
Paving the Way towards Sustainability of Polyurethanes: Synthesis and Properties of Terpene-Based Diisocyanate

 ,
, 

Abstract
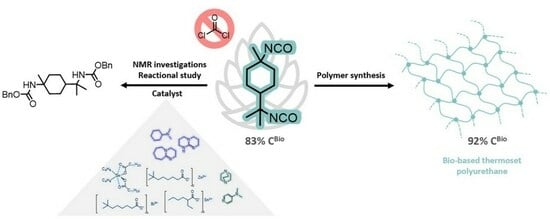
:
1. Introduction
2. Results and Discussion
2.1. PMDI Synthesis
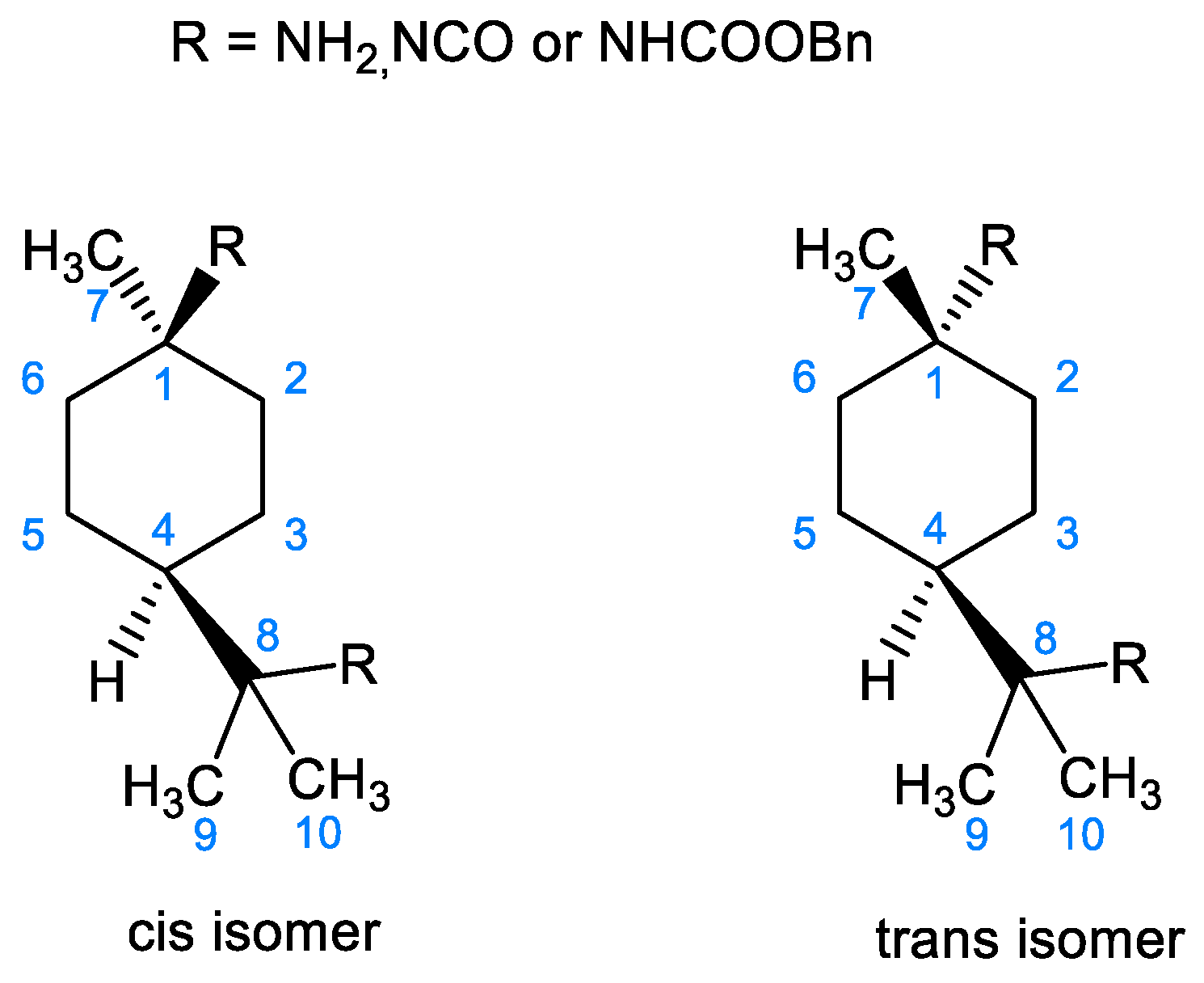
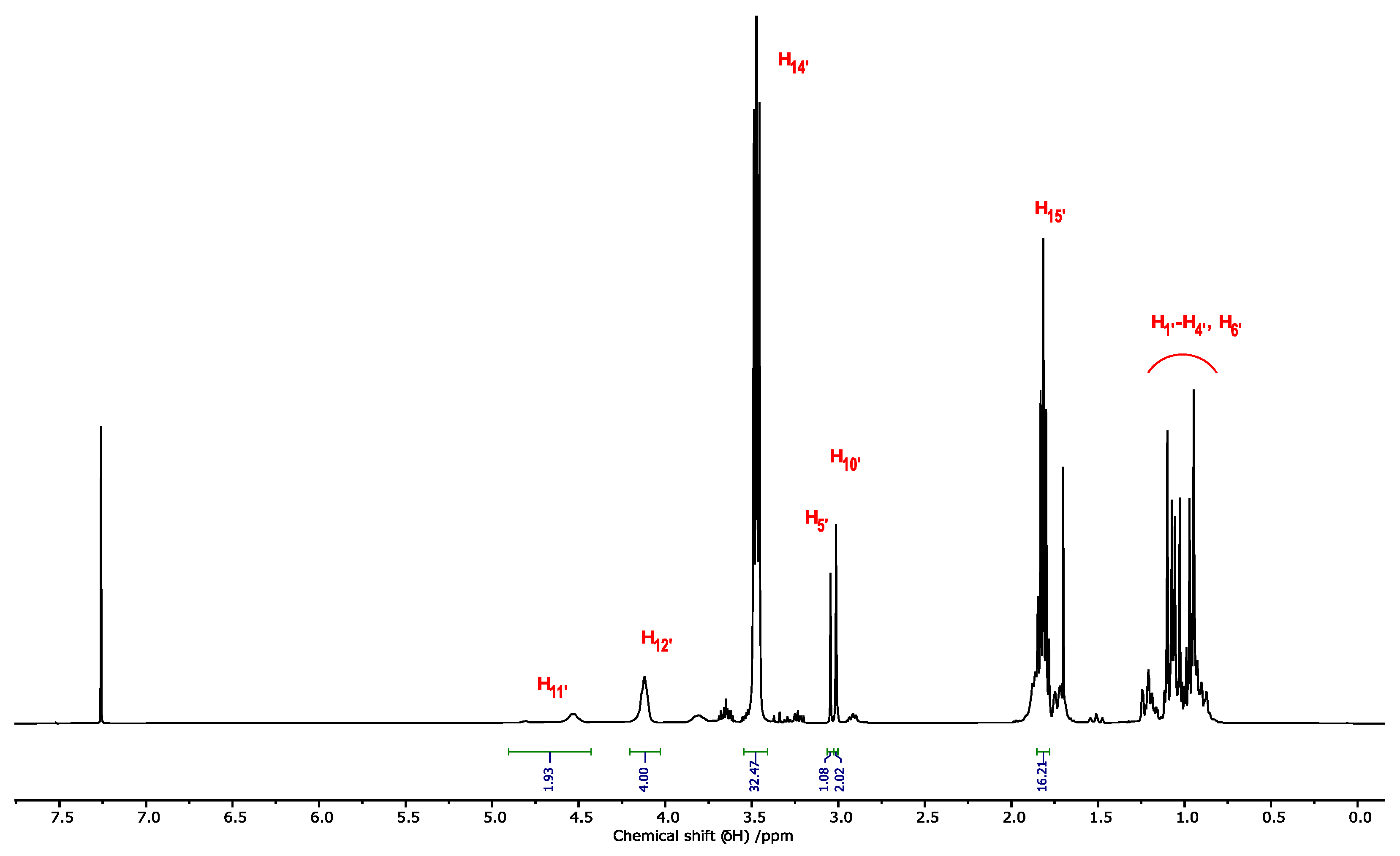
2.2. NMR Characterization of PMDA, PMDI, and p-Menthane-1,8-Dicarbamate

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1H | R = NH2 (Purified) | R = NCO | R = NHCOOBn | |||
|---|---|---|---|---|---|---|
| cis | trans | cis | trans | cis | trans | |
| 2/6 ax | 1.34 | 1.27 | 1.37 | 1.63 | 1.28 | 1.52 |
| 2/6 eq | 1.51 | 1.61 | 1.85 | 1.89 | 2.17 | 2.00 |
| 3/5 ax | 1.62 | 1.68 | 1.72 | 1.76 | 1.55 | 1.65 |
| 3/5 eq | 1.25 | 1.12 | 1.39 | 1.22 | 1.15 | 1.19 |
| 4 | 1.04 | 1.10 | 1.28 | 1.30 | 1.87 | 1.86 |
| 7 | 1.08 | 1.05 | 1.36 | 1.34 | 1.33 | 1.34 |
| 9/10 | 1.04 | 1.04 | 1.33 | 1.32 | 1.25 | 1.27 |
| 1-NH | 4.53 | 4.75 | ||||
| 8-NH | 4.67 | 4.68 | ||||
| 1-CH2 (Bn) | 5.06 | nd | ||||
| 8-CH2 (Bn) | 5.04 | nd | ||||
| 13C | R = NH2 (Purified) | R = NCO | R = NHCOOBn | |||
|---|---|---|---|---|---|---|
| cis | trans | cis | trans | cis | trans | |
| 1 | 47.8 | 49.0 | 58.1 | 57.6 | 52.0 | 52.6 |
| 2/6 | 40.1 | 41.9 | 39.5 | 40.1 | 36.8 | 37.3 |
| 3/5 | 22.8 | 24.5 | 23.4 | 24.0 | 22.6 | 23.4 |
| 4 | 49.1 | 49.2 | 47.8 | 47.5 | 44.3 | 44.9 |
| 7 | 33.1 | 25.7 | 31.6 | 25.5 | 28.0 | 21.9 |
| 8 | 51.3 | 51.2 | 61.0 | 60.7 | 55.5 | 55.5 |
| 9/10 | 28.5 | 28.5 | 28.1 | 28.5 | 24.5 | 24.5 |
| 1-NCO | 122.2 | 122.6/122.8 | 154.7 | nd | ||
| 8-NCO | 122.5 | 154.6 | nd | |||
| CH2 (Bn) | 66.1 | nd | ||||
| Cq (Bn) | 136.9 | nd | ||||
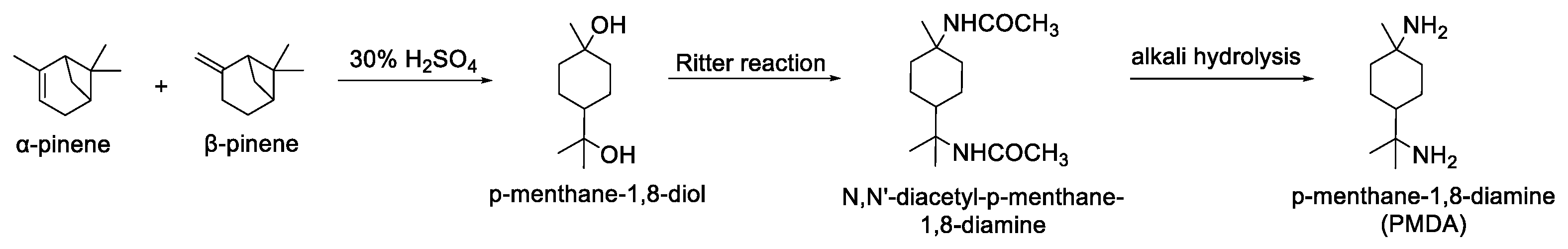
2.3. Influence of Catalyst Nature on the Reactivity of PMDI
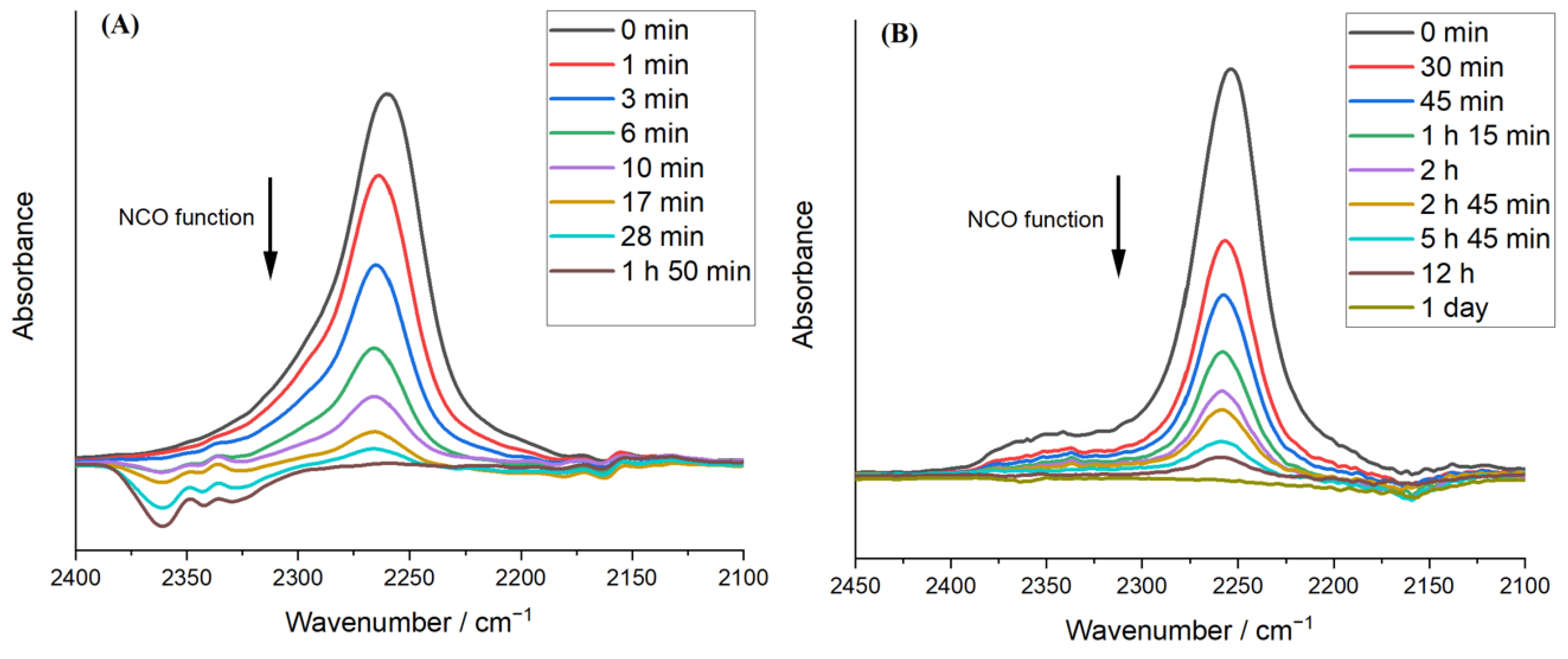
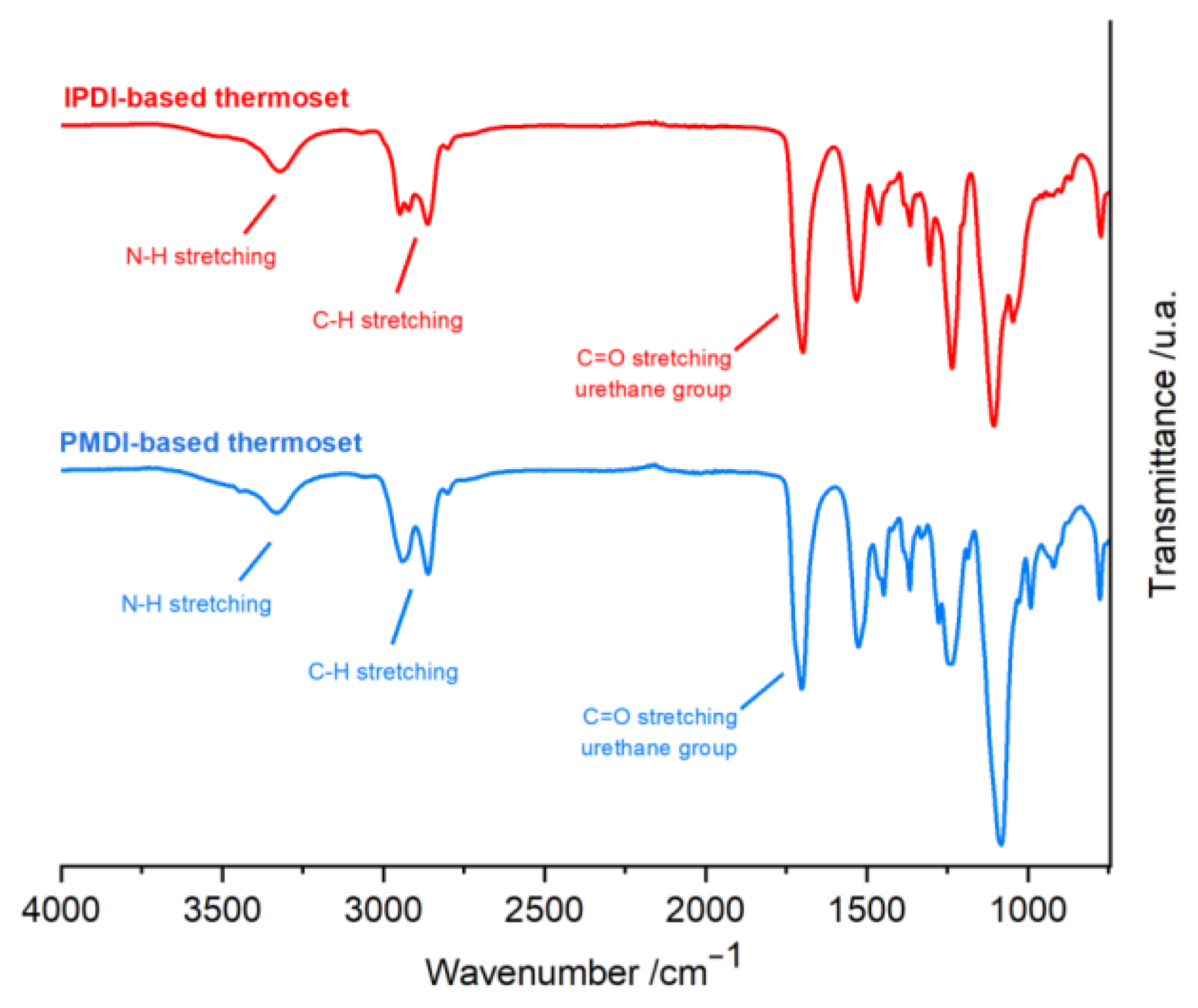
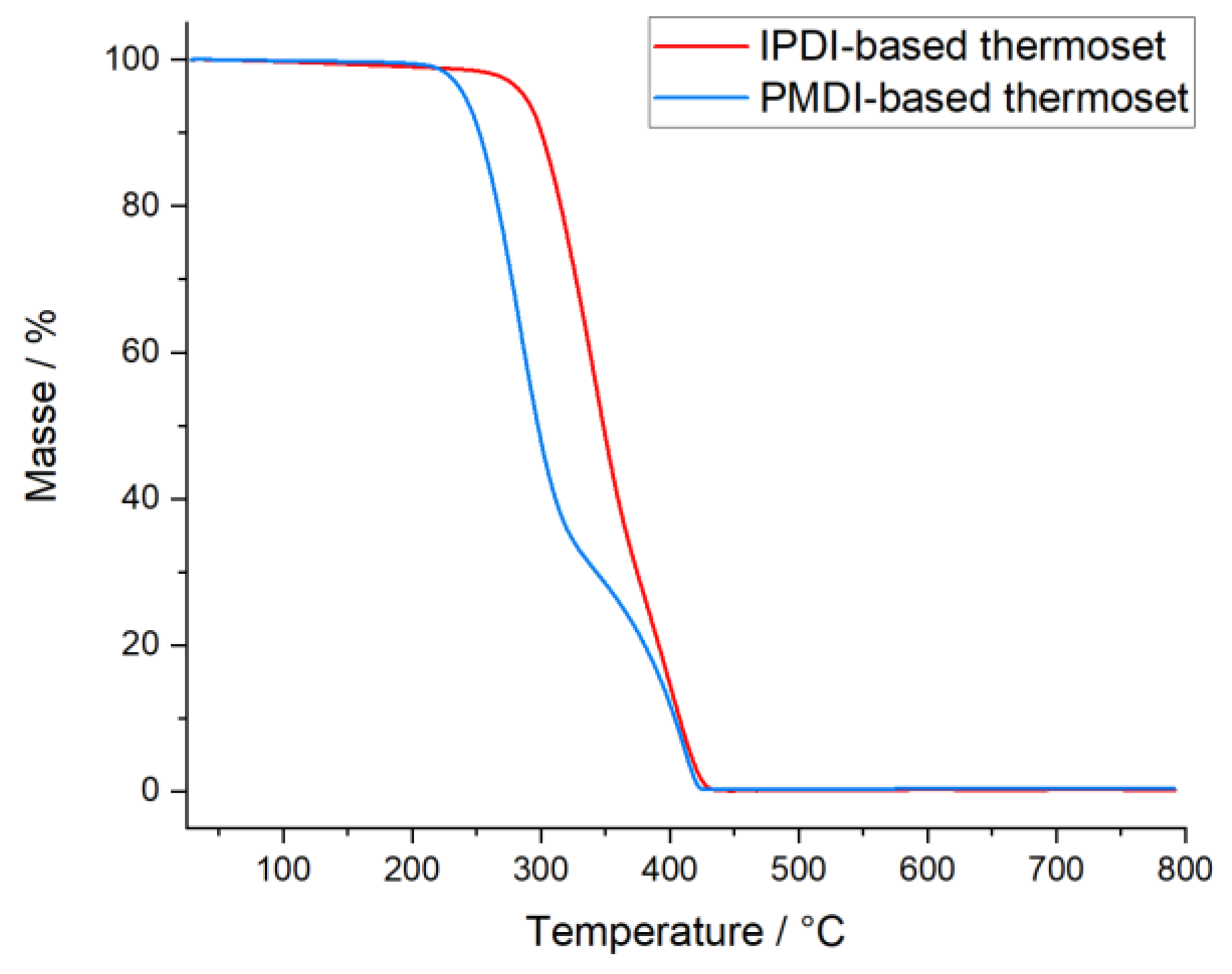
2.4. Synthesis, Characterization, and Reaction Study of PMDI- and IPDI-Based Polyurethanes
3. Materials and Methods
3.1. General Information
3.2. Experimental Section
3.2.1. General Procedure for the Purification of p-Menthane-1,8-diamine (PMDA)
3.2.2. General Procedure for the Synthesis of p-Menthane-1,8-diisocyanate (PMDI)
3.2.3. General Procedure for the Synthesis of p-Menthane-1,8-dicarbamate Monitoring by 1H NMR
3.2.4. General Procedure for the Synthesis of a Polyurethane Thermoset through a Two-Step Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Polyurethane Market Growth, Size, Global Industry Trend 2032. Available online: https://www.factmr.com/report/polyurethane-market (accessed on 12 May 2023).
- Final Report Summary—GRAIL (Glycerol Biorefinery Approach for the Production of High Quality Products of Industrial Value)|FP7|CORDIS|European Commission; European Commission: Luxembourg, 2017.
- Vaswani, S. Process Economics Program Report 283 Bio-Based 1,4-Butanediol; IHS: Santa Clara, CA, USA, 2012. [Google Scholar]
- Thengumpilil, N.B.K.; Penumarthy, V.; Ayyagari, A.L. Process for the Preparation of a Monoglyceride. U.S. Patent US6500974B2, 31 December 2002. [Google Scholar]
- Mutlu, H.; Meier, M.A.R. Castor Oil as a Renewable Resource for the Chemical Industry. Eur. J. Lipid Sci. Technol. 2010, 112, 10–30. [Google Scholar] [CrossRef]
- Montero De Espinosa, L.; Meier, M.A.R. Plant Oils: The Perfect Renewable Resource for Polymer Science?! Eur. Polym. J. 2011, 47, 837–852. [Google Scholar] [CrossRef]
- Petrovic, Z.S. Polyurethanes from Vegetable Oils. Polym. Rev. 2008, 48, 109–155. [Google Scholar] [CrossRef]
- Peyrton, J.; Chambaretaud, C.; Avérous, L. New Insight on the Study of the Kinetic of Biobased Polyurethanes Synthesis Based on Oleo-Chemistry. Molecules 2019, 24, 4332. [Google Scholar] [CrossRef]
- Gondaliya, A.; Nejad, M. Lignin as a Partial Polyol Replacement in Polyurethane Flexible Foam. Molecules 2021, 26, 2302. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sun, J.-T.; Wang, C.; Liu, S.; Yuan, D.; Zhou, X.; Tan, J.; Stubbs, L.; He, C. High Modulus, Strength, and Toughness Polyurethane Elastomer Based on Unmodified Lignin. ACS Sustain. Chem. Eng. 2017, 5, 7942–7949. [Google Scholar] [CrossRef]
- Sternberg, J.; Sequerth, O.; Pilla, S. Green Chemistry Design in Polymers Derived from Lignin: Review and Perspective. Prog. Polym. Sci. 2021, 113, 101344. [Google Scholar] [CrossRef]
- Henry, C.; Gondaliya, A.; Thies, M.; Nejad, M. Studying the Suitability of Nineteen Lignins as Partial Polyol Replacement in Rigid Polyurethane/Polyisocyanurate Foam. Molecules 2022, 27, 2535. [Google Scholar] [CrossRef]
- Tomaselli, S.; Bertini, F.; Cifarelli, A.; Vignali, A.; Ragona, L.; Losio, S. Antibacterial Properties of Polyurethane Foams Additivated with Terpenes from a Bio-Based Polyol. Molecules 2023, 28, 1966. [Google Scholar] [CrossRef]
- Debuissy, T.; Sangwan, P.; Pollet, E.; Avérous, L. Study on the Structure-Properties Relationship of Biodegradable and Biobased Aliphatic Copolyesters Based on 1,3-Propanediol, 1,4-Butanediol, Succinic and Adipic Acids. Polymer 2017, 122, 105–116. [Google Scholar] [CrossRef]
- Debuissy, T.; Pollet, E.; Avérous, L. Synthesis and Characterization of Fully Biobased Poly(Propylene Succinate-Ran-Propylene Adipate). Analysis of the Architecture-Dependent Physicochemical Behavior. J. Polym. Sci. A Polym. Chem. 2017, 55, 2738–2748. [Google Scholar] [CrossRef]
- Kluge, M.; Pérocheau Arnaud, S.; Robert, T. 1,3-Propanediol and Its Application in Bio-Based Polyesters for Resin Applications. Chem. Afr. 2019, 2, 215–221. [Google Scholar] [CrossRef]
- Sadavarte Nilakshi, V. Difunctional Monomers Starting from Cashew Nut Shell Liquid (CNSL) and High Performance Polymers Therefrom. Ph.D. Thesis, University of Pune, Pune, India, 2012. [Google Scholar]
- Chatterjee, D.; Sadavarte, N.V.; Shingte, R.D.; More, A.S.; Tawade, B.V.; Kulkarni, A.D.; Ichake, A.B.; Avadhani, C.V.; Wadgaonkar, P.P. Step-Growth Polymers from Cashew Nut Shell Liquid (CNSL)-Based Aromatic Difunctional Monomers. In Cashew Nut Shell Liquid; Springer: Berlin/Heidelberg, Germany, 2017; pp. 163–214. [Google Scholar]
- Kuhire, S.S.; Ichake, A.B.; Grau, E.; Cramail, H.; Wadgaonkar, P.P. Synthesis and Characterization of Partially Bio-Based Polyimides Based on Biphenylene-Containing Diisocyanate Derived from Vanillic Acid. Eur. Polym. J. 2018, 109, 257–264. [Google Scholar] [CrossRef]
- Neumann, C.N.D.; Bulach, W.D.; Rehahn, M.; Klein, R. Water-Free Synthesis of Polyurethane Foams Using Highly Reactive Diisocyanates Derived from 5-Hydroxymethylfurfural. Macromol. Rapid Commun. 2011, 32, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Sarkar, A.; Brindisi, M. The Curtius Rearrangement: Mechanistic Insight and Recent Applications in Natural Product Syntheses. Org. Biomol. Chem. 2018, 16, 2006–2027. [Google Scholar] [CrossRef] [PubMed]
- Lemouzy, S.; Delavarde, A.; Lamaty, F.; Bantreil, X.; Pinaud, J.; Caillol, S. Lignin-Based Bisguaiacol Diisocyanate: A Green Route for the Synthesis of Biobased Polyurethanes. Green Chem. J. 2023, 25, 4833–4839. [Google Scholar] [CrossRef]
- Knölker, H.-J.; Braxmeier, T.; Schlechtingen, G. A Novel Method for the Synthesis of Isocyanates Under Mild Conditions. Angew. Chem. Int. Ed. Engl. 1995, 34, 2497–2500. [Google Scholar] [CrossRef]
- Huang, D.; Zhu, S.; Lan, H.; Lin, Z.; Wang, X. Design, Synthesis and Herbicidal Activities of (3R,4R)-4,7,7-Trimethyl-6-Oxabicyclo[3.2.1]Octane-3,4-Diol Derivatives. Ind. Crops Prod. 2019, 129, 24–34. [Google Scholar] [CrossRef]
- Zhu, S.; Xu, S.; Yi, X.; Wang, J.; Zhao, Z.; Jiang, J. High Value-Added Application of Turpentine as a Potential Renewable Source for the Synthesis of Heterocyclic Schiff Base Derivatives of Cis-1,8-p-Menthane-Diamine Serving as Botanical Herbicides. Ind. Crops Prod. 2018, 115, 111–116. [Google Scholar] [CrossRef]
- Yi, X.; Xu, S.; Zhao, Z. Progress on Preparation and Application of P-Menthane-1,8-Diol Monohydrate. In Proceedings of the 3rd International Conference on Material, Mechanical and Manufacturing Engineering, Guangzhou, China, 27–28 June 2015; Atlantis Press: Paris, France, 2015. [Google Scholar]
- Kovals’skaya, S.S.; Kozlov, N.G.; Tikhonova, T.S. Stereoselective Synthesis of N,N′-Diacyl-p-Menthane-1,8-Diamines. Chem. Nat. Compd. 1989, 25, 552–557. [Google Scholar] [CrossRef]
- Dalling, D.K.; Grant, D.M. Carbon-13 Magnetic Resonance. XXI. Steric Interactions in the Methylcyclohexanes. J. Am. Chem. Soc. 1972, 94, 5318–5324. [Google Scholar] [CrossRef]
- Kapp, R.W. Isocyanates. In Encyclopedia of Toxicology; Elsevier: Amsterdam, The Netherlands, 2014; pp. 1112–1131. [Google Scholar]
- Kwon, J.-Y.; Yoo, H.-J.; Kim, H.-D. Effect of Chemical Structure on the Properties of UV-Cured Polyurethane Acrylates Films. Fibers Polym. 2001, 2, 141–147. [Google Scholar] [CrossRef]
- Zhang, Y.; Xia, Z.; Huang, H.; Chen, H. Thermal Degradation of Polyurethane Based on IPDI. J. Anal. Appl. Pyrolysis 2009, 84, 89–94. [Google Scholar] [CrossRef]
- Akram, N.; Zia, K.M.; Saeed, M.; Usman, M.; Khan, W.G. Role of Isophorone Diisocyanate in the Optimization of Adhesion Tendency of Polyurethane Pressure Sensitive Adhesives. J. Appl. Polym. Sci. 2019, 136, 47124. [Google Scholar] [CrossRef]
- Dasgupta, A.; van Ingen, Y.; Guerzoni, M.G.; Farshadfar, K.; Rawson, J.M.; Richards, E.; Ariafard, A.; Melen, R.L. Lewis Acid Assisted Brønsted Acid Catalysed Decarbonylation of Isocyanates: A Combined DFT and Experimental Study. Chem. A Eur. J. 2022, 28, e202201422. [Google Scholar] [CrossRef]
- Lhomme, J. Nouveaux Catalyseurs et Systèmes Catalytiques Appliqués à La Synthèse Du Polyuréthane via La Réaction Isocyanate—Alcool. Ph.D. Thesis, Institut National Des Sciences Appliquées de Lyon, Villeurbanne, France, 2013. [Google Scholar]
- Schwetlick, K.; Noack, R. Kinetics and Catalysis of Consecutive Isocyanate Reactions. Formation of Carbamates, Allophanates and Isocyanurates. J. Chem. Soc. Perkin Trans. 1995, 2, 395. [Google Scholar] [CrossRef]
- Luo, S.-G.; Tan, H.-M.; Zhang, J.-G.; Wu, Y.-J.; Pei, F.-K.; Meng, X.-H. Catalytic Mechanisms of Triphenyl Bismuth, Dibutyltin Dilaurate, and Their Combination in Polyurethane-Forming Reaction. J. Appl. Polym. Sci. 1997, 65, 1217–1225. [Google Scholar] [CrossRef]
- Van Maris, R.; Tamano, Y.; Yoshimura, H.; Gay, K.M. Polyurethane Catalysis by Tertiary Amines. J. Cell. Plast. 2005, 41, 305–322. [Google Scholar] [CrossRef]
- Thiele, L.; Becker, R. Catalytic Mechanisms of Polyurethane Formation. Adv. Urethane Sci. Technol. 1993, 12, 59–85. [Google Scholar]
- Alsarraf, J.; Ammar, Y.A.; Robert, F.; Cloutet, E.; Cramail, H.; Landais, Y. Cyclic Guanidines as Efficient Organocatalysts for the Synthesis of Polyurethanes. Macromolecules 2012, 45, 2249–2256. [Google Scholar] [CrossRef]
- Arnould, P.; Simon, F.; Fouquay, S.; Pardal, F.; Michaud, G.; Gajan, D.; Raynaud, J.; Monteil, V. Harnessing Catalysis Selectivity and Isophorone Diisocyanate Asymmetry for Tailored Polyurethane Prepolymers and Networks. Macromolecules 2022, 55, 3344–3352. [Google Scholar] [CrossRef]
- Schwetlick, K.; Noack, R.; Stebner, F. Three Fundamental Mechanisms of Base-Catalysed Reactions of Isocyanates with Hydrogen-Acidic Compounds. J. Chem. Soc. Perkin Trans. 1994, 2, 599. [Google Scholar] [CrossRef]
- Lomölder, R.; Plogmann, F.; Speier, P. Selectivity of Isophorone Diisocyanate in the Urethane Reaction Influence of Temperature, Catalysis, and Reaction Partners. J. Coat. Technol. 1997, 69, 51–57. [Google Scholar] [CrossRef]
- Gerard, J.-F.; Perchec, P.L.; Pham, Q.T. Polyuréthannes à Propriétés Emulsifiantes et Électrolytiques, 2. Cinétique de Polycondensation En Solution Des Alkylimino-2,2′ Diéthanols Avec Le Diisocyanate d’isophorone. Etude Par 1H et 13C NMR. Die Makromol. Chem. 1988, 189, 1719–1737. [Google Scholar] [CrossRef]
- Cunliffe, A.V.; Davis, A.; Farey, M.; Wright, J. The Kinetics of the Reaction of Isophorone Di-Isocyanate with Mono-Alcohols. Polymer 1985, 26, 301–306. [Google Scholar] [CrossRef]
- Lorenz, O.; Decker, H.; Rose, G. NCO-Prepolymere Aus Diisocyanaten Mit Unterschiedlich Reaktiven NCO-Gruppen. Angew. Makromol. Chem. 1984, 122, 83–99. [Google Scholar] [CrossRef]
- Hatada, K.; Ute, K.; Peter Pappas, S. E,Z Assignments of Isophorone Diisocyanate (IPDI) and Their Implications on the Relative Reactivity of the Isocyanate Groups. J. Polym. Sci. Part C Polym. Lett. 1987, 25, 477–480. [Google Scholar] [CrossRef]
- Bialas, N.; Höcker, H.; Marschner, M.; Ritter, W. 13C NMR Studies on the Relative Reactivity of Isocyanate Groups of Isophorone Diisocyanate Isomers. Die Makromol. Chem. 1990, 191, 1843–1852. [Google Scholar] [CrossRef]
- Blank, W.J.; He, Z.A.; Hessell, E.T. Catalysis of the Isocyanate-Hydroxyl Reaction by Non-Tin Catalysts. Prog. Org. Coat. 1999, 35, 19–29. [Google Scholar] [CrossRef]


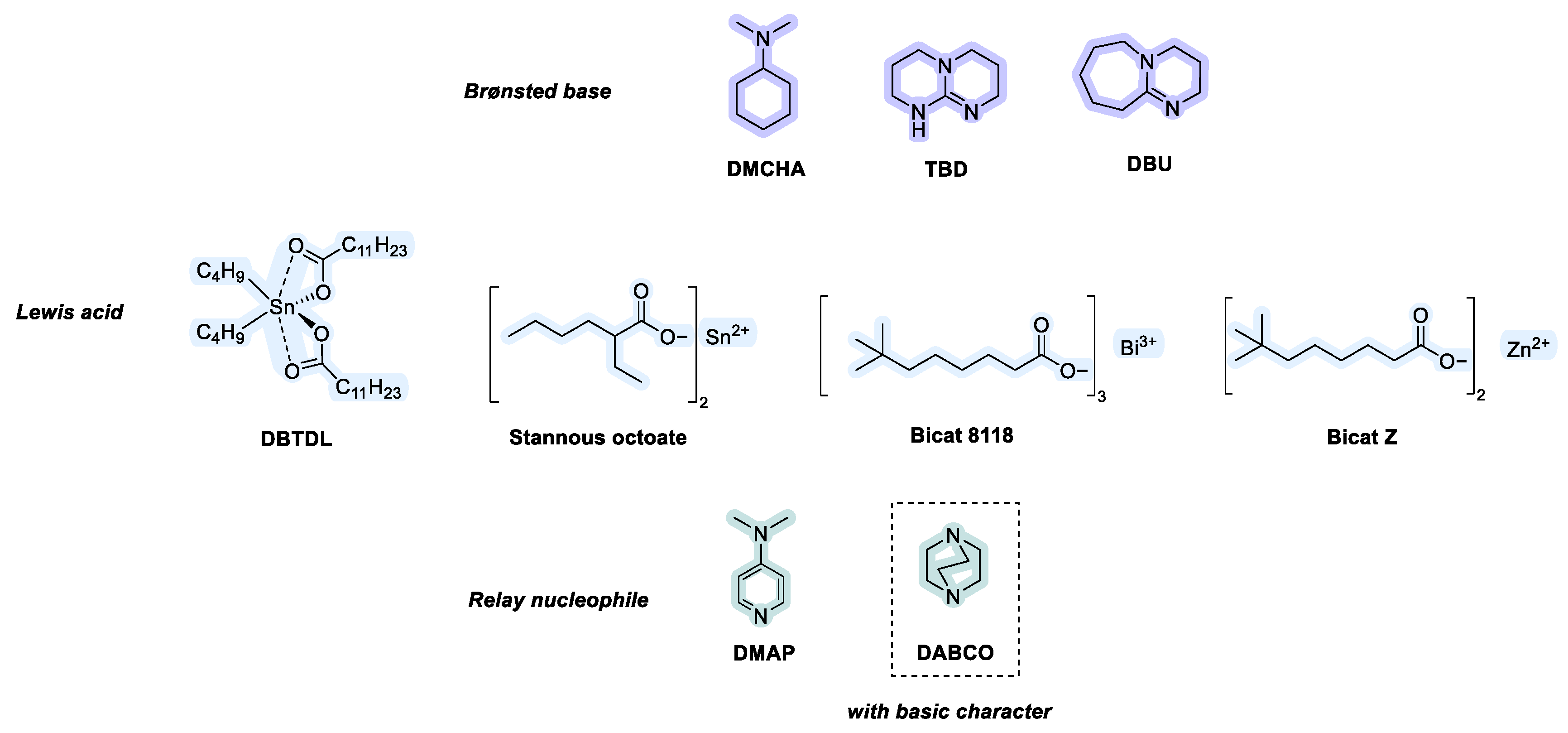
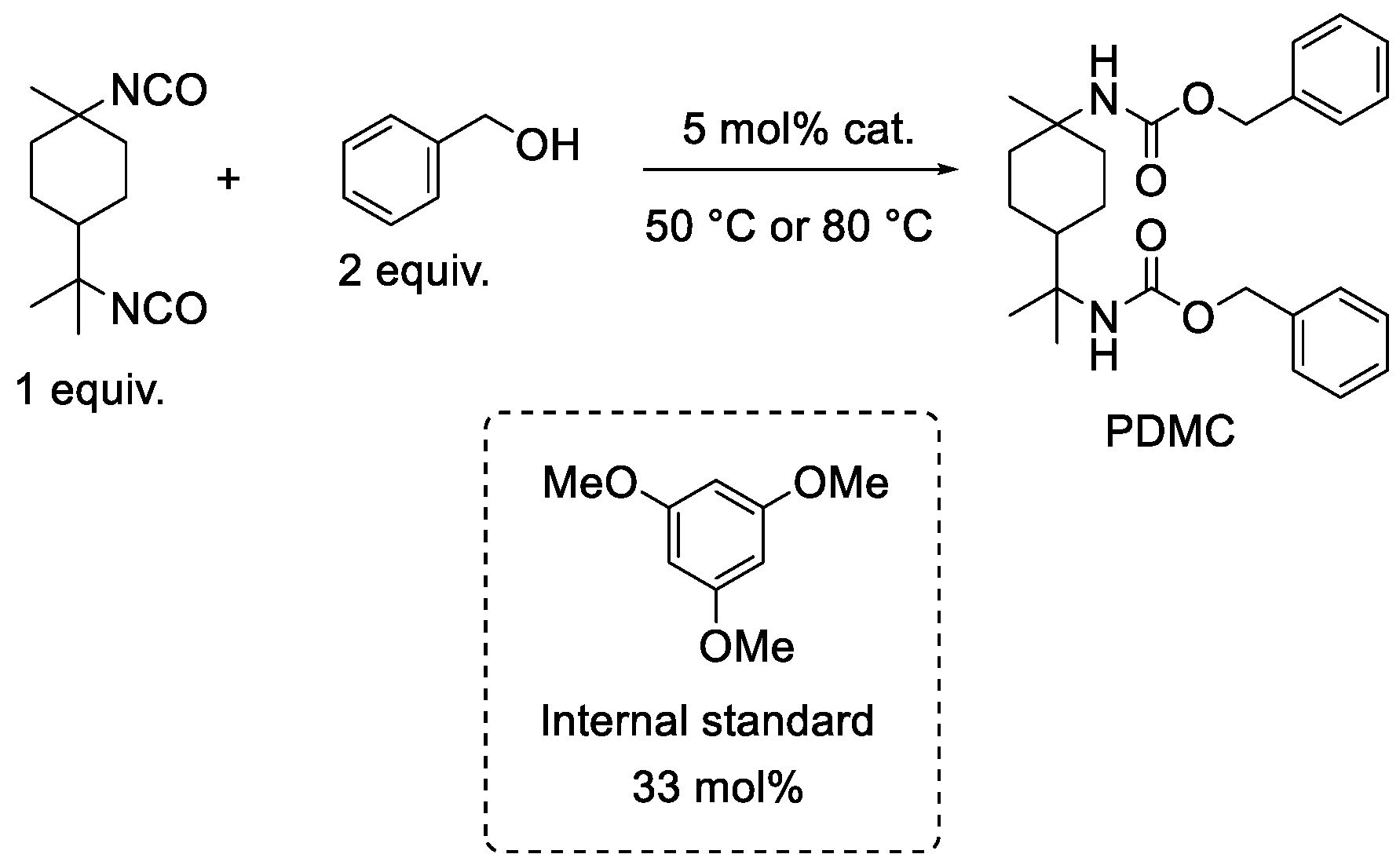
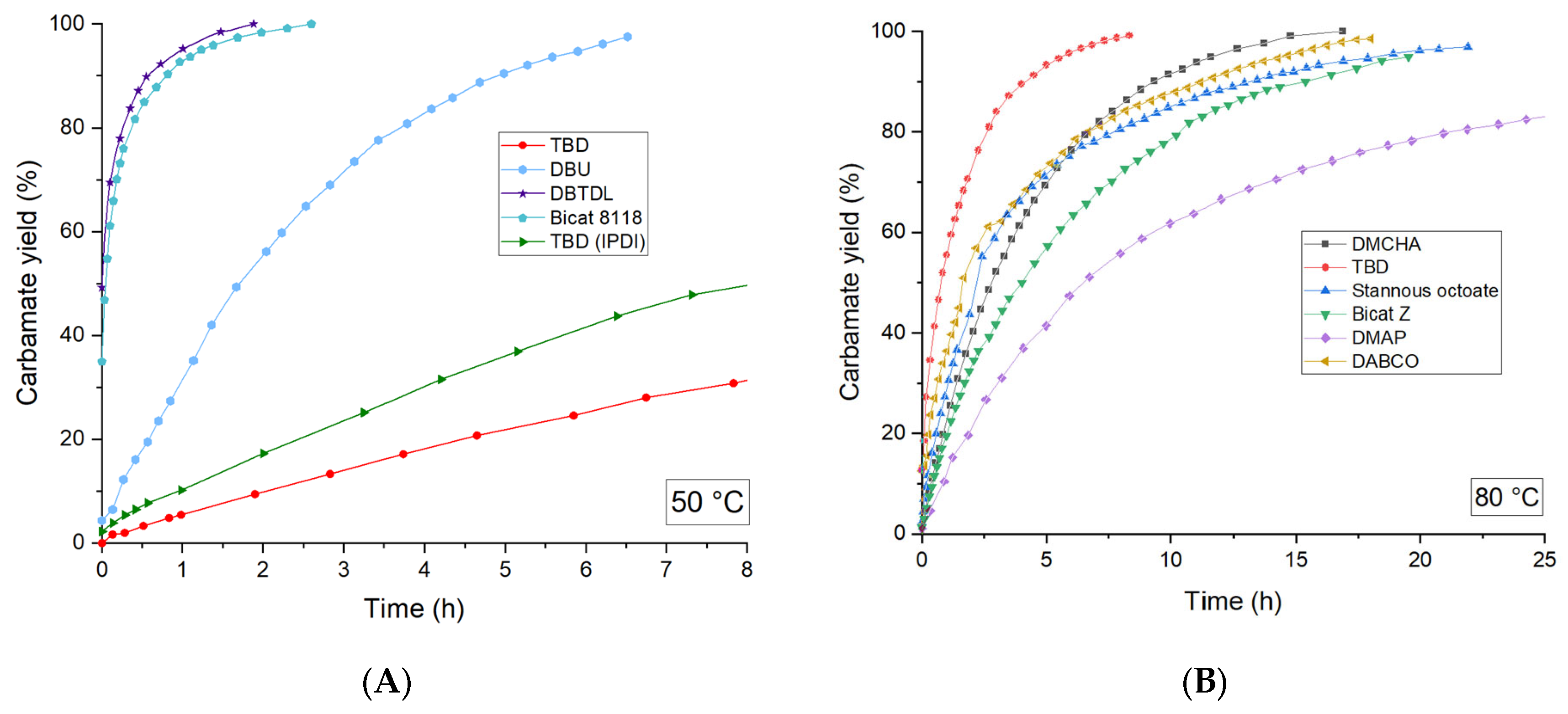
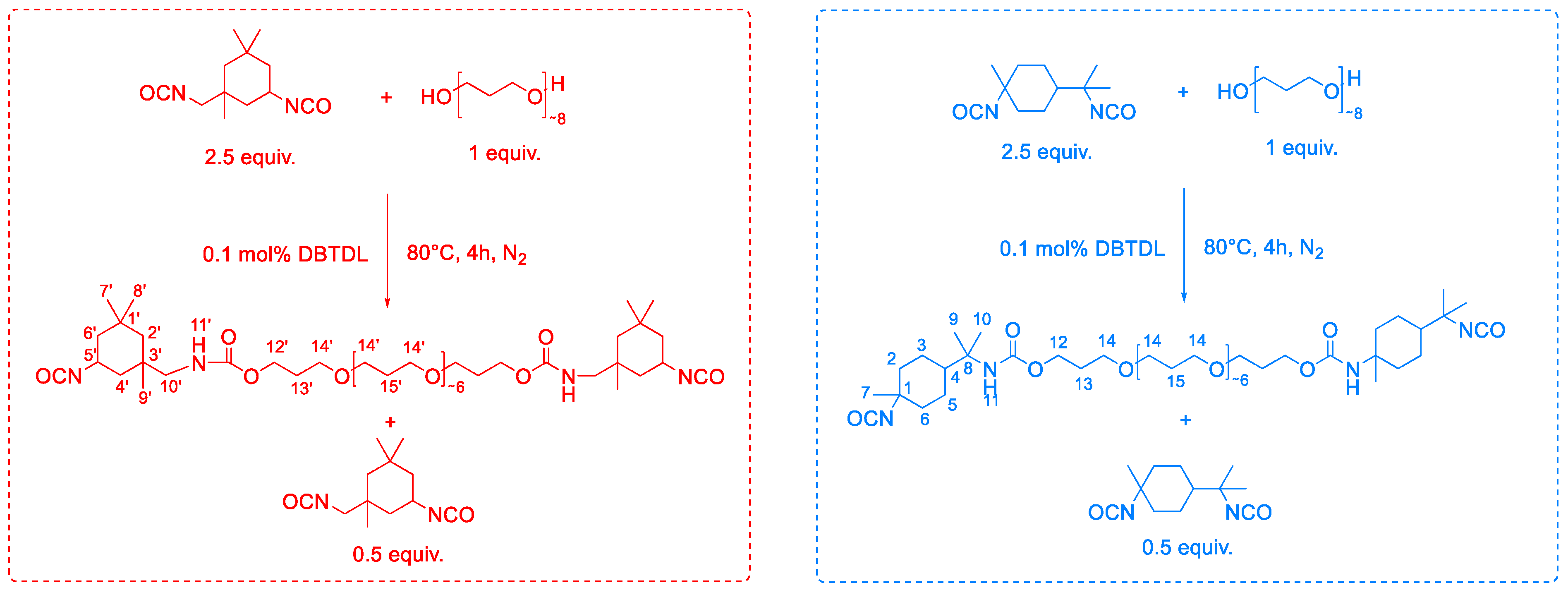


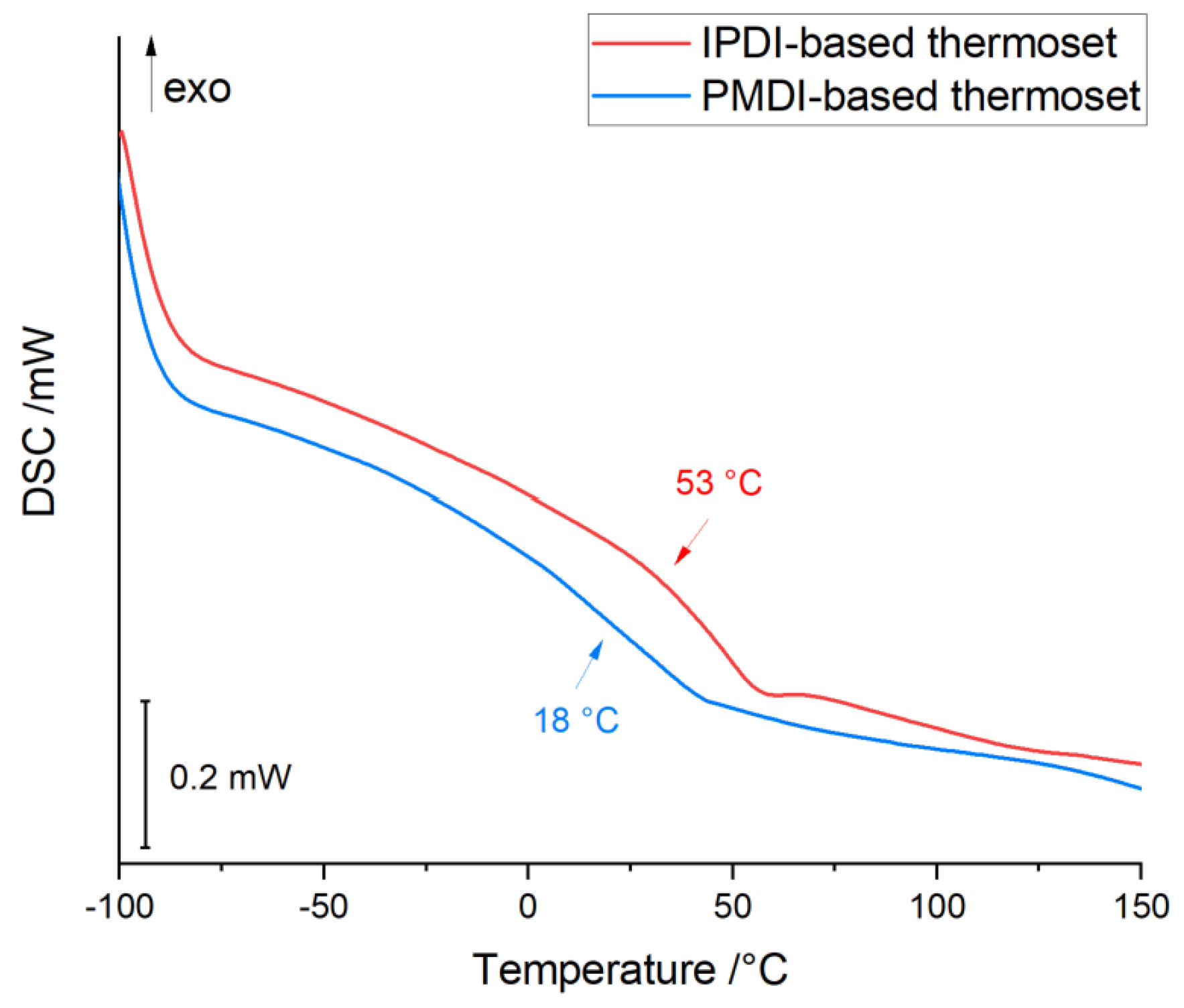
| Gelation Times (s) | ||
|---|---|---|
| Frequency (Hz) | IPDI-Based Formulation | PMDI-Based Formulation |
| 5 Hz | 430 | 4805 |
| 7.1 Hz | 424 | 4933 |
| 10 Hz | 424 | 5189 |
| Average | 426 ± 3 | 4975 ± 142 |
| Tg | SI | GC | ||||
|---|---|---|---|---|---|---|
| Unit | (°C) | % | ||||
| IPDI-based thermoset | 285 | 348 | 399 | 53 | 50 | 99 |
| PMDI-based thermoset | 240 | 297 | 393 | 18 | 75 | 98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delavarde, A.; Lemouzy, S.; Lebrun, A.; Pinaud, J.; Caillol, S. Paving the Way towards Sustainability of Polyurethanes: Synthesis and Properties of Terpene-Based Diisocyanate. Molecules 2023, 28, 7133. https://doi.org/10.3390/molecules28207133
Delavarde A, Lemouzy S, Lebrun A, Pinaud J, Caillol S. Paving the Way towards Sustainability of Polyurethanes: Synthesis and Properties of Terpene-Based Diisocyanate. Molecules. 2023; 28(20):7133. https://doi.org/10.3390/molecules28207133
Chicago/Turabian StyleDelavarde, Aliénor, Sebastien Lemouzy, Aurélien Lebrun, Julien Pinaud, and Sylvain Caillol. 2023. "Paving the Way towards Sustainability of Polyurethanes: Synthesis and Properties of Terpene-Based Diisocyanate" Molecules 28, no. 20: 7133. https://doi.org/10.3390/molecules28207133
APA StyleDelavarde, A., Lemouzy, S., Lebrun, A., Pinaud, J., & Caillol, S. (2023). Paving the Way towards Sustainability of Polyurethanes: Synthesis and Properties of Terpene-Based Diisocyanate. Molecules, 28(20), 7133. https://doi.org/10.3390/molecules28207133