Abstract
This work describes the synthesis of 3-hydroxy-3,4-dihydropyrido[2,1-c][1,4]oxazine-1,8-diones, their tautomerism, and reactivity towards binucleophiles. These molecules are novel and convenient building-blocks for the direct construction of biologically important polycyclic pyridones via an oxazinone ring-opening transformation promoted with ammonium acetate or acetic acid. In the case of o-phenylenediamine, partial aromatization of the obtained heterocycles proceeded to form polycyclic benzimidazole-fused pyridones (33–91%).
1. Introduction
4-Pyridones are important nitrogen-containing heterocycles, which have recently attracted much attention as biologically active [1,2,3,4] and natural compounds [5,6]. Polycyclic structures, such as dolutegravir, bictegravir, and cabotegravir, are used as modern inhibitors of HIV integrase for antiretroviral therapy [1,3] (Figure 1). Baloxavir marboxil also belongs to this class of these compounds and is applied as the first cap-dependent endonuclease inhibitor for the treatment of influenza [2].

Figure 1.
Some important polycyclic 4-pyridones.
The chemistry of these heterocycles is actively developed not only for the design of biologically important compounds [7,8,9], but also for effective preparation in the industry [1,2,3,10,11,12,13,14,15]. At the same time, there is a need to search for new multifarious pyridone building blocks [16,17,18,19,20,21,22,23,24,25] and convenient synthetic tools for the construction of polycyclic pyridones [1,2,3,15], including CH functionalization [26].
The general approach is well known in the literature based on the reaction of alkyl 1-(2,2-dimethoxyethyl)-4-oxo-1,4-dihydropyridine-2-carboxylates with binucleophiles to obtain the fused heterocycles [1,3,10,11,12,13,14] (Scheme 1). In this case, the acid-catalyzed deprotection of the dimethyl acetal group led to 2-(4-oxopyridin-1(4H)-yl)acetaldehydes, which are usually considered as intermediates of polycyclic pyridone formation. Substituted 3-hydroxy-3,4-dihydropyrido[2,1-c][1,4]oxazine-1,8-dione was also detected as the result of hydrolysis of the carboethoxy and dimethylacetal groups as a by-product in the synthesis of dolutegravir [10]. Such structure also can be suggested as a possible intermediate for this heterocyclization [1,10]. To the best of our knowledge, only unsubstituted 3-hydroxy-3,4-dihydropyrido[2,1-c][1,4]oxazine-1,8-dione was prepared from comanic acid in pure form and used for the transformation with (R)-3-aminobutan-1-ol [3,11]. Moreover, there are data on the Ugi reaction of a pyridone-bearing aldoacid with isonitriles for the synthesis of various piperazinone-fused pyridones [27].
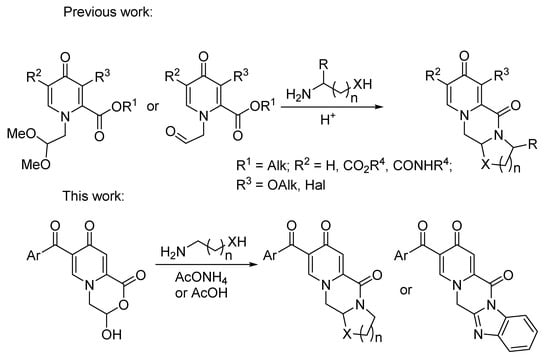
Scheme 1.
General strategy for the synthesis of polycyclic pyridones.
We decided to study 3-hydroxy-3,4-dihydropyrido[2,1-c][1,4]oxazine-1,8-diones in more detail in order to find new directions for the construction of polycyclic pyridones through morpholinone ring-opening reactions. These molecules bear the hidden aldehyde moiety, which can determine their high reactivity towards nucleophiles via the tautomeric equilibrium. This strategy based on the transformation with diamines can open access to new cyclic fused pyridones, which are of interest for the further design of biologically active compounds.
2. Results and Discussion
Synthesis of 3-hydroxy-3,4-dihydropyrido[2,1-c][1,4]oxazine-1,8-diones 3 and Their Chemical Properties
We started with the ANRORC reaction of 5-acyl-4-pyrone-2-carboxylate 1 with 2,2-dimethoxyethylamine as the effective method for the preparation of 4-pyridones [3,17,28,29] (Scheme 2, Table 1). The ring-opening transformations proceeded under reflux in toluene for 4 h to produce pyridones 2a–d,f in 30–90% yields. Pivaloyl-substituted pyrone 1e did not provide the desired product, and the reaction was carried in more polar MeCN, leading to pyridone 2e in 42% yield. Compounds 2a–e underwent the deprotection of the dimethyl acetal moiety in aqueous HCl to form 3-hydroxy-3,4-dihydropyrido[2,1-c][1,4]oxazine-1,8-diones 3a–e. For 2,5-dicarbethoxy-4-pyridone 2f, the heating in formic acid was used for the selective hydrolysis of the COOEt group at the C-2 position as a result of the promotion by the presence of the adjacent aldehyde fragment.
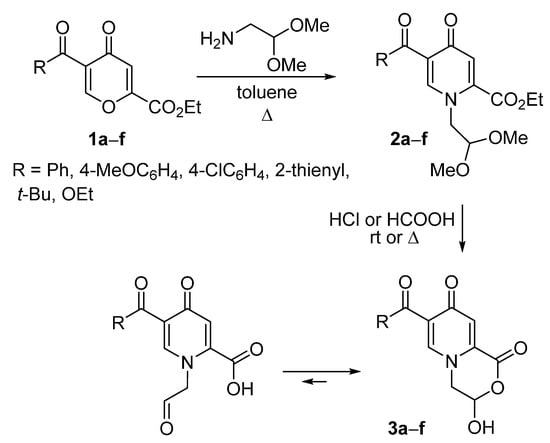
Scheme 2.
Synthesis of 3-hydroxy-3,4-dihydropyrido[2,1-c][1,4]oxazine-1,8-diones 3.

Table 1.
The scope of products 2 and 3.
Pyridones 3 can undergo aldehyde-lactol tautomerism [30] and exist as acyclic aldoacid or a cyclic lactol form (3-hydroxy-3,4-dihydropyrido[2,1-c][1,4]oxazine-1,8-diones) (Scheme 2). It is interesting to note that this type of the ring-chain tautomerism for morpholinones has not been studied before.
The 1H NMR spectra of products 3 in DMSO-d6 demonstrates the existence of only the lactol form. The spectral feature of the tautomer is the presence of a downfield signal of the OH group at δ 8.13–8.72 ppm and an ABX system of the morpholinone moiety. For the pivaloyl-substituted compound 3e, a singlet of the methylene group and a strongly broadened singlet of the CH proton were observed probably due to the rapid interconversion between different forms.
Pyridones 3 bear the carbonyl group at the C-5 position, which can be used for further modifications of the heterocyclic fragment. Therefore, the important task included the search for selective transformations on the morpholinone fragment. We have studied the detailed influence of conditions on the ring-opening reaction of 3-hydroxy-3,4-dihydropyrido[2,1-c][1,4]oxazine-1,8-diones (3b) with 3-aminopropan-1-ol (Table 2). A mixture of methanol–toluene was used as a solvent to increase the solubility of pyridone 3b. The reaction did not proceed without the use of catalysts even under the prolonged reflux. It was found that the transformation in the presence of acetic acid as an additive led to product 4a in 48% yield. We suggested that the formation of 3-hydroxypropylammonium acetate occurred, which acts as a nucleophile and activator of the morpholinone moiety. However, this transformation did not proceed at room temperature, as well as with the use of the 0.2 equiv. of acetic acid.
Table 2.
The optimization of reactions conditions for the synthesis of 4a from 3b and 3-aminopropan-1-ol a.
Taking into account the effect of AcOH, we tried to use AcONH4 as a bifunctional catalyst [31,32] for this process. To our delight, the product was obtained in a good yield (69%) under reflux for 12 h (TLC monitoring) (Table 2). The variation of the nature of the ammonium salt or temperature did not allow for the improvement of the reaction yield.
We tried to extend the optimized conditions with the use of ammonium acetate (Method A) for other 5-acylpyridones 3 and binucleophiles for the synthesis of polycyclic 4-pyridones (Table 3). In most cases, the acyl fragment strongly influenced the reaction selectivity, and an alternative method included the use of acetic acid (Method B). Pyridones 3b,c bearing para-substituted benzoyl fragments underwent the transformation in the presence of ammonium acetate and led to the formation of products 4a,b in 69–75% yields. Benzoyl- and thienoyl-substituted compounds 3a,d reacted more effectively in the conditions of method B and provided products 4c,d in 31–52% yields. When propane-1,3-diamine was used as a binucleophile, we were not able to isolate the desired polycyclic products in a pure form directly. The precipitates that formed always contained the starting diamine in significant amounts. Next, binucleophiles bearing two carbon atoms in the linker was used for the heterocyclization. The reaction with ethylenediamine proceeded in good yields and led to the formation of imidazo[1,2-a]pyrido[1,2-d]pyrazine-5,7-diones 5a,b in 78–84% yields (Method A). Thienoyl-substituted pyridone 3d underwent the ring-opening process in the presence of acetic acid (Method B) to produce compound 5c in 48% yield. At the same time, we failed to isolate any products in the pure form in the reaction with ethanolamine.
Table 3.
Reactions of compounds 3 with binucleophiles a.
The peculiarity of ammonium acetate is probably associated with its solubility in a methanol–toluene mixture and the ability to promote the ring-opening process of the morpholinone ring, which leads to the formation of the aldoacid (Scheme 3). Subsequent stages, including intermolecular attack of a binucleophile and intramolecular cyclization, can be catalyzed by both the ammonium cation and acetic acid. An experiment was carried out to study the reaction of ammonium acetate with pyridone 3b under reflux. According to the 1H NMR spectrum of the obtained precipitate, it was found that the formation of an open-chain structure occurred (see Supplementary Materials). Although we did not detect the aldehyde group, a singlet of the methylene group and absence of the 3-CH proton of the lactol form were observed in the 1H NMR spectrum.
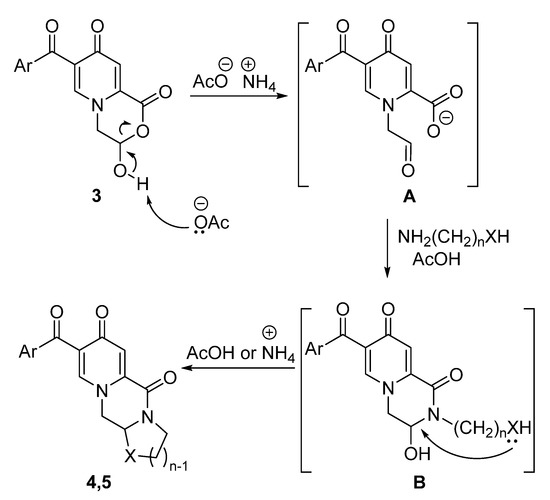
Scheme 3.
The proposed mechanism of the ring-opening transformation.
The reaction of pyridone 3 with o-phenylenediamine proceeded in the presence of ammonium acetate at room temperature or under reflux and was accompanied by aromatization under the action of atmospheric oxygen (Scheme 4, Table 4). The intermediate C was not isolated in pure form, but was detected as by-products in all cases. Carrying out the reaction under argon did not allow the selective formation of compound C. This result can indicate that the oxidation additionally promotes the reaction leading to the most stable product 6.
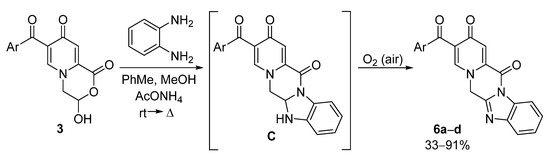
Scheme 4.
Reactions of compounds 3 with o-phenylenediamine.
Table 4.
The scope of products 6.
To obtain compounds 6 in a pure form directly, the reaction was carried out at room temperature for 12 h and subsequent reflux for 2 h. In these conditions, the aromatization proceeded completely and pyridones 6 bearing the benzimidazole fragment were isolated in 33–91% yields. The reaction turned out to be sensitive to the nature of the acyl moiety, which probably determined the occurrence of side reactions. Pivaloyl pyridone 3e led to degradation products, which did not bear the t-Bu group. In the 1H NMR spectra of compounds 6, the downfield singlet of methylene group was observed at δ 5.81–5.84 ppm due to the presence of two adjacent aromatic systems.
Thus, 3-hydroxy-3,4-dihydropyrido[2,1-c][1,4]oxazine-1,8-diones have been synthesized and demonstrated to exist predominantly in the lactol tautomeric form. The new and convenient approach has been developed for the preparation of polycyclic pyridones based on the pyridomorpholinones via ring-opening reactions. The binucleophile linker and the nature of nucleophile centers strongly influence the reaction outcome. The most active binucleophiles in this heterocyclization process are ethylenediamine and 3-aminopropan-1-ol. It has been demonstrated that the reaction with o-phenylenediamine is followed by oxidation and the formation of benzimidazole-fused 4-pyridones.
3. Materials and Methods
NMR spectra were recorded on Bruker DRX-400 (Bruker BioSpin GmbH, Ettlingen, Germany, work frequencies: 1H, 400 MHz; 13C, 101 MHz), Bruker Avance-400 (Bruker BioSpin GmbH, Rheinstetten, Germany, work frequencies: 1H, 400 MHz; 13C, 101 MHz), Bruker Avance III-500 (Bruker BioSpin GmbH, Rheinstetten, Germany, work frequencies: 1H, 500 MHz; 13C, 126 MHz), and Bruker Avance NEO (Bruker BioSpin GmbH, Rheinstetten, Germany, work frequencies: 1H, 600 MHz; 13C, 151 MHz) spectrometers in DMSO-d6 or CDCl3. The chemical shifts (δ) are reported in ppm relative to the internal standard TMS (1H NMR) and residual signals of the solvents (13C NMR). IR spectra were recorded on a Shimadzu IRSpirit-T (Shimadzu Corp., Kyoto, Japan) spectrometer using an attenuated total reflectance (ATR) unit (FTIR mode, diamond prism); the absorbance maxima (ν) are reported in cm–1. Mass spectra (ESI-MS) were measured with a Waters Xevo QTof instrument (Waters Corp., Milford, MA, USA). Elemental analyses were performed on an automatic analyzer PerkinElmer PE 2400 (Perkin Elmer Instruments, Waltham, MA, USA). Melting points were determined using a Stuart SMP40 melting point apparatus (Bibby Scientific Ltd., Stone, Staffordshire, UK). Column chromatography was performed on silica gel (Merck 60, 70–230 mesh). All solvents that were used were dried and distilled by standard procedures. 4-Pyrones 1 were prepared according to the literature methods [28,33,34].
3.1. General Procedure for the Preparation of 1-(2,2-dimethoxyethyl)-4-pyridones 2
Ethyl 5-acyl-4-oxo-4H-pyran-2-carboxylate 1 (0.330 mmol) was added to a cooled solution of 2,2-dimethoxyethanamine (0.0380 g, 0.361 mmol) in toluene (1 mL). The resulting mixture was stirred for 20 min at room temperature (the precipitation was observed) and heated under reflux for 4 h. The solvent was evaporated under reduced pressure, and the product was isolated by flash chromatography using ethyl acetate as an eluent. For pyridone 2e, acetonitrile was used instead of toluene (reflux for 3 h).
Ethyl 5-benzoyl-1-(2,2-dimethoxyethyl)-4-oxo-1,4-dihydropyridine-2-carboxylate (2a). Yield 0.1067 g (90%), yellow crystals. IR (ATR) ν 3053, 2932, 2836, 1720, 1652, 1630, 1475, 1294, 829, 701. 1H NMR (400 MHz, CDCl3) δ 1.41 (t, J = 7.1 Hz, 3H, Me), 3.42 (s, 6H, 2MeO), 4.34 (d, J = 4.5 Hz, 2H, CH2N), 4.40 (q, J = 7.1 Hz, 2H, CH2), 4.53 (t, J = 4.5 Hz, 1H, CH), 7.07 (s, 1H, H-3 Py), 7.43 (t, J = 7.6 Hz, 2H, H-3, H-5 Ph), 7.55 (t, J = 8.0 Hz, 2H, H-4 Ph), 7.79 (s, 1H, H-6 Py), 7.86 (dd, J = 8.0 Hz, J = 1.4 Hz, 2H, H-2, H-6 Ph). 13C NMR (126 MHz, DMSO-d6) δ 13.7, 54.2, 55.4, 62.5, 102.7, 122.3, 127.9, 128.4 (2C), 129.1 (2C), 133.2, 136.9, 141.6, 147.2, 162.0, 174.4, 193.2. HRMS (ESI) m/z [M + H]+. Calculated for C19H22NO6: 360.1453. Found: 360.1447.
Ethyl 1-(2,2-dimethoxyethyl)-5-(4-methoxybenzoyl)-4-oxo-1,4-dihydropyridine-2-carboxylate (2b). Yield 0.0828 g (63%), light yellow oil. 1H NMR (500 MHz, CDCl3) δ 1.41 (t, J = 7.1 Hz, 3H, Me), 3.42 (s, 6H, 2MeO), 4.36 (d, J = 4.5 Hz, 2H, CH2N), 4.40 (q, J = 7.1 Hz, 2H, CH2), 4.53 (t, J = 4.5 Hz, 1H, CH), 7.06 (s, 1H, H-3 Py), 7.40 (d, J = 8.7 Hz, 2H, H-3, H-5 Ar), 7.79 (d, J = 8.7 Hz, 2H, H-2, H-6 Ar), 7.82 (s, 1H, H-6 Py). 13C NMR (151 MHz, CDCl3) δ 14.0, 55.5, 55.7, 55.9, 62.8, 103.3, 113.6, 124.8, 129.4, 129.8, 132.3, 140.2, 147.3, 162.4, 163.8, 175.7, 191.6. Anal. Calculated for C20H23NO7·0.5H2O: C 60.29; H 6.07; N 3.52. Found: C 59.98; H 6.20; N 3.47.
Ethyl 5-(4-chlorobenzoyl)-1-(2,2-dimethoxyethyl)-4-oxo-1,4-dihydropyridine-2-carboxylate (2c). Yield 0.0949 g (73%), brown viscous liquid. IR (ATR) ν 3033, 2978, 2862, 1729, 1660, 1630, 1481, 1246, 851, 767. 1H NMR (400 MHz, CDCl3) δ 1.41 (t, J = 7.1 Hz, 3H, Me), 3.42 (s, 6H, 2MeO), 4.36 (d, J = 4.5 Hz, 2H, CH2N), 4.40 (q, J = 7.1 Hz, 2H, CH2), 4.53 (t, J = 4.5 Hz, 1H, CH), 7.06 (s, 1H, H-3 Py), 7.40 (d, J = 8.7 Hz, 2H, H-3, H-5 Ar), 7.79 (d, J = 8.7 Hz, 2H, H-2, H-6 Ar), 7.82 (s, 1H, H-6 Py). 13C NMR (126 MHz, CDCl3) δ 14.0, 55.7, 55.8, 62.8, 103.2, 125.3, 128.2, 128.5 (2C), 131.0 (2C), 135.4, 139.4, 140.3, 148.1, 162.2, 175.6, 192.1. Anal. Calculated for C19H20ClNO6: C 57.95; H 5.12; N 3.56. Found: C 58.35; H 5.42; N 3.48.
Ethyl 1-(2,2-dimethoxyethyl)-4-oxo-5-(thiophene-2-carbonyl)-1,4-dihydropyridine-2-carboxylate (2d). Yield 0.0868 g (72%), dark yellow viscous liquid. IR (ATR) ν 3066, 2985, 2933, 2841, 1637, 1620, 1478, 1250, 849, 719. 1H NMR (400 MHz, CDCl3) δ 1.41 (t, J = 7.1 Hz, 3H, Me), 3.42 (s, 6H, 2MeO), 4.34 (d, J = 4.5 Hz, 1H, CH2N), 4.40 (q, J = 7.1 Hz, 2H, CH2), 4.53 (t, J = 4.5 Hz, 1H, CH), 7.09 (s, 1H, H-3 Py), 7.12 (dd, J = 4.9 Hz, J = 3.9 Hz, 1H, H-4 Th), 7.68 (dd, J = 4.9, J = 1.1 Hz, 1H, H-5 Th), 7.81 (s, 1H, H-6 Py), 7.85 (dd, J = 3.9, J = 1.1 Hz, 1H, H-3 Th). 13C NMR (126 MHz, CDCl3) δ 13.9, 55.7, 55.8, 62.8, 103.2, 125.1, 128.1, 128.8, 134.7, 135.1, 140.0, 143.7, 147.4, 162.2, 175.3, 184.2. Anal. Calculated for C17H19NO5S: C 55.88; H 5.24; N 3.83. Found: C 56.09; H 5.15; N 3.90.
Ethyl 1-(2,2-dimethoxyethyl)-4-oxo-5-pivaloyl-1,4-dihydropyridine-2-carboxylate (2e). Yield 0.0470 g (42%), brown viscous liquid. IR (ATR) ν 2960, 2837, 1731, 1628, 1480, 1247, 952, 805. 1H NMR (400 MHz, CDCl3) δ 1.28 (s, 9H, t-Bu), 1.40 (t, J = 7.1 Hz, 3H, Me), 3.41 (s, 6H, 2MeO), 4.26 (d, J = 4.7 Hz, 2H, CH2N), 4.38 (q, J = 7.1 Hz, 2H, CH2), 4.50 (t, J = 4.7 Hz, 1H, CH), 6.97 (s, 1H, H-3 Py), 7.41 (s, 1H, H-6). 13C NMR (126 MHz, CDCl3) δ 13.9, 26.2, 44.7, 55.6, 55.8, 62.7, 103.4, 123.9, 132.1, 139.7, 144.1, 162.4, 175.4, 209.8. HRMS (ESI) m/z [M + H]+. Calculated for C17H26NO6: 340.1766. Found: 340.1760.
Diethyl 1-(2,2-dimethoxyethyl)-4-oxo-1,4-dihydropyridine-2,5-dicarboxylate (2f). Yield 0.0324 g (30%), brown viscous liquid. IR (ATR) ν 2978, 2839, 1725, 1664, 1631, 1474, 1299, 867, 776. 1H NMR (400 MHz, CDCl3) δ 1.38 (t, J = 7.1 Hz, 3H, Me), 1.39 (t, J = 7.1 Hz, 3H, Me), 3.41 (s, 6H, 2MeO), 4.33 (d, J = 4.7 Hz, 2H, CH2N), 4.37 (q, J = 7.1 Hz, 2H, CH2), 4.38 (q, J = 7.1 Hz, 2H, CH2), 4.49 (t, J = 4.7 Hz, 1H, CH), 7.07 (s, 1H, H-3 Py), 8.18 (s, 1H, H-6 Py). HRMS (ESI) m/z [M + H]+. Calculated for C15H22NO7: 328.1406. Found: 328.1396. The spectral data are in accordance with the patent literature [11].
3.2. General Method for the Preparation of dihydropyrido[2,1-c][1,4]oxazine-1,8-diones 3
1-(2,2-Dimethoxyethyl)-4-pyridone 2 (0.332 mmol) was stirred in hydrochloric acid (1: 1, 2 mL) for 24 h or, for 2b, in concentrated hydrochloric acid (2 mL) for 3 h at room temperature and for 3 h under reflux. The precipitate formed was filtered. For compound 2f, formic acid (85%, 2 mL) was used. The reaction mixture was stirred at room temperature for 3 h and heated at 85 °C for 4 h. After evaporation of the solvent, the product was isolated by flash chromatography using ethyl acetate as an eluent.
7-Benzoyl-3-hydroxy-3,4-dihydropyrido[2,1-c][1,4]oxazine-1,8-dione (3a). Yield 0.0506 g (53%), white powder, mp 187–188 °C. IR (ATR) ν 3228, 3078, 2489, 1665, 1637, 1470, 1213, 854, 748. 1H NMR (400 MHz, DMSO-d6) δ 4.29 (dd, J = 13.5 Hz, J = 3.3 Hz, 1H, CHH), 4.47 (dd, J = 13.5 Hz, J = 1.6 Hz, 1H, CHH), 6.08 (unresolved m, 1H, H-3), 6.99 (s, 1H, H-9), 7.50 (d, J = 7.7 Hz, 2H, H-3, H-5 Ph), 7.64 (t, J = 7.4 Hz, J = 1.0 Hz, 1H, H-4 Ph), 7.76 (dd, J = 8.4 Hz, J = 1.3 Hz, 2H, H-2, H-6 Ph), 8.28 (br.s, 1H, H-6), 8.72 (br.s, 1H, OH). 13C NMR (126 MHz, DMSO-d6) δ 52.4, 93.7, 121.1, 128.6 (2C), 129.3 (2C), 133.6, 136.5, 144.4, 158.1, 174.0, 192.9. HRMS (ESI) m/z [M + H]+. Calculated for C15H12NO5: 286.0714. Found: 286.0715.
3-Hydroxy-7-(4-methoxybenzoyl)-3,4-dihydropyrido[2,1-c][1,4]oxazine-1,8-dione (3b). Yield 0.0902 g (85%), beige powder, mp 205–206 °C. IR (ATR) ν 3060, 2934, 1684, 1599, 1268, 1153, 1021, 912, 843. 1H NMR (500 MHz, DMSO-d6) δ 3.84 (s, 3H, OMe), 4.25 (dd, J = 13.5 Hz, J = 3.6 Hz, 1H, CHH), 4.44 (dd, J = 13.5 Hz, J = 1.5 Hz, 1H, CHH), 6.07 (unresolved m, 1H, H-3), 6.94 (s, 1H, H-9), 7.03 (d, J = 8.9 Hz, 2H, H-3, H-5 Ar), 7.75 (d, J = 8.8 Hz, 2H, H-2, H-6 Ar), 8.18 (s, 1H, H-6), 8.66 (br.s, 1H, OH). 13C NMR (126 MHz, DMSO-d6) δ 52.1, 55.6, 93.6, 113.8 (2C), 121.2, 129.3, 129.9, 131.8 (2C), 135.9, 143.5, 158.2, 163.5, 174.2, 191.3. Anal. Calculated for C16H13NO6·0.25H2O: C 60.10; H 4.26; N 4.38. Found: C 60.17; H 3.98; 4.42.
7-(4-Chlorobenzoyl)-3-hydroxy-3,4-dihydropyrido[2,1-c][1,4]oxazine-1,8-dione (3c). Yield 0.0626 g (59%), white powder, mp 209–210 °C. IR (ATR) ν 3077, 1728, 1645, 1635, 1557, 1533, 1404, 1216, 1047, 732. 1H NMR (500 MHz, DMSO-d6) δ 4.32 (br.s, 1H, CHH), 4.45 (br.s, 1H, CHH), 6.10 (unresolved m, 1H, H-3), 6.90 (br.s, 1H, H-9), 7.57 (d, J = 8.9 Hz, 2H, H-3, H-5 Ar), 7.75 (d, J = 8.9 Hz, 2H, H-2, H-6 Ar), 8.28 (s, 1H, H-6), 8.67 (s, 1H, OH). 13C NMR (126 MHz, DMSO-d6) δ 52.0, 122.5, 128.6 (2C),128.7, 130.9 (2C), 135.5, 138.0, 144.6, 163.0, 174.8, 192.5 (2C were not observed). HRMS (ESI) m/z [M + H]+. Calculated for C15H11ClNO5: 320.0330. Found: 320.0326.
3-Hydroxy-7-(thiophene-2-carbonyl)-3,4-dihydropyrido[2,1-c][1,4]oxazine-1,8-dione (3d). Yield 0.0812 g (84%), beige powder, mp 206–207 °C. IR (ATR) ν 3071, 2904, 1730, 1638, 1407, 1209, 852, 749. 1H NMR (500 MHz, DMSO-d6) δ 4.26 (dd, J = 13.3 Hz, J =2.7 Hz, 1H, CHH), 4.46 (d, J = 13.3 Hz, 1H, CHH), 6.06 (s, 1H, H-3), 6.95 (s, 1H, H-9), 7.23 (dd, J = 4.9 Hz, J = 3.9 Hz, 1H, H-4 Th), 7.72 (dd, J = 3.9 Hz, J = 0.9 Hz, 1H, H-3 Th), 8.08 (dd, J = 4.9 Hz, J = 0.9 Hz, 1H, H-5 Th), 8.25 (s, 1H, H-6), 8.65 (d, J = 4.5 Hz, 1H, OH). 13C NMR (126 MHz, DMSO-d6) δ 51.8, 93.5, 122.1, 128.7, 129.3, 135.5, 135.7, 143.2, 143.3, 158.4, 174.4, 184.7 (1C was not observed). Anal. Calculated for C13H9NO5S: C 53.61; H 3.11; N 4.81. Found: C 53.64; H 3.39; N 4.90.
3-Hydroxy-7-pivaloyl-3,4-dihydropyrido[2,1-c][1,4]oxazine-1,8-dione (3e). Yield 0.0361 g (41%), brown liquid product. IR (ATR) ν 2972, 2935, 2909, 1697, 1479, 1364, 1216, 1060. 1H NMR (500 MHz, DMSO-d6) δ 1.18 (s, 9H, t-Bu), 4.33 (s, 2H, 4-CH2), 6.13 (br.s, 1H, H-3), 6.83 (s, 1H, H-9), 7.90 (s, 1H, H-6), 8.13 (s, 1H, OH). 13C NMR (126 MHz, DMSO-d6) δ 26.0, 44.0, 54.6, 88.4, 120.1, 131.9, 140.9, 159.7, 163.1, 174.9, 210.1. HRMS (ESI) m/z [M + H]+. Calculated for C13H16NO5: 266.1033. Found: 266.1028.
Ethyl 3-hydroxy-1,8-dioxo-1,3,4,8-tetrahydropyrido[2,1-c][1,4]oxazine-7-carboxylate (3f). Yield 0.0622 g (74%), brown powder, mp 193–194 °C. IR (ATR) ν 2985, 1734, 1695, 1575, 1450, 1306, 1200, 1112, 1012, 876, 797. 1H NMR (500 MHz, DMSO-d6) δ 1.26 (t, J = 7.1 Hz, 2H, Me), 4.21 (q, J = 7.1 Hz, 2H, CH2), 4.31 (br.s, 1H, CH), 4.41 (s, 1H, CH), 6.03 (s, 1H, H-3), 6.87 (s, 1H, H-9), 8.47 (s, 1H, H-6), 8.61 (s, 1H, OH). 13C NMR (126 MHz, DMSO-d6) δ 14.2, 52.0, 60.2, 93.6, 119.8, 123.1, 135.4, 146.5, 158.6, 163.8, 173.7. HRMS (ESI) m/z [M + H]+. Calculated for C11H12NO6: 254.0669. Found: 254.0665.
3.3. General Method for the Preparation of Compounds 4 and 5
Method A. 3-Hydroxy-7-acyl-3,4-dihydropyrido[2,1-c][1,4]oxazine-1,8-dione 3 (0.317 mmol) was stirred with amine (0.381 mmol) and ammonium acetate (0.0245 g, 0.317 mmol) in a mixture of toluene (1 mL) and methanol (1 mL) at 90 °C of an oil bath for 7–12 h. The precipitate obtained was filtered and recrystallized in a mixture of toluene and ethanol.
Method B. 3-Hydroxy-7-acyl-3,4-dihydropyrido[2,1-c][1,4]oxazine-1,8-dione 3 (0.317 mmol) was stirred with amine (0.381 mmol) and acetic acid (22.8 mg, 0.380 mmol) in a mixture of toluene (1 mL) and methanol (1 mL) at 90 °C of an oil bath for 7–12 h. The precipitate obtained was filtered and recrystallized in a mixture of toluene and ethanol.
9-(4-Methoxybenzoyl)-3,4,12,12a-tetrahydro-2H-pyrido[1’,2’:4,5]pyrazino[2,1-b][1,3]oxazine-6,8-dion (4a). The reaction was carried out for 12 h. Method A. Yield 0.0775 g (69%), yellow powder, mp 222–223 °C. IR (ATR) ν 2956, 2875, 1673, 1640, 1573, 1467, 1384, 783. 1H NMR (400 MHz, DMSO-d6) δ 1.61 (dm, J = 13.2 Hz, 1H, H-3’), 1.64–1.84 (m, 1H, H-3), 3.22 (td, J = 12.9 Hz, J = 3.2 Hz, 1H, CH), 3.84 (s, 3H, OMe), 3.90 (td, J = 11.8 Hz, J = 2.7 Hz, 1H, CH), 4.07 (dd, J = 11.3 Hz, J = 4.7 Hz, 1H, CH), 4.28 (dd, J = 14.1 Hz, J = 3.5 Hz, 1H, CH), 4.46 (dd, J = 14.0 Hz, J = 4.2 Hz, 2H, CH), 5.28 (t, J = 3.6 Hz, 1H, H-12a), 6.94 (s, 1H, H-7), 7.02 (d, J = 8.9 Hz, 2H, H-3, H-5 Ar), 7.75 (d, J = 8.9 Hz, 2H, H-2, H-6 Ar), 8.11 (s, 1H, H-10). 13C NMR (126 MHz, DMSO-d6) δ 25.0, 43.0, 50.2, 55.5, 67.1, 81.5, 113.7 (2C), 120.1, 129.6, 129.7, 131.7, 137.9 (2C), 142.9, 157.0, 163.3, 175.0, 192.0. HRMS (ESI) m/z [M + H]+. Calculated for C19H19N2O5: 355.1288. Found: 355.1294.
9-(4-Chlorobenzoyl)-3,4,12,12a-tetrahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-6,8-dione (4b). Method A. The reaction was carried out for 12 h. 3-Aminopropan-1-ol (47.6 mg, 0.634 mmol) was used. Yield 0.0853 g (75%), yellow powder, mp 259–261 °C. IR (ATR) ν 3020, 2950, 1674, 1627, 1572, 1452, 1357, 789. 1H NMR (500 MHz, DMSO-d6) δ 1.61 (dm, J = 12.5 Hz, 1H, H-3’), 1.70–1.82 (m, 1H, H-3), 3.22 (td, J = 13.0 Hz, J = 3.2 Hz, 1H, CH), 3.90 (td, J = 12.0 Hz, J = 2.4 Hz, 1H, CH), 4.07 (dd, J = 11.4 Hz, J = 4.8 Hz, 1H, CH), 4.32 (dd, J = 14.1 Hz, J = 3.5 Hz, 1H, CH), 4.43–4.50 (m, 2H, CH), 5.29 (t, J = 3.6 Hz, 1H, H-12a), 6.96 (s, 1H, H-7), 7.55 (d, J = 8.5 Hz, 2H, H-3, H-5 Ar), 7.75 (d, J = 8.5 Hz, 2H, H-2, H-6 Ar), 8.22 (s, 1H, H-10). 13C NMR (126 MHz, DMSO-d6) δ 25.0, 43.0, 50.3, 67.1, 81.4, 120.7, 128.4, 128.5 (2C), 131.0 (2C), 135.6, 137.9, 138.1, 144.2, 156.9, 175.0, 192.7. HRMS (ESI) m/z [M + H]+. Calculated for C18H15ClN2O4: 359.0789. Found: 359.0799.
9-(Thiophene-2-carbonyl)-3,4,12,12a-tetrahydro-2H-pyrido[1’,2’:4,5]pyrazino[2,1-b][1,3]oxazine-6,8-dione (4c). Method B. The reaction was carried out for 8h. Yield 0.0545 g (52%), yellow powder, mp 210–211 °C. IR (ATR) ν 2953, 1634, 1581, 1468, 1373, 1047, 714. 1H NMR (500 MHz, DMSO-d6) δ 1.61 (dm, J = 13.5 Hz, 1H, H-3’), 1.70–1.82 (m, 1H, H-3), 3.22 (td, J = 12.8 Hz, J = 3.2 Hz, 1H, CH), 3.89 (td, J = 11.9 Hz, J = 2.3 Hz, 1H, CH), 4.05 (dd, J = 11.3 Hz, J = 4.7 Hz, 1H, CH), 4.28 (dd, J = 14.2 Hz, J = 3.2 Hz, 1H, CH), 4.43–4.50 (m, 2H, CH), 5.27 (t, J = 3.6 Hz, 1H, H-12a), 6.97 (s, 1H, H-7), 7.22 (dd, J = 4.9 Hz, J = 4.0 Hz, 1H, H-4 Th), 7.74 (dd, J = 4.0 Hz, J = 0.7 Hz, 1H, H-3 Th), 8.05 (dd, J = 4.9 Hz, J = 0.7 Hz, 1H, H-5 Th), 8.19 (s, 1H, H-10). 13C NMR (126 MHz, DMSO-d6) δ 25.0, 43.0, 52.0, 67.1, 81.5, 120.4, 128.6, 128.9, 135.6, 137.9, 143.1, 143.4, 156.9, 174.6, 184.9 (1C was not observed). HRMS (ESI) m/z [M + H]+. Calculated for C16H15N2O4S: 331.0745. Found: 331.0753.
9-Benzoyl-3,4,12,12a-tetrahydro-2H-pyrido[1’,2’:4,5]pyrazino[2,1-b][1,3]oxazine-6,8-dione (4d). Method B. The reaction was carried out for 10 h. Yield 0.0319 g (31%), yellow powder, mp 245–246 °C. 1H NMR (400 MHz, DMSO-d6) δ 1.61 (dm, J = 14.6 Hz, 1H, H-3’), 1.67–1.84 (m, 1H, H-3), 3.22 (td, J = 13.0 Hz, J = 3.7 Hz, 1H, CH), 3.90 (td, J = 11.3 Hz, J = 2.5 Hz, 1H, CH), 4.07 (dd, J = 11.3 Hz, J = 4.5 Hz, 1H, CH), 4.31 (dd, J = 14.5 Hz, J = 3.5 Hz, 1H, CH), 4.42–4.51 (m, 2H, CH), 5.29 (t, J = 3.5 Hz, 1H, H-12a), 6.95 (s, 1H, H-7), 7.49 (t, J = 7.8 Hz, 2H, H-3, H-5 Ph), 7.62 (t, J = 7.8 Hz, 1H, H-4 Ph), 7.75 (d, J = 8.0 Hz, 2H, H-2, H-6 Ph), 8.18 (s, 1H, H-10). 13C NMR (126 MHz, DMSO-d6) δ 25.0, 50.2, 67.1, 81.5, 120.4, 128.4 (2C), 129.0, 129.1 (2C), 133.1, 136.8, 138.1, 143.6, 156.9, 175.0, 193.8. HRMS (ESI) m/z [M + H]+. Calculated for C18H17N2O4: 325.1188. Found: 325.1177.
8-(4-Methoxybenzoyl)-2,3,11,11a-tetrahydro-1H-imidazo[1,2-a]pyrido[1,2-d]pyrazine-5,7-dione (5a). Method A. The reaction was carried out for 12 h. Yield 0.0904 g (84%), brown powder, mp 249–250 °C;. IR (ATR) ν 2996, 2837, 1650, 1629, 1577, 1454, 1250, 786. 1H NMR (500 MHz, DMSO-d6) δ 2.99 (dt, J = 11.5 Hz, J = 7.4 Hz, 1H, CH), 3.20 (ddd, J = 11.2 Hz, J = 7.0 Hz, J = 3.9 Hz, 1H, CH), 3.39 (ddd, J = 11.1 Hz, J = 7.2 Hz, J = 3.8 Hz, 1H, CH), 3.54 (dt, J = 11.2 Hz, J = 7.3 Hz, 1H, CH), 3.84 (s, 3H, OMe), 3.92 (t, J = 11.8 Hz, 1H, CH), 4.52 (dd, J = 12.4 Hz, J = 3.7 Hz, 1H, CH), 4.75 (dd, J = 11.1 Hz, J = 3.6 Hz, 1H, CH), 6.79 (s, 1H, H-6), 7.01 (d, J = 8.8 Hz, 2H, H-3, H-5 Ar), 7.75 (d, J = 8.8 Hz, 2H, H-2, H-6 Ar), 8.09 (s, 1H, H-9) (NH was not observed). 13C NMR (126 MHz, DMSO-d6) δ 44.3, 44.8, 53.0, 55.5, 69.2, 113.6 (2C), 119.3, 129.0, 129.7, 131.7 (2C), 139.3, 143.3, 154.9, 163.2, 175.2, 192.2. HRMS (ESI) m/z [M + H]+. Calculated for C18H18N3O4: 340.1285. Found: 340.1297.
8-(4-Chlorobenzoyl)-2,3,11,11a-tetrahydro-1H-imidazo[1,2-a]pyrido[1,2-d]pyrazine-5,7-dione (5b). The reaction was carried out for 12 h. Method A. Yield 0.0850 g (78%), beige powder, mp 264–265 °C. IR (ATR) ν 2901, 1657, 1630, 1562, 1456, 1251, 769. 1H NMR (500 MHz, DMSO-d6) δ 2.95–3.03 (m, 1H, CH), 3.21 (ddd, J = 11.5 Hz, J = 7.1 Hz, J = 3.8 Hz, 1H, CH), 3.40 (ddd, J = 11.2 Hz, J = 7.1 Hz, J = 3.9 Hz, 1H, CH), 3.54 (dt, J = 11.4 Hz, J = 7.3 Hz, 1H, CH), 3.94 (t, J = 11.8 Hz, 1H, CH), 4.57 (dd, J = 12.4 Hz, J = 3.7 Hz, 1H, CH), 4.76 (dd, J = 11.1 Hz, J = 3.3 Hz, 1H, CH), 6.80 (s, 1H, H-6), 7.56 (d, J = 8.5 Hz, 2H, H-2, H-6 Ar), 7.75 (d, J = 8.5 Hz, 2H, H-2, H-6 Ar), 8.22 (s, 1H, H-9) (NH was not observed). 13C NMR (126 MHz, DMSO-d6) δ 44.3, 44.8, 53.1, 69.2, 119.9, 127.6, 128.5 (2C), 131.0 (2C), 135.8, 137.8, 139.5, 144.6, 154.7, 175.2, 192.8. HRMS (ESI) m/z [M + H]+. Calculated for C17H15ClN3O3: 344.0813. Found: 344.0802.
8-(Thiophene-2-carbonyl)-2,3,11,11a-tetrahydro-1H-imidazo[1,2-a]pyrido[1,2-d]pyrazine-5,7-dione (5c). Method B. The reaction was carried out for 7h. Yield 0.0480 g (48%), beige powder, mp 267–268 °C. 1H NMR (500 MHz, DMSO-d6) δ 2.99 (dt, J = 11.5 Hz, J = 7.4 Hz, 1H, CH), 3.20 (ddd, J = 11.1 Hz, J = 6.9 Hz, J = 3.9 Hz, 1H, CH), 3.39 (ddd, J = 11.2 Hz, J = 7.2 Hz, J = 3.8 Hz, 1H, CH), 3.54 (dt, J = 11.2 Hz, J = 7.3 Hz, 1H, CH), 3.92 (t, J = 11.8 Hz, 1H, CH), 4.51 (dd, J = 12.4 Hz, J = 3.7 Hz, 1H, CH), 4.75 (dd, J = 11.0 Hz, J = 3.6 Hz, 1H, CH), 6.81 (s, 1H, H-6), 7.22 (dd, J = 5.1 Hz, J = 3.9 Hz, 1H, H-4 Th), 7.76 (dd, J = 3.9 Hz, J = 0.8 Hz, 1H, H-3 Th), 7.76 (dd, J = 5.1 Hz, J = 0.8 Hz, 1H, H-5 Th), 8.18 (s, 1H, H-9). 13C NMR (126 MHz, DMSO-d6) δ 44.3, 44.8, 53.1, 69.2, 119.5, 128.2, 128.6, 135.5, 135.6, 139.3, 143.5, 154.8, 174.8, 185.0 (1C was not observed). HRMS (ESI) m/z [M + H]+. Calculated for C15H14N3O3S: 316.0756. Found: 316.0761.
3.4. General Method for the Preparation of Compound 6
3-Hydroxy-7-acyl-3,4-dihydropyrido[2,1-c][1,4]oxazine-1,8-dione 3 (0.317 mmol) was stirred with benzene-1,2-diamine (41.2 mg, 0.381 mmol) and ammonium acetate (0.0245 g, 0.317 mmol) in a mixture of toluene (1 mL) and methanol (1 mL) at room temperature for 12 h. Then, the reaction mixture was refluxed for 2 h. The precipitate obtained was filtered and washed with H2O and EtOH. The product was recrystallized in a mixture of toluene and ethanol.
3-Benzoyl-6H-benzo[4,5]imidazo[1,2-a]pyrido[1,2-d]pyrazine-2,13-dione (6a). Yield 0.0800 g (71%), beige powder, mp 252–253 °C. IR (ATR) ν 3037, 2808, 1685, 1443, 1211, 1114, 1027, 807. 1H NMR (500 MHz, DMSO-d6) δ 5.84 (s, 2H, CH2), 6.80 (s, 1H, H-1), 7.18 (dd, J = 6.0 Hz, J = 3.2 Hz, 2H, Ar), 7.50 (t, J = 7.7 Hz, 2H, H-3, H-5 Ph), 7.55 (dd, J = 6.0 Hz, J = 3.2 Hz, 2H, Ar), 7.63 (t, J = 7.4 Hz, 1H, H-4 Ph), 7.81 (d, J = 7.4 Hz, 2H, H-2, H-6 Ph), 8.33 (s, 1H, H-4). 13C NMR (126 MHz, DMSO-d6) δ 51.7, 114.9, 122.0 (2C), 122.6, 128.2, 128.3 (2C), 129.2 (2C), 133.0, 137.0, 138.2, 142.4, 147.0, 150.2, 168.1, 175.1, 193.5 (2C were not observed). HRMS (ESI) m/z [M + H]+. Calculated for C21H14N3O3: 356.1035. Found: 356.1048.
3-(4-Methoxybenzoyl)-6a,7-dihydro-6H-benzo[4,5]imidazo[1,2-a]pyrido[1,2-d]pyrazine-2,13-dione (6b). Yield 0.1112 g (91%), yellow powder, mp 234–235 °C. IR (ATR) ν 2970, 2751, 1632, 1556, 1466, 1329, 1214, 1028, 840. 1H NMR (500 MHz; DMSO-d6) δ 3.84 (s, 3H, OMe), 5.82 (s, 2H, CH2), 6.82 (s, 1H, H-1), 7.02 (d, J = 8.8 Hz, 2H, H-3, H-5 Ar), 7.18 (dd, J = 6.1 Hz, J = 3.1 Hz, 2H, Ar’), 7.54 (dd, J = 6.1 Hz, J = 3.1 Hz, Ar’), 7.80 (d, J = 8.8 Hz, 2H, H-2, H-6 Ar), 8.26 (s, 1H, H-4). 13C NMR (126 MHz, DMSO-d6) δ 51.5, 55.5, 113.6 (2C), 115.0, 121.9 (2C), 125.2, 128.1, 128.7, 128.8, 129.7, 131.7 (2C), 137.3, 138.3, 146.1, 150.2, 163.1, 163.2, 175.1, 191.8. HRMS (ESI) m/z [M + H]+. Calculated for C22H16N3O4: 386.1141. Found: 386.1141.
3-(4-Chlorobenzoyl)-6H-benzo[4,5]imidazo[1,2-a]pyrido[1,2-d]pyrazine-2,13-dione (6c). Yield 0.0408 g (33%), beige powder, mp 251–252 °C. IR (ATR) ν 2971, 2756, 1633, 1597, 1466, 1256, 779. 1H NMR (500 MHz, DMSO-d6) δ 5.84 (s, 2H, CH2), 6.82 (s, 1H, H-1), 7.18 (dd, J = 6.0 Hz, J = 3.2 Hz, 2H, Ar’), 7.54 (dd, J = 6.0 Hz, J = 3.2 Hz, 2H, Ar’), 7.57 (d, J = 8.5 Hz, 2H, H-3, H-5 Ar), 7.80 (d, J = 8.5 Hz, 2H, H-2, H-6 Ar), 8.37 (s, 1H, H-4). 13C NMR (126 MHz, DMSO-d6) δ 55.5, 113.5, 115.0, 122.1 (2C), 123.1, 125.2, 127.7, 128.5 (2C), 131.1 (2C), 135.8, 137.9, 138.0, 142.0, 147.9, 150.2, 163.0, 175.1, 192.5. Anal. Calculated for C21H12ClN3O3: C 64.71; H 3.10; N 10.78. Found: C 64.64; H 3.73; N 10.60.
3-(Thiophene-2-carbonyl)-6H-benzo[4,5]imidazo[1,2-a]pyrido[1,2-d]pyrazine-2,13-dione (6d). Yield 0.0481 g (42%), orange powder, mp 243–244 °C. IR (ATR) ν 2875, 2760, 1635, 1557, 1464, 1326, 1211, 1061, 878. 1H NMR (500 MHz, DMSO-d6) δ 5.81 (s, 2H, CH2), 6.81 (s, 1H, H-1), 7.18 (dd, J = 6.0 Hz, J = 3.2 Hz, 2H, Ar), 7.23 (dd, J = 4.9 Hz, J = 3.9 Hz, 1H, H-4 Th), 7.54 (dd, J = 6.0 Hz, J = 3.2 Hz, 2H, Ar), 7.84 (dd, J = 3.9 Hz, J = 1.1 Hz, 1H, H-3 Th), 8.05 (dd, J = 4.9 Hz, J = 1.1 Hz, 1H, H-5 Th), 8.35 (s, 1H, H-4). 13C NMR (126 MHz, DMSO-d6) δ 51.7, 115.0, 122.1 (2C), 122.8, 128.1, 128.6 (2C), 135.7 (2C), 138.0, 141.9, 143.6, 146.8, 150.3, 163.1, 174.7, 184.6 (1C was not observed). Anal. Calculated for C19H11N3O3S: C 63.15; H 3.07; N 11.63. Found: C 62.83; H 3.33; N 11.25.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/molecules28031285/s1, Full 1H, and 13C NMR spectra of all synthesized compounds; HRMS spectra of compounds 2a,e, 3a,c,e,f, 4a–d, 5a–c, 6a,b.
Author Contributions
Conceptualization and methodology were provided by V.Y.S. and D.L.O.; D.L.O. conceived and designed the experiments. The experimental work was conducted by V.V.V. and E.V.S.; D.L.O. and V.V.V. analyzed the results. D.L.O. studied and systemized the spectral data. Project administration and funding acquisition were carried out by V.Y.S. and D.L.O. All authors have read and agreed to the published version of the manuscript.
Funding
The research funding from the Ministry of Science and Higher Education of the Russian Federation (Ural Federal University Program of Development within the Priority-2030 Program) is gratefully acknowledged.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
Analytical studies were carried out using equipment at the Center for Joint Use ‘Spectroscopy and Analysis of Organic Compounds’ at the Postovsky Institute of Organic Synthesis of the Russian Academy of Sciences (Ural Branch) and the Laboratory of Complex Investigations and Expert Evaluation of Organic Materials of the Center for Joint Use at the Ural Federal University.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
The samples of the compounds are not available from the authors.
References
- Hughes, D.L. Review of synthetic routes and final forms of integrase inhibitors dolutegravir, cabotegravir, and bictegravir. Org. Process Res. Dev. 2019, 23, 716–729. [Google Scholar] [CrossRef]
- Hughes, D.L. Review of the patent literature: Synthesis and final forms of antiviral drugs tecovirimat and baloxavir marboxil. Org. Process Res. Dev. 2019, 23, 1298–1307. [Google Scholar] [CrossRef]
- Schreiner, E.; Richter, F.; Nerdinger, S. Development of synthetic routes to dolutegravir. Top. Heterocycl. Chem. 2016, 44, 187–208. [Google Scholar] [CrossRef]
- He, M.; Fan, M.; Peng, Z.; Wang, G. An overview of hydroxypyranone and hydroxypyridinone as privileged scaffolds for novel drug discovery. Eur. J. Med. Chem. 2021, 221, 113546. [Google Scholar] [CrossRef]
- Hayat, F.; Sonavane, M.; Makarov, M.V.; Trammell, S.A.J.; McPherson, P.; Gassman, N.R.; Migaud, M.E. The biochemical pathways of nicotinamide-derived pyridones. Int. J. Mol. Sci. 2021, 22, 1145. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-L.; Zhang, J.-Z.; Luo, D.-Q. The taxonomy, biology and chemistry of the fungal Pestalotiopsis genus. Nat. Prod. Rep. 2012, 29, 622–641. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kowalski, M.D.; Lakdawala, A.S.; Vogt, F.G.; Wu, L. An efficient and highly diastereoselective synthesis of GSK1265744, a potent HIV integrase inhibitor. Org. Lett. 2015, 17, 564–567. [Google Scholar] [CrossRef]
- Johns, B.A.; Kawasuji, T.; Weatherhead, J.G.; Taishi, T.; Temelkoff, D.P.; Yoshida, H.; Akiyama, T.; Taoda, Y.; Murai, H.; Kiyama, R.; et al. Carbamoyl pyridone HIV-1 integrase inhibitors 3. A diastereomeric approach to chiral nonracemic tricyclic ring systems and the discovery of dolutegravir (S/GSK1349572) and (S/GSK1265744). J Med. Chem. 2013, 56, 5901–5916. [Google Scholar] [CrossRef]
- Kawasuji, T.; Johns, B.A.; Yoshida, H.; Weatherhead, J.G.; Akiyama, T.; Taishi, T.; Taoda, Y.; Mikamiyama-Iwata, M.; Murai, H.; Kiyama, R.; et al. Carbamoyl pyridone HIV-1 integrase inhibitors. 2. Bi- and tricyclic derivatives result in superior antiviral and pharmacokinetic profiles. J. Med. Chem. 2013, 56, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Sankareswaran, S.; Mannam, M.; Chakka, V.; Mandapati, S.R.; Kumar, P. Identification and control of critical process impurities: An improved process for the preparation of dolutegravir sodium. Org. Process Res. Dev. 2016, 20, 1461–1468. [Google Scholar] [CrossRef]
- Sumino, Y.; Okamoto, K.; Masui, M.; Yamada, D.; Ikarashi, F. Process for Preparing Compound Having HIV Integrase Inhibitory Activity. WO Patent 018065, 9 February 2012. [Google Scholar]
- Maras, N.; Selic, L.; Cusak, A. Processes for Preparing Dolutegravir and Cabotegravir and Analogues Thereof. U.S. Patent 0368040, 11 July 2017. [Google Scholar]
- Srinivasachary, K.; Subbareddy, D.; Ramadas, C.; Balaji, S.K.K.; Somannavar, Y.S.; Ramadevi, B. Practical and efficient route to dolutegravir sodium via one-pot synthesis of key intermediate with controlled formation of impurities. Russ. J. Org. Chem. 2022, 58, 526–535. [Google Scholar] [CrossRef]
- Ziegler, R.E.; Desai, B.K.; Jee, J.-A.; Gupton, B.F.; Roper, T.D.; Jamison, T.F. 7-Step flow synthesis of the HIV integrase inhibitor dolutegravir. Angew. Chem. Int. Ed. 2018, 57, 7181–7185. [Google Scholar] [CrossRef] [PubMed]
- Dietz, J.-P.; Lucas, T.; Groß, J.; Seitel, S.; Brauer, J.; Ferenc, D.; Gupton, B.F.; Opatz, T. Six-step gram-scale synthesis of the human immunodeficiency virus integrase inhibitor dolutegravir sodium. Org. Process Res. Dev. 2021, 25, 1898–1910. [Google Scholar] [CrossRef]
- Kong, J.; Xia, H.; He, R.; Chen, H.; Yu, Y. Preparation of the key dolutegravir intermediate via MgBr2-promoted cyclization. Molecules 2021, 26, 2850. [Google Scholar] [CrossRef] [PubMed]
- Yasukata, T.; Masui, M.; Ikarashi, F.; Okamoto, K.; Kurita, T.; Nagai, M.; Sugata, Y.; Miyake, N.; Hara, S.; Adachi, Y.; et al. Practical synthetic method for the preparation of pyrone diesters: An efficient synthetic route for the synthesis of dolutegravir sodium. Org. Process Res. Dev. 2019, 23, 565–570. [Google Scholar] [CrossRef]
- Stojanović, M.; Bugarski, S.; Baranac-Stojanović, M. Synthesis of 2,3-dihydro-4-pyridones and 4-pyridones by the cyclization reaction of ester-tethered enaminones. J. Org. Chem. 2020, 85, 13495–13507. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, Y.; Xiao, L.-Y.; Peng, Q.-Q.; Zhao, Y.-L. Thermally induced formal [4+2] cycloaddition of 3-aminocyclobutenones with electron-deficient alkynes: Facile and efficient synthesis of 4-pyridones. Chem. Commun. 2018, 54, 8229–8232. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, J.; Lin, J.; Liu, F.; Liu, T.; Huang, C. Substituent-controlled chemoselective synthesis of multi-substituted pyridones via a one-pot three-component cascade reaction. Org. Biomol. Chem. 2020, 18, 1130–1134. [Google Scholar] [CrossRef]
- Fedin, V.V.; Usachev, S.A.; Obydennov, D.L.; Sosnovskikh, V.Y. Reactions of trifluorotriacetic acid lactone and hexafluorodehydroacetic acid with amines: Synthesis of trifluoromethylated 4-pyridones and aminoenones. Molecules 2022, 27, 7098. [Google Scholar] [CrossRef]
- Zantioti-Chatzouda, E.-M.; Kotzabasaki, V.; Stratakis, M. Synthesis of γ-pyrones and N-methyl-4-pyridones via the Au nanoparticle-catalyzed cyclization of skipped diynones in the presence of water or aqueous methylamine. J. Org. Chem. 2022, 87, 8525–8533. [Google Scholar] [CrossRef]
- Ropero, B.P.F.D.; Elsegood, M.R.J.; Fairley, G.; Pritchard, G.J.; Weaver, G.W. Pyridone functionalization: Regioselective deprotonation of 6-methylpyridin-2(1H)- and -4(1H)-one derivatives. Eur. J. Org. Chem. 2016, 2016, 5238–5242. [Google Scholar] [CrossRef]
- Obydennov, D.L.; El-Tantawy, A.I.; Sosnovskikh, V.Y. Synthesis of multifunctionalized 2,3-dihydro-4-pyridones and 4-pyridones via the reaction of carbamoylated enaminones with aldehydes. J. Org. Chem. 2018, 83, 13776–13786. [Google Scholar] [CrossRef] [PubMed]
- Obydennov, D.L.; Chernyshova, E.V.; Sosnovskikh, V.Y. Acyclic enaminodiones in the synthesis of heterocyclic compounds. Chem. Heterocycl. Comp. 2020, 56, 1241–1253. [Google Scholar] [CrossRef]
- Diesel, J.; Finogenova, A.M.; Cramer, N. Nickel-catalyzed enantioselective pyridone C−H functionalizations enabled by a bulky N-heterocyclic carbene ligand. J. Am. Chem. Soc. 2018, 140, 4489–4493. [Google Scholar] [CrossRef] [PubMed]
- Kea, D.; Wu, Y.; Zhang, L.; Shao, J.; Yu, Y.; Chen, W. Group-assisted-purification chemistry strategy for the efficient assembly of cyclic fused pyridinones. Synthesis 2022, 54, 1765–1774. [Google Scholar] [CrossRef]
- Obydennov, D.L.; Roeschenthaler, G.-V.; Sosnovskikh, V.Y. An improved synthesis and some reactions of diethyl 4-oxo-4H-pyran-2,5-dicarboxylate. Tetrahedron Lett. 2013, 54, 6545–6548. [Google Scholar] [CrossRef]
- Obydennov, D.L.; Khammatova, L.R.; Steben’kov, V.D.; Sosnovskikh, V.Y. Synthesis of novel polycarbonyl Schiff bases by ring-opening reaction of ethyl 5-acyl-4-pyrone-2-carboxylates with primary mono- and diamines. RSC Adv. 2019, 9, 40072–40083. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, L.; Zhou, Y.; Zha, D.; Hai, Y.; You, L. Dynamic covalent reactions controlled by ring-chain tautomerism of 2-formylbenzoic acid. Eur. J. Org. Chem. 2022, 2022, e202101461. [Google Scholar] [CrossRef]
- Osyanin, V.A.; Osipov, D.V.; Semenova, I.A.; Korzhenko, K.S.; Lukashenko, A.V.; Demidov, O.P.; Klimochkin, Y.N. Eco-friendly synthesis of fused pyrano[2,3-b]pyrans via ammonium acetate-mediated formal oxa-[3 + 3] cycloaddition of 4H-chromene-3-carbaldehydes and cyclic 1,3-dicarbonyl compounds. RSC Adv. 2020, 10, 34344–34354. [Google Scholar] [CrossRef]
- Zhang, Z.; Gao, X.; Wan, Y.; Huang, Y.; Huang, G.; Zhang, G. Ammonium acetate-promoted one-pot tandem aldol condensation/aza-addition reactions: Synthesis of 2,3,6,7-tetrahydro-1H-pyrrolo[3,2-c]pyridin-4(5H)-ones. ACS Omega 2017, 2, 6844–6851. [Google Scholar] [CrossRef]
- Obydennov, D.L.; Roeschenthaler, G.-V.; Sosnovskikh, V.Y. Synthesis of 6-aryl- and 5-aroylcomanic acids from 5-aroyl-2-carbethoxy-4-pyrones via a deformylative rearrangement and ring-opening/ring-closure sequence. Tetrahedron Lett. 2014, 55, 472–474. [Google Scholar] [CrossRef]
- Obydennov, D.L.; Goncharov, A.O.; Sosnovskikh, V.Y. Preparative synthesis of ethyl 5-acyl-4-pyrone-2-carboxylates and 6-aryl-, 6-alkyl-, and 5-acylcomanic acids on their basis. Russ. Chem. Bull. 2016, 65, 2233–2242. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).