
The Synthesis of Novel aza-Steroids and α, β-Unsaturated-Cyanoketone from Diosgenin

 ,
,
Abstract
:1. Introduction
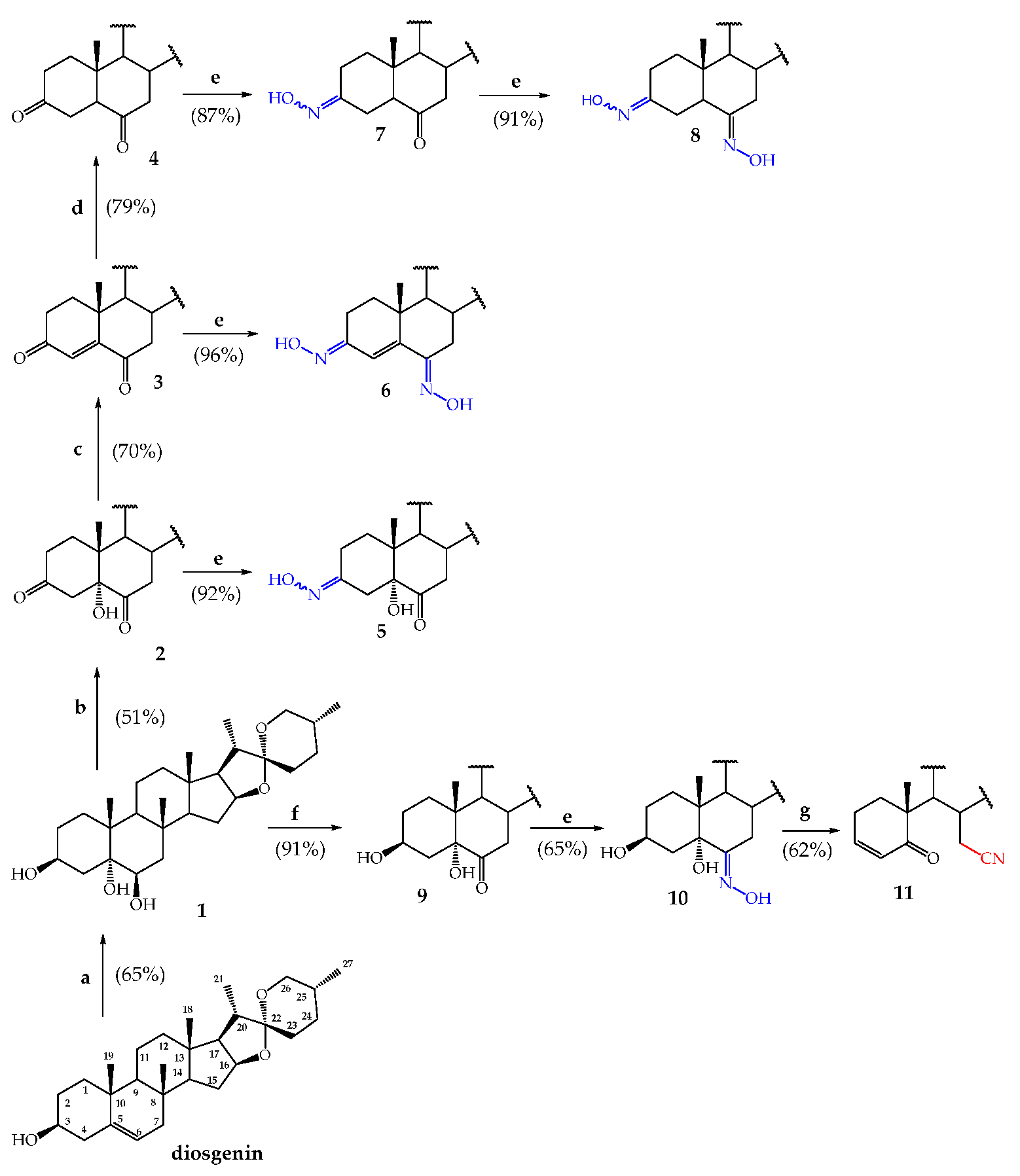
2. Results and Discussion
2.1. Synthesis
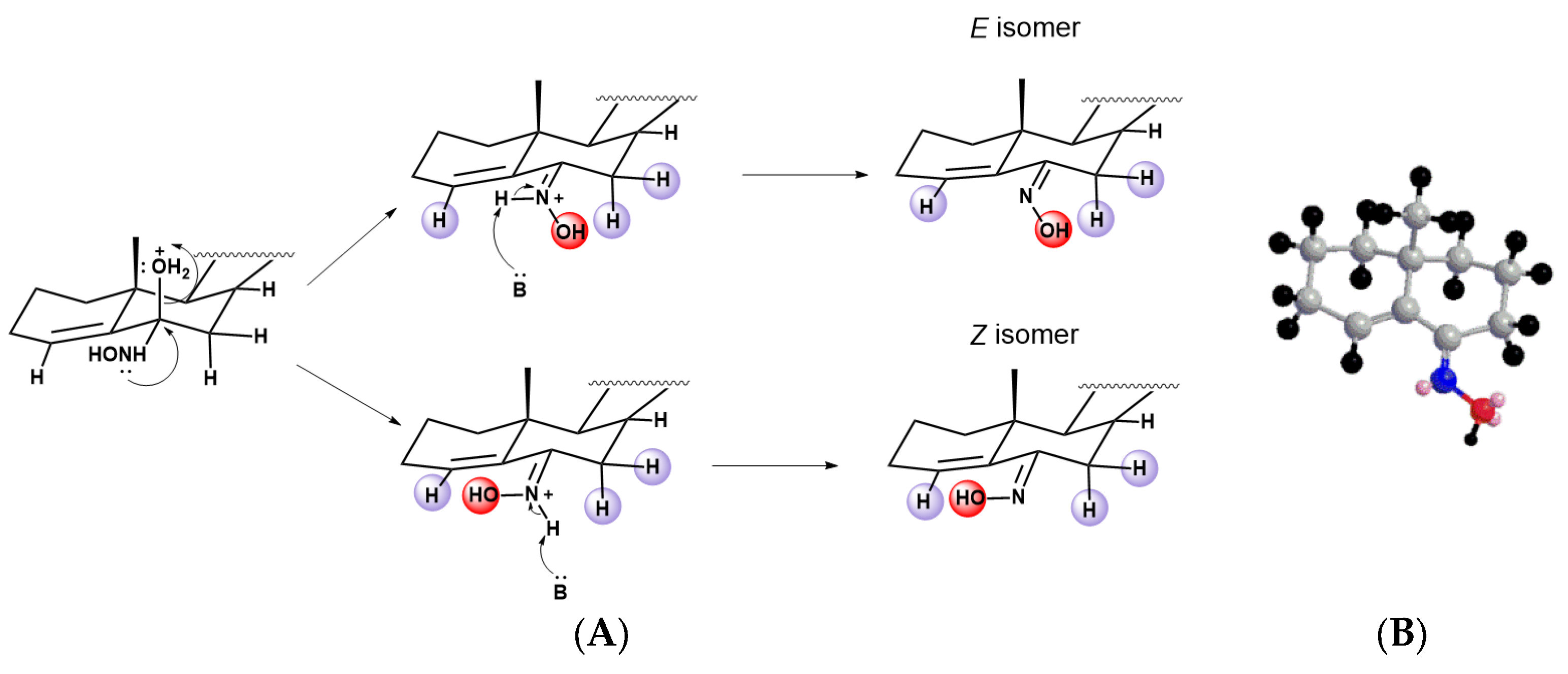
Synthesis of Steroidal Oximes
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Synthesis of Steroidal Ketones
3.2.1. (25R)-Spirost-5α-hydroxy-3,6-dione (2)
3.2.2. (25R)-Spirost-4-en-3,6-dione (3)
3.2.3. (25R)-5α-Spirost-3,6-dione (4)
3.3. General Procedure for the Synthesis of Steroidal Oximes
3.3.1. (25R)-(3E/Z)-Hydroximino-5α-spirost-5-hydroxy-6-ona (5)
3.3.2. (25R)-(3Z,6E)-Dihydroximinospirost-4-ene (6)
3.3.3. (25R)-(3E/Z)-Hydroximino-5α-spirost-6-ona (7)
3.3.4. (25R)-(3E/Z,6E)-dihydroximino-5α-spirostane (8)
3.3.5. (25R)-(6E)-Hydroximino-5α-spirost-3β,5-diol (10)
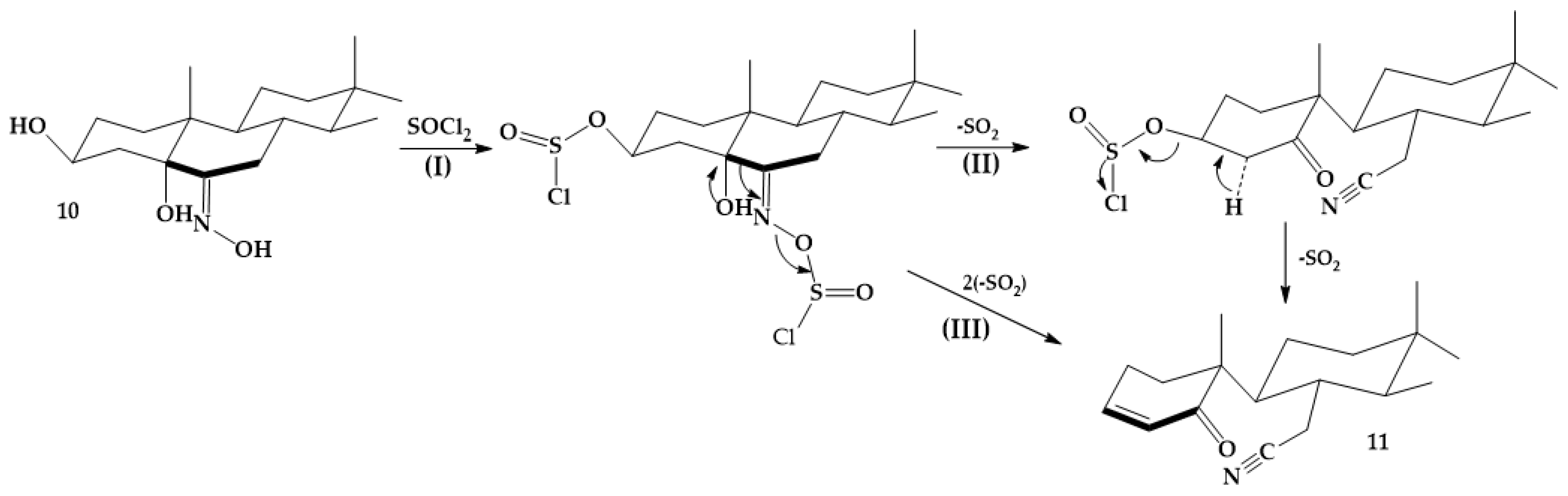
3.3.6. (25R)-5-Oxo-5,6-secospirost-3-en-6-nitrile (11)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Stenholm, S.; Kivimäki, M.; Jylhä, M.; Kawachi, I.; Westerlund, H.; Pentti, J.; Goldberg, M.; Zins, M.; Vahtera, J. Trajectories of self-rated health in the last 15 years of life by cause of death. Eur. J. Epidemiol. 2016, 31, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Necchi, A.; Miceli, R.; Bregni, M.; Bokemeyer, C.; Berger, L.A.; Oechsle, K.; Schumacher, K.; Kanfer, E.; Bourhis, J.H.; Massard, C.; et al. Prognostic impact of progression to induction chemotherapy and prior paclitaxel therapy in patients with germ cell tumors receiving salvage high-dose chemotherapy in the last 10 years: A study of the European Society for Blood and Marrow Transplantation Solid Tumors Working Party. Bone Marrow Transplant. 2016, 51, 384–390. [Google Scholar] [PubMed]
- Ulm, M.; Ramesh, A.V.; McNamara, K.M.; Ponnusamy, S.; Sasano, H.; Narayanan, R. Therapeutic advances in hormone-dependent cancers: Focus on prostate, breast and ovarian cancers. Endocr. Connect. 2019, 8, R10–R26. [Google Scholar] [CrossRef]
- Santen, R.J.; Manni, A.; Harvey, H.; Redmond, C. Endocrine Treatment of Breast Cancer in Women. Endocr. Rev. 1990, 11, 221–265. [Google Scholar] [CrossRef]
- Lumachi, F.; Luisetto, G.; Basso, S.M.; Basso, U.; Brunello, A.; Camozzi, V. Endocrine therapy of breast cancer. Curr. Med. Chem. 2011, 18, 513–522. [Google Scholar] [CrossRef]
- Łukasiewicz, S.; Czeczelewski, M.; Forma, A.; Baj, J.; Sitarz, R.; Stanisławek, A. Breast Cancer-Epidemiology, Risk Factors, Classification, Prognostic Markers, and Current Treatment Strategies-An Updated Review. Cancers 2021, 13, 4287. [Google Scholar] [CrossRef] [PubMed]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Martinkovich, S.; Shah, D.; Planey, S.L.; Arnott, J.A. Selective estrogen receptor modulators: Tissue specificity and clinical utility. Clin. Interv. Aging 2014, 9, 1437–1452. [Google Scholar]
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Impact of Natural Products on Developing New Anti-Cancer Agents. Chem. Rev. 2009, 109, 3012–3043. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Bosio, C.; Tomasoni, G.; Martinez, R.; Olea, A.F.; Carrasco, H.; Villena, J. Cytotoxic and apoptotic effects of leptocarpin, a plant-derived sesquiterpene lactone, on human cancer cell lines. Chem. Biol. Inter. 2015, 242, 415–421. [Google Scholar] [CrossRef]
- Montenegro, I.; Tomasoni, G.; Bosio, C.; Quinones, N.; Madrid, A.; Carrasco, H.; Olea, A.; Martinez, R.; Cuellar, M.; Villena, J. Study on the Cytotoxic Activity of Drimane Sesquiterpenes and Nordrimane Compounds against Cancer Cell Lines. Molecules 2014, 19, 18993–19006. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z. The modification of natural products for medical use. Acta Pharm. Sin. B 2017, 7, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Vázquez, J.M.V.; López-Muñoz, H.; Escobar-Sánchez, M.L.; Flores-Guzmán, F.; Weiss-Steider, B.; Hilario-Martínez, J.C.; Sandoval-Ramírez, J.; Fernández-Herrera, M.A.; Sánchez Sánchez, L. Apoptotic, necrotic, and antiproliferative activity of diosgenin and diosgenin glycosides on cervical cancer cells. Eur. J. Pharmacol. 2020, 871, 172942. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.G.; Elder, J.L.; Chang, P.R.; Richards, K.W. Microdetermination of Diosgenin from Fenugreek (Trigonella foenum-graecum) Seeds. J. Agric. Food Chem. 2000, 48, 5206–5210. [Google Scholar] [CrossRef] [PubMed]
- Hooker, E. Final report of the amended safety assessment of Dioscorea Villosa (Wild Yam) root extract. Int. J. Toxicol. 2004, 23 (Suppl. S2), 49–54. [Google Scholar]
- Singh, I.; Gautam, Y.; Vimala, Y. Detection and isolation of diosgenin from Costus speciosus callus raised from non-germinal seeds. Int. J. Chem. Life Sci. 2013, 2, 1240–1242. [Google Scholar]
- Chen, P.S.; Shih, Y.W.; Huang, H.C.; Cheng, H.W. Diosgenin, a steroidal saponin, inhibits migration and invasion of human prostate cancer PC-3 cells by reducing matrix metalloproteinases expression. PLoS ONE 2011, 6, e20164. [Google Scholar] [CrossRef]
- Moalic, S.; Liagre, B.; Corbière, C.; Bianchi, A.; Dauça, M.; Bordji, K.; Beneytout, J.L. A plant steroid, diosgenin, induces apoptosis, cell cycle arrest and COX activity in osteosarcoma cells. FEBS Lett. 2001, 506, 225–230. [Google Scholar] [CrossRef]
- Raju, J.; Mehta, R. Cancer chemopreventive and therapeutic effects of diosgenin, a food saponin. Nutr. Cancer 2009, 61, 27–35. [Google Scholar] [CrossRef]
- Sethi, G.; Shanmugam, M.K.; Warrier, S.; Merarchi, M.; Arfuso, F.; Kumar, A.P.; Bishayee, A. Pro-Apoptotic and Anti-Cancer Properties of Diosgenin: A Comprehensive and Critical Review. Nutrients 2018, 10, 645. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sánchez, L.; Hernández-Linares, M.G.; Escobar, M.L.; López-Muñoz, H.; Zenteno, E.; Fernández-Herrera, M.A.; Guerrero-Luna, G.; Carrasco-Carballo, A.; Sandoval-Ramírez, J. Antiproliferative, Cytotoxic, and Apoptotic Activity of Steroidal Oximes in Cervicouterine Cell Lines. Molecules 2016, 21, 1533. [Google Scholar] [CrossRef]
- Masood Ur, R.; Mohammad, Y.; Fazili, K.M.; Bhat, K.A.; Ara, T. Synthesis and biological evaluation of novel 3-O-tethered triazoles of diosgenin as potent antiproliferative agents. Steroids 2017, 118, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Yang, J.; Guo, L.; Wang, X.; Song, B.; Dong, W.; Wang, W. Novel diosgenin derivatives containing 1,3,4-oxadiazole/thiadiazole moieties as potential antitumor agents: Design, synthesis and cytotoxic evaluation. Eur. J. Med. Chem. 2020, 186, 111897. [Google Scholar] [CrossRef]
- Martínez-Pascual, R.; Meza-Reyes, S.; Vega-Baez, J.L.; Merino-Montiel, P.; Padrón, J.M.; Mendoza, Á.; Montiel-Smith, S. Novel synthesis of steroidal oximes and lactams and their biological evaluation as antiproliferative agents. Steroids 2017, 122, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Jindal, D.P.; Chattopadhaya, R.; Guleria, S.; Gupta, R. Synthesis and antineoplastic activity of 2-alkylaminoethyl derivatives of various steroidal oximes. Eur. J. Med. Chem. 2003, 38, 1025–1034. [Google Scholar] [CrossRef]
- Poza, J.; Rega, M.; Paz, V.; Alonso, B.; Rodríguez, J.; Salvador, N.; Fernández, A.; Jiménez, C. Synthesis and evaluation of new 6-hydroximinosteroid analogs as cytotoxic agents. Bioorg. Med. Chem. 2007, 15, 4722–4740. [Google Scholar] [CrossRef]
- Dawidar, A.; Saleh, A.; Abdel-Malek, M. Hydroxylation of Δ5-Steroids with N-Bromosuccinimide to 5α,6β-Diols. Z. Für Naturforschung B 1980, 35, 102–106. [Google Scholar] [CrossRef]
- García-Pupo, L.; Zaldo-Castro, A.; Exarchou, V.; Tacoronte-Morales, J.E.; Pieters, L.; Vanden Berghe, W.; Nuñez-Figueredo, Y.; Delgado-Hernández, R. In Vitro Neuroprotective and Anti-Inflammatory Activities of Natural and Semi-Synthetic Spirosteroid Analogues. Molecules 2016, 21, 992. [Google Scholar] [CrossRef]
- Wijnberg, J.B.P.A.; Vader, J.; De Groot, A. Stereospecific synthesis of selectively C-7-acetalized substituted 4a.beta.-methyl-3,4,4a,5,6,8a.alpha.-hexahydronaphthalene-1(2H),7(8H)-diones. A short total synthesis of (.+-.)-.beta.-eudesmol, (.+-.)-.beta.-selinene, and (.+-.)-.beta.-dictyopterol. J. Org. Chem. 1983, 48, 4380–4387. [Google Scholar] [CrossRef]
- Greene, T.W.; Wuts, P.G.M. Protective Groups in Organic Synthesis, 2nd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1991. [Google Scholar]
- Sandler, S.R.; Karo, W. Organic Functional Group Preparation; John Wiley & Sons, Inc.: New York, NY, USA, 1983. [Google Scholar]
- Krstić, N.; Bjelaković, M.; Dabovic, M.; Lorenc, L.; Pavlovic, V. Photochemical and Beckmann rearrangement of (Z)-cholest-4-en-6-one oxime. J. Serb. Chem. Soc. 2004, 69, 413–420. [Google Scholar] [CrossRef]
- Shoppee, C.W.; Roy, S.K. Aza-steroids. Part VI. Beckmann rearrangement of some α-hydroxy-ketoximes. J. Chem. Soc. 1963, 3774–3777. [Google Scholar] [CrossRef]
- Pejanovic, V.M.; Petrovtć, J.A.; Csanádi, J.J.; Stanković, S.M.; Miljkovic, D.A. Synthesis and unusual beckmann fragmentation reaction of syn-3-Methoxy-6α,17β-Dihydroxyestra-1,3,5(10)-trien-7-one oxime. Tetrahedron 1995, 51, 13379–13384. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | C-3 | C-6 | C-2 | C-4 | C-7 | |
|---|---|---|---|---|---|---|
| 2 | 210.0 | 210.7 | 36.8 | 44.2 | 41.1 | |
| 5 | E | 159.6 | 212.0 | 20.1 | 29.8 | 42.2 |
| Z | 159.2 | 211.6 | 25.7 | 27.7 | 42.4 | |
| 3 | 199.3 | 201.8 | 34.1 | 125.7 | 47.0 | |
| 6 | E | 156.8 | 156.9 | 31.5 | 119.4 | 29.9 |
| 4 | 211.1 | 208.8 | 37.5 | 37.1 | 48.4 | |
| 7 | E | 159.9 | 209.7 | 19.8 | 27.1 | 46.6 |
| Z | 158.1 | 209.8 | 26.9 | 19.9 | 46.6 | |
| 8 | E | 159.8 | 158.8 | 20.0 | 28.3 | 29.5 |
| Z | 160.0 | 159.1 | 21.2 | 21.1 | 29.6 | |
| H-2eq | H-2eq | H-7eq | |
|---|---|---|---|
| 4 | 2.11 | 2.41 | |
| 7 | E | 3.27 | 2.38 |
| Z | 1.98 | 2.38 | |
| 8 | E/E | 3.33 | 3.35 |
| Z/E | 1.57 | 3.35 |
| Entry | Starting Material | Products | Yields (%) | Reaction Times (h) |
|---|---|---|---|---|
| 1 | 2 | 5 | 92 a | 6 |
| 2 | 3 | 6 | 96 b | 8 |
| 3 | 4 | 7 | 87 b | 2 |
| 4 | 8 | 91 a | 3 | |
| 5 | 9 | 10 | 65 b | 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mesa, D.; Augusto, Y.E.; Hernández, G.; Figueroa-Macías, J.P.; Coll, F.; Olea, A.F.; Núñez, M.; Campo, H.A.; Coll, Y.; Espinoza, L. The Synthesis of Novel aza-Steroids and α, β-Unsaturated-Cyanoketone from Diosgenin. Molecules 2023, 28, 7283. https://doi.org/10.3390/molecules28217283
Mesa D, Augusto YE, Hernández G, Figueroa-Macías JP, Coll F, Olea AF, Núñez M, Campo HA, Coll Y, Espinoza L. The Synthesis of Novel aza-Steroids and α, β-Unsaturated-Cyanoketone from Diosgenin. Molecules. 2023; 28(21):7283. https://doi.org/10.3390/molecules28217283
Chicago/Turabian StyleMesa, Dayana, Yarelys E. Augusto, Giselle Hernández, Juan P. Figueroa-Macías, Francisco Coll, Andrés F. Olea, María Núñez, Hernán Astudillo Campo, Yamilet Coll, and Luis Espinoza. 2023. "The Synthesis of Novel aza-Steroids and α, β-Unsaturated-Cyanoketone from Diosgenin" Molecules 28, no. 21: 7283. https://doi.org/10.3390/molecules28217283
APA StyleMesa, D., Augusto, Y. E., Hernández, G., Figueroa-Macías, J. P., Coll, F., Olea, A. F., Núñez, M., Campo, H. A., Coll, Y., & Espinoza, L. (2023). The Synthesis of Novel aza-Steroids and α, β-Unsaturated-Cyanoketone from Diosgenin. Molecules, 28(21), 7283. https://doi.org/10.3390/molecules28217283