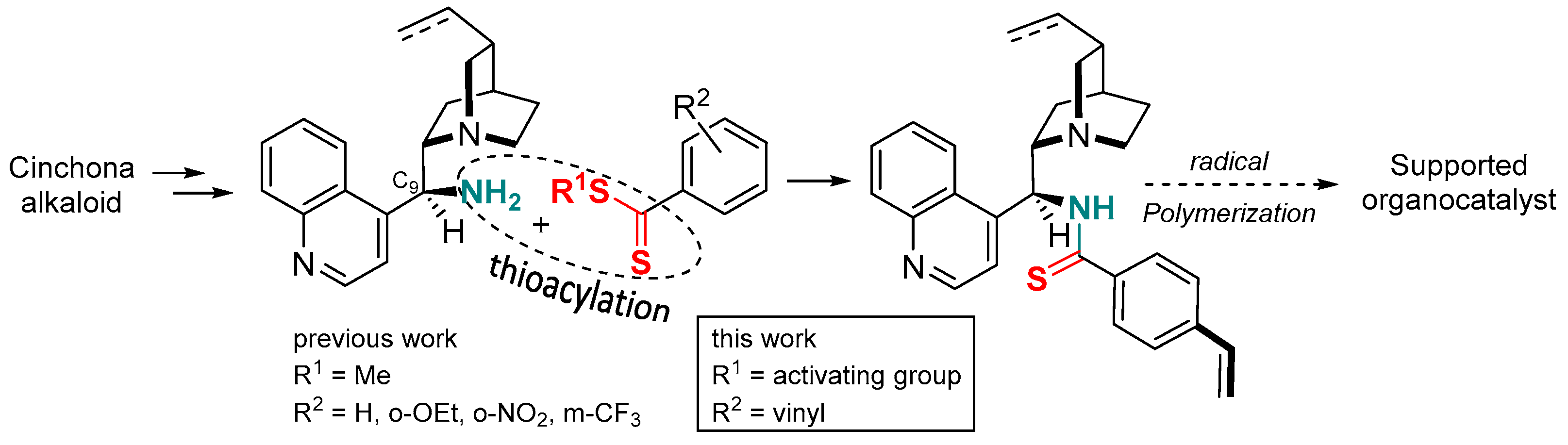
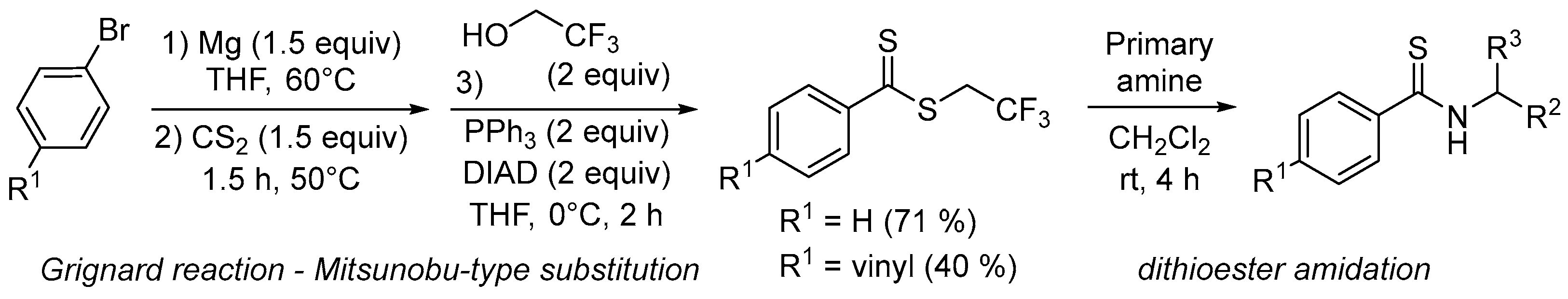
Dithiocarboxylic acids [1] or dithioesters [2] are common precursors of thioamides [3], a functional group that has found increasing uses over the last decade. In peptide chemistry, the thioamide function is a promising isostere of the amide group leading to interesting biological activities [4,5,6]. In organic chemistry, thioamide is particularly effective for the preparation of thiazoles or thiazolines and leads to higher yields compared to oxygen analogues [7,8,9]. In asymmetric synthesis, several groups including ours have described an improvement in the efficiency of thioamide organocatalysts in terms of yield and stereoselectivity compared to the amide analogues. For example, a thioamide-based catalyst has been successfully used in the addition of indoles to nitroalkenes [10], whereas proline-bearing thioamide functionality has led to a better result in various aldol reactions [11,12,13,14,15,16]. For the first time, we have used thioamide-substituted cinchona alkaloids for highly stereoselective decarboxylative Mannich or protonation reactions of malonic acid half-oxyesters affording α,β- and α-amino acid derivatives, respectively [17]. To prepare these organocatalysts, it should be noted that direct oxygen-sulfur exchange by the thionation of cinchona alkaloids bearing an amide function is ineffective and does not lead to the corresponding thioamides. For these specific scaffolds, the reaction of a methyldithioester with the 9-amino analogue of cinchona alkaloids led to thioamide-based organocatalysts in long reaction times (3–4 days) and moderate to good yields. In this context, we envisaged to synthesize (4-styrenyl)thioamide cinchona derivative which could be further polymerized to give a recyclable macromolecular catalyst [18]. Due to the sensitivity of the 4-styrenyl thioamide moiety and to ensure the larger-scale synthesis necessary for polymerization, we needed to optimize the synthesis of the dithioester precursor (Figure 1).
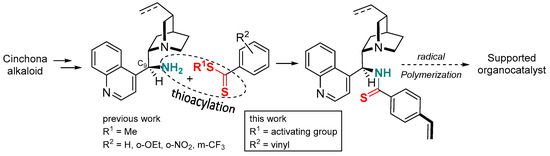
Figure 1.
Synthesis and evaluation of dithioesters to access an organocatalyst based on a 4-styrenyl thioamide alkaloid monomer.
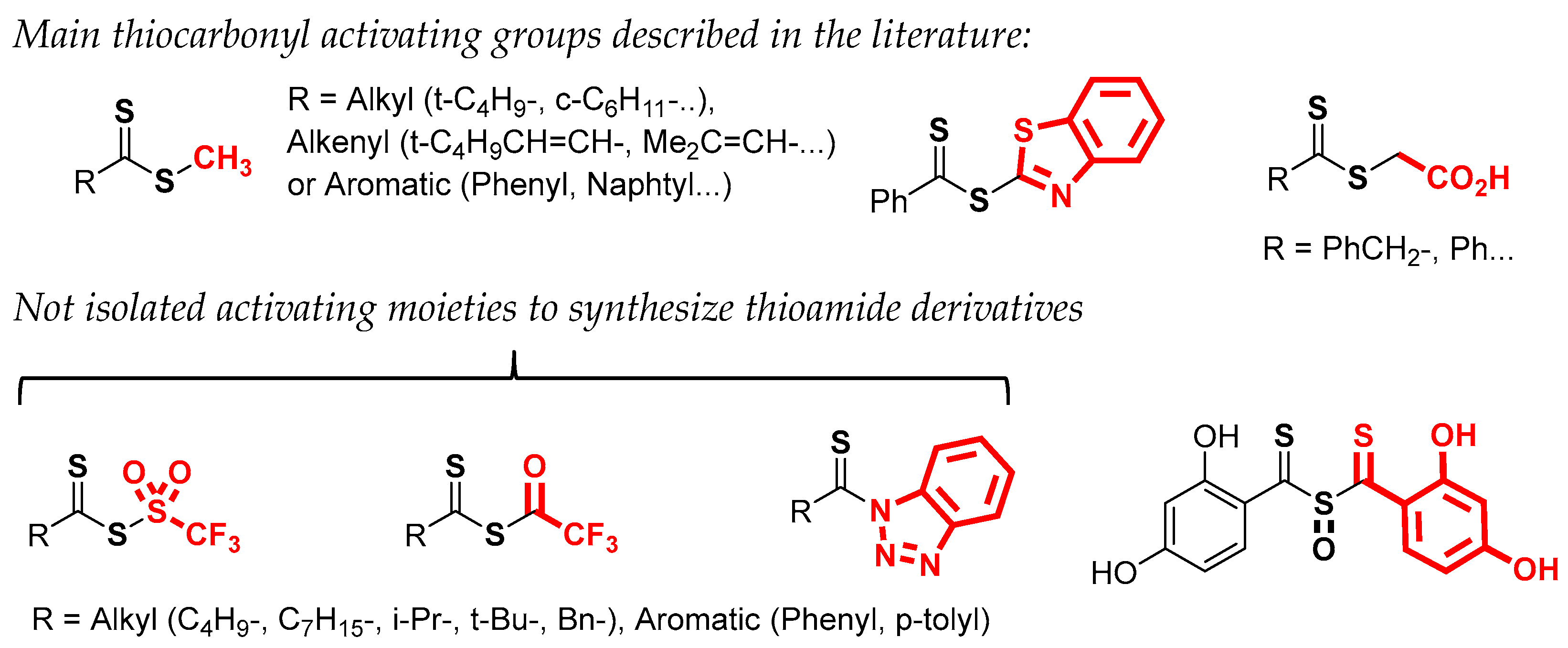
Among various synthetic routes, the main method described to synthesize a dithioester uses a Grignard reagent which reacts with carbon disulfide to give a dithiocarboxylic acid as a key precursor [19,20,21]. After acid treatment, the recovery of the desired product will depend on the stability of the dithiocarboxylic acid. Very recently, Mitzel et al. reported the behavior of certain aryl compounds carrying this functionality [22]. These reactive species often generate different types of oligomers or oxidative decomposition products. In the case of aryl dithiocarboxylic acids, the ortho substitution facilitates its isolation in pure form due to the steric hindrance which limits the formation of oligomers. Lampkins et al. synthesized various 4-alkoxy-dithionaphthoic acids in good yield and confirmed their stability [23]. The same authors used these dithiocarboxylic acids to prepare methyl dithioesters as precursors of thioamides. Nevertheless, the aminolysis with primary or secondary amines in the presence of DMAP was not possible for these compounds. To improve the reactivity of such aliphatic dithioesters (methyl or ethyl), various activating moieties have been tested (Figure 2) [24,25,26,27,28], mention should be made of benzothiazolyl [24], sulfinyl bisthiobenzoyl [25,26], or thioglycolic acid [7,8,27]. Among these, S-thiobenzoylglycolic acid represents an interesting choice. The corresponding dithioester can react with a primary amine in the presence of pyridine and triethylamine to generate the desired thioamide.
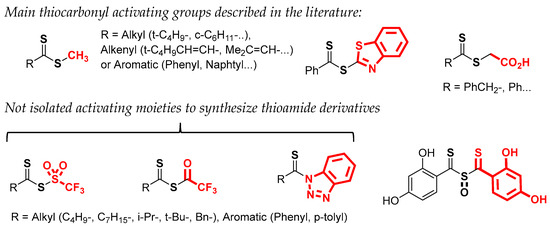
Figure 2.
Activated dithioesters and derivatives.
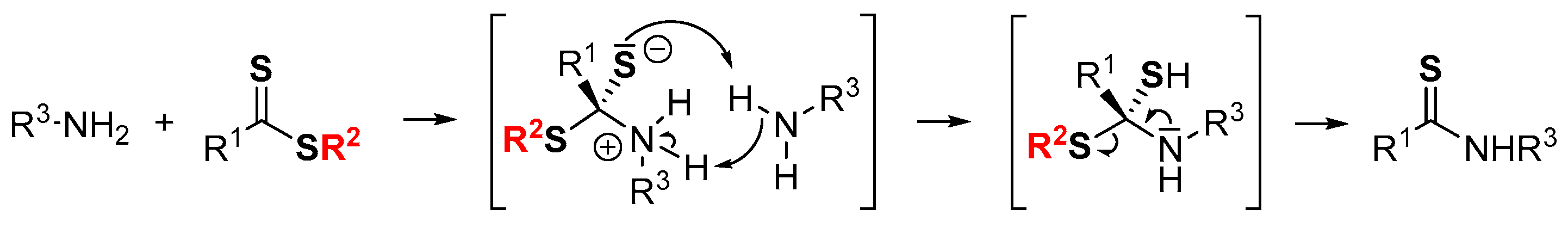
A strong point of these activating reagents is their electron-withdrawing character. Indeed, the proposed mechanism involves first the reversible nucleophilic addition of the primary amine to give a zwitterionic intermediate (Scheme 1). Depending on the nature of the R2 group, this zwitterionic intermediate is able to block or accelerate the thioamide formation process. The addition of a tertiary amine has no real influence on the reaction rate, unlike the amount of primary amine. Thus, an excess of primary amine is required to protonate the thiolate and deprotonate the ammonium moiety simultaneously, according to Scheme 1 [29,30,31].
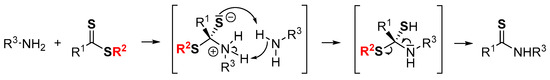
Scheme 1.
Proposed mechanism for the formation of thioamides from dithioesters and amines.
Therefore, according to this mechanism, amine hindrance plays a crucial role in thioamide formation. Thus, as part of our research in organocatalysis, this work aims to study the reactivity of newly activated aryldithioesters and styrenyldithioesters with more complex chiral primary amines such as cinchona alkaloids. In these cases, their original structure and the presence of a basic site (quinuclidine) accentuate the difficulty of developing efficient and rapid access to functionalized and supported organocatalysts.
2. Results
2.1. Synthesis of Dithioesters and Thionoesters
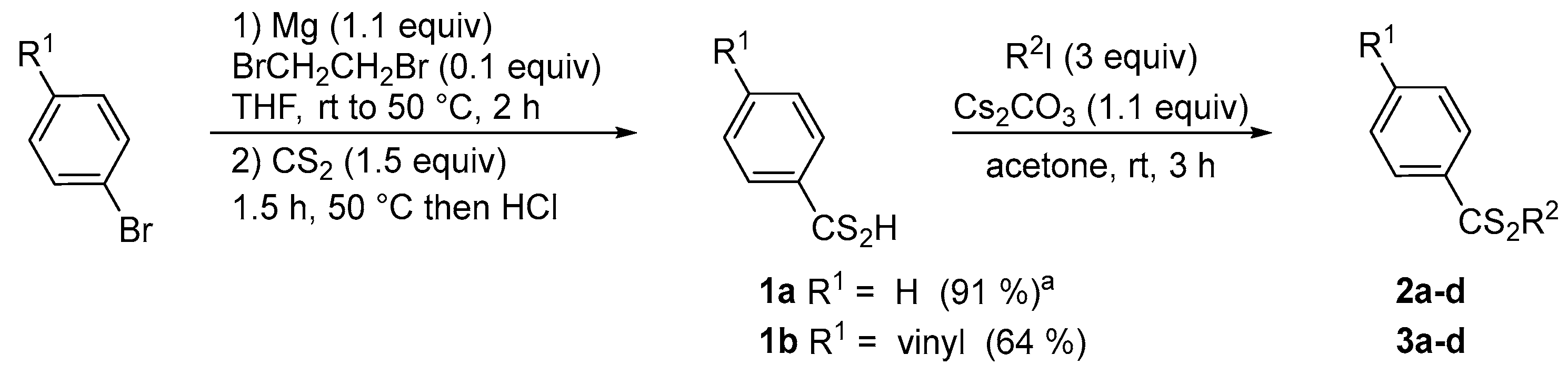
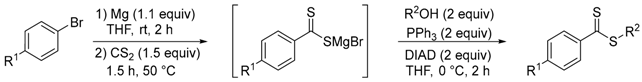
Firstly, the synthesis of dithioesters was studied using two different strategies. Under standard conditions, Grignard reagents were directly used from commercially available (R = H) or prepared from an aryl bromide and generated in situ (R = vinyl). Subsequently, the organomagnesium compound reacted with carbon disulfide (1.5 equiv) to form magnesium dithiocarboxylate. After acid treatment, the corresponding dithiocarboxylic acids 1a and 1b were isolated in 91% and 64% yield, respectively (Scheme 2). By the presence of a suitable base, these dithioacids reacted with an excess of iodomethane to generate the dithioesters 2a–3a. Several bases were tested such as K2CO3, Na2CO3, Cs2CO3, and Et3N. After optimization, the best yield was obtained using a stoichiometric quantity of cesium carbonate and iodomethane (3 equiv), at room temperature in acetone (or butanone). Under similar conditions, different alkylating agents (R2I) were used with dithioacids 1a–b, which made it possible to synthesize several dithioesters 2b–2d and 3b–3d to compare the effectiveness of this sequence (Table 1).
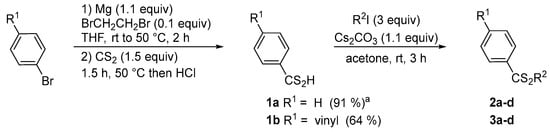
Scheme 2.
Synthesis of dithioesters 2a–d and 2b–d by Grignard reaction. a From PhMgBr.

Table 1.
Substrate scopes of dithioesters a.
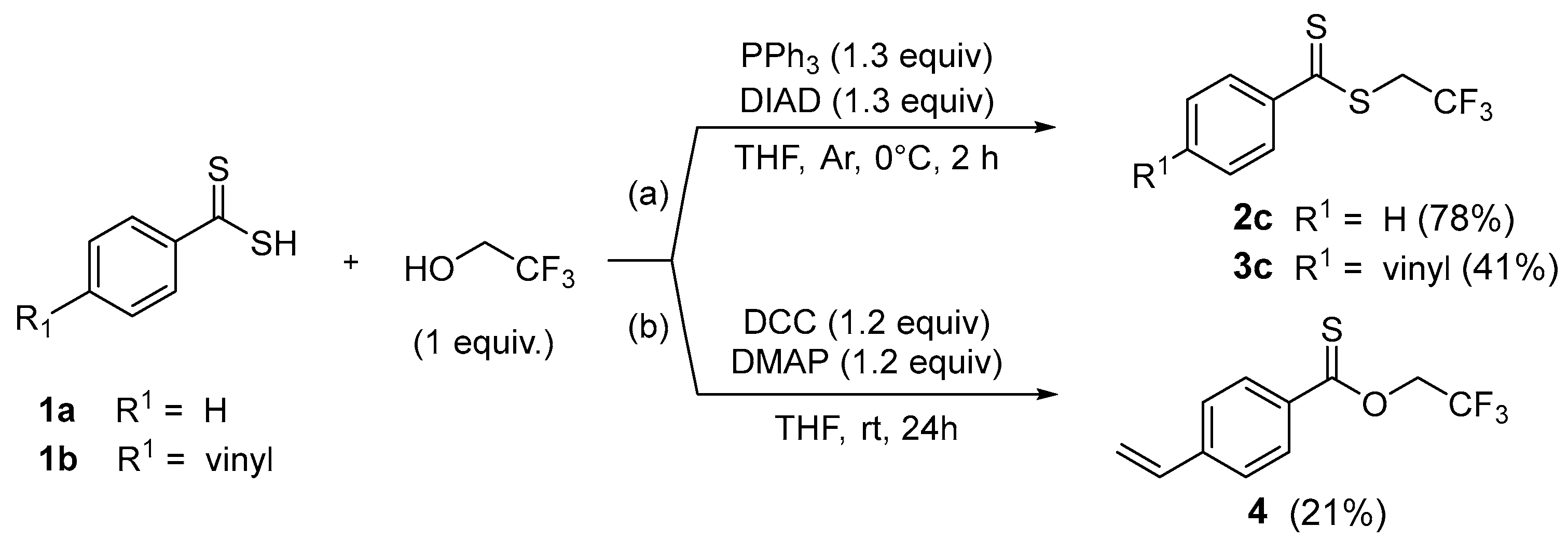
From dithiocarboxylic acid 1a, yields close to 70–80% were obtained with aliphatic (methyl or ethyl) and perfluorinated agents. Comparatively, the presence of a vinyl unit, which is more difficult to handle, causes a logical decrease in yield after acid treatment. After purification, the dithioesters 3a–d were isolated in moderate yields from 21% to 53%. Consequently to the low yield obtained for dithioester 3c, we evaluated the standard Mitsunobu reaction conditions to alkylate dithiocarboxylic acids 1a–b with trifluoroethanol (Scheme 3(a)) [32].
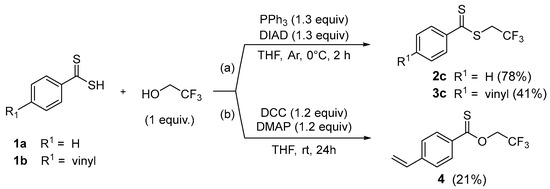
Scheme 3.
Preparation of dithioesters 2c–3c or thionoester 4 by standard Mitsunobu or Steglich reaction from dithiocarboxylic acids 1a–1b.
Thus, we used a stoichiometric amount of a dithiocarboxylic acid and 2,2,2-trifluoroethanol in the presence of a slight excess of PPh3 and DIAD in THF at 0 °C. After 2 h, we observed a good yield of 78% in dithioester 2c which is slightly higher than the alkylation reaction (72%). With the more sensitive vinylic unit, a significant proportion of dithioester 3c was isolated to reach 41% yield (instead of 21% for the alkylation reaction) confirming the potential of this procedure. In parallel, we prepared thionoester 4 as a potential precursor of thioamides using DCC (1.2 equiv) and DMAP (1.2 equiv) in THF at room temperature (Scheme 3(b)). After 24 h, dithiocarboxylic acid 1b afforded desired compound 4 in 21% yield.
Dithiocarboxylic acids are particularly delicate to store and handle with the rapid appearance of by-products consistent with the literature [22]. Thus, we evaluated a one-pot reaction starting from 4-vinylbenzene bromide, an easy-to-store commercial reagent. The use of an organometallic reagent as a nucleophile in a Mitsunobu-type substitution is a poorly described method. A few examples have described the use of a zinc [33,34] or lithium [35] salt but the reactivity of an organomagnesium reagent remains unknown (Table 2).
Table 2.
Mitsunobu-type substitution from organomagnesium reagents.
In this sequence, we exploited the previously optimized reaction to generate the Grignard reagent which was engaged directly with the 2,2,2-trifluoroethanol without acid treatment. With two equivalents of PPh3 and DIAD, the magnesium salts (R = H or vinyl) gave dithioesters 2c and 3c in 71% and 40%, respectively. Ethyl and benzyl alcohols were tested in this sequence with similar efficiency affording the corresponding dithioester 2b and 2e in good yield. Compared to isolated dithiocarboxylic acids which required acid treatment and tedious extraction, inefficient purification to be avoided by column chromatography, and two working days to generate the corresponding dithioester, we observed similar yields in less than one day for these two steps of the one-pot reaction. Having in hand an efficient and easy procedure to access dithioesters 2a–d and 3a–d and thionoester 4, we started the aminolysis of these model substrates.
2.2. Aminolysis of Dithioesters and Thionoesters
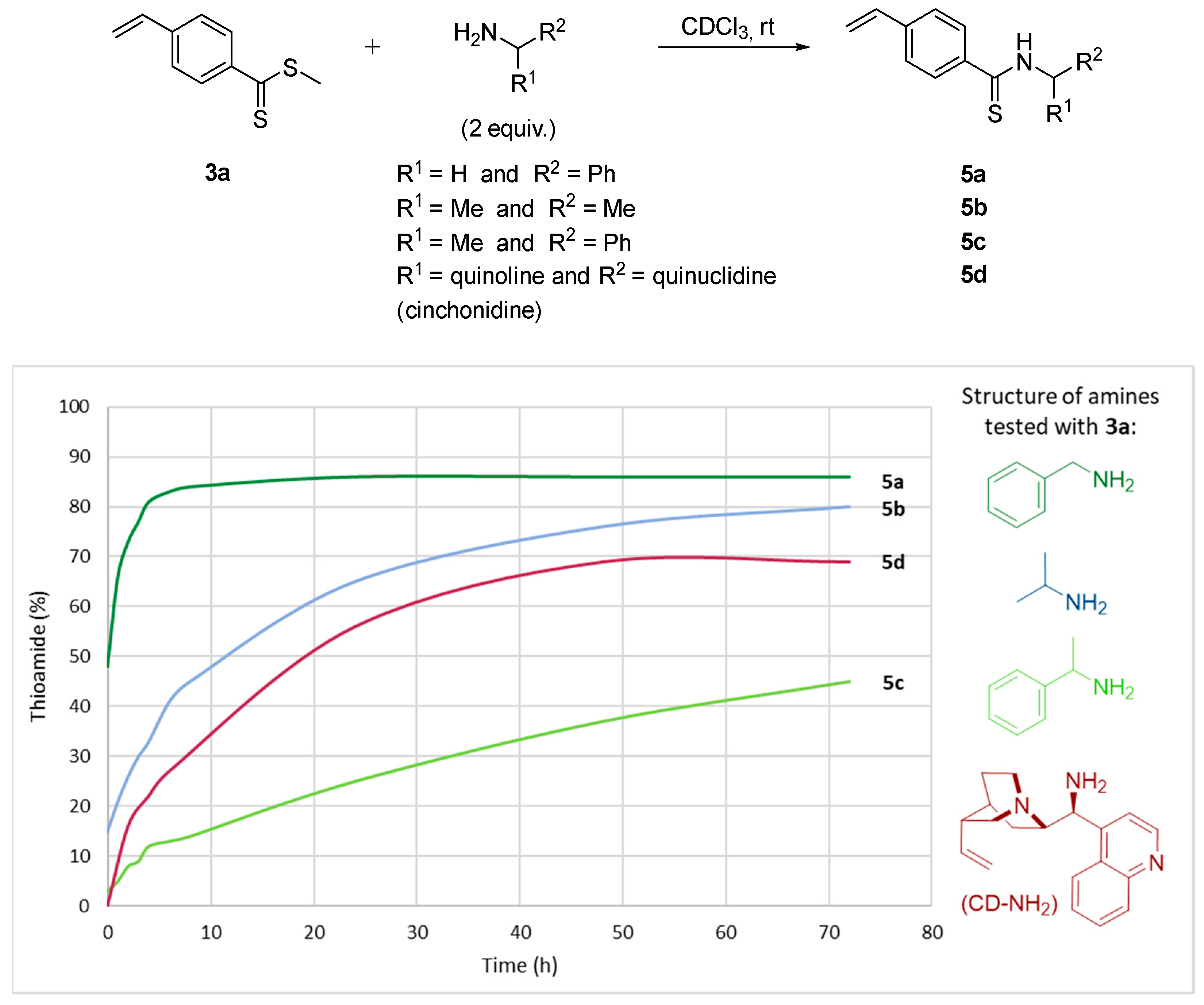
As a reference reaction, the reactivity of the methylated dithioester 3a was first evaluated through various nucleophiles carrying a primary amine, the 1H NMR monitoring is presented in Figure 3. First, benzylamine was compared to α-branched primary amines such as 2-propylamine and two chiral amines, α-methylbenzylamine and 9-aminocinchonidine. In the presence of an excess of benzylamine (2 equiv), a very rapid conversion to thioamide 5a was observed, reaching 80% after 4 h of reaction. On the contrary, isopropylamine reacts more slowly to give 60% of thioamide 5b after 20 h and 80% after 72 h of reaction. The use of chiral α-methylbenzylamine confirmed this trend with a clear loss of reactivity to generate only 45% of thioamide 5c after 72 h. Thus, Figure 3 reveals poor reactivity of amines having an alpha substitution of nitrogen. Nevertheless, the primary amine derivative of cinchonidine showed very similar kinetics to isopropylamine with 50% of thioamide 5d after 20 h to reach a maximum of 70% after 50 h. This difference may be related to the presence of the quinuclidine moiety carrying a basic tertiary amine nearby.
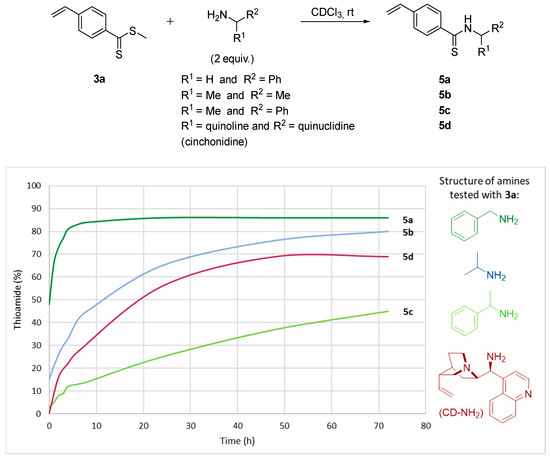
Figure 3.
1H NMR monitoring reaction progress between a primary amine on the dithioester 3a in chloroform-d.
The reaction conditions were then optimized using one equivalent of α-methylbenzylamine up to 4 equivalents relative to the dithioester 3a. With a stoichiometric ratio, the reactivity decreases significantly compared to the same reaction using two equivalents of amine. After 50 h, a low conversion of 30% was observed which is consistent with the mechanism proposed in Scheme 1. Beyond the use of two equivalents, the reaction rate increases only slightly, the addition of a large excess of amine does not represent any real synthetic interest. Finally, the influence of concentration and temperature was also studied. Thus, the most effective concentration of dithioester 3a was established at 0.45 mol/L in a concentration range between 0.9 mol/L and 0.09 mol/L, while non-isolated by-products were observed when the temperature was increased to 60 °C.
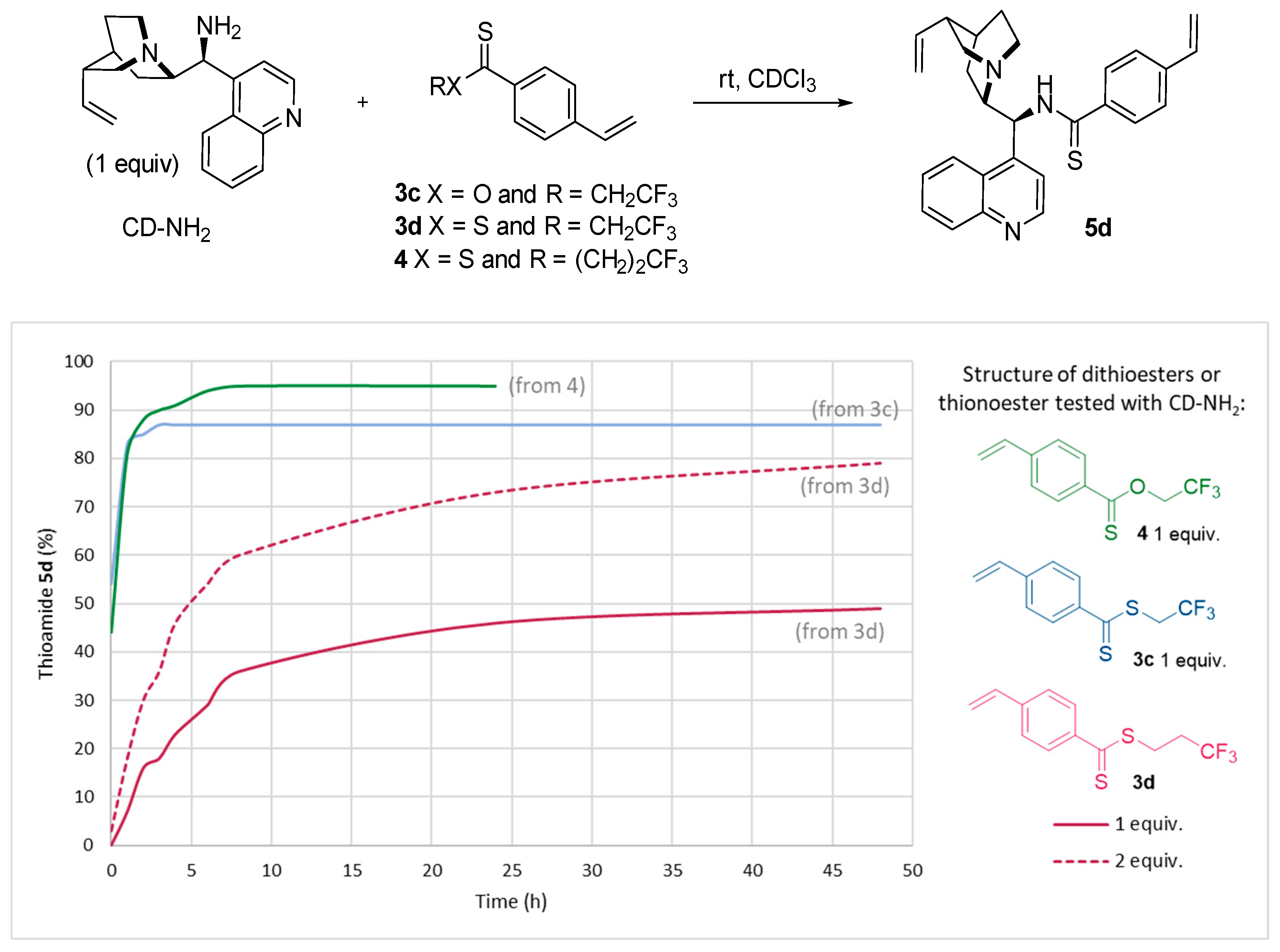
Under optimized conditions, dithioesters 3c–d and thionoester 4 were reacted with the amine derivative of cinchonidine (Figure 4). Here, the goal was to establish the most effective (and easily accessible) reagent. In this context, we also wanted to develop a simple method consuming a smaller quantity of nucleophiles. To this end, we evaluated the use of one equivalent of cinchonidine at room temperature.
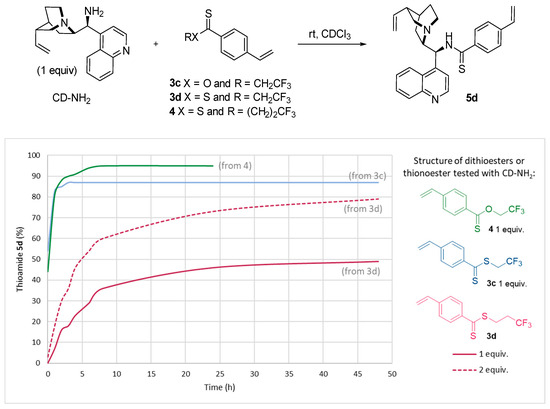
Figure 4.
1H NMR monitoring reaction progress between CD-NH2 and dithioesters 3c–d or 4.
Compared to a simple methyl, the electronic effects caused by a terminal CF3 unit is clearly positive in the addition of cinchonidine (Figure 4). A stoichiometric amount of the dithioester 3c led to an excellent yield of 83% after only one hour of reaction at room temperature, probably due to the higher electrophilicity of the thiocarbonyl and the proximity of electronegative fluorine atoms. A significant difference was observed with dithioester 3d which presents a longer flexible chain between the thiocarbonyl group and the fluorine atoms to lead to only 7% conversion after 1 h. Under these conditions, a maximum of 50% was detected by 1H NMR after 2 days of reaction. To confirm this result, excess of the dithioester 3d (2 equiv) was added to the cinchondine-NH2 to provide 50% of thioamide after 5 h at room temperature. After 2 days, a conversion of 80% was observed. Interestingly, thionoester 4 incorporating a trifluoroethyl group showed a very similar reactivity to the dithioester 3c, a very rapid substitution of 83% was calculated after 1 h of reaction at room temperature reaching 95% after 8 h.
3. Discussion
To establish the potential of the dithioester 3c in comparison to previous studies, we evaluated a commercial dithioester regularly cited in the literature for the synthesis of thioamides, namely S-thiobenzoylglycolic acid [27]. This reagent was reacted with α-methylbenzylamine or 9-aminocinchonidine in chloroform or pyridine (Scheme 4).
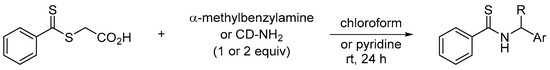
Scheme 4.
Reactivity of S-thiobenzoylglycolic acid towards a chiral primary amine.
In pyridine, a low conversion of 49% was calculated for α-methylbenzylamine after 5 h at room temperature to reach 72% after 24 h. In chloroform, the addition of this amine caused a precipitate very quickly. In this case, the addition of 2 equivalents of α-methylbenzylamine was necessary to observe a conversion of 46% after 4 h and 85% after 20 h (the concentration of the medium is very important to obtain a good conversion). From the same stoichiometry, thioamide 5c was isolated with a yield of 84% after 3 days of reaction with dithioester 3a while a yield of 78% and 69% was observed for 5a and 5b, respectively (see experimental section).
In contrast, cinchonidine-NH2 (1 equiv) did not react with S-thiobenzoylglycolic acid in chloroform or pyridine at room temperature. A very low conversion of 4% to thioamide was observed after 24 h of reaction. The low reactivity of this chiral primary amine with S-thiobenzoylglycolic acid confirmed the potential of the trifluorinated dithioester 3c evaluated in this study. Indeed, from a one-pot sequence, halogenated aromatic compounds provided fluorinated dithioesters in a few hours and their reactions with a (chiral) primary amine efficiently led to a thioamide (Scheme 5). Thus, the reaction sequence developed herein is well-suited particularly when direct thionation methods of amides failed.

Scheme 5.
One-pot sequence optimized in this study.
Fluorinated dithioesters showed excellent reactivity in an aminolysis reaction with a stoichiometric amount of 9-amino-9-deoxy-cinchonidine. Precisely, two equivalents of methylated dithioester 3a are necessary to prepare thioamide 5d with a maximum yield of 70% after 48 h while one equivalent of fluorinated dithioester 3c provides 90% in approximately 2 h. From these results, the overall sequence was used to synthesize 4-styrenylthioamide cinchonidine as a reactive monomer necessary for our ongoing project on polymer-supported organocatalysis.
4. Materials and Methods
4.1. General Information
All reagents were purchased from Acros Organics, Sigma Aldrich, Alfa Aesar, TCI, or Fluka and were used without further purification and used as received. Solvents were used in RPE grade without further purification. Anhydrous solvents were obtained from a PURESOLV SPS400 apparatus developed by Innovative Technology Inc. (Oldham, UK). 1H and 13C NMR spectra were recorded on a Bruker Avance III 500 MHz. Samples were dissolved in an appropriate deuterated solvent (CDCl3). The chemical shifts (δ) are expressed in ppm and coupling constants are indicated in Hz. Abbreviations for signal coupling are as follows: s = singlet; d = doublet; t = triplet; q = quartet; m = multiplet; br = broad signal). To assign the signals to the different proton and carbon atoms, additional 2D NMR experiments (COSY, HSQC, HMBC) and NOESY experiments were performed. 1,3,5-trimethoxybenzene was used as an internal standard to determine 1H NMR yield. High-resolution mass spectra (HRMS) were performed on Acquity UPLC HClass Xevo G2-XS QTof (WATERS) by electrospray ionization (ESI). Infrared (IR) spectra were recorded with a Perkin Elmer 16 PC FTIR ATR spectrometer, using the pure product (oil or solid). Thin Layer Chromatography (TLC) was run on pre-coated aluminum plates of silica gel 60 F-254 (Merck). Flash chromatography was performed on a silica gel column (Merck silica gel, 40–63 µm) using air pressure.
4.2. Experimental
4.2.1. General Procedure for the Preparation of Dithioesters 2a–d by Alkylation of Dithiocarboxylic Acid 1a
To an oven-dried flask purged with argon was added a solution of phenylmagnesium bromide 1M in THF (8.27 mL, 8.27 mmol) and diluted with dry THF (80 mL). Carbon disulfide (0.75 mL, 12.41 mmol) was slowly added at room temperature and the reaction mixture was stirred at 50 °C for 1.5 h. The resulting mixture was allowed to cool down to room temperature and 10 mL of water was added to quench the reaction. The mixture was evaporated under reduced pressure to remove the THF. The aqueous solution was diluted with water (50 mL) and washed with DCM twice, then acidified with 1M HCl. Once acidified, a pink-colored precipitate appeared. The mixture was extracted three times with DCM and the solvent was evaporated under reduced pressure. Freshly prepared dithiocarboxylic acid (1.16 g, 7.52 mmol) was dissolved in acetone (26 mL). CsCO3 (2.70 g, 8.27 mmol), the corresponding alkyl halide (3 equiv.) was then added and the mixture was stirred at room temperature overnight. The resulting dithioester was purified by column chromatography using pentane as solvent.
4.2.2. Characterization of Dithioesters
Methyl dithiobenzoate (2a) (CAS: 2168-78-7). Dithioester 2a was obtained as a red liquid. Yield: 62%. 1H NMR (500 MHz, CDCl3): δ = 8.03–8.02 (m, 2H), 7.54 (tt, J = 7.4, 1.2 Hz, 1H), 7.42–7.38 (m, 2H), 2.79 (3H, s). 13C NMR (125 MHz, CDCl3): δ = 228.6, 144.7, 132.1, 128.1, 126.6, 20.5. HMRS (ASAP+) C8H9S2 [(M+H)+]: calculated: 169.0146, found: 169.0145. IR: 1229, 686 cm−1.
Ethyl dithiobenzoate (2b) (CAS: 936-63-0). Dithioester 2b was obtained as a red liquid. Yield: 72%. 1H NMR (500 MHz, CDCl3): δ = 8.01–7.99 (m, 2H), 7.51 (tt, J = 7.4, 1.2 Hz, 1H), 7.40–7.37 (m, 2H), 3.37 (q, J = 7.6 Hz, 2H), 1.42 (t, J = 7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ = 228.7, 145.3, 132.3, 128.4, 126.9, 31.6, 12.4. HMRS (ASAP+) C9H11S2 [(M+H)+]: calculated: 183.0302, found: 183.0294. IR: 1205, 688 cm−1.
2,2,2-trifluoroethyl dithiobenzoate (2c). Dithioester 2c was obtained as a red liquid. Yield: 72%. 1H NMR (500 MHz, CDCl3): δ = 8.04–8.02 (m, 2H), 7.59 (tt, J = 7.4, 1.2 Hz, 1H), 7.45–7.41 (m, 2H), 4.26 (q, J = 9.9 Hz, 2H). 13C NMR (125 MHz, CDCl3): δ = 223.6, 144.3, 133.2, 128.6, 127.3, 124.8 (q, J = 276.8 Hz), 38.0 (q, J = 38.0 Hz). 19F NMR (471 MHz, CDCl3): δ = −64.42 (t, J = 9.9 Hz). HMRS (ASAP+) C9H8F3S2 [(M+H)+]: calculated: 237.0020, found: 237.0019. IR: 1305, 1269, 1235, 1210, 1129, 1048, 646 cm−1.
2,2,2-trifluoropropyl dithiobenzoate (2d). Dithioester 2d was obtained as a red liquid. Yield: 78%. 1H NMR (500 MHz, CDCl3): δ = 8.04–8.02 (m, 2H), 7.57 (tt, J = 7.3, 1.3 Hz, 1H), 7.43–7.40 (m, 2H), 3.60–3.57 (m, 2H), 2.62–2.53 (m, 2H). 13C NMR (125 MHz, CDCl3): δ = 226.8, 144.75, 132.9, 128.6, 127.0, 126.1 (q, J = 277.3 Hz), 32.5 (q, J = 29.3 Hz), 28.7 (q, J = 3.2 Hz). 19F NMR (471 MHz, CDCl3): δ = −66.15 (t, J = 9.9 Hz). HMRS (ASAP+) C10H10F3S2 [(M+H)+]: calculated: 251.0176, found: 251.0176. IR: 1241, 1206, 1092, 687 cm−1.
Benzyl dithiobenzoate (2e). Dithioester 2e was obtained as a red liquid. Yield: 59%. 1H NMR (600 MHz, CDCl3): δ = 8.02 (d, J = 8.4; 1.1 Hz, 2H), 7.56 (tt, J = 7.4, 1.1 Hz, 1H), 7.41–7.38 (m, 4H), 7.37–7.34 (m, 2H), 7.31 (tt, J = 7.2, 1.1 Hz, 1H), 4.65 (s, 2H). 13C NMR (150 MHz, CDCl3): δ = 227.8, 144.8, 135.1, 132.5, 129.4, 128.8, 128.5, 127.9, 127.0, 42.4. HMRS (ASAP+) C14H13S2 [(M+H)+]: calculated: 245.0459, found: 245.0451. IR: 1225, 685 cm−1.
4.2.3. General Procedure for the Preparation of Dithioesters 3a, b, d by Alkylation of Dithiocarboxylic Acid 1b
To an oven-dried flask purged with argon, magnesium turnings were added (291 mg, 12 mmol) with 2 mL of dry THF. Then, 1,2-dibromoethane (205 mg, 1.09 mmol) was added. A solution of 4-bromostyrene (2 g, 10.9 mmol) in dry THF (5 mL) was added dropwise at room temperature. The reaction mixture was then diluted with 16 mL of dry THF and stirred at 60 °C for 2 h. The reaction mixture was allowed to cool to room temperature and carbon disulfide (0.98 mL, 16.35 mmol) was slowly added. The reaction mixture was stirred at 50 °C for 1.5 h. The resulting mixture was allowed to cool to room temperature and 10 mL of water was added to quench the reaction. The mixture was evaporated under reduced pressure to remove the THF. The aqueous solution was diluted with water (50 mL) and washed with DCM twice, then acidified with 1M HCl. After the acidification of the mixture, a pink-colored precipitate appeared. The mixture was extracted three times with DCM and the solvent was evaporated under reduced pressure. Freshly prepared dithiocarboxylic acid (1.26 g, 6.98 mmol) was dissolved in acetone (24 mL). CsCO3 (2.50 g, 7.68 mmol), hydroquinone (77.08 mg, 0.70 mmol), and the corresponding alkyl halide (3 equiv.) were then added and the mixture was stirred at room temperature overnight. The resulting dithioester was purified by column chromatography using pentane as solvent.
4.2.4. Characterization of 4-Vinyldithioesters
Methyl 4-vinyldithiobenzoate (3a). Dithioester 3a was obtained as a red solid. Yield: 35%. Mp: 25 °C. 1H NMR (500 MHz, CDCl3): δ = 8.04 (d, J = 8.5 Hz, 2H), 7.41 (d, J = 8.0 Hz, 2H), 6.74 (dd, J = 17.6, 11.0 Hz, 1H), 5.88 (d, J = 17.6 Hz, 2H), 5.40 (d, J = 11.0 Hz, 2H), 2.78 (s, 3H). 13C NMR (125 MHz, CDCl3): δ = 228.1, 144.1, 141.6, 136.0, 127.3, 126.2, 116.4, 20.7. HMRS (ASAP+) C10H11S2 [(M+H)+]: calculated: 195.0302, found: 195.0299. IR: 1237, 637cm−1.
Ethyl 4-vinyldithiobenzoate (3b). Dithioester 3b was obtained as a red liquid. Yield: 49%. 1H NMR (500 MHz, CDCl3): δ = 8.01 (d, J = 8.4 Hz, 2H), 7.40 (d, J = 8.4 Hz, 2H), 6.73 (dd, J = 17.6, 10.9 Hz, 1H), 5.87 (d, J = 17.6 Hz, 1H), 5.39 (d, J = 10.9 Hz, 1H), 3.37 (q, J = 7.5 Hz, 2H), 1.42 (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ = 227.3, 144.3, 141.6, 136.1, 127.3, 126.1, 116.3, 31.5, 12.4. HMRS (ASAP+) C11H13S2 [(M+H)+]: calculated: 209.0459, found: 209.0458. IR: 2953, 2922, 2852, 1212 cm−1.
2,2,2-trifluoropropyl 4-vinyldithiobenzoate (3d). Dithioester 3d was obtained as a red solid. Yield: 53%. Mp: 50 °C. 1H NMR (500 MHz, CDCl3): δ = 8.01 (d, J = 8.4 Hz, 2H), 7.42 (d, J = 8.4 Hz, 2H), 6.74 (dd, J = 17.6, 10.9 Hz, 1H), 5.88 (d, J = 17.6 Hz, 1H), 5.41 (d, J = 10.9 Hz, 1H), 3.59–3.56 (dt, J = 7.6, 1.2 Hz, 2H), 2.65–2.52 (m, 2H). 13C NMR (125 MHz, CDCl3): δ = 225.4, 143.7, 142.1, 135.9, 127.4, 126.3, 126.1 (q, J = 277.4 Hz) 116.8, 32.5 (q, J = 28.9 Hz) 28.5 (q, J = 3.2 Hz). 19F NMR (471 MHz, CDCl3): δ = −66.15 (t, J = 10.4 Hz). HMRS (ASAP+) C12H12F3S2 [(M+H)+]: calculated: 277.0333, found: 277.0339. IR: 2921, 1235, 1217, 1134, 639 cm−1.
4.2.5. Synthesis of 2,2,2-Trifluoroethyl 4-Vinyldithiobenzoate 3c by Cascade Reaction
Grignard reaction: Magnesium turnings (0.44 g, 18.30 mmol) were added to an oven-dried flask. The flask was dried at 200 °C (with a heat gun) and purged with argon. Next, 2 mL of dry THF and 1,2-dibromoethane (0.14 mL, 1.64 mmol) were added and the mixture was vigorously stirred. A solution of 4-bromostyrene (2.14 mL, 16.39 mmol) in dry THF (6 mL) was prepared in an oven-dried flask and 2 mL of this solution was added to the magnesium at room temperature. The rest of the solution was added dropwise after the beginning of the reaction (gentle bubbling). The reaction mixture was then diluted with 24 mL of dry THF and stirred at 60 °C for 2 h. The reaction mixture was allowed to cool to room temperature and carbon disulfide (1.48 mL, 24.59 mmol) was added dropwise. The reaction mixture was stirred at 50 °C for 1.5 h.
Mitsunobu reaction: A solution of PPh3 (8.60 g, 32.78 mmol), DIAD (6.44 mL, 32.78 mmol), 2,2,2-trifluoroethanol (2.48 mL, 32.78 mmol), and hydroquinone (0.18 g, 1.64 mmol) was prepared in 35 mL of dry THF at 0 °C. The solution was stirred for 30 min at 0 °C and the crude mixture containing the Grignard product was added. The solution was stirred for 2 h at 0 °C and the product was purified by column chromatography using pentane as solvent. Dithioester 3c was obtained as a red solid. Yield: 2.02 g (47%). Mp: 40 °C.
1H NMR (500 MHz, CDCl3): δ = 8.03 (d, J = 8.4 Hz, 2H), 7.44 (d, J = 8.4 Hz, 2H), 6.75 (dd, J = 17.6, 10.9 Hz, 1H), 5.90 (d, J = 17.6 Hz, 1H), 5.39 (d, J = 10.9 Hz, 1H), 4.25 (q, J = 9.9 Hz, 2H). 13C NMR (125 MHz, CDCl3): δ = 222.2, 143.3, 142.6, 135.9, 127.8, 126.4, 124.9 (q, J = 276.7 Hz), 117.1, 38.0 (q, J = 32.9 Hz). 19F NMR (471 MHz, CDCl3): δ = −64.41 (t, J = 9.9 Hz). HMRS (ASAP+) C11H10F3S2 [(M+H)+]: calculated: 263.0176, found: 263.0184. IR: 2911, 1302, 1231, 1054, 638 cm−1.
4.2.6. Synthesis of 2,2,2-Trifluoropropyl 4-Vinylthiobenzoate 4
Freshly prepared dithiocarboxylic acid 1b (500 mg, 2.77 mmol), trifluoroethanol (0.42 mL, 5.55 mmol), DMAP (407 mg, 3.33 mmol), and DCC (687 mg, 3.33 mmol), were dissolved in THF (8.21 mL) and the mixture was stirred at room temperature for 24 h. The product was purified by flash chromatography (pentane). Thionoester 4 was obtained as a yellow liquid. Yield: 125 mg (18%).
1H NMR (500 MHz, CDCl3): δ = 8.17 (d, J = 8.6 Hz, 2H), 7.43 (d, J = 8.6 Hz, 2H), 6.76 (dd, J = 17.6, 10.9 Hz, 1H), 5.90 (d, J = 17.6 Hz, 1H), 5.43 (d, J = 10.9 Hz, 1H), 4.99 (q, J = 8.3 Hz, 2H). 13C NMR (125 MHz, CDCl3): δ = 208.3, 142.4, 136.3, 135.9, 129.5, 126.1, 123.2 (q, J = 275.5 Hz), 117.1, 67.1 (q, J = 36.4 Hz). 19F NMR (471 MHz, CDCl3): δ = −72.79 (t, J = 8.3 Hz). HMRS (ASAP+) C11H10OF3S [(M+H)+]: calculated: 247.0404, found: 247.0401. IR: 2953, 1276, 1199, 1164 cm−1.
4.2.7. General Procedure for Thioamide 5a–c Synthesis
Dithioester 3a (176 mg, 0.91 mmol,) was dissolved in DCM (2 mL) with the corresponding amine (1.81 mmol) and the mixture was stirred at room temperature for three days. The thioamide obtained was purified by column chromatography (cyclohexane:AcOEt, 90:10).
N-benzyl-4-vinylbenzothioamide (5a). Thioamide 5a was obtained as a yellow solid. Yield: 78%. Mp: 81 °C. 1H NMR (500 MHz, CDCl3): δ = 7.83–7.76 (br, 1H), 7.73 (d, J = 8.4 Hz, 2H), 7.41–7.33 (m, 7H), 6.71 (dd, J = 17.6, 10.9 Hz, 1H), 5.82 (d, J = 17.6 Hz, 1H), 5.35 (d, J = 10.9 Hz, 1H), 4.98 (d, J = 5.2 Hz, 2H). 13C NMR (125 MHz, CDCl3): δ = 198.4, 140.5, 136.3, 135.9, 129.1, 128.4, 128.3, 127.1, 126.3, 116.0, 51.0. HMRS (ASAP+) C16H16NS [(M+H)+]: calculated: 254.1003, found: 254.1002. IR: 3308, 1521, 1500, 1326 cm−1.
4-vinyl-N-propan-2-ylbenzothioamide (5b). Thioamide 5b was obtained as a yellow solid. Yield: 69%. Mp: 76 °C. 1H NMR (500 MHz, CDCl3): δ = 7.63 (d, J = 8.5 Hz, 2H), 7.70–7.49 (br, 1H), 7.33 (d, J = 8.6 Hz, 2H), 6.68 (dd, J = 17.6, 10.9 Hz, 1H), 5.78 (d, J = 17.6 Hz, 1H), 5.32 (d, J = 10.9 Hz, 1H), 4.76 (sextet, J = 6.6 Hz, 1H), 1.33 (d, J = 6.7 Hz, 6H). 13C NMR (125 MHz, CDCl3): δ = 197.2, 141.2, 140.2, 135.9, 127.0, 126.2, 115.9, 48.1, 21.6. HMRS (ASAP+) C12H16NS [(M+H)+]: calculated: 206.1003, found: 206.1006. IR: 3191, 1521, 1502, 1363 cm−1.
4-vinyl-N-(1-phenylethyl)benzothioamide (5c). Thioamide 5c was obtained as a yellow oil. Yield: 84%. Mp 76 ˚C. 1H NMR (500 MHz, CDCl3): δ = 7.78–7.72 (br, 1H), 7.70 (d, J = 8.7 Hz, 2H), 7.43–7.37 (m, 6H), 7.33 (tt, J = 7.1, 1.5 Hz, 1H), 6.71 (dd, J = 17.6, 10.9 Hz, 1H), 5.92 (quintuplet, J = 7.2 Hz, 1H), 5.82 (d, J = 17.6 Hz, 1H), 5.35 (d, J = 10.9 Hz, 1H), 1.71 (d, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ = 197.4, 141.5, 141.0, 140.4, 135.9, 129.0, 128.4, 128.0, 127.1, 126.7, 126.3, 116.0, 55.2, 20.3. HMRS (ASAP+) C17H18NS [(M+H)+]: calculated: 268.1160, found: 268.1162. IR: 3235, 3028, 2974, 1516, 1493, 1367 cm−1.
4.2.8. Synthesis of N-(9-Deoxyepicinchonidin-9-yl)-4-vinylbenzothioamide 5d
Dithioester 3c (200 mg, 0.76 mmol) was dissolved in DCM (1.7 mL) and 9-amino-(9-deoxy)epi-cinchonidine (224 mg, 0.76 mmol) was added. The mixture was stirred at room temperature for four hours. The crude was purified by column chromatography (DCM:MeOH, 95:5) to yield thioamide 5d as a yellow solid. Yield: 313 mg (93%). mp: 92 ˚C. 1H NMR (500 MHz, CDCl3): δ = 9.42–9.01 (br, 1H), 8.87 (d, J = 4.6 Hz, 1H), 8.45 (d, J = 8.2 Hz, 1H), 8.15 (dd, J = 8.5, 1.0 Hz, 1H), 7.79 (d, J = 8.4 Hz, 2H), 7.73 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 7.64 (t, J = 7.3 Hz, 1H), 7.45 (d, J = 4.5 Hz, 1H), 7.42 (d, J = 8.4 Hz, 2H), 6.72 (dd, J = 17.6, 10.9 Hz, 1H), 5.82 (d, J = 17.6 Hz, 1H), 5.79–5.71 (br, 1H), 5.67 (ddd, J = 17.3, 10.2, 7.3 Hz, 1H), 5.34 (d, J = 11.0 Hz, 1H), 4.96 (dt, J = 17.1, 1.3 Hz, 1H), 4.92 (dt, J = 10.4, 1.3 Hz, 1H ), 3.27 (dd, J = 13.9, 10.1 Hz, 1H), 3.24–3.15 (br, 1H), 3.09–2.98 (m, 1H), 2.80–2.69 (m, 2H), 2.36–2.27 (br, 1H), 1.77–1.68 (m, 2H), 1.68–1.58 (m, 1H), 1.45–1.36 (m, 1H), 1.17 (dd, J = 13.7, 6,7 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ = 197.8, 150.2, 148.6, 146.0, 141.1, 140.5, 140.4, 136.0, 130.6, 129.2, 127.5, 127.3, 126.7, 126.3, 123.8, 119.5, 115.99, 114.9, 61.5, 58.6, 55.9, 41.0, 39.7, 27.9, 27.3, 26.1. HMRS (ASAP+) C28H30N3S [(M+H)+]: calculated: 440.2160, found: 440.2159. IR: 3256, 2937, 1480 cm−1.
5. Conclusions
In conclusion, this work reported a complete study to synthesize fluorinated dithioesters. After optimization, an unprecedented one-pot reaction was developed corresponding to a Grignard reaction—Mitsunobu type substitution. Subsequently, the reactivity of these fluorinated dithioesters was evaluated with several primary amines which led to excellent reactivity compared to usual dithioesters, i.e., the methyl dithioester or the S-thiobenzoylglycolic acid. For the trifluoroethyl dithioester 3c, the addition of one equivalent of amine is sufficient to achieve a quasi-quantitative conversion in accordance with green chemistry. This work revealed the potential of the trifluoroethyl group to activate a dithioester function for the synthesis of a 4-styrenylthioamide cinchonidine monomer in a very short time. This new access to functionalized chiral thioamides is currently used in our laboratory for an application in polymer-supported organocatalysis.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28217333/s1. Methyl dithiobenzoate 2a;Ethyl dithiobenzoate 2b; 2,2,2-trifluoroethyl dithiobenzoate 2c;2,2,2-trifluoropropyl dithiobenzoate 2d; Benzyl dithiobenzoate 2e; Methyl 4-vinyldithiobenzoate 3a; 2,2,2-trifluoroethyl 4-vinyldithiobenzoate 3c; 2,2,2-trifluoropropyl 4-vinyldithiobenzoate 3d; 2,2,2-trifluoroethyl 4-vinylthiobenzoate 4; N-benzyl-4-vinylbenzothioamide 5a; 4-vinyl-N-propan-2-ylbenzothioamide 5b; 4-vinyl-N-(1-phenylethyl)benzothioamide 5c; N-(9-Deoxyepicinchonidin-9-yl)-4-vinylbenzothioamide 5d.
Author Contributions
Conceptualization, methodology, supervision, project administration and funding acquisition, B.L., J.R. and J.B.; formal analysis, investigation and data curation, A.G.-B.; writing—original draft preparation, writing—review and editing, J.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Normandy Region (fellowships to A.G.-B.). The authors acknowledge the Ministery of Higher Education and Research, the LABEX SynOrg (ANR-11-LABX-0029) and the European FEDER.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the Supplementary Material.
Acknowledgments
We thank Rémi Legay and Karine Jarsalé for technical assistance and discussions regarding NMR and Mass spectrometry analyses.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Boreen, M.A.; Parker, B.F.; Hohloch, S.; Skeel, B.A.; Arnold, J. f-Block complexes of a m-terphenyl dithiocarboxylate ligand. Dalton Trans. 2018, 47, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Moad, G. Dithioesters in RAFT Polymerization; Wiley: Clayton, Australia, 2021. [Google Scholar]
- Murai, T. Chemistry of Thioamides; Springer: Gifu, Japan, 2019. [Google Scholar]
- Kumari, S.; Carmona, A.V.; Tiwari, A.K.; Trippier, P.C. Amide Bond Bioisosteres: Strategies, Synthesis, and Successes. J. Med. Chem. 2020, 63, 12290–12358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, H.; Li, G.; Zhu, X.; Shang, L.; He, Y.; Liu, X.; Ma, Y.; Szostak, M. Transamidation of thioamides with nucleophilic amines: Thioamide N–C(S) activation by ground-state-destabilization. Org. Biomol. Chem. 2022, 20, 5981–5988. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, A.; Raines, R.T. An Evaluation of Peptide-Bond Isosteres. ChemBioChem 2011, 12, 1801–1807. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Hao, J.; Reddy, M.V.; Rao, P.B.; Rassias, G.; Snyder, S.A.; Huang, X.; Chen, D.Y.-K.; Brenzovich, W.E.; Giuseppone, N.; et al. Chemistry and Biology of Diazonamide A: Second Total Synthesis and Biological Investigations. J. Am. Chem. Soc. 2004, 126, 12897–12906. [Google Scholar] [CrossRef] [PubMed]
- Taily, I.M.; Saha, D.; Banerjee, P. Aza-Oxyallyl Cation Driven 3-Amido Oxetane Rearrangement to 2-Oxazolines: Access to Oxazoline Amide Ethers. J. Org. Chem. 2022, 87, 2155–2166. [Google Scholar] [CrossRef]
- Wang, C.; Han, C.; Yang, J.; Zhang, Z.; Zhao, Y.; Zhao, J. Ynamide-Mediated Thioamide and Primary Thioamide Syntheses. J. Org. Chem. 2022, 87, 5617–5629. [Google Scholar] [CrossRef]
- Ganesh, M.; Seidel, D. Catalytic Enantioselective Additions of Indoles to Nitroalkenes. J. Am. Chem. Soc. 2008, 130, 16464–16465. [Google Scholar] [CrossRef]
- Wang, B.; Liu, X.-W.; Liu, L.-Y.; Chang, W.-X.; Li, J. Highly Efficient Direct Asymmetric Aldol Reactions Catalyzed by a Prolinethioamide Derivative in Aqueous Media. Eur. J. Org. Chem. 2010, 2010, 5951–5954. [Google Scholar] [CrossRef]
- Almasi, D.; Alonso, D.A.; Nájera, C. Prolinamides versus Prolinethioamides as Recyclable Catalysts in the Enantioselective Solvent-Free Inter- and Intramolecular Aldol Reactions. Adv. Synth. Catal. 2008, 350, 2467–2472. [Google Scholar] [CrossRef]
- Gryko, D.; Lipiński, R. L-Prolinethioamides—Efficient Organocatalysts for the Direct Asymmetric Aldol Reaction. Adv. Synth. Catal. 2005, 347, 1948–1952. [Google Scholar] [CrossRef]
- Gryko, D.; Lipiński, R. Asymmetric Direct Aldol Reaction Catalysed by L-Prolinethioamides. Eur. J. Org. Chem. 2006, 2006, 3864–3876. [Google Scholar] [CrossRef]
- Alemán, C. On the Ability of Modified Peptide Links to Form Hydrogen Bonds. J. Phys. Chem. A 2001, 105, 6717–6723. [Google Scholar] [CrossRef]
- Lee, H.-J.; Choi, Y.-S.; Lee, K.-B.; Park, J.; Yoon, C.-J. Hydrogen Bonding Abilities of Thioamide. J. Phys. Chem. A 2002, 106, 7010–7017. [Google Scholar] [CrossRef]
- Singjunla, Y.; Pigeaux, M.; Laporte, R.; Baudoux, J.; Rouden, J. Thioamide-Substituted Cinchona Alkaloids as Efficient Organocatalysts for Asymmetric Decarboxylative Reactions of MAHOs. Thioamide-Substituted Cinchona Alkaloids as Efficient Organocatalysts for Asymmetric Decarboxylative Reactions of MAHOs. Eur. J. Org. Chem. 2017, 2017, 4319–4323. [Google Scholar] [CrossRef]
- Lepoittevin, B.; Baudoux, J.; Bray, D.; Gonzalo-Barquero, A.; Rouden, J. Polymer-supported thioamide-derived cinchona alkaloids as efficient organocatalysts for the enantioselective decarboxylative protonation. React. Funct. Polym. 2023, 192, 105731. [Google Scholar] [CrossRef]
- Cortez-Lemus, N.A.; Salgado-Rodriguez, R.; Licea-Claverie, A. Preparation of α,ω-telechelic hexyl acrylate polymers with -OH, -COOH, and -NH2 functional groups by RAFT. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 3033–3051. [Google Scholar] [CrossRef]
- Thuillier, A. Dithioesters in Organic Synthesis. Phosphorus Sulfur Relat. Elem. 1985, 23, 253–276. [Google Scholar] [CrossRef]
- Westmijze, H.; Kleijn, H.; Meijer, J.; Vermeer, P. Synthesis of Dithioesters from Organocopper(I) Compounds. Synthesis 1979, 1979, 432–434. [Google Scholar] [CrossRef]
- Grote, J.; Friedrich, F.; Berthold, K.; Hericks, L.; Neumann, B.; Stammler, H.-G.; Mitzel, N.W. Dithiocarboxylic Acids: An Old Theme Revisited and Augmented by New Preparative, Spectroscopic and Structural Facts. Chem. Eur. J. 2018, 24, 2626–2633. [Google Scholar] [CrossRef]
- Woth, A.C.; Needham, C.E.; Franklin, D.B.; Lampkins, A.J. Facile Synthesis of Lipophilic δ-Amino Acid Conjugates from 4-Alkoxy-dithionaphthoic Acids. Synth. Commun. 2012, 42, 2694–2706. [Google Scholar]
- Yeo, S.K.; Choi, B.G.; Kim, J.D.; Lee, J.H. A Convenient Method for the Synthesis of Thiobenzamide Derivatives and O-Thiobenzoates by Use of 2-Benzothiazolyl Dithiobenzoate as Effective Thiobenzoylation Reagent. Bull. Korean Chem. Soc. 2002, 23, 1029–1030. [Google Scholar] [CrossRef]
- Matysiak, J.; Niewiadomy, A. Application of Sulfinyl bis(2,4-dihydroxythiobenzoyl) in the Synthesis of N-Substituted 2-Amino-5-(2,4-dihydroxyphenyl)-1,3,4-thiadiazoles. Synth. Commun. 2006, 36, 1621–1630. [Google Scholar] [CrossRef]
- Matysiak, J.; Niewiadomy, A. Synthesis and antimycotic activity of N-azolyl-2,4-dihydroxythiobenzamides. Bioorg. Med. Chem. 2003, 11, 2285–2291. [Google Scholar] [CrossRef]
- Guan, X.; Drake, M.R.; Tan, Z. Total Synthesis of Human Galanin-Like Peptide through an Aspartic Acid Ligation. Org. Lett. 2013, 15, 6128–6131. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Moutou, J.-L.; Yang, Z. A New Versatile One-Pot Synthesis of Functionalized Thioamides from Grignards, Carbon Disulfide and Amines. Synthesis 1995, 12, 1497–1505. [Google Scholar] [CrossRef]
- Delêtre, M.; Levesque, G. Kinetics and Mechanism of Polythioamidation in Solution. 1. Reaction of Mono- and Bis(dithioester)s with Excess Amine. Macromolecules 1990, 23, 4733–4741. [Google Scholar] [CrossRef]
- Oh, H.K.; Woo, S.Y.; Shin, C.H.; Park, Y.S.; Lee, I. Kinetics and Mechanism of the Aminolysis of Phenyl Dithioacetates in Acetonitrile. J. Org. Chem. 1997, 62, 5780–5784. [Google Scholar] [CrossRef]
- Oh, H.K.; Shin, C.H.; Lee, I. Kinetics and mechanism of the aminolysis of phenyl dithiobenzoates. J. Chem. Soc. Perkin Trans 2 1995, 6, 1169–1173. [Google Scholar] [CrossRef]
- Kpegba, K.; Metzner, P. Synthesis of Dithioic Acid Esters by a Mitsunobu-Type Reaction of Alkanedithioic Acids and Alcohols. Synthesis 1989, 48, 137–139. [Google Scholar] [CrossRef]
- Ho, P.-T.; Davies, N. Reaction of alcohols with zinc halide, diethyl azodicarboxylate, and triphenylphosphine. An effective method for the preparation of halides. J. Org. Chem. 1984, 49, 3027–3029. [Google Scholar] [CrossRef]
- Viaud, M.C.; Rollin, P. Zinc Azide Mediated Mitsunobu Substitution. An Expedient Method for the One-Pot Azidation of Alcohols. Synthesis 1990, 1990, 130–132. [Google Scholar] [CrossRef]
- Manna, S.; Falck, J.R. A convenient preparation of alkyl halides and cyanides from alcohols by modification of the Mitsunobu procedure. Synth. Commun. 1985, 15, 663–668. [Google Scholar] [CrossRef]
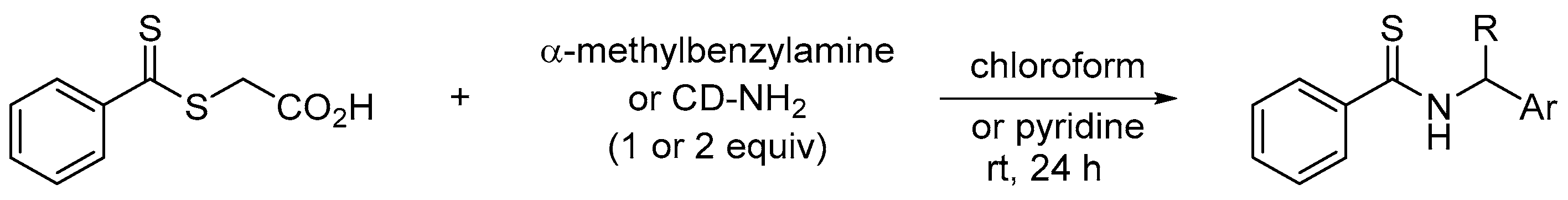
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).