
Selective Schiff Base Formation of Group 9 Organometallic Complexes with Functionalized Spirobifluorene Ligands



Abstract
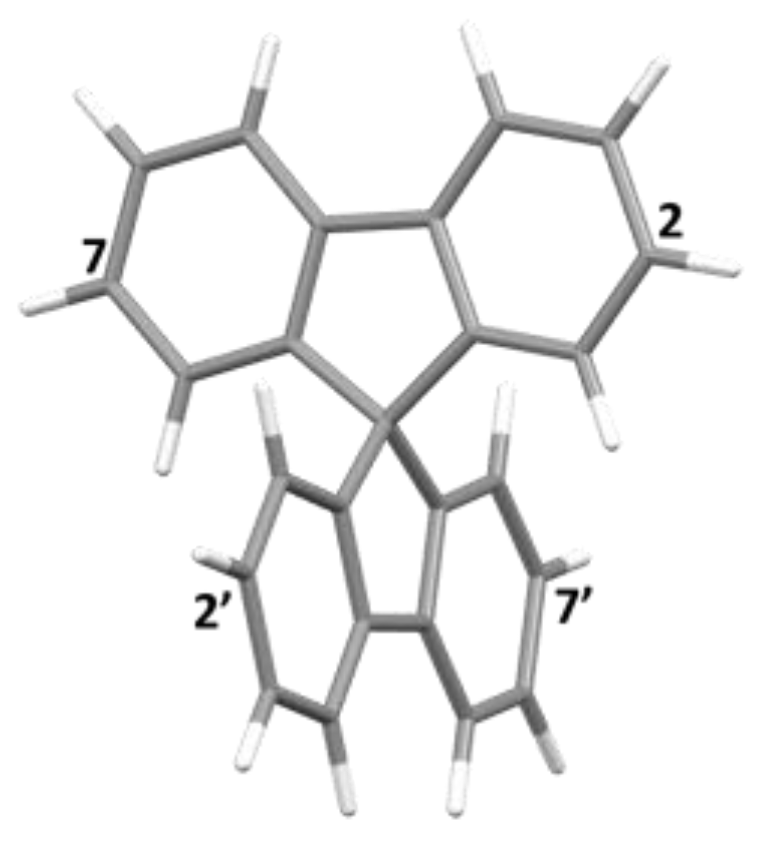
:1. Introduction
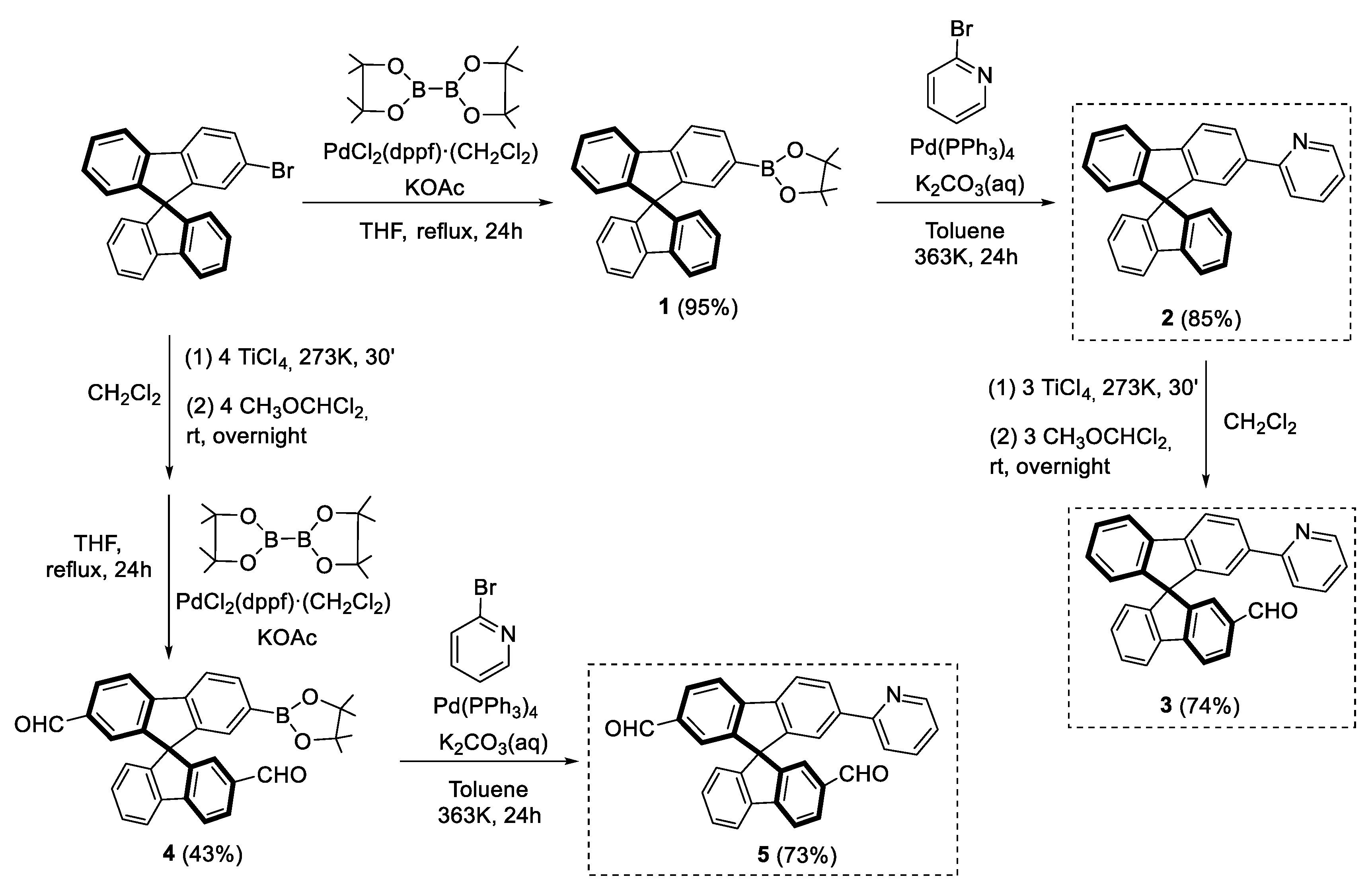
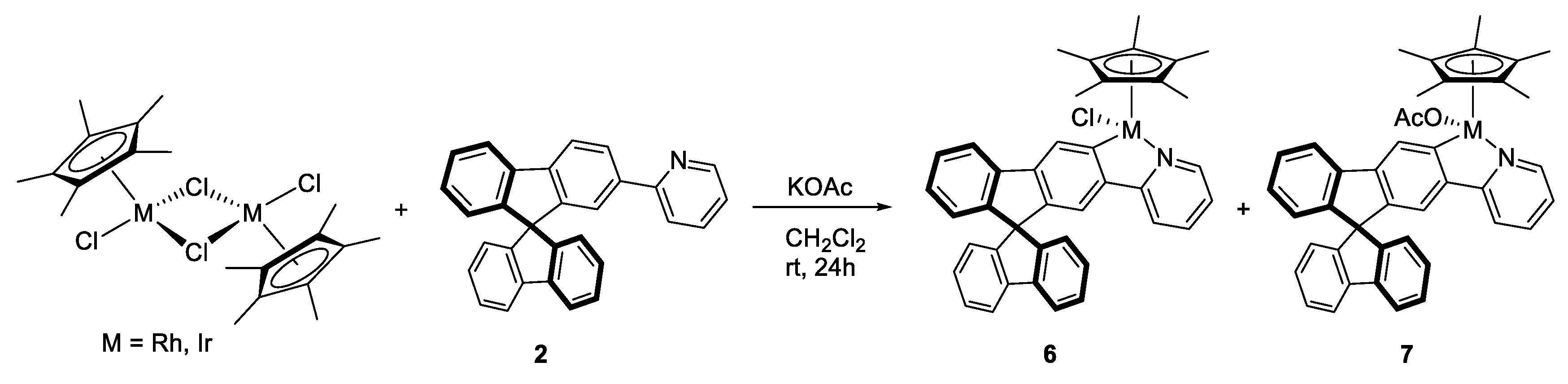
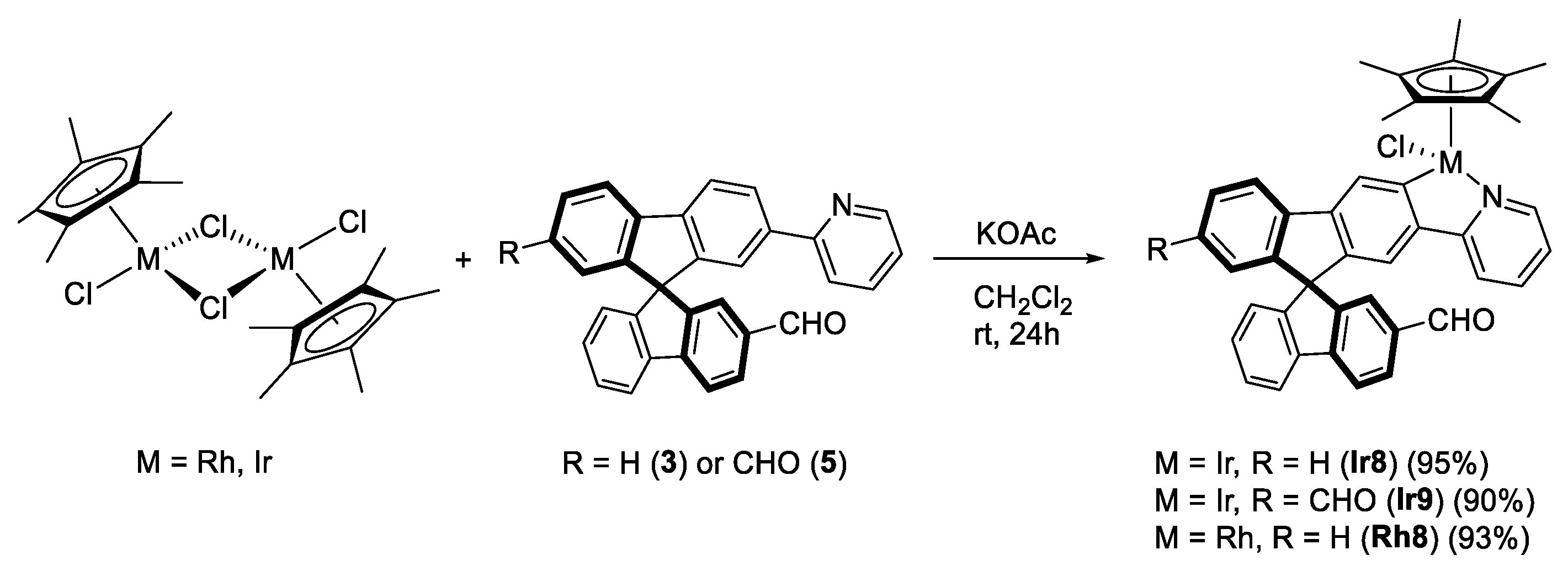
2. Results and Discussion
3. Experimental Section
3.1. Materials and Methods
3.2. Synthesis of the Organic Compounds
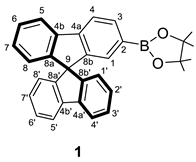
3.2.1. Synthesis of 2-(9,9′-spirobi[fluoren]-2-yl)-4,4,5,5-tetramethyl-3,2-dioxa-1-boronate (1)
3.2.2. Synthesis of 2-(9,9′-spirobi[fluoren]-2-yl)pyridine (2)

3.2.3. Synthesis of (±) 2′-(pyridin-2-yl)-9,9′-spirobifluorene-2-carbaldehyde (3)

3.2.4. Synthesis of (±) 7-(4,4,5,5-tetramethyl-3,2-dioxa-1-boronate)-9,9′-spirobifluorene-2,2′-dicarbaldehyde (4)

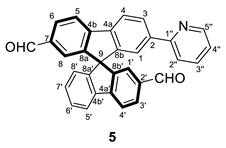
3.2.5. Synthesis of (±) 7′-(pyridin-2-yl)-9,9′-spirobifluorene-2,2′-dicarbaldehyde (5)
3.3. Synthesis of the Organometallic Complexes
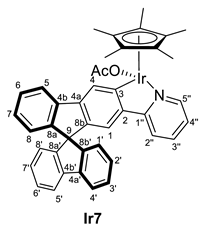
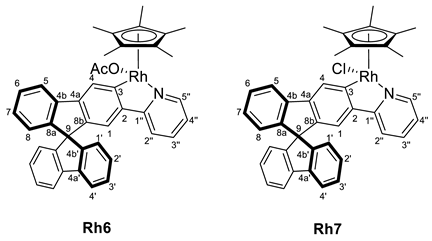
3.3.1. Synthesis of [MCp*Cl(2-py-SBF)] (M = Rh (Rh6), Ir (Ir6)), and [MCp*(OCOCH3)(2-py-SBF)] (M = Rh (Rh7), Ir (Ir7))

3.3.2. Synthesis of (R,M)/(R,P)-[MCp*Cl(2‘-CHO-2-py-SBF)] (M = Rh (Rh8), Ir (Ir8))
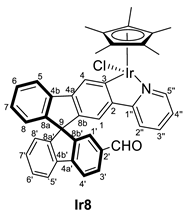

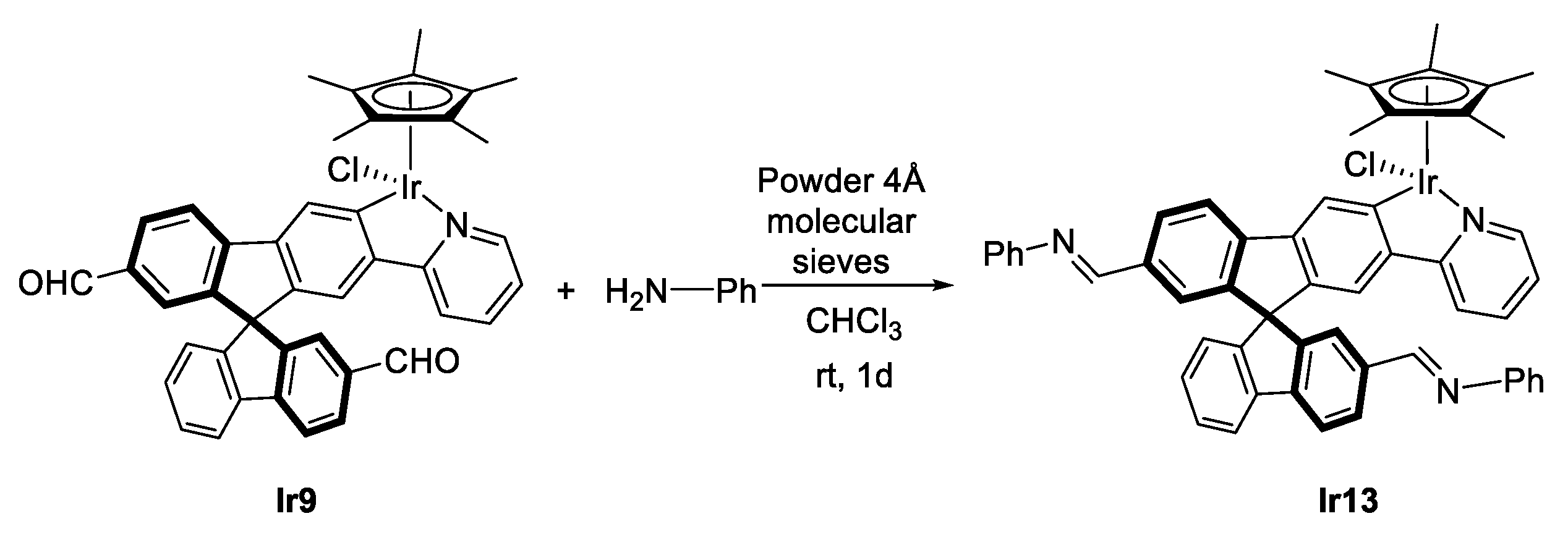
3.3.3. Synthesis of (R,M)/(R,P)-[IrCp*Cl(2‘,7-diCHO-2-py-SBF)] (Ir9)

3.4. Reactivity of Complexes Ir8, Rh8 and Ir9 towards Amines
3.4.1. General Procedure for the Reactivity of (R,M)/(R,P)-[MCp*Cl(2‘-CHO-2-py-SBF)] (M = Rh (Rh8), Ir (Ir8)) or (R,M)/(R,P)-[IrCp*Cl(2‘,7-diCHO-2-py-SBF)] (Ir9) with amines
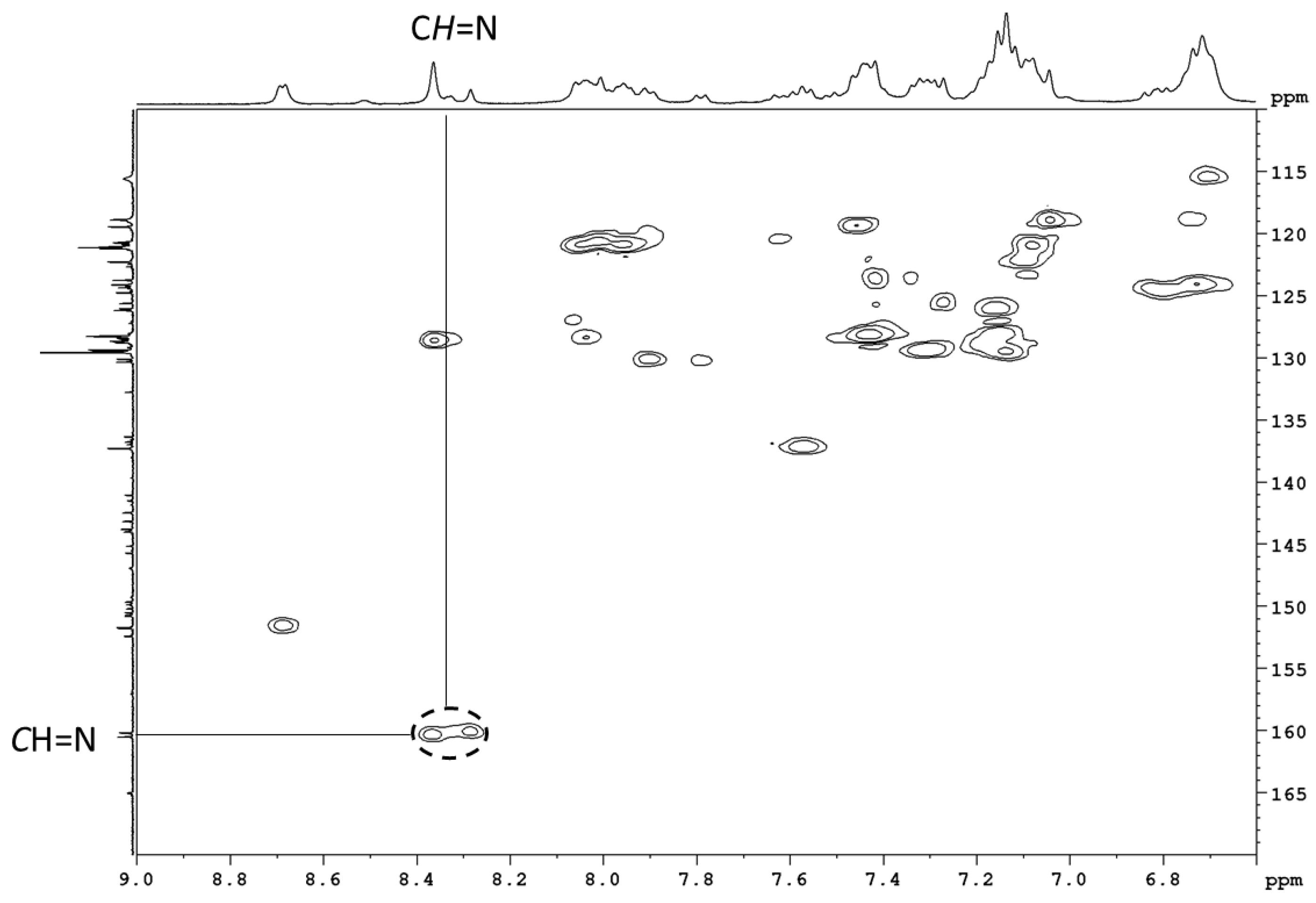
3.4.2. Selected Characterization Data of Imine Complexes
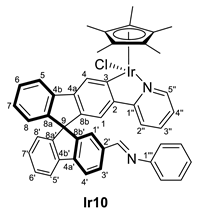
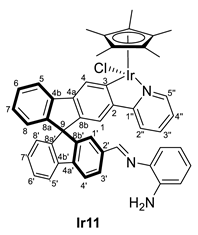

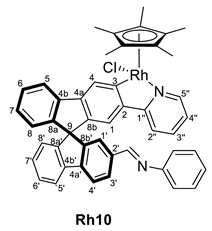

3.5. Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Lawrence, S.A. Amines: Synthesis, Properties and Applications; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- García-Calvo, V.; García-Calvo, J.; Fernández-Espinosa, I.; Carbayo, A.; Rojo, M.J.; Rodríguez, M.T.; García-Herbosa, G.; Torroba, T.; Cuevas, J.V. Luminescent complexes of iridium(iii) with aliphatic amines and detection of biogenic amines. Inorg. Chim. Acta 2020, 504, 119409. [Google Scholar] [CrossRef]
- Almeida, C.M.R.; Magalhães, J.M.C.S.; Barroso, M.F.; Durães, L. Latest advances in sensors for optical detection of relevant amines: Insights into lanthanide-based sensors. J. Mater. Chem. C 2022, 10, 15263–15276. [Google Scholar] [CrossRef]
- Lee, B.; Scopelliti, R.; Severin, K. A molecular probe for the optical detection of biogenic amines. Chem. Commun. 2011, 47, 9639–9641. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-P.; Peng, H.-L.; Ye, B.-H. Chiral sensor for enantiomeric purity of amines, amino alcohols and amino esters based on bis-cyclometalated Ir(III) complex using 1H NMR spectroscopy. Inorg. Chim. Acta 2018, 482, 691–697. [Google Scholar] [CrossRef]
- De los Santos, Z.A.; Ding, R.; Wolf, C. Quantitative chirality sensing of amines and amino alcohols via Schiff base formation with a stereodynamic UV/CD probe. Org. Biommol. Chem. 2016, 14, 1934–1939. [Google Scholar] [CrossRef]
- Pilicer, S.L.; Mancinelli, M.; Mazzanti, A.; Wolf, C. Predictive chirality sensing via Schiff base formation. Org. Biommol. Chem. 2019, 17, 6699–6705. [Google Scholar] [CrossRef]
- Wang, W.; Xia, X.; Bian, G.; Song, L. A chiral sensor for recognition of varied amines based on 19F NMR signals of newly designed rhodium complexes. Chem. Commun. 2019, 55, 6098–6101. [Google Scholar] [CrossRef]
- Wang, J.; Liu, H.-B.; Tong, Z.; Ha, C.-S. Fluorescent/luminescent detection of natural amino acids by organometallic systems. Coord. Chem. Rev. 2015, 303, 139–184. [Google Scholar] [CrossRef]
- Pei, J.; Ni, J.; Zhou, X.-H.; Cao, X.-Y.; Lai, Y.-H. Head-to-Tail Regioregular Oligothiophene-Functionalized 9,9‘-Spirobifluorene Derivatives. 1. Synthesis. J. Org. Chem. 2002, 67, 4924–4936. [Google Scholar] [CrossRef]
- Saragi, T.P.I.; Spehr, T.; Siebert, A.; Fuhrmann-Lieker, T.; Salbeck, J. Spiro Compounds for Organic Optoelectronics. Chem. Rev. 2007, 107, 1011–1065. [Google Scholar] [CrossRef]
- Guo, X.; Lv, F.; Zhao, Z.; Gu, J.; Qu, B.; Xiao, L.; Chen, Z. Spirobifluorene-based oligopyridine derivatives as electron-transporting materials for green phosphorescent organic light-emitting diodes. Org. Electron. 2020, 77, 105498. [Google Scholar] [CrossRef]
- Rajasekar, M. Spirobifluorene derivatives and their biomaterial applications: Current trends. J. Mol. Struct. 2022, 1255, 132406. [Google Scholar] [CrossRef]
- Vaghi, L.; Rizzo, F. The Future of Spirobifluorene-Based Molecules as Hole-Transporting Materials for Solar Cells. Solar RRL 2023, 7, 2201108. [Google Scholar] [CrossRef]
- Poriel, C.; Sicard, L.; Rault-Berthelot, J. New generations of spirobifluorene regioisomers for organic electronics: Tuning electronic properties with the substitution pattern. Chem. Commun. 2019, 55, 14238–14254. [Google Scholar] [CrossRef] [PubMed]
- Talavera, M.; Pereira-Cameselle, R.; Peña-Gallego, Á.; Vázquez-Carballo, I.; Prieto, I.; Alonso-Gómez, J.L.; Bolaño, S. Optical and electrochemical properties of spirobifluorene iridanaphthalene complexes. Dalton Trans. 2023, 52, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Ozcelik, A.; Peña-Gallego, M.d.l.Á.; Pereira-Cameselle, R.; Alonso-Gómez, J.L. Design and synthesis of chiral spirobifluorenes. Chirality 2020, 32, 464–473. [Google Scholar] [CrossRef]
- Ventura, B.; Barbieri, A.; Degli Esposti, A.; Seneclauze, J.B.; Ziessel, R. Spirobifluorene Bridged Ir(III) and Os(II) Polypyridyl Arrays: Synthesis, Photophysical Characterization, and Energy Transfer Dynamics. Inorg. Chem. 2012, 51, 2832–2840. [Google Scholar] [CrossRef]
- Ozcelik, A.; Pereira-Cameselle, R.; Peña-Gallego, Á.; Alonso-Gómez, J.L. A Tetracyanobutadiene Spirobifluorene: Synthesis, Enantiomeric Resolution and Chiroptical Properties. Eur. J. Org. Chem. 2022, 2022, e202101333. [Google Scholar] [CrossRef]
- Wei, C.; Wu, J.; Feng, X.; Yang, Z.; Zhang, J.; Ji, H. A spirobifluorene-based water-soluble imidazolium polymer for luminescence sensing. New J. Chem. 2021, 45, 13021–13028. [Google Scholar] [CrossRef]
- Tang, G.; Chen, S.S.Y.; Shaw, P.E.; Hegedus, K.; Wang, X.; Burn, P.L.; Meredith, P. Fluorescent carbazole dendrimers for the detection of explosives. Polym. Chem. 2011, 2, 2360–2368. [Google Scholar] [CrossRef]
- Douthwaite, R.E.; Guillaume, N.; Whitwood, A.C. Synthesis and structures of metallocene complexes of iron(II) and ruthenium(II) derived from 9,9′-spirobifluorene. J. Organomet. Chem. 2006, 691, 711–716. [Google Scholar] [CrossRef]
- Nahaei, A.; Mandegani, Z.; Chamyani, S.; Fereidoonnezhad, M.; Shahsavari, H.R.; Kuznetsov, N.Y.; Nabavizadeh, S.M. Half-Sandwich Cyclometalated RhIII Complexes Bearing Thiolate Ligands: Biomolecular Interactions and In Vitro and In Vivo Evaluations. Inorg. Chem. 2022, 61, 2039–2056. [Google Scholar] [CrossRef] [PubMed]
- Lamansky, S.; Djurovich, P.; Murphy, D.; Abdel-Razzaq, F.; Kwong, R.; Tsyba, I.; Bortz, M.; Mui, B.; Bau, R.; Thompson, M.E. Synthesis and Characterization of Phosphorescent Cyclometalated Iridium Complexes. Inorg. Chem. 2001, 40, 1704–1711. [Google Scholar] [CrossRef] [PubMed]
- Benavent, L.; Boudreault, P.-L.T.; Esteruelas, M.A.; López, A.M.; Oñate, E.; Tsai, J.-Y. Phosphorescent Iridium(III) Complexes with a Dianionic C,C′,N,N′-Tetradentate Ligand. Inorg. Chem. 2020, 59, 12286–12294. [Google Scholar] [CrossRef]
- Talavera, M.; Bolaño, S.; Bravo, J.; Castro, J.; García-Fontán, S. Cyclometalated Iridium Complexes from Intramolecular C–H Activation of [IrCp*Cl{═C(OMe)CH═C(CH3)R}L] (R = CH3, Ph; L = PPh2Me, PMe3). Organometallics 2013, 32, 7241–7244. [Google Scholar] [CrossRef]
- Talavera, M.; Pereira-Cameselle, R.; Bolaño, S. Rhodafuran from a methoxy(alkenyl)carbene by the rhoda-1,3,5-hexatriene route. Dalton Trans. 2018, 47, 9064–9071. [Google Scholar] [CrossRef]
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals, 3rd ed.; Butterworth/Heinemann: London, UK; Oxford, UK, 1988. [Google Scholar]
- White, C.; Yates, A.; Maitlis, P.M.; Heinekey, D.M. (η5-Pentamethylcyclopentadienyl)Rhodium and -Iridium Compounds. In Inorganic Syntheses; Grimes, R.N., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1992; pp. 228–234. [Google Scholar]
- Shen, Y.; Zhang, Z.; Liu, H.; Yan, Y.; Zhang, S.; Yang, B.; Ma, Y. Highly Efficient Orange-Red/Red Excimer Fluorescence from Dimeric π–π Stacking of Perylene and Its Nanoparticle Applications. J. Phys. Chem. C 2019, 123, 13047–13056. [Google Scholar] [CrossRef]
- Liang, J.; Dong, L.; Qian, F.; Kong, Y.; Wang, M.; Xu, X.; Shao, X.; Li, Z. A bench-stable reagent for C-4 selective deuteriodifluoromethylation of azines. Chin. Chem. Lett. 2022, 33, 4817–4821. [Google Scholar] [CrossRef]
- SAINT, Data Integration Software Package; Version 6.01; Bruker Analytical X-ray Systems Inc.: Madison, WI, USA, 1997.
- Bruker, AXS. SADABS, Program for Empirical Absorption Correction of Area Detector Data; Bruker Analytical X-ray Systems Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | Ir6 | Rh6 | Angle | Ir6 | Rh6 |
|---|---|---|---|---|---|
| a M-CT | 1.823 | 1.833 | C(22)-M(1)-N(1) | 77.96(13) | 78.83(9) |
| C(15)-C(21) | 1.446(5) | 1.459(3) | C(21)-C(22)-M(1) | 115.6(2) | 115.32(17) |
| M(1)-Cl(1) | 2.3873(9) | 2.3881(7) | C(22)-M(1)-Cl(1) | 87.92(9) | 89.29(7) |
| C(21)-C(22) | 1.413(5) | 1.418(3) | N(1)-M(1)-Cl(1) | 88.58(8) | 91.46(6) |
| M(1)-C(22) | 2.047(3) | 2.027(2) | C(15)-N(1)-M(1) | 116.8(2) | 116.23(16) |
| M(1)-N(1) | 2.097(3) | 2.094(2) | N(1)-C(15)-C(21) | 114.1(3) | 113.9(2) |
| N(1)-C(15) | 1.361(4) | 1.358(3) |

| Entry | Metal Complex | R | Complex: Amine Ratio | Temperature (K) | Product | Conversion (%) a |
|---|---|---|---|---|---|---|
| 1 | Ir8 | Ph | 1:4 | 298 | Ir10 | 99 |
| 2 | Ir8 | o-NH2-C6H4 | 1:1.2 | 298 | Ir11 | 98 |
| 3 | Ir8 | CH2CH2NH2 | 1:2 | 298 | Ir12 | 0 |
| 4 | Ir8 | CH2CH2NH2 | 1:2 | 343 | Ir12 | 0 |
| 5 | Rh8 | Ph | 1:3 | 298 | Rh10 | 0 |
| 6 | Rh8 | Ph | 1:3 | 343 | Rh10 | 99 |
| 7 | Rh8 | CH2CH2NH2 | 1:2 | 298 | Rh12 | 99 |
| Complex | 1H NMR (ppm) a | 13C{1H} NMR (ppm) a | IR (cm−1) b | ||
|---|---|---|---|---|---|
| δ (CH=N) | δ (CH=N) | δ (C-CH=N) | δ (Ir-C) | ν (C=N) | |
| Ir10 | 8.40, 8.29 | 160.5, 160.2 | 136.7 | 164.4, 164.2 | 1601 |
| Ir11 | 8.41 (major) 8.37 (minor) | 157.8 | 137.2 | 164.1 | 1601 |
| Ir13 | 8.37, 8.36 (2H), 8.31 | 160.5, 160.3 (2C), 160.1 | 136.8, 136.5, 36.4 (C2′ + C7) | 164.4, 164.3 | 1600 |
| Rh10 | 8.36, 8.28 | 160.5, 160.2 | 136.8 and 136.4 | 179.7, 176.6 (both d, J = 32 Hz) | 1600 |
| Rh12 | 8.10, 8.03 | 162.1 | 136.7 | 179.8 (br) | 1599 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cid-Seara, K.M.; Pereira-Cameselle, R.; Bolaño, S.; Talavera, M. Selective Schiff Base Formation of Group 9 Organometallic Complexes with Functionalized Spirobifluorene Ligands. Molecules 2023, 28, 7155. https://doi.org/10.3390/molecules28207155
Cid-Seara KM, Pereira-Cameselle R, Bolaño S, Talavera M. Selective Schiff Base Formation of Group 9 Organometallic Complexes with Functionalized Spirobifluorene Ligands. Molecules. 2023; 28(20):7155. https://doi.org/10.3390/molecules28207155
Chicago/Turabian StyleCid-Seara, Krystal M., Raquel Pereira-Cameselle, Sandra Bolaño, and Maria Talavera. 2023. "Selective Schiff Base Formation of Group 9 Organometallic Complexes with Functionalized Spirobifluorene Ligands" Molecules 28, no. 20: 7155. https://doi.org/10.3390/molecules28207155
APA StyleCid-Seara, K. M., Pereira-Cameselle, R., Bolaño, S., & Talavera, M. (2023). Selective Schiff Base Formation of Group 9 Organometallic Complexes with Functionalized Spirobifluorene Ligands. Molecules, 28(20), 7155. https://doi.org/10.3390/molecules28207155