

Rational Design of Photocontrolled Rectifier Switches in Single-Molecule Junctions Based on Diarylethene

Abstract
:1. Introduction
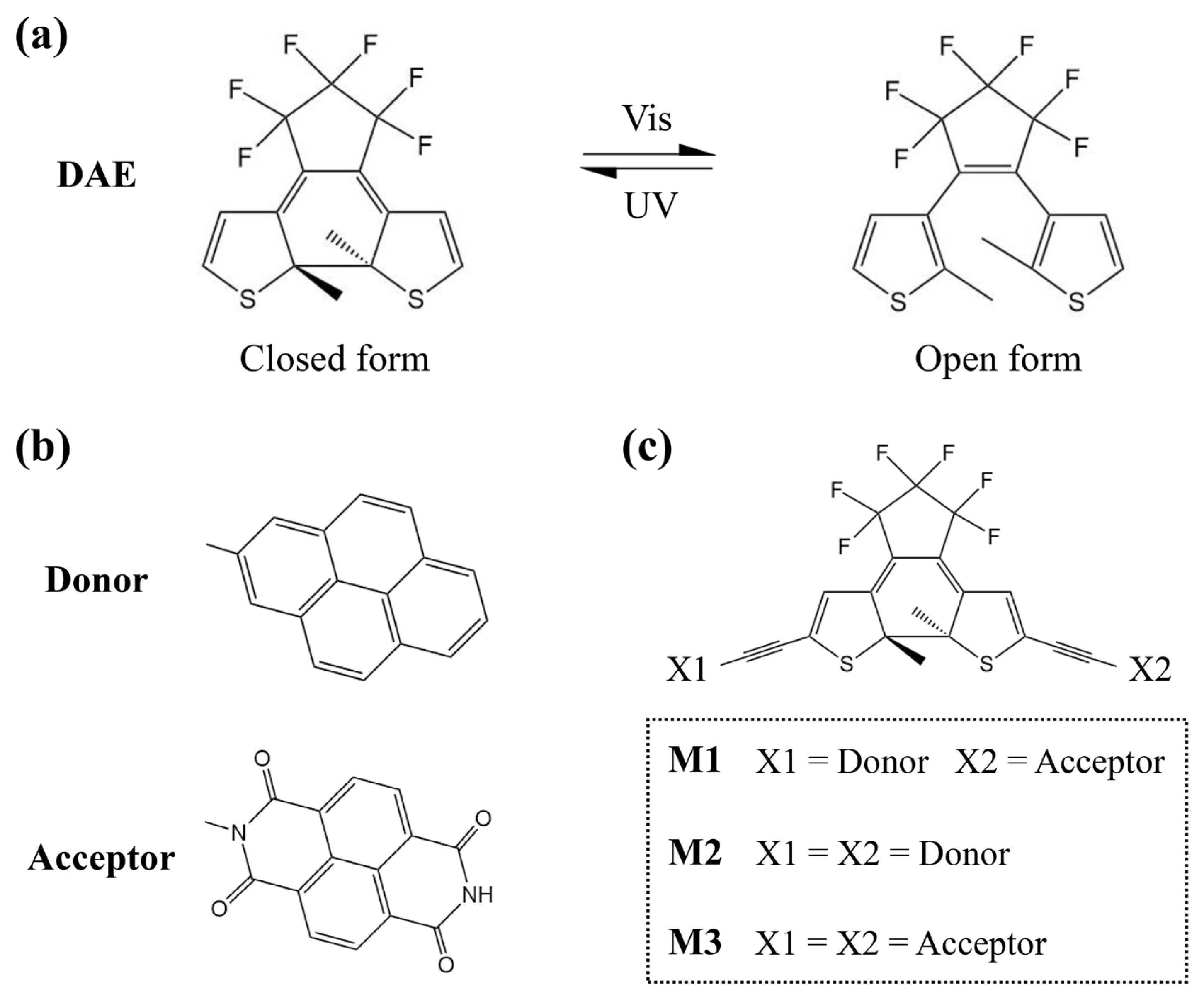
2. Results and Discussion
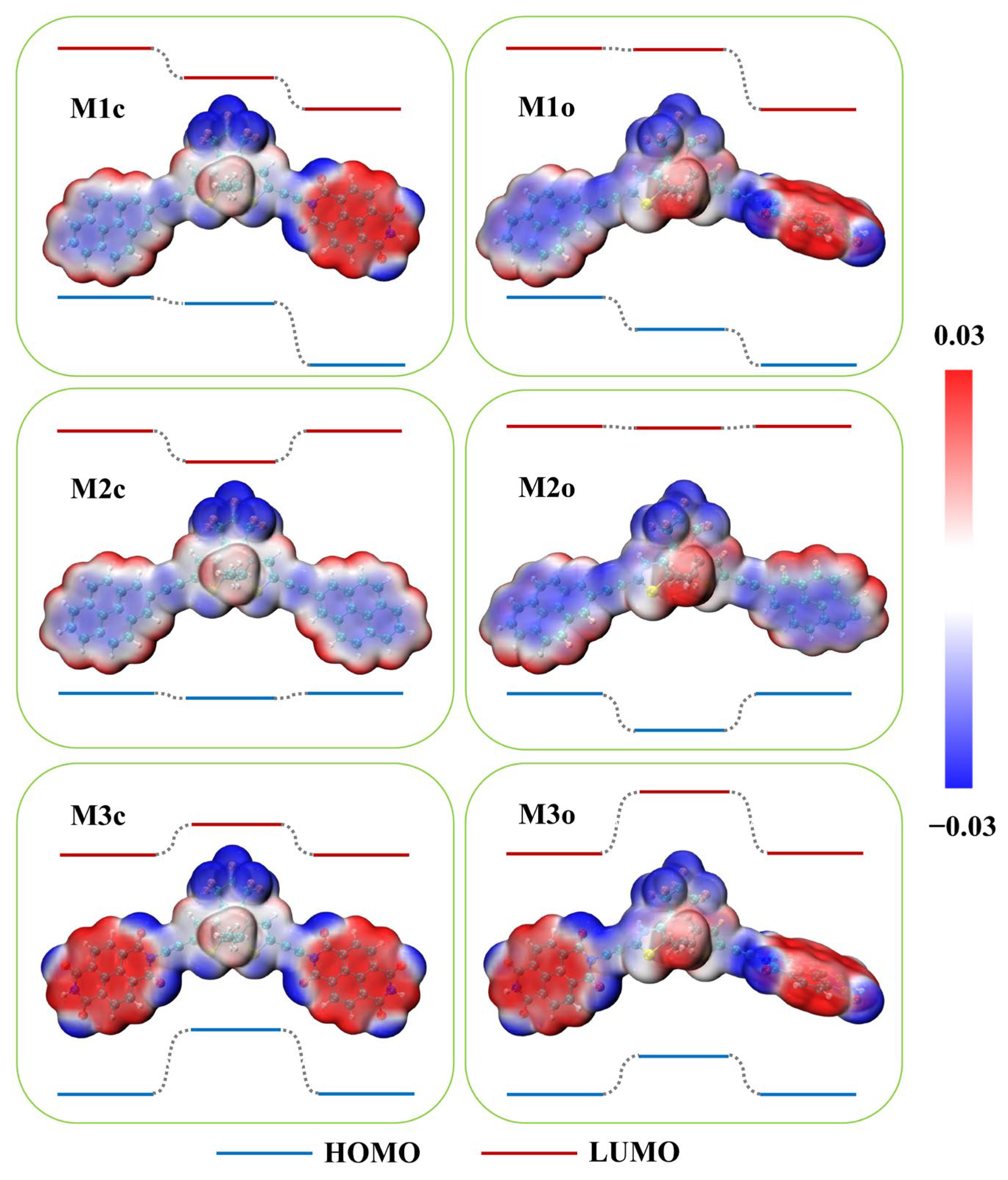
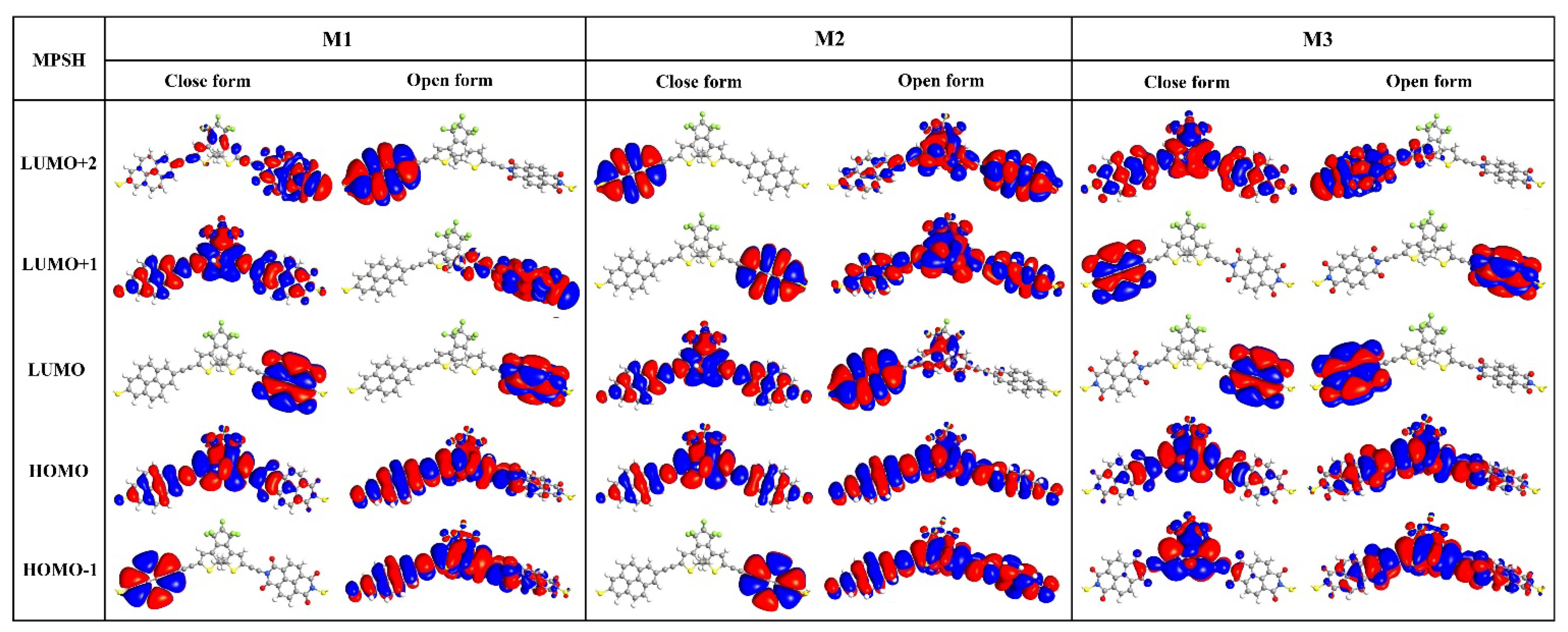
2.1. Electronic Properties of Single Molecules
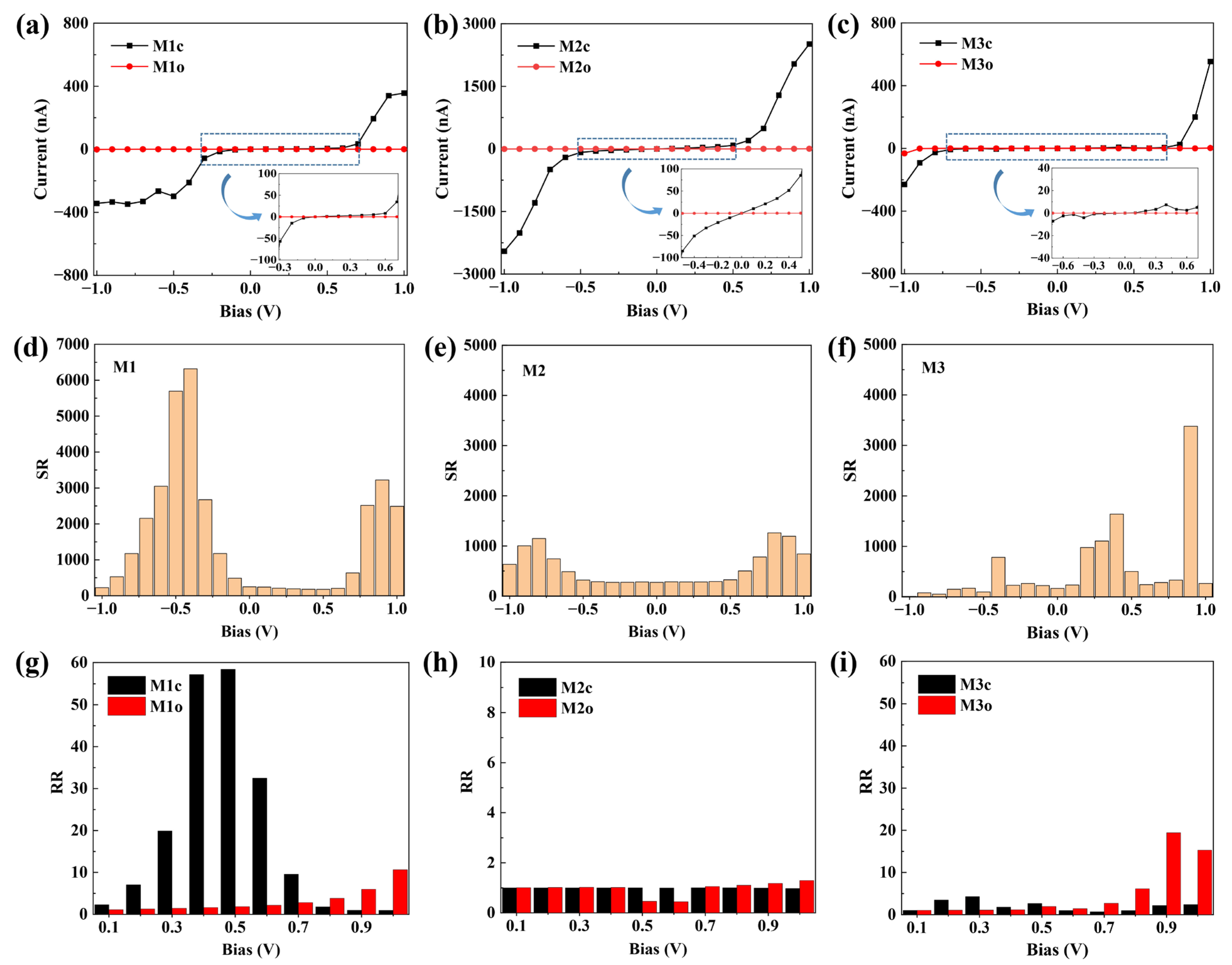
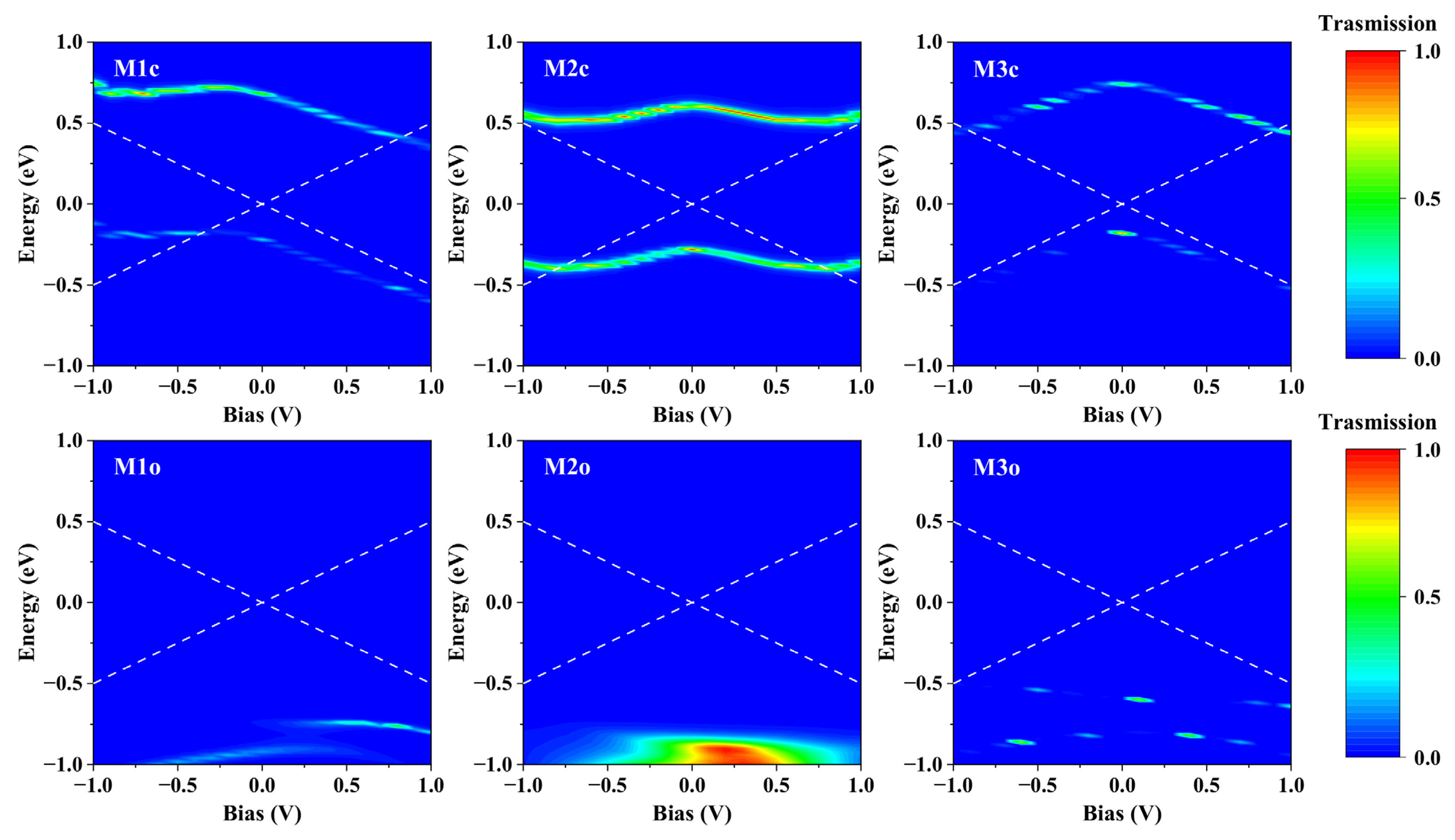
2.2. Current–Voltage (I–V) Characteristics
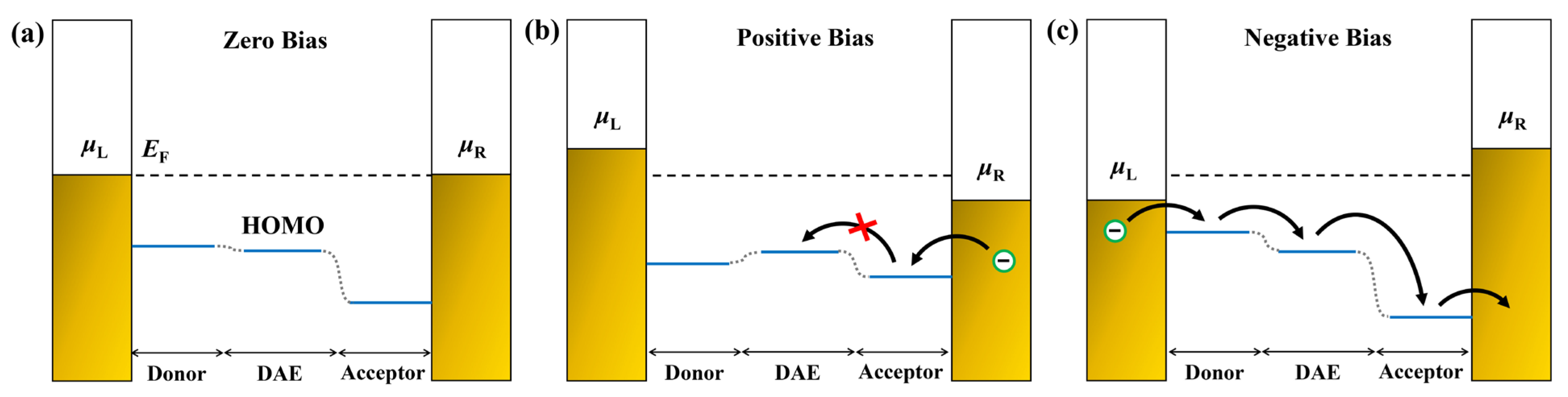
2.3. Explanations of I−V Characteristics
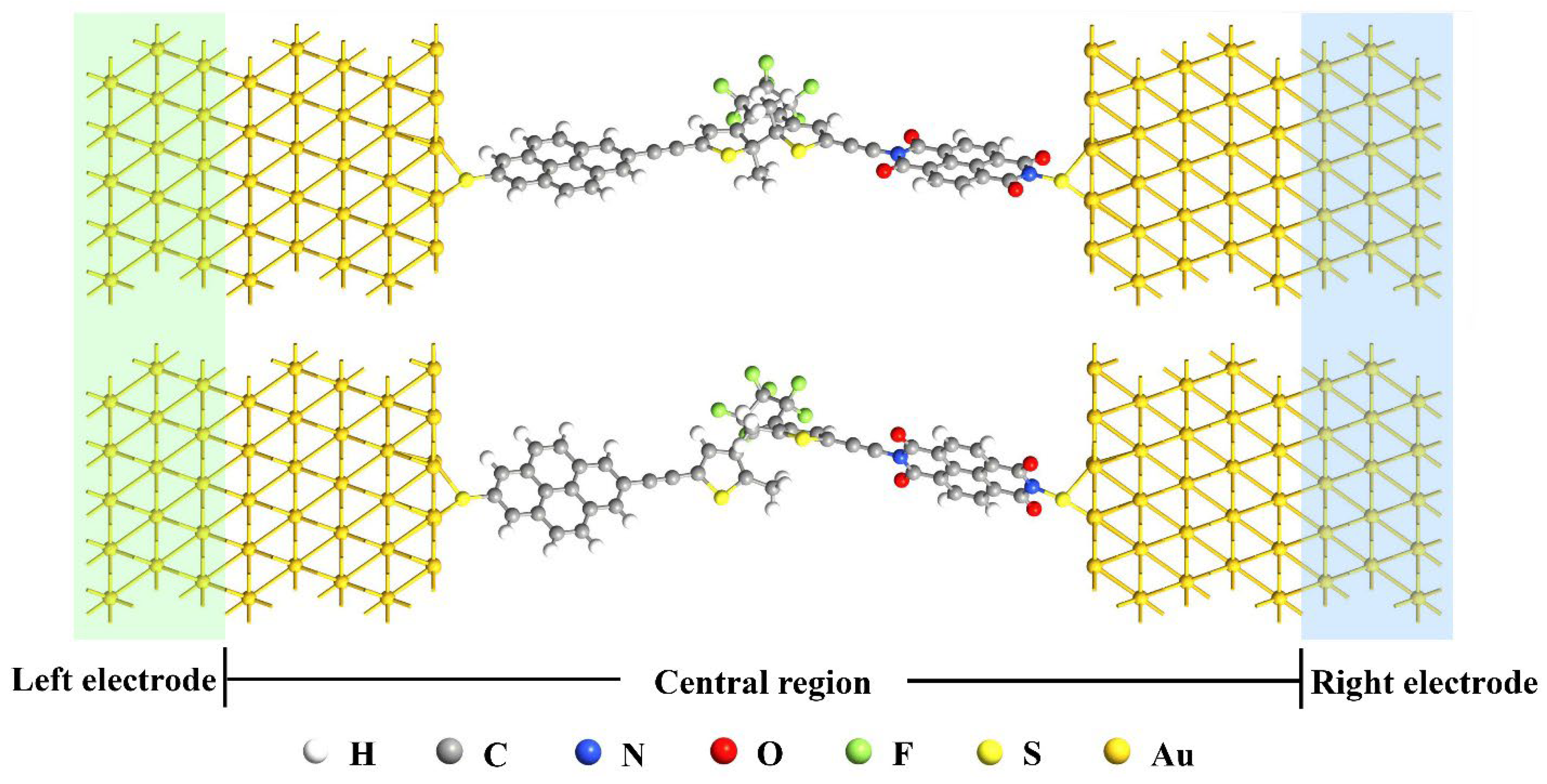
3. Models and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Xie, X.; Li, P.; Xu, Y.; Zhou, L.; Yan, Y.; Xie, L.; Jia, C.; Guo, X. Single-molecule junction: A reliable platform for monitoring molecular physical and chemical processes. ACS Nano 2022, 16, 3476–3505. [Google Scholar] [CrossRef]
- Tang, C.; Shiri, M.; Zhang, H.; Ayinla, R.T.; Wang, K. Light-driven charge transport and optical sensing in molecular junctions. Nanomaterials 2022, 12, 698. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.J. Recent experimental advances and prospects for the next decade of single-molecule electronics: From devices to applications. Chin. Sci. Bull. 2023, 68, 2197–2212. (In Chinese) [Google Scholar] [CrossRef]
- Liberka, M.; Zychowicz, M.; Hooper, J.; Nakabayashi, K.; Ohkoshi, S.I.; Chorazy, S. Synchronous switching of dielectric constant and photoluminescence in cyanidonitridorhenate-based crystals. Angew. Int. Ed. Chem. 2023, 62, e202308284. [Google Scholar] [CrossRef] [PubMed]
- Aviram, A.; Ratner, M.A. Molecular rectifiers. Chem. Phys. Lett. 1974, 29, 277–283. [Google Scholar] [CrossRef]
- Jaroš, A.; Bonab, E.F.; Straka, M.; Foroutan-Nejad, C. Fullerene-based switching molecular diodes controlled by oriented external electric fields. J. Am. Chem. Soc. 2019, 141, 19644–19654. [Google Scholar] [CrossRef]
- Chen, X.; Roemer, M.; Yuan, L.; Du, W.; Thompson, D.; del Barco, E.; Nijhuis, C.A. Molecular diodes with rectification ratios exceeding 105 driven by electrostatic interactions. Nat. Nanotechnol. 2017, 12, 797–803. [Google Scholar] [CrossRef]
- Capozzi, B.; Xia, J.L.; Adak, O.; Dell, E.J.; Liu, Z.F.; Taylor, J.C.; Neaton, J.B.; Campos, L.M.; Venkataraman, L. Single-molecule diodes with high rectification ratios through environmental control. Nat. Nanotechnol. 2015, 10, 522–527. [Google Scholar] [CrossRef]
- Hnid, I.; Frath, D.; Lafolet, F.; Sun, X.; Lacroix, J.-C. Highly efficient photoswitch in diarylethene-based molecular junctions. J. Am. Chem. Soc. 2020, 142, 7732–7736. [Google Scholar] [CrossRef]
- Zhang, J.L.; Zhong, J.Q.; Lin, J.D.; Hu, W.P.; Wu, K.; Xu, G.Q.; Wee, A.T.S.; Chen, W. Towards single molecule switches. Chem. Soc. Rev. 2015, 44, 2998–3022. [Google Scholar] [CrossRef]
- Kuang, G.; Shi, Z.C.; Yan, L.; Chen, K.Q.; Shang, X.; Liu, P.N.; Lin, N. Negative differential conductance in polyporphyrin oligomers with nonlinear backbones. J. Am. Chem. Soc. 2018, 140, 570–573. [Google Scholar] [CrossRef]
- Yang, X.; Tan, F.; Dong, Y.; Yu, H.; Liu, Y. Transition metal-containing molecular devices: Controllable single-spin negative differential thermoelectric resistance effects under gate voltages. Phys. Chem. Chem. Phys. 2019, 21, 5243–5252. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Daaoub, A.; Sangtarash, S.; Li, X.; Tang, Y.; Zou, Q.; Sadeghi, H.; Liu, S.; Huang, X.; Tan, Z.; et al. Anti-resonance features of destructive quantum interference in single-molecule thiophene junctions achieved by electrochemical gating. Nat. Mater. 2019, 18, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Buerkle, M.; Li, G.; Rostamian, A.; Wang, H.; Wang, Z.; Bowler, D.R.; Miyazaki, T.; Xiang, L.; Asai, Y.; et al. Gate controlling of quantum interference and direct observation of anti-resonances in single molecule charge transport. Nat. Mater. 2019, 18, 357–363. [Google Scholar] [CrossRef] [PubMed]
- McCreery, R.L. Carbon-based molecular junctions for practical molecular electronics. Acc. Chem. Res. 2022, 55, 2766–2779. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Fu, H.; Wang, B.; Cheng, J.; Hu, W.; Yin, B.; Peng, P.; Zhou, S.; Gao, X.; Jia, C.; et al. Dipole-modulated charge transport through PNP-type single-molecule junctions. J. Am. Chem. Soc. 2022, 144, 20797–20803. [Google Scholar] [CrossRef] [PubMed]
- Pei, L.Q.; Horsley, J.R.; Seng, J.W.; Liu, X.; Yeoh, Y.Q.; Yu, M.X.; Wu, X.H.; Abell, A.D.; Zheng, J.F.; Zhou, X.S.; et al. Mechanically induced switching between two discrete conductance states: A potential single-molecule variable resistor. ACS Appl. Mater. Interfaces 2021, 13, 57646–57653. [Google Scholar] [CrossRef]
- Wu, X.; Xiao, S.; Quan, J.; Tian, C.; Gao, G. Perylene-based molecular device: Multifunctional spintronic and spin caloritronic applications. Phys. Chem. Chem. Phys. 2023, 25, 7354–7365. [Google Scholar] [CrossRef]
- Song, Y.; Wang, C.-K.; Chen, G.; Zhang, G.-P. A first-principles study of phthalocyanine-based multifunctional spintronic molecular devices. Phys. Chem. Chem. Phys. 2021, 23, 18760–18769. [Google Scholar] [CrossRef]
- Mu, Y.; Yu, J.; Hu, R.; Wang, C.-H.; Cheng, C.; Hou, B.-P. Ab initio study revealing remarkable oscillatory effects and negative differential resistance in the molecular device of silicon carbide chains. Phys. Chem. Chem. Phys. 2023, 25, 13265–13274. [Google Scholar] [CrossRef]
- Zhang, N.; Lo, W.Y.; Cai, Z.; Li, L.; Yu, L. Molecular rectification tuned by through-space gating effect. Nano Lett. 2017, 17, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Perrin, M.L.; Galan, E.; Eelkema, R.; Thijssen, J.M.; Grozema, F.; van der Zant, H.S. A gate-tunable single-molecule diode. Nanoscale 2016, 8, 8919–8923. [Google Scholar] [CrossRef] [PubMed]
- Xin, N.; Hu, C.; Sabea, H.A.; Zhang, M.; Zhou, C.; Meng, L.; Jia, C.; Gong, Y.; Li, Y.; Ke, G.; et al. Tunable symmetry-breaking-induced dual functions in stable and photoswitched single-molecule junctions. J. Am. Chem. Soc. 2021, 143, 20811–20817. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, T. Recent progress in the development of molecular-scale electronics based on photoswitchable molecules. J. Mater. Chem. C 2020, 8, 821–848. [Google Scholar] [CrossRef]
- Liu, W.; Yang, S.; Li, J.; Su, G.; Ren, J.C. One molecule, two states: Single molecular switch on metallic electrodes. WIREs Comput. Mol. Sci. 2021, 11, e1511. [Google Scholar] [CrossRef]
- Goulet-Hanssens, A.; Eisenreich, F.; Hecht, S. Enlightening materials with photoswitches. Adv. Mater. 2020, 32, 1905966. [Google Scholar] [CrossRef] [PubMed]
- Irie, M.; Fukaminato, T.; Matsuda, K.; Kobatake, S. Photochromism of diarylethene molecules and crystals: Memories, switches, and actuators. Chem. Rev. 2014, 114, 12174–12277. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Jeong, H.; Hwang, W.-T.; Jang, Y.; Sysoiev, D.; Scheer, E.; Huhn, T.; Min, M.; Lee, H.; Lee, T. Reversible switching phenomenon in diarylethene molecular devices with reduced graphene oxide electrodes on flexible substrates. Adv. Funct. Mater. 2015, 25, 5918–5923. [Google Scholar] [CrossRef]
- Koo, J.; Jang, Y.; Martin, L.; Kim, D.; Jeong, H.; Kang, K.; Lee, W.; Kim, J.; Hwang, W.-T.; Xiang, D.; et al. Unidirectional real-time photoswitching of diarylethene molecular monolayer junctions with multilayer graphene electrodes. ACS Appl. Mater. Interfaces 2019, 11, 11645–11653. [Google Scholar] [CrossRef]
- Kudernac, T.; van der Molen, S.J.; van Wees, B.J.; Feringa, B.L. Uni- and bi-directional light-induced switching of diarylethenes on gold nanoparticles. Chem. Commun. 2006, 34, 3597–3599. [Google Scholar] [CrossRef]
- Jia, C.; Migliore, A.; Xin, N.; Huang, S.; Wang, J.; Yang, Q.; Wang, S.; Chen, H.; Wang, D.; Feng, B.; et al. Covalently bonded single-molecule junctions with stable and reversible photoswitched conductivity. Science 2016, 352, 1443–1445. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-B.; Zhang, S.; Bai, E.; Gao, X.; Wang, J.; Qi, J.; Liu, J.; Zhao, J.; Zhang, L.; Yoon, J. Future-oriented advanced diarylethene photoswitches: From molecular design to spontaneous assembly systems. Adv. Mater. 2022, 34, 2108289. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Xin, N.; Hu, C.; Sabea, H.A.; Zhang, M.; Jiang, H.; Ji, Y.; Jia, C.; Yan, Z.; Zhang, Q.; et al. Dual-gated single-molecule field-effect transistors beyond Moore’s law. Nat. Commun. 2022, 13, 1410. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Mennucci, B.; Tomasi, J.; Cammi, R.; Cheeseman, J.R.; Frisch, M.J.; Devlin, F.J.; Gabriel, S.; Stephens, P.J. Polarizable continuum model (PCM) calculations of solvent effects on optical rotations of chiral molecules. J. Phys. Chem. A 2002, 106, 6102–6113. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, T. Efficient evaluation of electrostatic potential with computerized optimized code. Phys. Chem. Chem. Phys. 2021, 23, 20323–20328. [Google Scholar] [CrossRef] [PubMed]
- Lu, T. Multiwfn; Version 3.8 (dev); Beijing Kein Research Center for Natural Science: Beijing, China, 2023; p. 252. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- QuantumATK, Version P-2019.03. Available online: https://www.synopsys.com/silicon/quantumatk.html (accessed on 20 August 2019).
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. II. Operators for fast iterative diagonalization. Phys. Rev. B 1991, 43, 8861–8869. [Google Scholar] [CrossRef]
- Büttiker, M.; Imry, Y.; Landauer, R.; Pinhas, S. Generalized many-channel conductance formula with application to small rings. Phys. Rev. B 1985, 31, 6207–6215. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Transferred Electrons a (e−) | |||
|---|---|---|---|
| Molecule | X1→Switch b | Switch→X2 b | X1→X2 b |
| M1c | 0.353 | 0.958 | 1.032 |
| M1o | 0.033 | 1.505 | 1.338 |
| M2c | 0.279 | −0.273 | −0.000 |
| M2o | 0.020 | −0.025 | −0.002 |
| M3c | −0.973 | 0.973 | −0.001 |
| M3o | −0.807 | 1.168 | 0.005 |
| M1 | M2 | M3 | |
|---|---|---|---|
| T0 (closed form) | 1.68 × 10−4 | 1.26 × 10−3 | 2.03 × 10−5 |
| T0 (open form) | 6.76 × 10−7 | 4.60 × 10−6 | 1.22 × 10−7 |
| SRmax | 6319 | 1261 | 3381 |
| RRmax (closed form) | 58.4 | 1.0 | 4.3 |
| RRmax (open form) | 10.6 | 1.3 | 19.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Z.; Cui, P.; Deng, M. Rational Design of Photocontrolled Rectifier Switches in Single-Molecule Junctions Based on Diarylethene. Molecules 2023, 28, 7158. https://doi.org/10.3390/molecules28207158
Wu Z, Cui P, Deng M. Rational Design of Photocontrolled Rectifier Switches in Single-Molecule Junctions Based on Diarylethene. Molecules. 2023; 28(20):7158. https://doi.org/10.3390/molecules28207158
Chicago/Turabian StyleWu, Ziye, Peng Cui, and Mingsen Deng. 2023. "Rational Design of Photocontrolled Rectifier Switches in Single-Molecule Junctions Based on Diarylethene" Molecules 28, no. 20: 7158. https://doi.org/10.3390/molecules28207158
APA StyleWu, Z., Cui, P., & Deng, M. (2023). Rational Design of Photocontrolled Rectifier Switches in Single-Molecule Junctions Based on Diarylethene. Molecules, 28(20), 7158. https://doi.org/10.3390/molecules28207158