Abstract
The possibility of functionalization of 2-(polyfluorophenyl)-4H-chromen-4-ones, with them having different numbers of fluorine atoms, with 1,2,4-triazole or imidazole under conditions of base-promoted nucleophilic aromatic substitution has been shown. A high selectivity of mono-substitution was found with the use of an azole (1.5 equiv.)/NaOBut(1.5 equiv.)/MeCN system. The structural features of fluorinated mono(azolyl)-substituted flavones in crystals were established using XRD analysis. The ability of penta- and tetrafluoroflavones to form persubstituted products with triazole under azole (6 equiv.)/NaOBut(6 equiv.)/DMF conditions was found in contrast to similar transformations with imidazole. On the basis of mono(azolyl)-containing polyfluoroflavones in reactions with triazole and pyrazole, polynuclear hybrid compounds containing various azole fragments were obtained. For poly(pyrazolyl)-substituted flavones, green emission in the solid state under UV-irradiation was found, and for some derivatives, weak fungistatic activity was found.
1. Introduction
Flavones based on a 2-phenylchromen-4-one backbone are important heteroaromatic scaffolds in organic chemistry due to their availability and significant synthetic and biological potential [1,2,3,4,5]. The uniqueness of this heterocyclic backbone is also due to the fact that its derivatives are widely represented in the plant world, which often determines their diverse biological action [6,7,8,9,10,11]. Isolation, identification, and chemical modification of flavones of plant origin is one of the rapidly developing areas in drug design [12,13]. Another equally important area of progress in the chemistry of flavones is the development of synthetic strategies for their modification. In addition, here, certain successes have recently been achieved, for example, its modification has been proposed by the Buchwald–Hartwig reaction [14], metal-catalyzed cascade rearrangements [15,16], electrochemical dimerization [17], CH-functionalization [18], etc. However, all of these transformations most often require expensive catalysts or complex installations.
In turn, fluorinated flavones offer extra possibilities for their functionalization in reactions with nucleophilic reagents. Fluoroaromatic compounds are well known to be perspective frameworks for their modification by different methods for formation of new C–C and C–heteroatom bonds [19,20,21], including C–N bond formation in reactions with azole-type heterocycles [22,23,24]. There are known approaches to the synthesis of chromone–azole dyads [25]; however, data on direct functionalization of fluorine-containing flavones with azoles under SNAr reaction conditions have only been only in publications from our research team [26,27], although the reaction of nucleophilic aromatic substitution of fluorine atoms is a quite simple, economically and environmentally friendly process, which offers the possibility of substitution for fluorinated substrates with advantages over reactions that use expensive catalysts [28]. To date, we have considerable practice in the synthesis and modification of polyfluoroflavones [26,27,29,30,31,32]. Previously, we proposed a convenient and efficient method for the synthesis of B-ring polyfluorinated flavones (2-(polyfluorophenyl)chromen-4-ones) [29], which can be involved in SNAr reactions to obtain polynuclear heterocyclic compounds based on flavones and azoles. We have shown the possibility of controlling the fluorine atoms substitution of 2-(polyfluorophenyl)chromen-4-ones by pyrazole and assumed the mechanism for sequential fluorine substitution on an example of 2-pentafluorophenyl-4H-chromen-4-one [26].
Within this work in continuation of our research in this area, the features of the functionalization of 2-(polyfluoroaryl)-4H-chromen-4-ones 1–3, with them having different numbers of fluorine atoms in the aryl substituent (Figure 1), by 1H-1,2,4-triazole and imidazole under conditions of base-promoted nucleophilic aromatic substitution were studied.

Figure 1.
Polyfluorinated flavones in the SNAr reaction with the 1H-1,2,4-triazole and imidazole under study in this work.
The introduction of azole fragments has a wide perspective due to their ability to form various noncovalent interactions with different therapeutic targets, which is valuable for drug design. Different azole derivatives have significant potential for medicinal chemistry [33,34,35,36,37,38,39,40]. Of particular interest are 1H-1,2,4-triazole and imidazole derivatives, which are known to possess therapeutic effect against drug-resistant pathogens [40,41,42]. The way to more effective medicines is through the synthesis of azole hybrids with other pharmacophores [27,43,44,45], which might be flavones, with them having a pyran framework that determines their great potential as antiviral antibacterial agents. Polyazole hybrid derivatives are also given special attention due to their potential applications as electron-transporting materials, emitters, and host materials in OLEDs, the most attractive products of organic electronics [46,47,48]. In this regard, the synthesis of hybrid compounds based on the flavone and azole cycles seems to be a prominent problem, which can be solved by the SNAr reaction of polyfluorinated flavones with azoles.
2. Results
The study of the reaction of polyfluoroflavones 1–3 with 1H-1,2,4-triazole and imidazole under base-promoted nucleophilic aromatic substitution was carried out according to three synthetic protocols developed during the research of transformations with pyrazole [26]. The application of the Cs2CO3-promoted conditions will allow for observation of the spectrum of possible substituted products, while the application of the NaOBut-promoted conditions should facilitate for selective mono- and persubstitution of fluorine, which depends on variation of the nucleophile and base loading. The most convenient method for identifying fluorine-containing compounds in their mixtures is 19F NMR spectroscopy data.
The reaction of flavone 1 with 3 equiv. of triazole and Cs2CO3 in MeCN led to a mono-, tri-, tetra-, and penta(1H-1,2,4-triazol-1-yl)-substituted products mixture, from which only mono- and penta-substituted flavones 4 and 7 were isolated by column chromatography (Scheme 1). The 19F NMR data of fluorine-containing products 4–7 and their ratio in the reaction mixture are shown in Table 1. Under optimized conditions, selective synthesis of mono- and penta(1H-1,2,4-triazol-1-yl)-substituted products 4 and 7 with a good preparative yield was achieved (Scheme 1).

Scheme 1.
Reaction of 2-(pentafluorophenyl)-4H-chromen-4-one 1 with 1H-1,2,4-triazole.

Table 1.
19F NMR data of mixture of Cs2CO3-promoted reaction of flavone 1 with 1H-1,2,4-triazole.
In contrast to the transformations with triazole, the reaction of flavone 1 with 3 equiv. of imidazole under the same conditions resulted in the formation of mono-, di-, and tri(1H-imidazol-1-yl)-substituted products 8–10, which were isolated with a poor preparative yield. The 19F NMR data of fluorine-containing products 8–10 and their ratio in the mixture are shown in Table 2. Under conditions preferable to monosubstitution, 2-[2,3,5,6-tetrafluoro-4-(1H-imidazol-1-yl)phenyl]-4H-chromen-4-one 8 was synthesized with a good yield (Scheme 2). In addition, under conditions conducive to the formation of a persubstituted product, the reaction of these reagents is extremely nonselective.
Table 2.
19F NMR data of a mixture of Cs2CO3-promoted reaction of flavone 1 with imidazole.
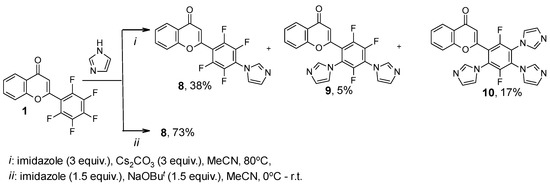
Scheme 2.
Reaction of 2-(pentafluorophenyl)-4H-chromen-4-one 1 with imidazole.
It should be noted that for 2-[2,5-difluoro-3,4,6-tri(1H-imidazol-1-yl)phenyl]-4H-chromen-4-one 10, an isomeric structure—2-[3,5-difluoro-2,4,6-tri(1H-imidazol-1-yl)phenyl]-substituted product can be proposed. The structures of 9 and 10 were solved on the basis of a comparative analysis of 1H, 19F NMR data of these compounds 9 and 10, and NMR data of formerly synthesized [26] tri(1H-pyrazol-1-yl)-substituted analogue A (Appendix A, Table A1).
The crystal structures of 2-[2,3,5,6-tetrafluoro-4-(1H-azol-1-yl)phenyl]-4H-chromen-4-ones flavones 4 and 8 were confirmed by XRD analysis (Figure 2 and Figure 3). Both homologues 4 and 8 have similar structural characteristics, in contrast to the pyrazolyl-substituted analogue [26], which does not have intramolecular interactions coordinating both pyrone and aryl moieties, which are therefore aplanar. In addition, the introduction of triazole and imidazole fragments has a critical effect on the structure of their unit cell in the crystal. Thus, a cell of compound 4 has a rhombic syngony and compound 8 has a monoclinic syngony. It is important to note that for two systems of atoms C16C11C2C3 and C17N1C14C15 of product 4 and similar systems C3C2C11C12 and C13C14N1C17 of analogue 8, the torsion angles have values close in absolute value, but pairwise opposite in sign, equal to 42.70(0.76), −47.50(0.85), and −58.89(0.31), 56.50(0.34) degrees for 4 and 8, respectively. The unit cell of the crystal 4 consists of four molecules due to the formation of O…H, F…F, and C…C sp2 short intermolecular contacts (O3…H17 2.373, O3…H18 2.634, C5…C14 3.331(8), C9…C12 3.303(7), F2…F4 2.889(5) Å) (Figure 2). The unit cell of crystal 8 also consists of four molecules stabilized by C…H, C…F, and C…C sp2 short intermolecular contacts (C18…H5 2.80(2), C7…F3 3.117(3), C9…C16 3.396(3) Å) (Figure 3).
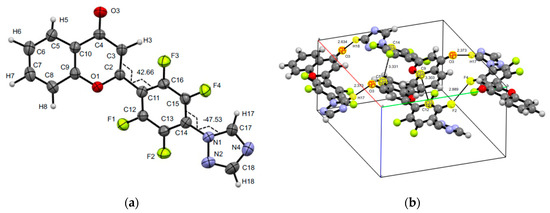
Figure 2.
Molecular structure and selected torsions (a), and unit cell (b) of compound 4 with atoms represented as thermal ellipsoids of thermal vibrations with a 50% probability.
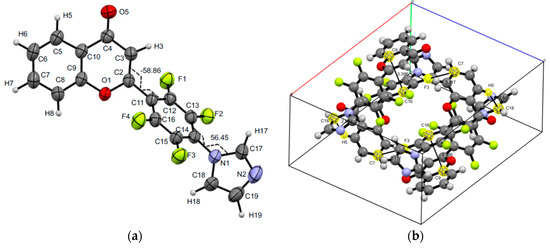
Figure 3.
Molecular structure and selected torsions (a), and unit cell (b) of compound 8 with atoms represented as thermal ellipsoids of thermal vibrations with a 50% probability.
Furthermore, we introduced flavone 2, with it having 2,3,4,5-tetrafluorophenyl substituent, in the SNAr reaction with triazole and imidazole. The reaction of compound 2 with 3 equiv. of triazole (Scheme 3) and imidazole (Scheme 4) in the presence of Cs2CO3 in MeCN led to mono-, di-, and tri(azol-1-yl)-substituted products mixture 11–13 and 15–17, respectively. The 19F NMR data of fluorine-containing flavones 11–13 and 15–17 and their ratio in the mixture are shown in Table 3. Under optimized conditions, selective synthesis of mono- 11 and tetra(triazol-1-yl)-substituted products 14 was conducted with a good preparative yield (Scheme 3). However, under similar conditions, only mono(imidazolyl)-substituted flavone 15 was obtained in a good yield from the reaction with imidazole, and it was not possible to isolate the persubstituted product (Scheme 4) as in the reaction of flavone 1 (Scheme 2). This may indicate a lower reactivity of imidazole compared to triazole and the previously studied pyrazole under the conditions used [26].
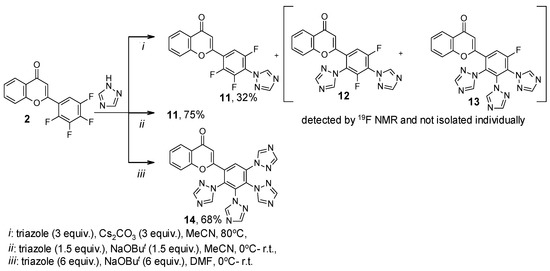
Scheme 3.
Reaction of 2-(2,3,4,5-tetrafluorophenyl)-4H-chromen-4-one 2 with 1H-1,2,4-triazole.
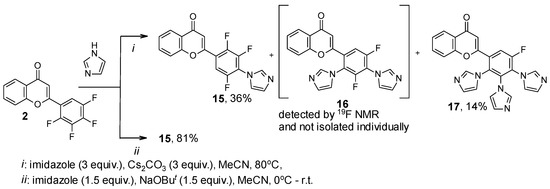
Scheme 4.
Reaction of 2-(2,3,4,5-tetrafluorophenyl)-4H-chromen-4-one 2 with imidazole.
Table 3.
19F NMR data of mixtures of Cs2CO3-promoted reactions of flavone 2 with 1H-1,2,4-triazole and imidazole.
The structure of 2-[2,3,5-trifluoro-4-(1H-imidazol-1-yl)phenyl]-4H-chromen-4-one 15 was confirmed by XRD analysis (Figure 4). As well as products 4 and 8, compound 15 has no intramolecular interactions, which coordinated both pyrone and aryl moieties. However, the replacement of fluorine at the C6′ site by hydrogen allows the mutual position of these two moieties of flavone 15 in crystal close to coplanar. Torsions C3C2C11C12 and C1N1C14C13 are −7.10(0.30) and −48.14(0.27) degrees. The unit cell of the crystal 15 is monoclinic, consists of four molecules, stabilized by pairs of O…H, C…O short intermolecular contacts (O4…H17 2.33(3), C17…O4 3.148(3), and C2…O4 3.127(2) Å).
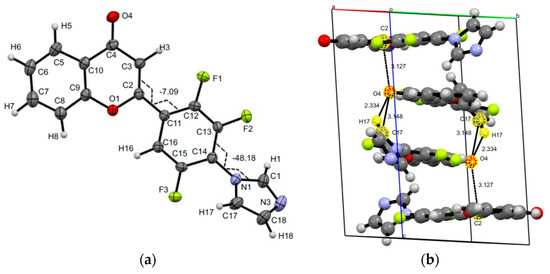
Figure 4.
Molecular structure and selected torsions (a), and unit cell (b) of compound 15 with atoms represented as thermal ellipsoids of thermal vibrations with a 50% probability.
The reaction of 2-(3,4,5-trifluoro-2-methoxy-phenyl)-4H-chromen-4-one 3 with 3 equiv. of triazole in the presence of Cs2CO3 in MeCN led to the formation of mono- and di(1H-1,2,4-triazol-1-yl)-substituted products 18, 19, which were isolated from the mixture (Scheme 5). In the 19F NMR spectrum of the mixture, in addition to the signals corresponding to products 18 and 19, two pairs of signals were recorded, presumably assigned by us to compounds 20 and 21 (Table 4). Due to the low intensity and insufficient resolution of these signals, the possibility of their detection is difficult. We believe that the formation of product 20 is possible due to the demethylation of flavone 18 under the reaction conditions used. The formed phenolic group in compound 20 can be in equilibrium between keto and enol forms, and keto form may undergo a nucleophilic addition reaction with triazole followed by aromatization to provide flavone 21.

Scheme 5.
Reaction of 2-(3,4,5-trifluoro-2-methoxyphenyl)-4H-chromen-4-one 3 with 1H-1,2,4-triazole.
Table 4.
19F NMR data of mixture of Cs2CO3-promoted reaction of flavone 3 and 1H-1,2,4-triazole.
Under NaOBut-promoted conditions for selective monosubstitution corresponding 2-[2,3,5,6-tetrafluoro-4-(1H-1,2,4-triazol-1-yl)phenyl]-4H-chromen-4-one 18 was synthesized with a good preparative yield. The reaction with four-fold excess of triazole and NaOBut led to flavone 19 and 2-[2-hydroxy-3,4-di(1H-1,2,4-triazol-1-yl)phenyl]-4H-chromen-4-one 22, which was obtained by demethylation of 2-methoxy-3,4-di(1H-1,2,4-triazol-1-yl)-substituted precursor 19, with poor yields, similar to pyrazolyl-substituted analogues [26]. The use of six-fold excess of triazole and NaOBut gave the same result as above with four-fold excess.
From the reaction of flavone 3 with 3 equiv. of imidazole in the presence of Cs2CO3 in MeCN, only mono-substituted product 23 was isolated individually. The 19F NMR spectrum of the mixture (Table 5) also contained signals assigned to flavones 24 and 25 based on the conclusions given above for similar transformations with triazole. Under optimized conditions, 2-[3,5-difluoro-2-methoxy-4-(1H-imidazol-1-yl)phenyl]-4H-chromen-4-one 23 was synthesized (Scheme 6). Using an excess of this azole has not been successful in isolating any individual products.
Table 5.
19F NMR data of mixture of Cs2CO3-promoted reaction of flavone 3 and imidazole.
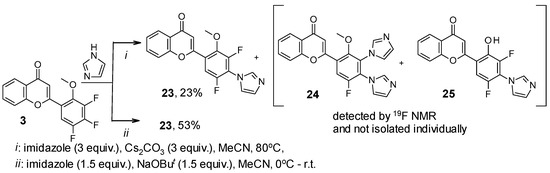
Scheme 6.
Reaction of 2-(3,4,5-trifluoro-2-methoxyphenyl)-4H-chromen-4-one 3 with imidazole.
The structures of 2-[5-fluoro-2-methoxy-3,4-di(1H-1,2,4-triazol-1-yl)phenyl]-4H-chromen-4-one 19 and 2-[4,5-difluoro-2-methoxy-4-(1H-1,2,4-imidazol-1-yl)phenyl]-4H-chromen-4-one 23 were confirmed by XRD analysis (Figure 5 and Figure 6). Compounds 19 and 23 also do not have any intramolecular interactions. It was found that the introduction of one or two azole fragments affects the geometric parameters of crystals 19 and 23. The unit cell of compound 19 has a triclinic syngony and consists of two molecules stabilized by a pair of short intramolecular contacts C…F (C10…F1 3.159(6) Å). The cell of compound 23 of the monoclinic system consists of four molecules forming short intermolecular contacts O…H, C…F (O3…H8 2.568, O3…H16 2.32(3), C3…F001 3.148(3) Å). It should be noted that the torsion angles of two similar systems of atoms C16C11C2C3 and C3C2C11C12 of di(triazolyl)- and mono(imidazolyl)-substituted flavones 19 and 23 have a significant difference both in absolute value and in sign and are equal to −22.81(0.72) and 8.64(0.33) degrees, respectively. The geometry of the azole fragments of molecules 19 and 23 does not reveal fundamental differences. The torsion angles of the atomic systems C18N1C15C16, C20N4C14C15, and C17N1C14C13 are 56.12(0.58), 56.96(3.50), and 49.82(0.37) degrees, respectively.

Figure 5.
Molecular structure and selected torsions (a) and unit cell (b) of compound 19 with atoms represented as thermal ellipsoids of thermal vibrations with a 50% probability.
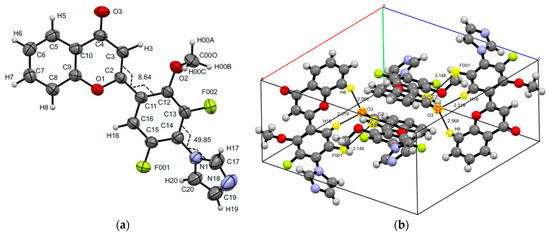
Figure 6.
Molecular structure and selected torsions (a) and unit cell (b) of compound 23 with atoms represented as thermal ellipsoids of thermal vibrations with a 50% probability.
It should be noted that a common characteristic of the crystals of both mono(1H-imidazol-1-yl)-substituted products 15 and 23 is the formation of a unit cell of the monoclinic system. However, the torsion angles of the systems of atoms C3C2C11C12, C1N1C14C13 of trifluoroflavone 15 and C3C2C11C12 and C17N1C14C13 of difluoroflavone 23 have practically equal absolute values and opposite values (Figure 4 and Figure 6).
In this work, we also studied the possibility of the synthesis of 2-[poly(1H-azol-1-yl)phenyl]-4H-chromen-4-ones, involving two different azole-type heterocycles in their structure by the NaOBut-promoted SNAr reaction of mono(1H-azol-1-yl)-substituted flavones with azoles.
Thus, the reaction of mono(triazolyl)-substituted flavone 4 with four-fold excess of pyrazole and NaOBut led to 2-[2,5-difluoro-3,6-di(1H-pyrazol-1-yl)-4-(1H-1,2,4-triazol-1-yl)phenyl]- and 2-[2,3,5,6-tetra(1H-pyrazol-1-yl)-4-(1H-1,2,4-triazol-1-yl)phenyl]-4H-chromen-4-ones 26 and 27. Flavone 26 was shown to react with two-fold excess of pyrazole and NaOBut to form product of complete substitution 27 (Scheme 7). Under conditions preferable for persubstitution, flavone 8 reacts with pyrazole and triazole, leading to the formation of compounds 28 and 29. Similarly, 2-(2,3,5,6-tetrafluoro-4-(1H-pyrazol-1-yl)phenyl)-4H-chromen-4-one 30 obtained earlier [26] forms product 31 with triazole (Scheme 8). Unfortunately, we have not yet succeeded in growing a suitable single crystal for XRD due to the limited solubility of polynuclear products 27–29 and 31 in organic solvents.
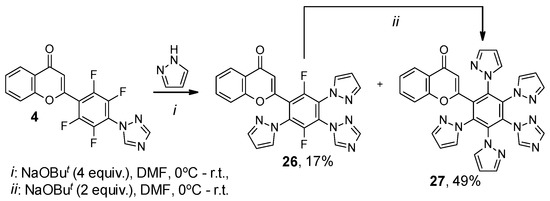
Scheme 7.
Reaction of flavone 4 with pyrazole.
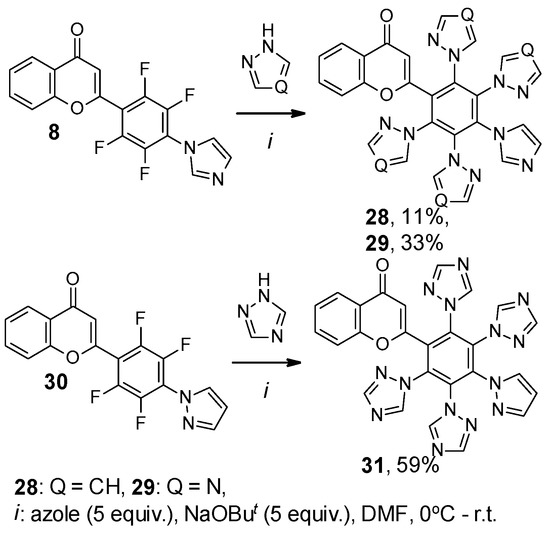
Scheme 8.
Reaction of monoazolyl-substituted flavone 4, 30 with azoles.
As is known, aryl-azole scaffolds are considered as promising materials for OLEDs [46,47,48,49]; therefore, we recorded the photoluminescence spectra for poly(azolyl)-substituted flavones 28–31 and formerly synthesized penta(pyrazolyl)-substituted analogue 32 [26]. It was found that 2-[4-(imidazolyl)-2,3,5,6-tetra(pyrazolyl)- and penta(pyrazolyl)-substituted flavones 28 and 32 exhibit emission in the solid state under UV-irradiation, in contrast to poly(triazolyl)-containing derivatives 29 and 31. The emission spectra of these compounds were recorded, and the data are presented in Table 6 and Figure 7. Flavones 28 and 32 possess green emission with maxima 504 nm. The commission international de L’Eclairage (CIE) coordinates were (0.255; 0.528) and (0.255; 0.526) for 28 and 32, respectively. Substitution of pyrazole fragment at C4′ by imidazole results in a 1.6-fold increase in quantum yield (0.18 and 0.29 for 28 and 32, correspondingly). Detailed data of the fluorescence lifetime measurements of flavones 28 and 32 are given in Appendix B.
Table 6.
Photoluminescent data for compounds 28 and 32 in powder at r.t.
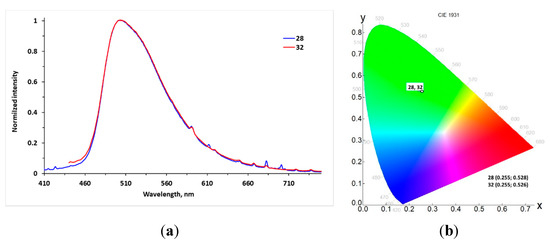
Figure 7.
Emission (–) spectra (a) and CIE 1931 chromaticity diagram (b) for chromophores 28 and 32 in solid state.
Azolyl-substituted flavones are of great interest in the search for the bioactive compounds among them, and, in particular, for antimycotic agents [50]; therefore, we screened the fungistatic activity of a number of compounds (Appendix C) obtained in this work and earlier [29] in relation to four control strains of clinically significant species of pathogenic fungi Trichophyton rubrum, Epidermophyton floccosum, Microsporum canis, and Candida parapsilosis. It was found that flavones 8 and 14 have a weak inhibitory effect against T. rubrum and M. canis (MIC 100 mg/mL), and derivatives 23 and 33, combined methoxy and azole substituents, in the absence of fungistatic activity (MIC > 100 µg/mL) showed high and moderate activity in inhibiting the growth of 50% fungal culture (MIC50 1.56–12.5 µg/mL).
3. Materials and Methods
3.1. Chemistry: General Information and Synthetic Techniques
Solvents and reagents except fluorine-containing flavones are commercially available and were used without purification. The NMR spectra of the synthesized compounds (see Supplementary Materials) were recorded on Bruker DRX-400 and Bruker AVANCE III 500 spectrometers (1H, 400.13 (DRX400) and 500.13 (AV500) MHz, 13C, 125.76 MHz, Me4Si as an internal standard, 19F, 376.44 (DRX400) and 470.52 (AV500) MHz, C6F6 as an internal standard, chemical shifts were not converted to CCl3F)). IR spectra were recorded on a Perkin Elmer Spectrum Two FT-IR spectrometer (UATR) in the range of 4000–400 cm–1. Elemental (C, H, N) analysis was performed on a Perkin Elmer PE 2400 Series II CHNS-O EA 1108 elemental analyzer. The melting points were measured on a Stuart SMP3 in open capillaries. The reaction progress was monitored by TLC on ALUGRAM Xtra SIL G/UV254 sheets. The starting flavones (1–3) were synthesized by a procedure [29].
Synthetic technique A for the synthesis of azolyl-substituted flavones using Cs2CO3. flavone (0.5 or 1 mmol), azole (3 equiv.), and Cs2CO3 (3 equiv.) were suspended in 10 mL of MeCN. The reaction mixture was heated to 80 °C. The reaction progress was monitored by TLC. At the end of the reaction, the mixture was diluted with water (10 mL) and extracted with CHCl3 or DCM (2 × 10 mL). Organic layers were combined, and the solvent was removed. The residue was immobilized on silica gel and purified by column chromatography using an appropriate eluent mixture (2:1 v/v).
Synthetic technique B for the synthesis of azolyl-substituted flavones using NaOBut. flavone (0.5 or 1 mmol) was dissolved in dry DMF (5 mL), placed in a sealed vial and cooled to 0 °C. Azole (from 1.5 to 6 equiv.) and NaOBut (from 1.5 to 6 equiv.) were suspended in dry DMF (5 mL) and stirred at room temperature for 5 min. The mixture was cooled to 0 °C and was added to a flavone solution in DMF while stirring. After 10 min. in a cooling bath (0 °C), the vialwas removed and the reaction continued at room temperature. The reaction progress was monitored by TLC. At the end, the reaction mixture was diluted with water (10 mL), stirred, and cooled. The formed precipitate was filtered off and washed with water. The water solution was neutralized with 0.1M HCl and extracted with CHCl3 or DCM. The organic layer was separated, and the solvent was removed. Organic residues were combined, immobilized on silica gel and purified by column chromatography using an appropriate eluent mixture (2:1 v/v).
3.2. Spectral and Elemental Analysis Data of Synthesized Compounds
2-[2,3,5,6-Tetrafluoro-4-(1H-1,2,4-triazol-1-yl)phenyl]-4H-chromen-4-one (4). Yield 202 mg (56% according to technique A), 256 mg (71% according to technique B); white powder; mp 192–194 °C; IR ν 3108, 3043 (C–HAr), 1639 (C=O), 1528, 1485, 1381 (C=CAr, C–HAr, C–N), 1139, 1110 (C–F) cm−1; 1H NMR (500.13 MHz, CDCl3) δ 6.68 (s, 1H, CHPyranone), 7.50 (m, 1H, CHAr), 7.53 (d, J = 8.6 Hz, 1H, CHAr), 7.76 (ddd, J = 8.6, 7.1, 1.7 Hz, 1H, CHAr), 8.26 (dd, J = 8.0, 1.5 Hz, 1H, CHAr), 8.29 (s, 1H, CHTriazole), 8.52 (s, 1H, CHTriazole) ppm; 13C NMR (125.76 MHz, CDCl3) δ 113.7 (t, J = 15 Hz, CAr), 116.1 (t, J = 3 Hz, CAr), 118.2 (s, CAr), 118.8 (s, CAr), 123.9 (s, CPyranone), 125.9 (s, CAr), 126.0 (s, CAr), 134.5 (s, CAr), 141.7 (m, 2CArF), 144.7 (ddt, J = 257, 14, 5 Hz, 2CArF), 145.3 (t, J = 3 Hz, CTriazole), 151.7 (s, CAr), 153.5 (s, CTriazole), 156.6 (s, CPyranone), 177.0 (s, CPyranone) ppm; 19F NMR (470.52 MHz, CDCl3) δ 16.66 (m, 2F), 24.83 (m, 2F) ppm; Anal. calcd. for C17H7F4N3O2: C 56.52, H 1.95, N 11.63, found: C 56.26, H 1.84, N 11.38.
2-[2,3,4,5,6-Penta(1H-1,2,4-triazol-1-yl)phenyl]-4H-chromen-4-one (7). Yield 39 mg (7% according to technique A), 335 mg (60% according to technique B); light-yellow powder; mp 322–323 °C; IR ν 3118, 3086 (C–HAr), 1651 (C=O), 1514, 1459, 1379, 1272 (C=CAr, C–HAr, C–N) cm−1; 1H NMR (500.13 MHz, CDCl3) δ 6.20 (s, 1H, CHPyranone), 7.16 (d, J = 8.4 Hz, 1H, CHAr), 7.42 (m, 1H, CHAr), 7.65 (ddd, J = 8.5, 7.2, 1.7 Hz, 1H, CHAr), 7.87 (s, 1H, CHTriazole), 7.88 (s, 2H, CHTriazole), 7.90 (s, 2H, CHTriazole), 8.08 (dd, J = 8, 1.6 Hz, 1H, CHAr), 8.10 (s, 1H, CHTriazole), 8.15 (s, 2H, CHTriazole), 8.22 (s, 2H, CHTriazole) ppm; 13C NMR (125.76 MHz, CDCl3) δ 114.9, 117.4 (s, 2CAr), 123.2, 126.1, 126.5, 133.1, 134.3 (s, 2CAr), 134.9, 135.3, 136.1, 145.3 (s, 2CTriazole), 145.7, 146.0 (s, 2CTriazole), 153.4, 153.7 (s, 2CTriazole), 153.7 (s, 2CTriazole), 155.7, 175.8 ppm; Anal. Calcd. For C25H15N15O2: C 53.86, 2.71, N 37.69, found: C 53.60, H 2.68, N 37.69.
2-[2,3,5,6-Tetrafluoro-4-(1H-imidazol-1-yl)phenyl]-4H-chromen-4-one (8). Yield 137 mg (38% according to technique A), 263 mg (73% according to technique B); white powder, mp 172–174 °C; IR ν 3110 (C–HAr), 1653 (C=O), 1489, 1459, 1376 (C=CAr, C–HAr, C–N), 1224 (C–F) cm−1; 1H NMR (400.13 MHz, CDCl3) δ 6.67 (s, 1H, CHPyranone), 7.31 (br.s., 1H, CHImidazole), 7.33 (br.s., 1H, CHImidazole), 7.48–7.53 (m, 2H, 2CHAr), 7.76 (ddd, J = 8.6, 7.3, 1.5 Hz, 1H, CHAr), 7.88 (br.s., 1H, CHImidazole), 8.27 (dd, J = 7.9, 1.5 Hz, 1H, CHAr) ppm; 13C NMR (125.76 MHz, CDCl3) δ 112.0 (t, J = 15 Hz, CAr), 115.9 (t, J = 3 Hz, CAr), 118.2, 119.3 (t, J = 13 Hz, CAr), 119.7 (t, J = 2 Hz, CImidazole), 123.9, 125.9, 126.0, 130.6, 134.5, 137.5 (t, J = 4 Hz, CImidazole), 140.1–142.3 (m, 2CArF), 144.9 (ddt, J = 257, 14, 5 Hz, 2CArF), 152.0, 156.6, 177.1 ppm; 19F NMR (376.44 MHz, CDCl3) δ 14.88 (dqd, J = 6.0, 4.4, 2.4 Hz, 2F), 24.51 (dq, J = 7.3, 4.2 Hz, 2F) ppm; Anal. calcd. for C18H8F4N2O2: C 60.01, H 2.24, N 7.78, found: C 59.89, H 2.30, N 7.96.
2-[2,3,5-Trifluoro-4,6-di(1H-imidazol-1-yl)phenyl]-4H-chromen-4-one (9). Yield 20 mg (5%); light-yellow powder, mp 200–203 °C; 1H NMR (400.13 MHz, CDCl3) δ 6.52 (d, J = 1.5 Hz, 1H, CHPyranone), 7.05 (br.s., 1H, CHImidazole), 7.13 (br.s., 1H, CHImidazole), 7.18 (d, J = 8.4 Hz, 1H, CHAr), 7.33 (br.s., 1H, CHImidazole), 7.35 (br.s., 1H, CHImidazole), 7.41–7.45 (m, 1H, CHAr), 7.65 (br.s., 1H, CHImidazole), 7.65 (dd, J = 15.7, 1.7 Hz, 1H, CHAr), 7.90 (br.s., 1H, CHImidazole), 8.17 (dd, J = 7.9, 1.5 Hz, 1H, CHAr) ppm; 19F NMR (376.44 MHz, CDCl3) δ 23.60 (d, J = 22.2 Hz, 1F), 26.33 (ddd, J = 22.6, 13.0, 1.3 Hz, 1F), 30.57 (d, J = 13.0 Hz, 1F) ppm. Anal. calcd. for C21H11F3N4O2: C 61.77, H 2.72, N 13.72, found: C 61.89, H 2.79, N 13.66.
2-[2,5-Difluoro-3,4,6-tri(1H-imidazol-1-yl)phenyl]-4H-chromen-4-one (10). Yield 95 mg (17%); light-yellow powder; mp 227–228 °C; 1H NMR (400.13 MHz, CDCl3) δ 6.51 (d, J = 1.3 Hz, 1H, CHPyranone), 6.81 (d, J = 1.3 Hz, 1H, CHImidazole), 6.83 (d, J = 1.2 Hz, 1H, CHImidazole), 7.10 (d, J = 1.2 Hz, 1H, CHImidazole), 7.15–7.20 (m, 1H, CHAr), 7.21–7.23 (m, 3H, 3CHImidazole), 7.43–7.47 (m, 1H, CHAr), 7.50 (br.s, 2H, 2CHImidazole), 7.65–7.69 (m, 1H, CHAr), 7.70 (br.s, 1H, 1CHImidazole), 8.18 (dd, J = 8.0, 1.6 Hz, 1H, CHAr) ppm; 13C NMR (125.76 MHz, CDCl3) δ 115.8 (d, J = 3 Hz, CHPyranone), 117.8, 119.0, 119.2, 120.0, 120.5 (d, J = 17 Hz, CArF), 123.3 (d, J = 16 Hz, CArF), 123.6, 125.6 (dd, J = 15, 2 Hz, CArF), 125.8–126.0 (m, CArF), 125.9, 126.2, 131.0, 131.46, 131.47, 134.7, 136.9 (d, J = 3 Hz, CArF), 137.0 (d, J = 2 Hz, CImidazole), 137.5 (d, J = 2 Hz, CImidazole), 148.3 (dd, J = 256, 4 Hz, CArF), 150.9 (dd, J = 257, 4 Hz, CArF), 152.7 (d, J = 2 Hz, CImidazole), 156.2, 176.6 ppm; 19F NMR (376.44 MHz, CDCl3) δ 31.47 (dm, J = 14.4 Hz, 1F), 41.18 (dd, J = 14.7, 1.2 Hz, 1F) ppm. Anal. calcd. for C24H14F2N6O2: C 63.16, H 3.09, N 18.41, found: C 62.90, H 2.82, N 18.24.
2-[2,3,5-Trifluoro-4-(1H-1,2,4-triazol-1-yl)phenyl]-4H-chromen-4-one (11). Yield 257 mg (75%); white powder; mp 210–211 °C; IR ν 3138, 3118, 3070 (C–HAr), 1634 (C=O), 1531, 1462, 1369 (C=CAr, C–HAr, C–N), 1040 (C–F) cm−1; 1H NMR (500.13 MHz, CDCl3) δ 7.02 (s, 1H, CHPyranone), 7.49 (t, J = 7.5 Hz, 1H, CHAr), 7.58 (d, J = 8.4 Hz, 1H, CHAr), 7.74–7.79 (m, 2H, 2CHAr), 8.25 (dd, J = 8.0, 1.6 Hz, 1H, CHAr), 8.27 (s, 1H, CHTriazole), 8.49 (s, 1H, CHTriazole) ppm; 13C NMR (125.76 MHz, CDCl3) δ 110.6 (dd, J = 24, 4 Hz, CAr), 113.9, 114.0, 118.0 (m, CAr, CTriazole), 122.7 (t, J = 9 Hz, CAr), 123.8, 125.9, 134.5, 144.8–145.1 (m, CArF), 145.3 (t, J = 3 Hz, CTriazole), 146.9–147.2 (m, CArF), 151.7 (ddd, J = 254, 3, 2 Hz, CArF), 153.3, 154.9 (td, J = 4, 2 Hz, CPyranone), 156.1, 177.6 ppm; 19F NMR (470.52 MHz, CDCl3) δ 23.72 (d, J = 20.1 Hz, 1F), 24.92 (ddd, J = 20.3, 14.5, 5.9 Hz, 1F), 39.24 (dd, J = 14.6, 10.8 Hz, 1F) ppm; Anal. calcd. for C17H8F3N3O2: C 59.48, H 2.35, N 12.24, found: C 54.40, H 2.32, N 12.29.
2-[2,3,4,5-Tetra(1H-1,2,4-triazol-1-yl)phenyl]-4H-chromen-4-one (14). Yield 333 mg (68%); light-yellow powder; mp 295–296 °C; ; IR ν 3134, 3108 (C–HAr), 1644 (C=O), 1508, 1467 (C=CAr, C–HAr, C–N) cm−1; 1H NMR (500.13 MHz, (CD3)2SO) δ 6.87 (s, 1H, CHPyranone), 7.22 (d, J = 8.3 Hz, 1H, CHAr), 7.49–7.53 (m, 1H, CHAr), 7.80 (ddd, J = 8.7, 7.4, 1.6 Hz, 1H, CHAr), 8.00 (s, 1H, CHTriazole), 8.03 (dd, J = 7.9, 1.5 Hz, 1H, CHAr), 8.05 (s, 1H, CHTriazole), 8.10 (s, 1H, CHTriazole), 8.17 (s, 1H, CHTriazole), 8.61 (s, 1H, CHTriazole), 8.68 (s, 1H, CHTriazole), 8.86 (s, 1H, CHTriazole), 8.87 (s, 1H, CHTriazole), 9.04 (s, 1H, CHTriazole) ppm; 13C NMR (125.76 MHz, CDCl3) δ 112.8, 118.1 (s, 2CAr), 122.9, 124.9, 126.1, 129.3, 131.1, 132.9 (d, J = 2 Hz, CAr), 133.6, 134.9, 135.2, 146.0, 146.8, 146.9, 147.2, 152.6, 152.7, 152.8, 153.1, 155.4, 158.6, 176.5 ppm; Anal. calcd. for C23H14N12O2: C 56.33, H 2.88, N 34.27, found: C 56.11, H 2.90, N 34.22.
2-[2,3,5-Trifluoro-4-(1H-imidazol-1-yl)phenyl]-4H-chromen-4-one (15). Yield 277 mg (81%); white powder; mp 204–206 °C; IR ν 3160, 3133, 3077 (C–HAr), 1625, 1606 (C=O), 1515, 1465, 1351 (C=CAr, C–HAr, C–N), 1007 (C–F) cm−1; 1H NMR (500.13 MHz, CDCl3) δ 7.00 (s, 1H, CHPyranone), 7.31 (br.s., 2H, 2CHImidazole), 7.47–7.50 (m, 1H, 1CHAr), 7.58 (d, J = 8.2 Hz, 1H, CHAr), 7.71–7.74 (m, 1H, 1CHAr), 7.75–7.78 (m, 1H, 1CHAr), 7.88 (br.s., 1H, CHImidazole), 8.25 (dd, J = 8.0, 1.6 Hz, 1H, CHAr) ppm; 13C NMR (125.76 MHz, CDCl3) δ 110.6 (dd, J = 25, 4 Hz, CAr), 113.5, 113.6, 118.0, 118.5 (dd, J = 17, 12 Hz, CArF), 119.8 (t, J = 2 Hz, CImidazole), 120.7 (t, J = 9 Hz, CArF), 123.7, 125.9, 130.2, 134.4, 137.6 (t, J = 4 Hz, CImidazole), 145.3 (ddd, J = 256, 17, 4 Hz, CArF), 146.2 (ddd, J = 258, 14, 4 Hz, CArF), 151.2 (dt, J = 251, 3 Hz, CArF), 155.1 (m, CArF), 156.1, 177.7 ppm; 19F NMR (470.52 MHz, CDCl3) δ 21.78 (dd, J = 19.8, 1.2 Hz, 1F), 24.83 (ddd, J = 20.1, 14.3, 1.2 Hz, 1F), 38.13–38.18 (m, 1F) ppm; Anal. calcd. for C18H9F3N2O2: C 63.16, H 2.65, N 8.18, found: C 63.16, H 2.81, N 8.29.
2-[5-Fluoro-2,3,4-tri(1H-imidazol-1-yl)phenyl]-4H-chromen-4-one (17). Yield 31 mg (14%); yellow powder; mp 168–170 °C; 1H NMR (400.13 MHz, CDCl3) δ 6.40 (s, 1H, CHPyranone), 6.57 (t, J = 1.3 Hz, 1H, CHImidazole), 6.73 (d, J = 1.1 Hz, 1H, CHAr), 6.78 (t, J = 1.3 Hz, 1H, CHImidazole), 7.03–7.04 (m, 2H, CHImidazole), 7.13 (br.s, 1H, CHImidazole), 7.15–7.17 (m, 2H, CHImidazole), 7.39 (br.s, 1H, CHImidazole), 7.40–7.44 (m, 1H, CHAr), 7.47 (br.s, 1H, CHImidazole), 7.65 (ddd, J = 8.7, 7.3, 1.6 Hz, 1H, CHAr), 7.85 (d, J = 9.1 Hz, 1H, CHAr), 8.15 (dd, J = 8.0, 1.5 Hz, 1H, CHAr) ppm; 13C NMR (125.76 MHz, CDCl3) δ 112.8, 117.9, 118.5 (d, J = 23 Hz, CArF), 119.0, 119.1, 120.0, 123.4, 125.7–125.8 (m, CArF, CAr), 126.1, 129.6 (d, J = 4 Hz, CArF), 131.0, 131.1, 131.5, 132.8 (d, J = 2 Hz, CArF), 132.9 (d, J = 9 Hz, CArF), 134.7, 136.5, 137.0 (d, J = 2 Hz, CImidazole), 137.4, 155.9, 156.2 (d, J = 259 Hz, CArF), 158.6 (d, J = 2Hz, CPyranone), 177.1 ppm; 19F NMR (376.44 MHz, CDCl3) δ 47.37 (d, J = 9.2 Hz, 1F) ppm; Anal. calcd. for C24H15FN6O2: C 65.75, H 3.45, N 19.17, found: C 65.95, H 3.32, N 18.84.
2-[3,5-Difluoro-4-(1H-1,2,4-triazol-1-yl)-2-methoxyphenyl]-4H-chromen-4-one (18). Yield 35 mg (20% according to technique A), 123 mg (69% according to technique B); white powder; mp 172–174 °C; IR ν 3143, 3108, 3063 (C–HAr, C–HAlk), 1665 (C=O), 1523, 1464, 1377 (C=CAr, C–HAr, C–N), 1034 (C–F) cm−1; 1H NMR (400.13 MHz, CDCl3) δ 4.06 (d, J = 1.9 Hz, 3H, OCH3), 7.15 (s, 1H, CHPyranone), 7.45–7.49 (m, 1H, CHAr), 7.56 (dd, J = 8.4, 0.5 Hz, CHAr), 7.66 (dd, J = 10.4, 2.3 Hz, CHAr), 7.75 (ddd, J = 8.7, 7.2, 1.7 Hz, CHAr), 8.25 (s, 1H, CHTriazole), 8.26 (dd, J = 7.9, 1.6 Hz, CHAr), 8.45 (br.s, 1H, CHTriazole) ppm; 13C NMR (125.76 MHz, CDCl3) δ 62.1 (d, J = 6 Hz, COMe), 111.2 (dd, J = 23, 4 Hz, CArF), 113.6, 117.6 (dd, J = 16, 14 Hz, CArF), 118.0, 123.8, 125.6, 125.8, 127.3 (dd, J = 9, 4 Hz, CArF), 134.2, 144.0 (dd, J = 12, 4 Hz, CArF), 145.4 (br.s, CTriazole), 150.5 (dd, J = 257, 4 Hz, CArF), 151.4 (dd, J = 252, 3 Hz, CArF), 153.1, 156.2, 157.2 (dd, J = 4, 2 Hz, CPyranone), 178.2 ppm; 19F NMR (376.44 MHz, CDCl3) δ 29.29–29.30 (m, 1F), 37.19 (dd, J = 10.5, 1.3 Hz, 1F) ppm; Anal. calcd. for C18H11F2N3O3: C 60.85, H 3.12, N 11.83, found: C 60.92, H 3.26, N 11.72.
2-[5-Fluoro-2-methoxy-3,4-di(1H-1,2,4-triazol-1-yl)phenyl]-4H-chromen-4-one (19). Yield 24 mg (12%); white powder; mp 203–204 °C; IR ν 3113, 3100, 2953, 2924 (C–HAr, C–HAlk), 1640 (C=O), 1510, 1467, 1374 (C=CAr, C–HAr, C–N), 1008 (C–F) cm−1; 1H NMR (400.13 MHz, CDCl3) δ 3.49 (s, 3H, OCH3), 7.16 (s, 1H, CHPyranone), 7.48–7.52 (m, 1H, CHAr), 7.59 (d, J = 8.4 Hz, 1H, CHAr), 7.76–7.80 (m, 1H, CHAr), 7.96–8.01 (m, 3H, CHTriazole), 8.27 (dd, J = 8.0, 1.4 Hz, 1H, CHAr), 8.37 (d, J = 1.5 Hz, 1H, CHAr), 8.42 (s, 1H, CHTriazole) ppm; 13C NMR (125.76 MHz, CDCl3) δ 62.4 (s, COMe), 113.4, 118.0 (d, J = 24 Hz, CArF), 118.0, 123.8, 124.9 (d, J = 15 Hz, CArF), 125.9, 126.0, 129.0 (d, J = 8 Hz, CArF), 129.6 (d, J = 2 Hz, CTriazole), 134.5, 145.7 (d, J = 2 Hz, CTriazole), 146.3, 151.0 (d, J = 4 Hz, CArF), 152.6 (d, J = 253 Hz, CArF), 152.8, 152.9, 156.3, 157.1 (d, J = 2 Hz, CPyranone), 177.9 ppm; 19F NMR (376.44 MHz, CDCl3) δ 38.81 (dd, J = 9.9, 1.6 Hz, 1F) ppm; Anal. calcd. for C20H13FN6O3: C 59.41, H 3.24, N 20.78, found: C 59.51, H 3.23, N 20.79.
2-[5-Fluoro-2-hydroxy-3,4-di(1H-1,2,4-triazol-1-yl)phenyl]-4H-chromen-4-one (22). Yield 22 mg (11%); yellow powder; mp 313–316 °C; 1H NMR (400.13 MHz, (CD3)2SO) δ 7.04 (s, 1H, CHPyranone), 7.52–7.56 (m, 1H, CHAr), 7.78–7.80 (m, 1H, CHAr), 7.88 (ddd, J = 8.6, 7.2, 1.6 Hz, 1H, CHAr), 8.07 (s, 1H, CHTriazole), 8.09 (dd, J = 8.1, 1.5 Hz, 1H, CHAr), 8.11 (s, 1H, CHTriazole), 8.26 (d, J = 10.4 Hz, 1H, CHAr), 8.76 (s, 1H, CHTriazole), 8.79 (s, 1H, CHTriazole), 10.98 (s, 1H, OH) ppm; 13C NMR (125.76 MHz, (CD3)2SO) δ 112.6, 118.3 (d, J = 23 Hz, CArF), 118.7, 123.3 (d, J = 9 Hz, CArF), 124.0, 124. 7, 124.8 (d, J = 15 Hz, CArF), 125.7, 134.5, 146.6, 147.3, 148.7 (d, J = 2 Hz, CTriazole), 149.1 (d, J = 244 Hz, CArF), 152.3, 152.5, 156.0, 159.0, 177.1 ppm; 19F NMR (376.44 MHz, (CD3)SO) δ 30.74 (d, J = 9.6 Hz, 1F) ppm.
2-[3,5-Difluoro-4-(1H-imidazol-1-yl)-2-methoxyphenyl]-4H-chromen-4-one (23). Yield 39 mg (23% according to technique A), 94 mg (53% according to technique B); white powder; mp 176–177 °C; IR ν 3108, 3043, 2999, 2950 (OMe, C–HAr), 1630 (C=O), 1524, 1480, 1370 (C=CAr, C–HAr, C–O, C–N), 1112 (C–F) cm−1; °C; 1H NMR (400.13 MHz, CDCl3) δ 4.04 (d, J = 1.7 Hz, 3H, OMe), 7.14 (s, 1H, CHPyranone), 7.27–7.29 (m, 2H, 2CHImidazole), 7.45–7.49 (m, 1H, 1CHAr), 7.55–7.57 (m, 1H, 1CHAr), 7.64 (dd, J = 10.8, 2.2 Hz, 1H, CHAr), 7.75 (ddd, J = 8.6, 7.2, 1.7 Hz, 1H, CHAr), 7.83 (br.s., 1H, CHImidazole), 8.25 (dd, J = 8.0, 1.5 Hz, 1H, CHAr) ppm; 13C NMR (125.76 MHz, CDCl3) δ 62.1 (d, J = 6 Hz, COMe), 111.2 (dd, J = 24, 4 Hz, CAr), 113.3, 118.0, 118.2 (dd, J = 14, 2 Hz, CAr), 120.0 (t, J = 2 Hz, CImidazole), 123.8, 125.5–125.6 (m, CAr,CImidazole), 125.8, 129.9, 134.2, 137.7 (t, J = 3 Hz, CImidazole), 144.2 (dd, J = 12, 4 Hz, CAr), 150.1 (dd, J = 254, 4 Hz, CArF), 151.1 (dd, J = 249, 4 Hz, CArF), 156.2, 157.4 (dd, J = 4, 2 Hz, CPyranone), 178.2 ppm; 19F NMR (376.44 MHz, CDCl3) δ 27.59–27.60 (m, 1F), 36.45–36.48 (m, 1F)ppm; Anal. calcd. for C19H12F2N2O3: C 64.41, H 3.41, N 7.91, found: C 64.37, H 3.40, N 8.02.
2-[2,5-Difluoro-3,6-di(1H-pyrazol-1-yl)-4-(1H-1,2,4-triazol-1-yl)phenyl]-4H-chromen-4-one (26). Yield 39 mg (17%); white powder; mp 256–258 °C; IR ν 3130, 3106, 3071 (C–HAr), 1642 (C=O), 1529, 1483, 1390 (C=CAr, C–HAr, C–N), 1139, 1127 (C–F) cm−1; 1H NMR (400.13 MHz, CDCl3) δ 6.44 (d, J = 1.2 Hz, 1H, CPyranone), 6.45–6.46 (m, 1H, CHImidazole), 6.48–6.49 (m, 1H, CHImidazole), 7.19–7.21 (m, 1H, CHAr), 7.40–7.44 (m, 1H, CHAr), 7.58 (d, J = 1.6 Hz, 1H, CAr), 7.63–7.66 (m, 3H, CHImidazole), 7.84 (t, J = 2.6 Hz, 1H, CImidazole), 8.05 (s, 1H, CHTriazole), 8.19 (dd, J = 8.0, 1.5 Hz, 1H, CAr), 8.24 (s, 1H, CHTriazole) ppm; 13C NMR (125.76 MHz, CDCl3) δ 108.4, 108.6, 115.0 (d, J = 3 Hz, CPyrazole), 118.0, 121.4 (d, J = 17 Hz, CArF), 123.7, 125.5 (dd, J = 14, 3 Hz, CArF), 125.6, 125.8, 126.3 (d, J = 17 Hz, CArF), 128.7 (dd, J = 14, 3 Hz, CArF), 132.0 (d, J = 4 Hz, CPyrazole), 132.3 (d, J = 2 Hz, CPyrazole), 134.1, 142.7, 142.9, 145.7 (d, J = 1 Hz, CPyranone), 148.0 (dd, J = 258, 4 Hz, CArF), 151.1 (dd, J = 257, 4 Hz, CArF), 153.1, 154.3 (d, J = 2 Hz, CPyranone), 177.2 ppm; 19F NMR (376.44 MHz, CDCl3) δ 30.62 (dd, J = 14.4, 1.8 Hz, 1F), 30.75–39.80 (m, 1F) ppm; Anal. calcd. for C23H13F2N7O2: C 60.40, 2.86, N 21.44, found: C 60.15, H 2.68, N 21.54.
2-[2,3,5,6-Tetra(1H-pyrazol-1-yl)-4-(1H-1,2,4-triazol-1-yl)phenyl]-4H-chromen-4-one (27). Yield 136 mg (49%); yellow powder; mp 299–301 °C; IR ν 3126, 3092 (C–HAr), 1646 (C=O), 1525, 1468, 1389 (C=CAr, C–HAr, C–N) cm−1; 1H NMR (400.13 MHz, CDCl3) δ 5.30 (s, 1H, CHPyrazole), 6.08 (s, 1H, CHPyrazole), 6.16–6.18 (m, 5H, CHPyranone, CHPyrazole), 7.11 (d, J = 8.4 Hz, 1H, CHAr), 7.24 (d, J = 2.5 Hz, 1H, CHPyrazole), 7.29 (d, J = 2.5 Hz, 1H, CHPyrazole), 7.32–7.36 (m, 1H, CHAr), 7.43 (d, J = 1.6 Hz, 2H, CHPyrazole), 7.49 (d, J = 1.6 Hz, 2H, CHPyrazole), 7.54–7.59 (m, 1H, CHAr), 7.74 (s, 1H, CHTriazole), 8.05 (s, 1H, CHTriazole), 8.06–8.08 (m, 1H, CHAr) ppm; 13C NMR (125.76 MHz, CDCl3) δ 108.0 (s, 2CPyrazole), 108.2 (s, 2CPyrazole), 113.3, 117.8, 123.3, 125.4, 125.7 (s, 2CAr), 131.8 (s, 2CPyrazole), 132.1 (s, 2CPyrazole), 132.7, 133.8 (s, 2CAr), 135.2, 135.9, 138.2, 142.2 (s, 2CPyrazole), 142.3 (s, 2CPyrazole), 145.9, 152.3, 156.0, 156.6, 177.0 ppm; Anal. calcd. for C29H19N11O2: C 62.92, 3.46, N 27.83, found: C 63.15, H 3.68, N 27.69.
2-[4-(1H-Imidazol-1-yl)-2,3,5,6-tetra(1H-pyrazol-1-yl)phenyl]-4H-chromen-4-one (28). Yield 31 mg (11%); yellow powder; mp 320–321 °C; IR ν 3127 (C–HAr), 1649 (C=O), 1525, 1467, 1388 (C=CAr, C–HAr, C–N) cm−1; 1H NMR (400.13 MHz, CDCl3) δ 6.03 (s, 1H, CHPyranone), 6.12–6.13 (m, 1H, CHPyrazole), 6.15 (dd, J = 4.3, 2.2 Hz, 4H, CHPyrazole), 7.11 (d, J = 8.3 Hz, 1H, CHAr), 7.20–7.23 (m, 3H, CHPyrazole, CHImidazole), 7.31–7.34 (m, 3H, CHAr, CHPyrazole, CHImidazole), 7.39 (d, J = 1.6 Hz, 2H, CHPyrazole), 7.41 (d, J = 1.6 Hz, 1H, CHImidazole), 7.47 (d, J = 1.5 Hz, 2H, CHPyrazole), 7.55 (ddd, J = 8.6, 7.3, 1.6 Hz, 1H, CHAr), 8.06 (dd, J = 7.9, 1.5 Hz, 1H, CHAr) ppm; 13C NMR (125.76 MHz, CDCl3) δ 107.3, 107.5 (s, 2CPyrazole), 107.8 (s, 2CPyrazole), 113.2, 117.9, 123.3, 125.3, 125.6, 131.5, 131.9 (s, 2CAr), 132.0 (s, 2CPyrazole), 132.2 (s, 2CPyrazole), 133.7, 135.8, 138.3, 138.4, 141.7 (s, 2CAr), 141.7 (s, 2CPyrazole), 141.9 (s, 2CPyrazole), 156.0, 157.1, 177.1 ppm; Anal. calcd. for C30H20N10O2: C 65.21, 3.65, N 25.35, found: C 65.15, H 3.57, N 25.55.
2-[4-(1H-Imidazol-1-yl)-2,3,5,6-tetra(1H-1,2,4-triazol-1-yl)phenyl]-4H-chromen-4-one (29). Yield 92 mg (33%); pale pink powder; mp 299–301 °C; IR ν 3123, 3105 (C–HAr), 1647 (C=O), 1510, 1464, 1378 (C=CAr, C–HAr, C–N) cm−1; 1H NMR (500.13 MHz, (CD3)2SO) δ 6.30 (s, 1H, CHPyranone), 6.84 (s, 1H, CHImidazole), 7.01 (t, J = 1.3 Hz, 1H, CHImidazole), 7.34 (d, J = 8.2 Hz, 1H, CHAr), 7.47–7.49 (m, 1H, CHAr), 7.52 (s, 1H, CHImidazole), 7.80 (ddd, J = 8.7, 7.2, 1.7 Hz, 1H, CHAr), 7.92 (dd, J = 8.0, 1.6 Hz, 1H, CHAr), 8.08 (s, 2H, CHTriazole), 8.11 (s, 2H, CHTriazole), 8.63 (s, 2H, CHTriazole), 8.82 (s, 2H, CHTriazole) ppm; 13C NMR (125.76 MHz, CDCl3) δ 113.7, 117.9, 121.0, 122.4, 124.9, 126.3, 129.3, 130.6, 134.0 (s, 2CAr), 135.0, 135.5 (s, 2CAr), 136.0, 137.9, 146.7 (s, 2CTriazole), 146.8 (s, 2CTriazole), 152.9 (s, 2CTriazole), 152.9 (s, 2CTriazole), 154.8, 155.4, 175.4 ppm; Anal. calcd. for C26H16N14O2: C 56.11, 2.90, N 35.24, found: C 56.30, H 2.98, N 35.04.
2-[4-(1H-Pyrazol-1-yl)-2,3,5,6-tetra(1H-1,2,4-triazol-1-yl)phenyl]-4H-chromen-4-one (31). Yield 159 mg (59%); pale pink powder; mp 309–310 °C; IR ν 3113 (C–HAr), 1651 (C=O), 1511, 1466, 1386 (C=CAr, C–HAr, C–N) cm−1; 1H NMR (500.13 MHz, (CD3)2SO) δ 6.31–6.32 (m, 1H, CHPyrazole), 6.32 (s, 1H, CHPyranone), 7.34 (d, J = 8.2 Hz, 1H, CHAr), 7.46–7.49 (m, 1H, CHAr), 7.56 (d, J = 1.7 Hz, 1H, CHPyrazole), 7.67 (d, J = 2.5 Hz, 1H, CHPyrazole), 7.79 (ddd, J = 8.7, 7.2, 1.7 Hz, 1H, CHAr), 7.92 (dd, J = 8.0, 1.6 Hz, 1H, CHAr), 8.04 (s, 2H, CHTriazole), 8.05 (s, 2H, CHTriazole), 8.53 (s, 2H, CHTriazole), 8.82 (s, 2H, CHTriazole) ppm; 13C NMR (125.76 MHz, (CD3)2SO) δ 107.9, 113.7, 118.0, 122.4, 124.9, 126.3, 130.8, 133.1, 133.9 (s, 2CAr), 135.1, 135.4 (s, 2CAr), 137.6, 142.5, 146.8 (s, 2CTriazole), 146.9 (s, 2CTriazole), 152.7 (s, 2CTriazole), 152.9 (s, 2CTriazole), 155.0, 155.4, 175.5 ppm; Anal. calcd. for C26H16N14O2: C 56.11, 2.90, N 35.24, found: C 56.26, H 2.99, N 35.11.
3.3. XRD Experiments
The X-ray studies were performed on an Xcalibur 3 CCD (Oxford Diffraction Ltd., Abingdon, UK) diffractometer with a graphite monochromator, λ(MoKα) 0.71073 Å radiation and T 295(2) K. An empirical absorption correction was applied. Using Olex2 [51], the structure was solved with the Superflip [52] structure solution program using charge flipping and refined with the ShelXL [53] refinement package using Least Squares minimization. All non-hydrogen atoms were refined in the anisotropic approximation; H-atoms at the C–H bonds were refined in the “rider” model with dependent displacement parameters. An empirical absorption correction was carried out through spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm by the program “CrysAlisPro” (Rigaku Oxford Diffraction).
The main crystallographic data for 4: C17H7F4N3O2, M 361.26, orthorhombic, a 15.8944(12), b 12.7694(11), c 7.3245(6) Å, V 1486.6(2) Å3, space group Pna21, Z 4, μ(Mo Kα) 0.125 mm–1, 256 refinement parameters, 3609 reflections measured, and 2493 unique (Rint = 0.0617), which were used in all calculations. CCDC 2225826 contains the supplementary crystallographic data for this compound.
The main crystallographic data for 8: C18H8F4N2O2, M 360.26, monoclinic, a 15.0164(11), b 7.9494(7), c 12.8592(10) Å, β 99.256(7)°, V 1515.0(2) Å3, space group P21/c, Z 4, μ(Mo Kα) 0.125 mm–1, 268 refinement parameters, 4162 reflections measured, and 2246 unique (Rint = 0.0633), which were used in all calculations. CCDC 2,225,827 contains the supplementary crystallographic data for this compound.
The main crystallographic data for 15: C18H9F3N2O2, M 342.27, monoclinic, a 13.3565(10), b 7.8477(5), c 14.7075(12) Å, β 113.251(9)°, V 1416.4(2) Å3, space group P21/n, Z 4, μ(Mo Kα) 0.125 mm–1, 262 refinement parameters, 3865 reflections measured, and 2692 unique (Rint = 0.0597), which were used in all calculations. CCDC 2,225,828 contains the supplementary crystallographic data for this compound.
The main crystallographic data for 19: C20H13FN6O3, M 404.36, triclinic, a 13.3565(10), b 7.8477(5), c 14.7075(12) Å, α 111.535(14), β 94.311(13), γ 101.586(12)°, V 1416.4(2) Å3, space group P, Z 2, μ(Mo Kα) 0.125 mm–1, 288 refinement parameters, 3662 reflections measured, 1504 unique (Rint = 0.0711) which were used in all calculations. CCDC 2,225,829 contains the supplementary crystallographic data for this compound.
The main crystallographic data for 23: C19H12F2N2O3, M 354,31, monoclinic, a 14.0716(10), b 7.7151(5), c 14.5964(12) Å, β 100.453(7)°, V 1553.0(2) Å3, space group P21/c, Z 4, μ(Mo Kα) 0.125 mm–1, 257 refinement parameters, 4236 reflections measured, and 2349 unique (Rint = 0.0568) which were used in all calculations. CCDC 2,225,830 contains the supplementary crystallographic data for this compound.
3.4. Fungistatic Activity Evaluation
The following dermatophyte fungal strains were used: Trichophyton rubrum (RCPF F 1408), Epidermophyton floccosum (RCPF F 1659/17), and Microsporum canis (RCPF F 1643/1585), as well as yeast-like fungus Candida parapsilosis (RCPF 1245/ ATCC 22019). The fungi cultures were obtained from the Russian Collection of Pathogenic Fungi (Kashkin Research Institute of Medical Mycology; Mechnikov Northwest State Medical University, St.-Petersburg). Saburo agar and Saburo broth were used for the fungi. The microorganisms were identified as matrix-extracted bacterial proteins with an accuracy of 99.9% using a BioMerieux VITEK MS MALDI-TOF analyzer. The test cultures were prepared to an optical density of 0.5 according to McFarland (1.5 × 108 CFU/ mL) using a BioMerieux DensiCHEK densimeter. The suspensions of C. parapsilosis were prepared from 24 h cultures, and dermatophyte inocula were prepared after incubation for 2 weeks and preliminary homogenization in sterile saline. The fungi were inoculated at a concentration of 105 CFU/mL. The antimycotic activity was evaluated by a micro method [54]. The agar nutrient medium was maintained in liquid by heating to 52 °C. The chemical compounds to be tested were dissolved in DMSO to a concentration of 1000 μg/ mL, and the stock solutions were diluted with distilled sterilized water; serial dilutions (from 250–200 μg/ mL) were made using nutrient media. Dermatophytes were incubated at 27 °C for up to 7–10 days and C. parapsilosis for 24 h in a moist 5.0% CO2 chamber. In each case, positive and negative controls were used. The minimum inhibitory concentration was determined visually as the lowest concentration at which a test culture no longer grows. Chemically pure fluconazole was used as a reference drug.
4. Conclusions
The data obtained in this work and earlier in the study of transformations with pyrazole [29] thus indicate that base-promoted reactions of nucleophilic aromatic substitution are a convenient method for the functionalization of polyfluoroflavones 1–3 with azoles with different numbers of nitrogen atoms. At the same time, it was found that monosubstitution of the para-fluorine atom successfully and selectively occurs while using the system (azole (1.5 equiv.)/NaOBut (1.5 equiv.)/MeCN) regardless of the structure and properties of the used polyfluorinated substrates and nucleophilic reagents, since in all cases, mono(azolyl)-substituted flavones were obtained in good yields. Under the conditions (azole (6 equiv.)/NaOBut (6 equiv.)/DMF), which promote the formation of persubstituted products, the interactions of polyfluoroflavones 1–3 with pyrazole are distinguished by high selectivity [26], while similar reactions with triazole produced productively only for penta- and tetrafluoroflavones 1 and 2, and the same transformations with imidazole in general are extremely non-selective.
Comparing the conversion of polyfluoroflavones 1–3 in reactions with azoles under conditions that do not provide selective substitution (azole (3 equiv.)/Cs2CO3 (3 equiv.)/MeCN)), it can be noted that transformations with pyrazole [26] are characterized by easier formation of polysubstituted products, and under conditions conducive to persubstitution, it is possible to build the following series of azoles according to reactivity: pyrazole ≥ triazole > imidazole. Obviously, in both cases, the reactivity of azoles does not correspond to their basicity, and therefore to some extent, nucleophilicity, since imidazole is known to be the strongest base among them [55]. According to the literature data [56,57], polyfluoroaromatic compounds generally react with nucleophiles via Meisenheimer complexes. For flavones 1–3, we assume an analogous mechanism [26]; first nucleophile attack occurs on the activated C4′ site of flavones 1–3 with the generation of an intermediate of a stable quinoid structure, followed by formation of a mono(azolyl)-substituted product. Sequential substitution is coordinated by the joint activating effect of the substituents, and the resulting intermediate complexes are stabilized both by O,N-bidentate coordination between the azole and the pyrone fragment of the molecule with Na+ and by the coordination of neighboring azole moieties with Na+. It is likely that the higher reactivity of pyrazole, triazole, and their intermediates compared to imidazole and its derivatives in SNAr poly- and per-substitution reactions is due to the possibility of participation of their imine nitrogen atoms in the N–N=C function in coordination during the formation of transitional complexes.
In addition, it was shown that the per-substitution conditions can be successfully used for the synthesis of polynuclear hybrid compounds containing two different azole fragments by the reaction of mono(azolyl)-substituted flavones with pyrazole and triazole.
Using XRD data, the structural features of triazolyl- and imidazolyl-substituted flavones in crystal form were established. For example, in contrast to previously synthesized pyrazole analogues [26], new azole derivatives do not contain an intramolecular H-bond.
In terms of possible practical applications, it has been established that the resulting poly(pyrazolyl)-substituted flavones have luminescent properties, which makes further development of research in this area promising. In addition, weak antimycotic activity was found for some azolyl-containing flavones.
Supplementary Materials
NMR data of the synthesized compounds can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28020869/s1. Figure S1. 1H NMR spectrum of compound 4; Figure S2. 13C NMR spectrum of compound 4; Figure S3. 19F NMR spectrum of compound 4; Figure S4. 1H NMR spectrum of compound 7; Figure S5. 13C NMR spectrum of compound 7; Figure S6. 1H NMR spectrum of compound 8; Figure S7. 13C NMR spectrum of compound 8; Figure S8. 19F NMR spectrum of compound 8; Figure S9. 1H NMR spectrum of compound 9; Figure S10. 19F NMR spectrum of compound 9; Figure S11. 1H NMR spectrum of compound 10; Figure S12. 13C NMR spectrum of compound 10; Figure S13. 19F NMR spectrum of compound 10; Figure S14. 1H NMR spectrum of compound 11; Figure S15. 13C NMR spectrum of compound 11; Figure S16. 19F NMR spectrum of compound 11; Figure S17. 1H NMR spectrum of compound 14; Figure S18. 13C NMR spectrum of compound 14; Figure S19. 1H NMR spectrum of compound 15; Figure S20. 13C NMR spectrum of compound 15; Figure S21. 19F NMR spectrum of compound 15; Figure S22. 1H NMR spectrum of compound 17; Figure S23. 13C NMR spectrum of compound 17; Figure S24. 19F NMR spectrum of compound 17; Figure S25. 1H NMR spectrum of compound 18; Figure S26. 13C NMR spectrum of compound 18; Figure S27. 19F NMR spectrum of compound 18; Figure S28. 1H NMR spectrum of compound 19; Figure S29. 13C NMR spectrum of compound 19; Figure S30. 19F NMR spectrum of compound 19; Figure S31. 1H NMR spectrum of compound 22; Figure S32. 13C NMR spectrum of compound 22; Figure S33. 19F NMR spectrum of compound 22; Figure S34. 1H NMR spectrum of compound 23; Figure S35. 13C NMR spectrum of compound 23; Figure S36. 19F NMR spectrum of compound 23; Figure S37. 1H NMR spectrum of compound 26; Figure S38. 13C NMR spectrum of compound 26; Figure S39. 19F NMR spectrum of compound 26; Figure S40. 1H NMR spectrum of compound 27; Figure S41. 13C NMR spectrum of compound 27; Figure S42. 1H NMR spectrum of compound 28; Figure S43. 13C NMR spectrum of compound 28; Figure S44. 1H NMR spectrum of compound 29; Figure S45. 13C NMR spectrum of compound 29; Figure S46. 1H NMR spectrum of compound 31; Figure S47. 13C NMR spectrum of compound 31.
Author Contributions
Conceptualization Y.V.B.; methodology and synthesis, validation, and interpretation of analysis data, K.V.S. and M.A.P.; analytical experiments E.F.Z.; writing—original draft preparation, K.V.S. and M.A.P.; writing—review and editing, Y.V.B.; supervision, V.I.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the Ministry of Science and Higher Education of the Russian Federation (State task AAAA-A19-119012290115-2).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The analytical data analysis (IR and NMR spectroscopy, elemental analysis, XRD analysis, and photoluminescent data) was performed on equipment belonging to the Center for Joint Use of Scientific Equipment “Spectroscopy and Analysis of Organic Compounds” IOS UB RAS. Fungistatic activity evaluation was performed on equipment belonging to the Ural Research Institute of Dermatovenereology and Immunopathology, Yekaterinburg, Russian Federation. The authors acknowledge Natalia A. Gerasimova for her contribution.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors.
Appendix A
Table A1.
NMR data of characteristic F and H nuclei of flavones 9, 10, and A.
Table A1.
NMR data of characteristic F and H nuclei of flavones 9, 10, and A.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | H3 | F2 | F3 | F5 | ||||
| δ | J | δ | J | δ | J | δ | J | |
| 9 | 6.52, d | 5JHF 1.4 | 26.33 ddd | 3JFF 22.6 5JFF 13.0 5JFH 1.4 | 23.60 d | 3JFF 22.4 | 30.57 d | 5JFF 13.1 |
| 10 | 6.51, d | 5JHF 1.3 | 41.18 dd | 5JFF 14.7 5JFH 1.3 | – | – | 31.47 d | 5JFF 14.7 |
| A | 6.44, d | 5JHF 1.1 | 29.51 dd | 5JFF 14.4 5JFH 1.1 | – | – | 39.83 d | 5JFF 14.3 |
Appendix B
Table A2.
Detailed data of the fluorescence lifetime measurements of 28 and 32: τ—lifetime, f—fractional contribution, τ avg—average lifetime, and χ2—chi-squared distribution.
Table A2.
Detailed data of the fluorescence lifetime measurements of 28 and 32: τ—lifetime, f—fractional contribution, τ avg—average lifetime, and χ2—chi-squared distribution.
| Compound | Solid | |||||
|---|---|---|---|---|---|---|
| τ1, ns | f1, % | τ2, ns | f2, % | τ avg, ns | χ2 | |
| 28 | 3.57 | 16.5 | 8.53 | 83.5 | 7.72 | 1.109 |
| 32 | 2.84 | 21.3 | 7.88 | 78.7 | 6.81 | 1.206 |

Figure A1.
Time−resolved fluorescence lifetime decay profile of solid powder 28 (green) and instrumental response function (IRF, blue). λex = 375 nm and λem = 504 nm.
Figure A1.
Time−resolved fluorescence lifetime decay profile of solid powder 28 (green) and instrumental response function (IRF, blue). λex = 375 nm and λem = 504 nm.


Figure A2.
Time−resolved fluorescence lifetime decay profile of solid powder 32 (green), instrumental response function (IRF, blue). λex = 375 nm and λem = 504 nm.
Figure A2.
Time−resolved fluorescence lifetime decay profile of solid powder 32 (green), instrumental response function (IRF, blue). λex = 375 nm and λem = 504 nm.

Appendix C
Table A3.
Fungistatic activity of fluorinated flavones and their azolyl-substituted derivatives.
Table A3.
Fungistatic activity of fluorinated flavones and their azolyl-substituted derivatives.
| No | Compounds | MIC (μg/mL)/MIC50 (μg/mL) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | R5 | T. rubrum | E. floccosum | M. canis | C. parapsilosis | |
| 4 | F | F | trz | F | F | >200 | >200 | >200 | >200 |
| 8 | F | F | imz | F | F | 100/25 | 200/100 | 100/50 | >200 |
| 11 | H | F | trz | F | F | >200 | >200 | >200 | >200 |
| 14 | H | trz | trz | trz | trz | 100/25 | 200/100 | 100/50 | >200 |
| 18 | OMe | F | trz | F | H | >200 | >200 | >200 | >200 |
| 19 | OMe | trz | trz | F | H | - | >200/50 | >200/200 | >200/200 |
| 23 | OMe | F | imz | F | H | >200/12.5 | >200/12.5 | >200/6.25 | >200 |
| 26 | F | pz | trz | F | pz | >200 | >200 | >200 | >200 |
| 33 | OMe | pz | pz | F | H | >100/1.56 | >100/1.56 | >100 | >100 |
| 34 | F | pz | pz | F | pz | >100 | >100 | >100 | >100 |
| 35 | pz | pz | pz | pz | pz | >100 | >100 | >100 | >100 |
| Fluconazole | 3.12 | 1.56 | 3.12 | 0.5–2 | |||||
References
- Tian, S.; Luo, T.; Zhu, Y.; Wan, J.-P. Recent advances in diversification of chromones and flavones by direct C–H bond activation or functionalization. Chin. Chem. Lett. 2020, 31, 3073–3082. [Google Scholar] [CrossRef]
- Kshatriya, R.; Jejurkar, V.P.; Saha, S. Recent advances in the synthetic methodologies of flavones. Tetrahedron 2018, 74, 811–833. [Google Scholar] [CrossRef]
- Spagnuolo, C.; Moccia, S.; Russo, G.L. Anti-inflammatory effects of flavonoids in neurodegenerative disorders. Eur. J. Med. Chem. 2018, 153, 105–115. [Google Scholar] [CrossRef]
- Singh, M.; Kaur, M.; Silakari, O. Flavones: An important scaffold for medicinal chemistry. Eur. J. Med. Chem. 2014, 84, 206–239. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.K.; Singh, H.; Satyanarayana, M.; Srivastava, S.P.; Tiwari, P.; Singh, A.B.; Dwivedi, A.K.; Singh, S.K.; Srivastava, M.; Nath, C.; et al. Flavone-based novel antidiabetic and antidyslipidemic agents. J. Med. Chem. 2012, 55, 4551–4567. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.-M.; Benfodda, Z.; Dunyach-Rémy, C.; Bénimélis, D.; Roulard, R.; Fontaine, J.-X.; Mathiron, D.; Quéro, A.; Molinié, R.; Meffre, P. Isolation and identification of flavones responsible for the antibacterial activities of Tillandsia bergeri extracts. ACS Omega 2022, 7, 35851–35862. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Wu, M.; Xiang, S.; Song, T.; Li, Y.; Long, B.; Feng, C.; Shi, Z. Total syntheses and antibacterial evaluations of neocyclomorusin and related flavones. J. Nat. Prod. 2022, 85, 2217–2225. [Google Scholar] [CrossRef] [PubMed]
- Rubin, D.; Sansom, C.E.; Lucas, N.T.; McAdam, J.C.; Simpson, J.; Lord, J.M.; Perry, N.B. O-Acylated flavones in the alpine daisy Celmisia viscosa: Intraspecific variation. J. Nat. Prod. 2022, 85, 1904–1911. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cao, Y.; Chen, S.; Lin, J.; Yang, X.; Huang, D. Structure–activity relationship (SAR) of flavones on their anti-inflammatory activity in murine macrophages in culture through the NF-κB pathway and c-Src kinase receptor. J. Agric. Food Chem. 2022, 70, 8788–8798. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-Y.; Chen, M.-Y.; Hsu, C.; Kuan, K.-Y.; Chang, C.-F.; Wang, C.-W.; Hsu, C.-P.; Su, N.-W. Luteolin phosphate derivatives generated by cultivating Bacillus subtilis var. Natto BCRC 80517 with luteolin. J. Agric. Food Chem. 2022, 70, 8738–8745. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Wu, M.; Li, Y.; Lu, L.; Qin, J.; He, Y.; Shi, Z. Total syntheses and anti-inflammatory evaluations of pongamosides A–C, natural furanoflavonoid glucosides from fruit of Pongamia pinnata (L.) Pierre. J. Nat. Prod. 2022, 85, 1118–1127. [Google Scholar] [CrossRef]
- Li, J.; Tan, L.-H.; Zou, H.; Zou, Z.-X.; Long, H.-P.; Wang, W.-X.; Xu, P.-S.; Liu, L.-F.; Xu, K.-P.; Tan, G.-S. Palhinosides A–H: Flavone glucosidic truxinate esters with neuroprotective activities from Palhinhaea cernua. J. Nat. Prod. 2020, 83, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Koh, J.-J.; Aung, T.T.; Sin, W.L.W.; Lim, F.; Wang, L.; Lakshminarayanan, R.; Zhou, L.; Tan, D.T.H.; Cao, D.; et al. Semisynthetic flavone-derived antimicrobials with therapeutic potential against methicillin-resistant Staphylococcus aureus (MRSA). J. Med. Chem. 2017, 60, 6152–6165. [Google Scholar] [CrossRef]
- Pajtás, D.; Kónya, K.; Kiss-Szikszai, A.; Džubák, P.; Pethő, Z.; Varga, Z.; Panyi, G.; Patonay, T. Optimization of the synthesis of flavone–amino acid and flavone–dipeptide hybrids via Buchwald–Hartwig reaction. J. Org. Chem. 2017, 82, 4578–4587. [Google Scholar] [CrossRef] [PubMed]
- Byun, Y.; Moon, K.; Park, J.; Ghosh, P.; Mishra, N.K.; Kim, I.S. Methylene thiazolidinediones as alkylation reagents in catalytic C–H functionalization: Rapid access to glitazones. Org. Lett. 2022, 24, 8578–8583. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Schaus, S.E.; Porco, J.A., Jr. Metal-catalyzed cascade rearrangements of 3-alkynyl flavone ethers. Org. Lett. 2013, 15, 1962–1965. [Google Scholar] [CrossRef]
- Hosseini, S.; Thapa, B.; Medeiros, M.J.; Pasciak, E.M.; Pence, M.A.; Twum, E.B.; Karty, J.A.; Gao, X.; Raghavachari, K.; Peters, D.G.; et al. Electrosynthesis of a baurone by controlled dimerization of flavone: Mechanistic insight and large-scale application. J. Org. Chem. 2020, 85, 10658–10669. [Google Scholar] [CrossRef]
- Tang, Q.; Bian, Z.; Wu, W.; Wang, J.; Xie, P.; Pittman, C.U., Jr.; Zhou, A. Making flavone thi-oethers using halides and powdered sulfur or Na2S2O3. J. Org. Chem. 2017, 82, 10617–10622. [Google Scholar] [CrossRef]
- Tokárová, Z.; Balogh, R.; Tisovský, P.; Hrnčariková, K.; Végh, D. Direct nucleophilic substitution of polyfluorobenzenes with pyrrole and 2,5-dimethylpyrrole. J. Fluorine Chem. 2017, 204, 59–64. [Google Scholar] [CrossRef]
- Gerencsér, J.; Balázs, A.; Dormán, G. Synthesis and modification of heterocycles by metal-catalyzed cross-coupling reactions. In Topics in Heterocyclic Chemistry; Patonay, T., Kónya, K., Eds.; Springer International Publishing AG: Cham, Switzerland, 2016; Volume 45. [Google Scholar]
- Kong, X.; Zhang, H.; Cao, C.; Shi, Y.; Pang, G. Effective transition metal free and selective C–F activation under mild conditions. RSC Adv. 2015, 5, 7035–7048. [Google Scholar] [CrossRef]
- Boelke, A.; Sadat, S.; Lork, E.; Nachtsheim, B.J. Pseudocyclic bis-N-heterocycle-stabilized iodanes—Synthesis, characterization and applications. Chem. Commun. 2021, 57, 7434–7437. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Wang, L.; Qin, D.; Zhou, J.; Duan, H. Two novel fluorescent probes as systematic sensors for multiple metal ions: Focus on detection of Hg2+. ACS Omega 2020, 5, 24285–24295. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Hong, X.; Cui, H.-Z.; Huang, S.; Yi, Y.; Hou, X.-F. The construction of C–N, C–O, and C(sp2)–C(sp3) bonds from fluorine-substituted 2-aryl benzazoles for direct synthesis of N-, O-, C-functionalized 2-aryl benzazole derivatives. J. Org. Chem. 2018, 83, 6363–6372. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.M.M.; Silva, V.L.M.; Silva, A.M.S. Synthesis of chromone-related pyrazole compounds. Molecules 2017, 22, 1665–1713. [Google Scholar] [CrossRef] [PubMed]
- Shcherbakov, K.V.; Panova, M.A.; Burgart, Y.V.; Saloutin, V.I. Selective nucleophilic aromatic substitution of 2-(polyfluorophenyl)-4H-chromen-4-ones with pyrazole. J. Fluorine Chem. 2022, 263, 110034. [Google Scholar] [CrossRef]
- Shcherbakov, K.V.; Artemyeva, M.A.; Burgart, Y.V.; Saloutin, V.I.; Volobueva, A.S.; Misiurina, M.A.; Esaulkova, Y.L.; Sinegubova, E.O.; Zarubaev, V.V. 7-Imidazolylsubstituted 4’-methoxy and 3’,4’-dimethoxy-containing polyfluoroflavones as promising antiviral agents. J. Fluorine Chem. 2020, 240, 109657. [Google Scholar] [CrossRef]
- Podlech, J. Elimination of fluorine to form C–N bonds. In Organo-Fluorine Compounds, 4th ed.; Baasner, B., Hagemann, H., Tatlow, J.C., Eds.; Georg Thieme Verlag: Stuttgart, Germany, 1999; pp. 449–464. [Google Scholar]
- Shcherbakov, K.V.; Panova, M.A.; Burgart, Y.V.; Zarubaev, V.V.; Gerasimova, N.A.; Evstigneeva, N.P.; Saloutin, V.I. The synthesis and biological evaluation of A- and B-ring fluorinated flavones and their key intermediates. J. Fluorine Chem. 2021, 249, 109857. [Google Scholar] [CrossRef]
- Shcherbakov, K.V.; Artemyeva, M.A.; Burgart, Y.V.; Evstigneeva, N.P.; Gerasimova, N.A.; Zilberberg, N.V.; Kungurov, N.V.; Saloutin, V.I.; Chupakhin, O.N. Transformations of 3-acyl-4H-polyfluorochromen-4-ones under the action of amino acids and biogenic amines. J. Fluorine Chem. 2019, 226, 109354. [Google Scholar] [CrossRef]
- Shcherbakov, K.V.; Burgart, Y.V.; Saloutin, V.I.; Chupakhin, O.N. Modification of polyfluoro-containing 3-(ethoxycarbonyl)flavones by biogenic amines and amino acids. Curr. Org. Synth. 2018, 15, 707–714. [Google Scholar] [CrossRef]
- Shcherbakov, K.V.; Burgart, Y.V.; Saloutin, V.I.; Chupakhin, O.N. Polyfluorinecontaining chromen-4-ones: Synthesis and transformations. Russ. Chem. Bull. 2016, 65, 2151–2162. [Google Scholar] [CrossRef]
- Ahmadi, A.; Mohammadnejadi, E.; Karami, P.; Razzaghi-Asi, N. Current status and structure activity relationship of privileged azoles as antifungal agents. Int. J. Antimicrob. Agents 2022, 59, 106518. [Google Scholar] [CrossRef]
- Seck, I.; Nguemo, F. Triazole, imidazole, and thiazole-based compounds as potential agents against coronavirus. Results Chem. 2021, 3, 100132. [Google Scholar] [CrossRef]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A review on recent advances in nitrogen-containing molecules and their biological applications. Molecules 2020, 25, 1909–1951. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Sumran, G. An insight on medicinal attributes of 1,2,4-triazoles. Eur. J. Med. Chem. 2020, 205, 112652. [Google Scholar] [CrossRef] [PubMed]
- Prasher, P.; Sharma, M. “Azole” as privileged heterocycle for targeting the inducible cyclooxygenase enzyme. Drug Dev. Res. 2020, 82, 167–197. [Google Scholar] [CrossRef]
- Hou, Y.; Shang, C.; Wang, H.; Yun, J. Isatin-azole hybrids and their anticancer activities. Arch. Pharm. Chem. Life Sci. 2019, 353, e1900272. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Peng, Y.; Wang, S.; Ji, J.; Rakesh, K.P. Triazole derivatives as inhibitors of Alzheimer’s disease: Current developments and structure-activity relationships. Eur. J. Med. Chem. 2019, 180, 656–672. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.-L.; Jin, X.-H.; Huang, Z.-P.; Yu, H.-F.; Zeng, Z.-G.; Gao, T.; Feng, L.-S. Resent advances of imidazole-containing derivatives as anti-tubercular agents. Eur. J. Med. Chem. 2018, 150, 347–365. [Google Scholar] [CrossRef]
- Yan, M.; Xu, L.; Wang, Y.; Wan, J.; Liu, T.; Liu, W.; Wan, Y.; Zhang, B.; Wang, R. Opportunities and challenges of using five-membered ring compounds as promising antitubercular agents. Drug Dev. Res. 2020, 81, 402–418. [Google Scholar] [CrossRef]
- Gao, F.; Wang, T.; Xiao, J.; Huang, G. Antibacterial activity study of 1,2,4-triazole derivatives. Eur. J. Med. Chem. 2019, 173, 274–281. [Google Scholar] [CrossRef]
- Teli, G.; Chawla, P.A. Hybridization of imidazole with various heterocycles in targeting cancer. ChemistrySelect 2021, 6, 4803–4836. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, S.; Ba, Y.; Xu, Z. 1,2,4-Triazole-quinoline/ quinolone hybrids as potential anti-bacterial agents. Eur. J. Med. Chem. 2019, 174, 1–8. [Google Scholar] [CrossRef]
- Fan, Y.-L.; Liu. M. Coumarin-triazole hybrids and their biological activities. J. Heterocycl. Chem. 2018, 55, 791–802. [Google Scholar] [CrossRef]
- Zhang, T.; Zhu, M.; Li, J.; Zhang, Y.; Wang, X. Bipolar host materials comprising carbazole, pyridine and triazole moieties for efficient and stable phosphorescent OLEDs. Dyes Pigm. 2021, 192, 109426. [Google Scholar] [CrossRef]
- Ye, S.; Zhuang, S.; Pan, B.; Guo, R.; Wang, L. Imidazole derivatives for efficient organic light-emitting diodes. J. Inf. Disp. 2020, 21, 173–196. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, Y.; Zhang, J.; Zhang, D.; Miao, Y.; Shinar, J.; Shinar, R.; Wang, H.; Xu, B. Low efficiency rol-off phosphorescent organic light-emitting devices using thermally activated delayed fluorescence hosts materials based 1,2,4-triazole acceptor. Org. Electron. 2019, 74, 13–22. [Google Scholar] [CrossRef]
- Tao, Y.; Yang, C.; Qin, J. Organic host materials for phosphorescent organic light-emitting diodes. Chem. Soc. Rev. 2011, 40, 2943–2970. [Google Scholar] [CrossRef]
- Emami, L.; Faghih, Z.; Ataollahi, E. Azole derivatives: Recent advances as potent antibacterial and antifungal agents. Curr. Med. Chem. 2022, 129, 220–249. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Palatinus, L.; Chapuis, G. SUPERFLIP–A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2007, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Kubanova, A.A.; Stepanova, Z.V.; Gus’kova, T.A.; Pushkina, T.V.; Krylova, L.Y.; Shilova, I.B.; Trenin, A.S. Metodicheskie rekomendacii po izucheniyu protivogribkovoi aktivnosti lekarstvennykh sredstv (Guidelines for the study of the antifungal activity of medicines). In Rukovodstvo po Provedeniyu Doklinicheskikh Issledovanii Lekarstvennykh Sredstv (A Guide to Preclinical Trials of Medicines); Mironov, A.N., Ed.; Grif i K: Moscow, Russia, 2012; pp. 578–586. [Google Scholar]
- Koldobskii, G.I.; Ostrovskii, V.A. Acid-base properties of five-membered nitrogen-containing heterocycles. Chem. Heterocycl. Compds. 1988, 24, 469–480. [Google Scholar] [CrossRef]
- Krishnan, R.; Parthiban, A. Regioselective preparation of functional aryl ethers and esters by stepwise nucleophilic aromatic substitution reaction. J. Fluorine Chem. 2014, 162, 17–25. [Google Scholar] [CrossRef]
- Chambers, R.D.; Martin, P.A.; Sandford, G.; Williams, L.H. Mechanisms of reactions of halogenated compounds: Part 7. Effects of fluorine and other groups as substituents on nucleophilic aromatic substitution. J. Fluorine Chem. 2008, 129, 998–1002. [Google Scholar] [CrossRef]















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).