

Computational Studies of Auto-Active van der Waals Interaction Molecules on Ultra-Thin Black-Phosphorus Film

Abstract
1. Introduction
2. Outcomes and Discussion
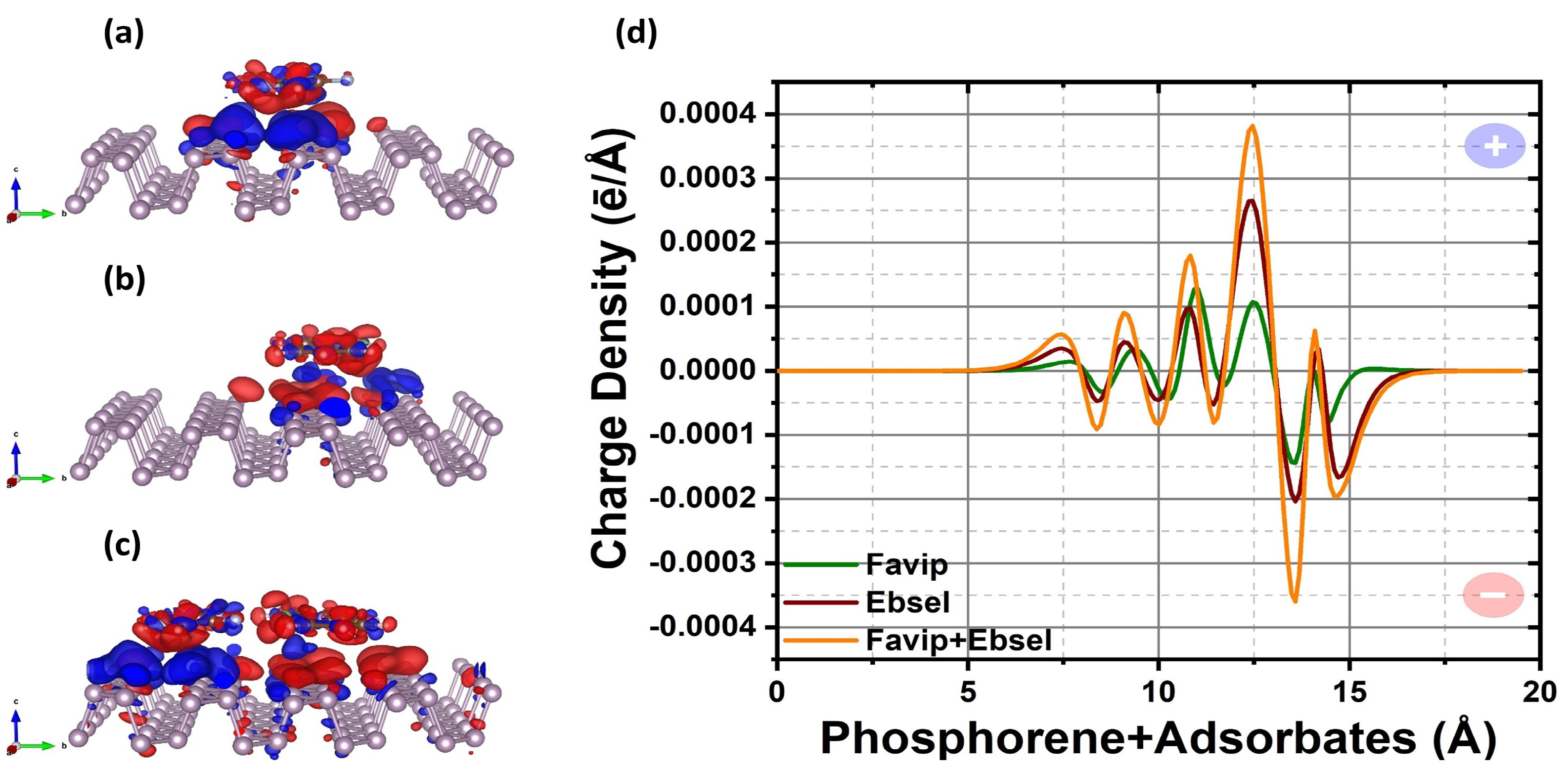
2.1. Structural Properties
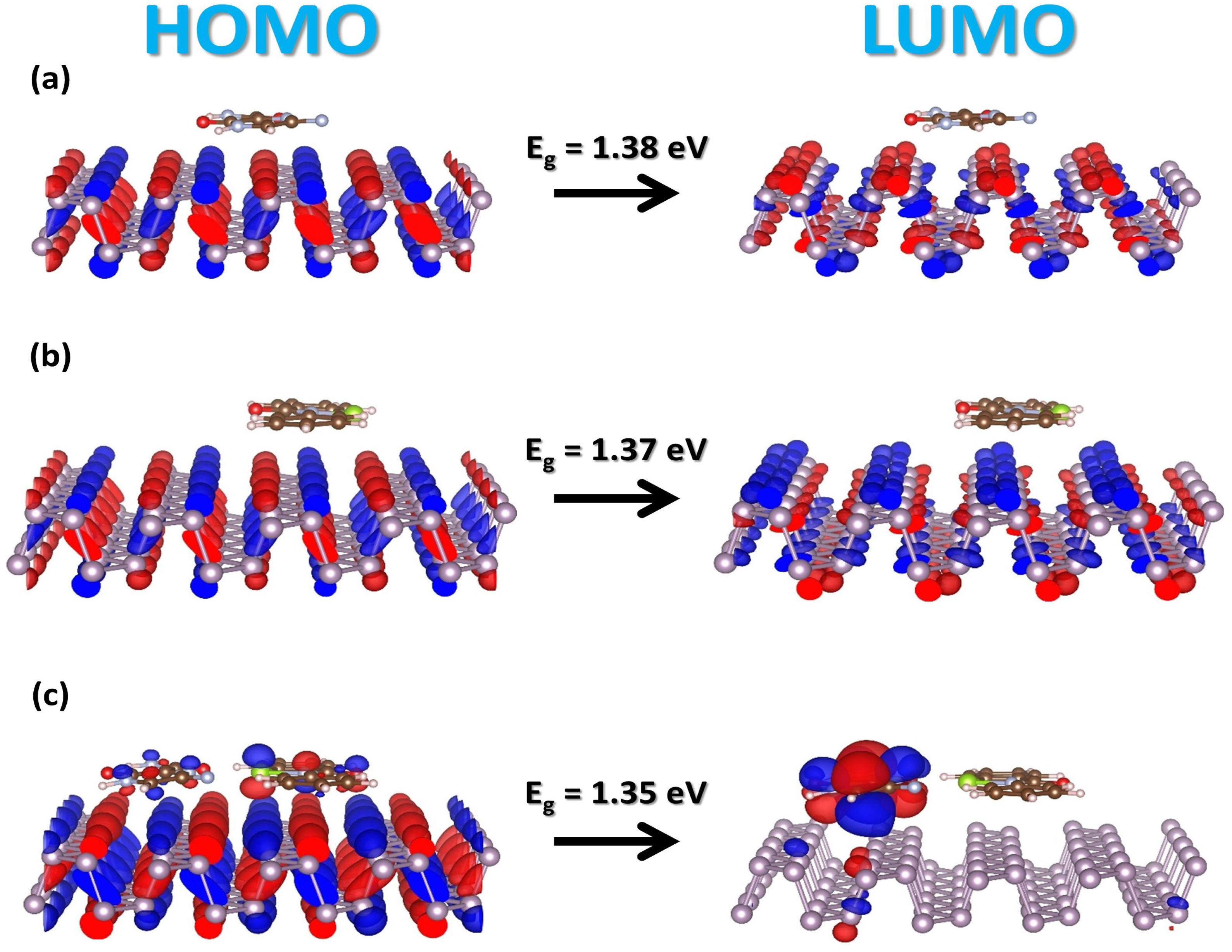
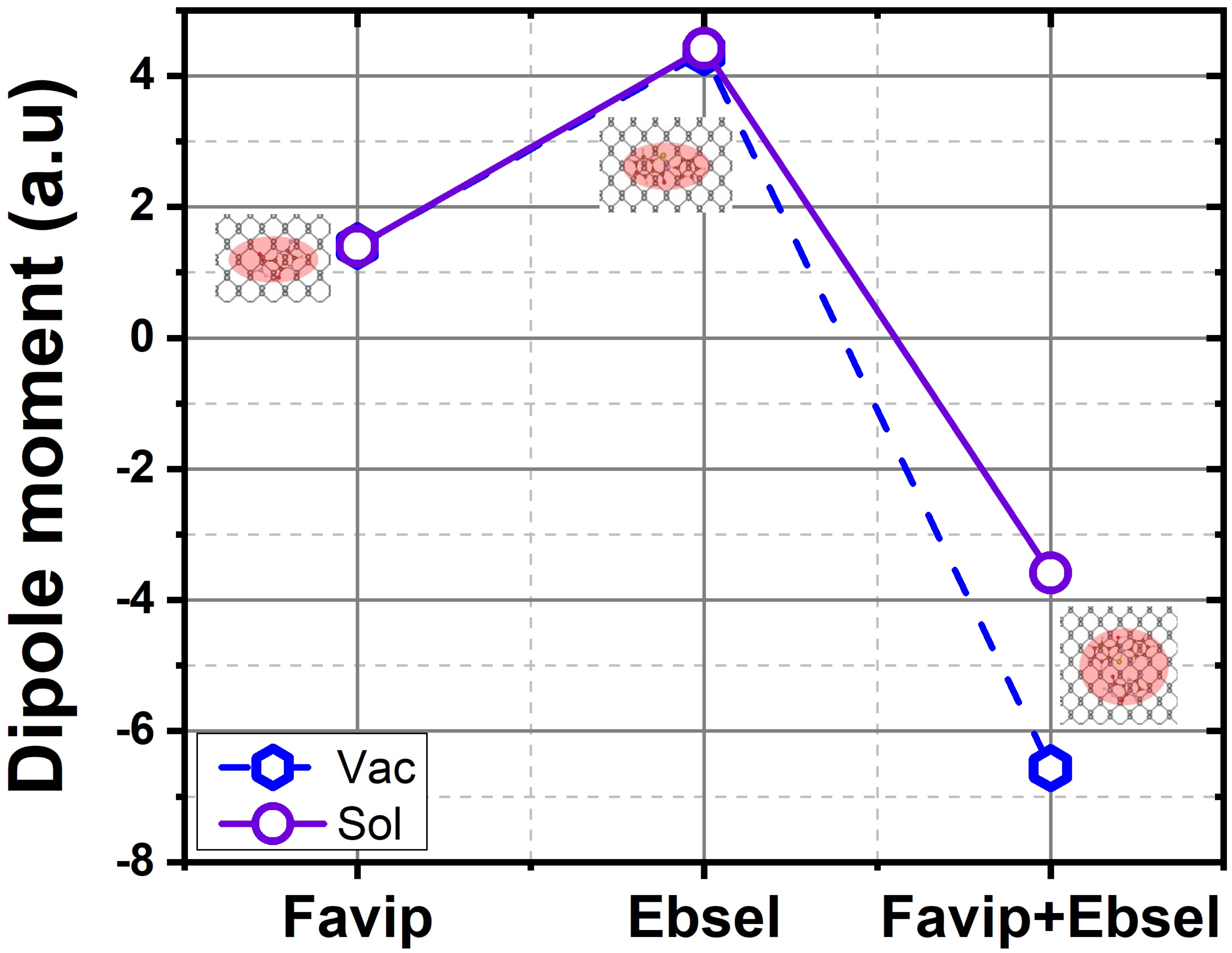
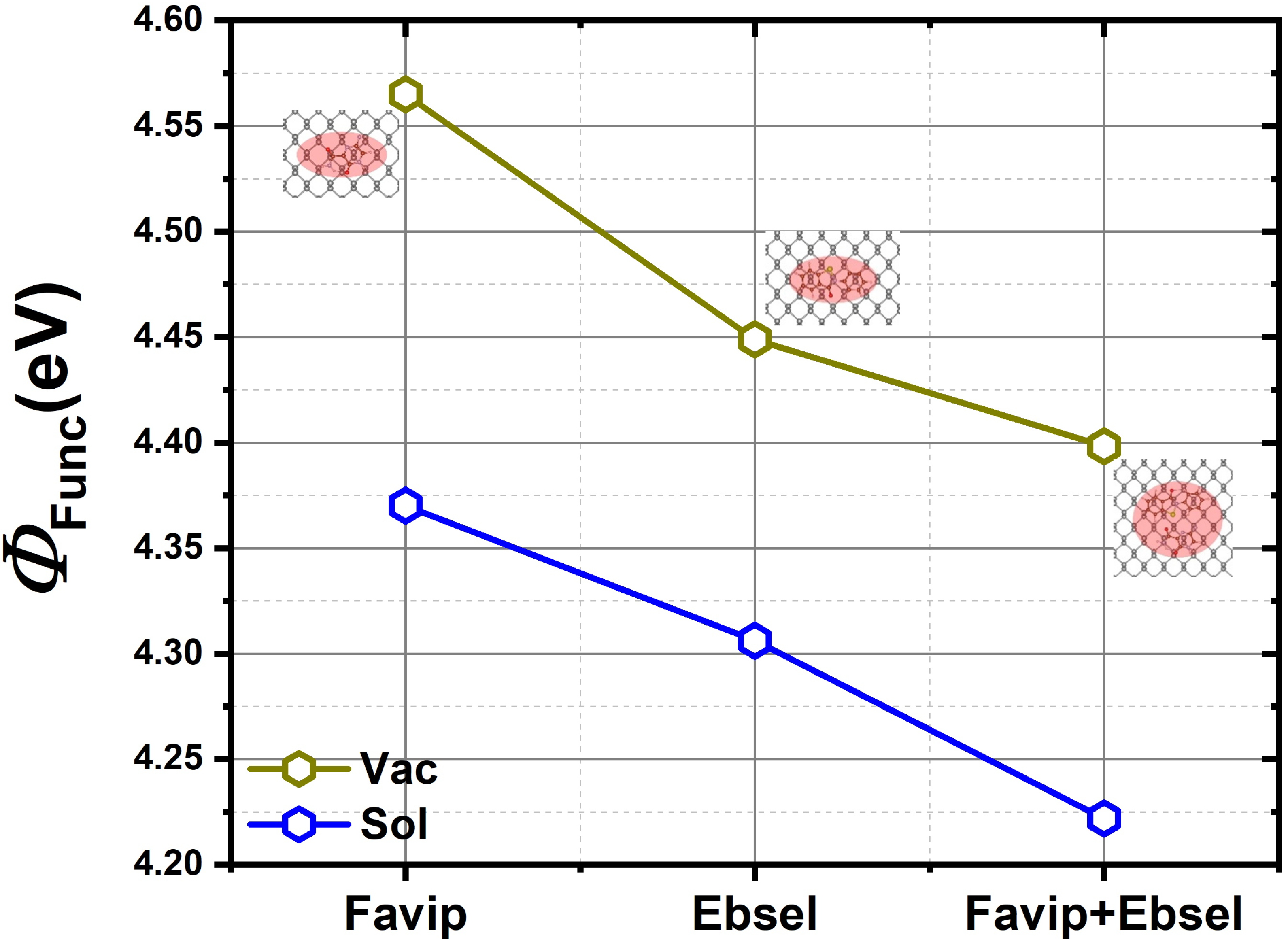
2.2. Electronic Properties
3. Thermodynamic Properties
4. Methodology
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Renson, M.; Etschenberg, E.; Winkelmann, J. 2–Phenyl–1,2–benzisoselenazol–3 (2H)–one Containing Pharmaceutical Preparations and Process for the Treatment of Rheumatic Diseases. U.S. Patent No. 4,352,799, 1982. [Google Scholar]
- Chen, L.; Gui, C.; Luo, X.; Yang, Q.; Gunther, S.; Scandella, E.; Drosten, C.; Bai, D.; He, X.; Ludewig, B.; et al. Cinanserin is an inhibitor of the 3C–like proteinase of severe acute respiratory syndrome coronavirus and strongly reduces virus replication in vitro. J. Virol. 2005, 79, 7095–7103. [Google Scholar] [CrossRef]
- Carmo, A.; Pereira–Vaz, J.; Mota, V.; Mendes, A.; Morais, C.; da Silva, A.C.; Camilo, E.; Pinto, C.S.; Cunha, E.; Pereira, J.; et al. Clearance and persistence of SARS-CoV-2 RNA in patients with COVID-19. J. Med. Virol. 2020, 92, 2227–2231. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S.; et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci. 2020, 25, 315–331. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Pe, C. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Scavone, C.; Brusco, S.; Bertini, M.; Sportiello, L.; Rafaniello, C.; Zoccoli, A.; Berrino, L.; Racagni, G.; Rossi, F.; Capuano, A. Current Pharmacological Treatments for COVID-19: What’s Next? Br. J. Pharmacol. 2020, 177, 4813–4824. [Google Scholar] [CrossRef]
- Sreekanth Reddy, O.; Lai, W.-F. Tackling COVID-19 Using Remdesivir and Favipiravir as Therapeutic Options. Chembiochem 2021, 22, 939–948. [Google Scholar] [CrossRef]
- Santi, C.; Scimmi, C.; Sancineto, L. Ebselen and Analogues: Pharmacological Properties and Synthetic Strategies for Their Preparation. Molecules 2021, 26, 4230. [Google Scholar] [CrossRef]
- Guy, R.K.; Di Paola, R.S.; Romanelli, F.; Dutch, R.E. Rapid repurposing of drugs for COVID-19. Science 2020, 368, 829–830. [Google Scholar] [CrossRef]
- Delang, L.; Abdelnabi, R.; Neyts, J. Favipiravir as a potential countermeasure against neglected and emerging RNA viruses. Antivir. Res. 2018, 153, 85–94. [Google Scholar] [CrossRef]
- Furuta, Y.; Komeno, T.; Nakamura, T. Favipiravir (T–705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 449–463. [Google Scholar] [CrossRef]
- Furuta, Y.; Gowen, B.B.; Takahashi, K.; Shiraki, K.; Smee, D.F.; Barnard, D.L. Favipiravir (T–705), a novel viral RNA polymerase inhibitor. Antivir. Res. 2013, 100, 446–454. [Google Scholar]
- Lynch, E.; Kil, J. Development of ebselen, a glutathione peroxidase mimic, for the prevention and treatment of noise–induced hearing loss. Semin. Hear. 2009, 30, 047–055. [Google Scholar] [CrossRef]
- Kil, J.; Lobarinas, E.; Spankovich, C.; Griffiths, S.K.; Antonelli, P.J.; Lynch, E.D.; Le Prell, C.G. Safety and efficacy of ebselen for the prevention of noise–induced hearing loss: A randomised, double–blind, placebo–controlled, phase 2 trial. Lancet 2017, 390, 969–979. [Google Scholar] [CrossRef]
- Masaki, C.; Sharpley, A.L.; Cooper, C.M.; Godlewska, B.R.; Singh, N.; Vasudevan, S.R.; Harmer, C.J.; Churchill, G.C.; Sharp, T.; Rogers, R.D.; et al. Effects of the potential lithium–mimetic, ebselen, on impulsivity and emotional processing. Psychopharmacology 2016, 233, 2655–2661. [Google Scholar] [CrossRef]
- Singh, N.; Halliday, A.C.; Thomas, J.M.; Kuznetsova, O.V.; Baldwin, R.; Woon, E.; Aley, P.K.; Antoniadou, I.; Sharp, T.; Vasudevan, S.R.; et al. A safe lithium mimetic for bipolar disorder. Nat. Commun. 2013, 4, 1332. [Google Scholar] [CrossRef]
- Udwadia, Z.F.; Singh, P.; Barkate, H.; Patil, S.; Rangwala, S.; Pendse, A.; Kadam, J.; Wu, W.; Caracta, C.F.; Tandon, M. Efficacy and safety of favipiravir, an oral RNA-dependent RNA polymerase inhibitor, in mild-to-moderate COVID-19: A randomized, comparative, open-label, multicenter, phase 3 clinical trial. Int. J. Infect. Dis. 2021, 103, 62–71. [Google Scholar] [CrossRef]
- Dong, L.; Hu, S.; Gao, J. Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug Discov. Ther. 2020, 14, 58–60. [Google Scholar] [CrossRef]
- Yang, K.; Feng, L.; Shi, X.; Liu, Z. Nano–graphene in biomedicine: Theranostic applications. Chem. Soc. Rev. 2013, 42, 530. [Google Scholar] [CrossRef]
- Ferrari, M. Cancer nanotechnology: Opportunities and challenges. Nat. Rev. Cancer 2005, 5, 161. [Google Scholar] [CrossRef]
- Lin, H.; Chen, Y.; Shi, J.-L. Insights into 2D MXenes for Versatile Biomedical Applications: Current Advances and Challenges Ahead. Adv. Sci. 2018, 5, 1800518. [Google Scholar] [CrossRef]
- Gao, G.; Gao, W.; Cannuccia, E.; Taha-Tijerina, J.; Balicas, L.; Mathkar, A.; Narayanan, T.; Liu, Z.; Gupta, B.K.; Peng, J. Artificially Stacked Atomic Layers: Toward New van der Waals Solids. Nano Lett. 2012, 12, 3518–3525. [Google Scholar] [CrossRef]
- Backes, C.; Abdelkader, A.M.; Alonso, C.; Andrieux-Ledier, A.; Arenal, R.; Azpeitia, J.; Balakrishnan, N.; Banszerus, L.; Barjon, J.; Bartali, R. Production and processing of graphene and related materials. 2D Mater. 2020, 7, 022001. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Fal, V.; Colombo, L.; Gellert, P.; Schwab, M.; Kim, K. A roadmap for graphene. Nature 2012, 490, 192–200. [Google Scholar] [CrossRef]
- Kotagiri, N.; Sudlow, G.P.; Akers, W.J.; Achilefu, S. Breaking the depth dependency of phototherapy with Cerenkov radiation and low-radiance-responsive nanophotosensitizers. Nat. Nanotechnol. 2015, 10, 370. [Google Scholar] [CrossRef]
- Laref, A.; Alshammari, N.; Laref, S.; Luo, S.-J. Surface passivation effects on the electronic and optical properties of silicon quantum dots. Sol. Energy Mater. Sol. Cells 2014, 120, 622–630. [Google Scholar] [CrossRef]
- Wick, P.; Louw–Gaume, A.E.; Kucki, M.; Krug, H.F.; Kostarelos, K.; Fadeel, B.; Dawson, K.A.; Salvati, A.; Vázquez, E.; Ballerini, L. Classification Framework for Graphene–Based Materials. Angew. Chem. Int. Ed. 2014, 53, 7714–7718. [Google Scholar] [CrossRef]
- Laref, A.; Alsagri, M.; Alay-e-Abbas, S.M.; Laref, S.; Huang, H.M.; Xiong, Y.C.; Yang, J.T.; Khandy, S.A.; Rai, D.P.; Varshney, D.; et al. Electronic structure and optical characteristics of AA stacked bilayer graphene: A first principles calculations. Optik 2020, 206, 163755. [Google Scholar] [CrossRef]
- Smith, A.T.; La Chance, A.M.; Zeng, S.; Liu, B.; Sun, L. Synthesis, properties, and applications of graphene oxide/reduced graphene oxide and their nanocomposites. Nano Mater. Sci. 2019, 1, 31–47. [Google Scholar] [CrossRef]
- Huang, X.; Tang, S.; Mu, X.; Dai, Y.; Chen, G.; Zhou, Z.; Ruan, F.; Yang, Z.; Zheng, N. Freestanding palladium nanosheets with plasmonic and catalytic properties. Nat. Nanotechnol. 2011, 6, 28. [Google Scholar] [CrossRef]
- Tan, C.; Zhang, H. Two-dimensional transition metal dichalcogenide nanosheet-based composites. Chem. Soc. Rev. 2015, 44, 2713. [Google Scholar] [CrossRef]
- Sovizi, S.; Szoszkiewicz, R. Single atom doping in 2D layered MoS2 from a periodic table perspective. Surf. Sci. Rep. 2022, 77, 100567. [Google Scholar]
- Majed, A.; Kothakonda, M.; Wang, F.; Tseng, E.N.; Prenger, K.; Zhang, X.; Persson, P.O.Å.; Wei, J.; Sun, J.; Naguib, M. Transition Metal Carbo-Chalcogenide “TMCC”: A New Family of 2D Materials. Adv. Mater. 2022, 34, 2200574. [Google Scholar] [CrossRef]
- Kalantar-zadeh, K.; Ou, J.Z.; Daeneke, T.; Mitchell, A.; Sasaki, T.; Fuhrer, M.S. Two-dimensional and layered transition metal oxides. Appl. Mater. Today 2016, 5, 73. [Google Scholar] [CrossRef]
- Hong, G.; Lee, J.C.; Robinson, J.T.; Raaz, U.; Xie, L.; Huang, N.F.; Cooke, J.P.; Dai, H. Multifunctional in vivo vascular imaging using near–infrared II fluorescence. Nat. Med. 2012, 18, 1841. [Google Scholar] [CrossRef]
- Anasori, B.; Lukatskaya, M.R.; Gogotsi, Y. 2D metal carbides and nitrides (MXenes) for energy storage. Nat. Rev. Mater. 2017, 2, 16098. [Google Scholar] [CrossRef]
- Laref, A.; Alsagri, M.; Alay-e-Abbas, S.M.; Barakat, F.; Laref, S.; Huang, H.M.; Xiong, Y.C.; Yang, J.T.; Wu, X. Impact of phosphorous and sulphur substitution on Dirac cone modification and optical behaviors of monolayer graphene for nanoelectronic devices. Appl. Surf. Sci. 2019, 489, 358–371. [Google Scholar] [CrossRef]
- Laref, S.; Asaduzzaman, A.M.; Beck, W.; Deymier, P.A.; Runge, K.; Adamowicz, L.; Muralidharan, K. Characterization of graphene–fullerene interactions: Insights from density functional theory. Chem. Phys. Lett. 2013, 582, 115–118. [Google Scholar] [CrossRef]
- Ma, B.; Martín, C.; Kurapati, R.; Bianco, A. Degradation-by-design: How chemical functionalization enhances the biodegradability and safety of 2D materials. Chem. Soc. Rev. 2020, 49, 6224–6247. [Google Scholar] [CrossRef]
- Liu, H.; Du, Y.; Deng, Y.; Peide, D.Y. Semiconducting black phosphorus: Synthesis, transport properties and electronic applications. Chem. Soc. Rev. 2015, 44, 2732. [Google Scholar] [CrossRef]
- Tao, W.; Zhu, X.; Yu, X.; Zeng, X.; Xiao, Q.; Zhang, X.; Ji, X.; Wang, X.; Shi, J.; Zhang, H.; et al. Black Phosphorus Nanosheets as a Robust Delivery Platform for Cancer Theranostic. Adv. Mater. 2017, 29, 1603276. [Google Scholar] [CrossRef]
- Reina, G.; González-Domínguez, J.M.; Criado, A.; Vázquez, E.; Bianco, A.; Prato, M. Promises, facts and challenges for graphene in biomedical applications. Chem. Soc. Rev. 2017, 46, 4400. [Google Scholar] [CrossRef]
- Tao, W.; Ji, X.; Zhu, X.; Li, L.; Wang, J.; Zhang, Y.; Saw, P.E.; Li, W.; Kong, N.; Islam, M.A.; et al. Two-Dimensional Antimonene-Based Photonic Nanomedicine for Cancer Theranostics. Adv. Mater. 2018, 30, 1802061. [Google Scholar] [CrossRef]
- Xue, T.Y.; Liang, W.Y.; Li, Y.W.; Sun, Y.H.; Xiang, Y.J.; Zhang, Y.P.; Dai, Z.G.; Duo, Y.H.; Wu, L.M.; Qi, K.; et al. Ultrasensitive detection of miRNA with an antimonene–based surface plasmon resonance sensor. Nat. Commun. 2019, 10, 28. [Google Scholar] [CrossRef]
- Woolhouse, M.; Scott, F.; Hudson, Z.; Howey, R.; Chase-Topping, M. Human viruses: Discovery and emergence. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 2864–2871. [Google Scholar] [CrossRef]
- Laref, S.; Wang, B.; Inal, S.; Al-Ghamdi, S.; Gao, X.; Gojobori, T. A Peculiar Binding Characterization of DNA (RNA) Nucleobases at MoOS-Based Janus Biosensor: Dissimilar Facets Role on Selectivity and Sensitivity. Biosensors 2022, 12, 442. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.-Q.; Mu, J.-S.; Kargbo, D.; Song, Y.-B.; Niu, W.-K.; Nie, W.-M.; Kanu, A.; Liu, W.-W.; Wang, Y.-P.; Dafae, F.; et al. Clinical and Virological Characteristics of Ebola Virus Disease Patients Treated With Favipiravir (T–705)–Sierra Leone, 2014. Clin. Infect. Dis. 2016, 63, 1288–1294. [Google Scholar] [CrossRef]
- Rosenke, K.; Feldmann, H.; Westover, J.B.; Hanley, P.W.; Martellaro, C.; Feldmann, F.; Saturday, G.; Lovaglio, J.; Scott, D.P.; Furuta, Y.; et al. Use of Favipiravir to Treat Lassa Virus Infection in Macaques. Emerg. Infect. Dis. J. 2018, 24, 1696–1698. [Google Scholar] [CrossRef]
- Shen, L.; Li, B.; Qiao, Y. Fe3O4 Nanoparticles in Targeted Drug/Gene Delivery Systems. Materials 2018, 11, 324. [Google Scholar] [CrossRef]
- Rahimi, R.; Solimannejad, M. BC3 graphene–like monolayer as a drug delivery system for nitrosourea anticancer drug: A first–principles perception. Appl. Surf. Sci. 2020, 525, 146577. [Google Scholar] [CrossRef]
- Hashemzadeh, H.; Raissi, H. Covalent organic framework as smart and high efficient carrier for anticancer drug delivery: A DFT calculations and molecular dynamics simulation stud. J. Phys. D Appl. Phys. 2018, 51, 345401. [Google Scholar] [CrossRef]
- Mou, T.; Wang, B. Rational Surface Modification of Two–Dimensional Layered Black Phosphorus: Insights from First–Principles Calculations. ACS Omega 2018, 3, 2445–2451. [Google Scholar] [CrossRef]
- Su, J.; Mou, T.; Wen, J.; Wang, B. Nondestructive functionalization of monolayer black phosphorus using Lewis acids: A first–principles study. Appl. Surf. Sci. 2020, 518, 146210. [Google Scholar] [CrossRef]
- Sundararaman, M.; Weaver, L.; Hennig, A. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 2014, 140, 084106. [Google Scholar]
- Mathew, K.; Kolluru, V.S.C.; Mula, S.; Steinmann, S.N.; Hennig, R.G. Implicit self-consistent electrolyte model in plane-wave density-functional theory. J. Chem. Phys. 2019, 151, 234101. [Google Scholar] [CrossRef]
- Yasir, H.M.; Hanoon, F.H. DFT and TD–DFT Study of Favipiravir Tautomerism as RNA Polymerase Inhibitors: COVID-19. IOP Conf. Ser. Mater. Sci. Eng. 2020, 928, 072066. [Google Scholar] [CrossRef]
- Wendel, A.; Fausel, M.; Safayhi, H.; Tiegs, G.; Otter, R. A novel biologically active seleno–organic compound–II: Activity of PZ 51 in relation to Glutathione Peroxidase. Biochem. Pharmacol. 1984, 33, 3241–3245. [Google Scholar] [CrossRef] [PubMed]
- Ou, W.; Byeon, J.H.; Thapa, R.K.; Ku, S.K.; Yong, C.S.; Kim, J.O. Plug-and-Play Nanorization of Coarse Black Phosphorus for Targeted Chemo-Photoimmunotherapy of Colorectal Cancer. ACS Nano 2018, 12, 10061–10074. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab–initio total energy calculations for metals and semiconductors using a plane–wave basis set. Comput. Mater. Sci. 1996, 6, 15. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented–wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, S. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT–D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comp. Chem. 2011, 32, 1456. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
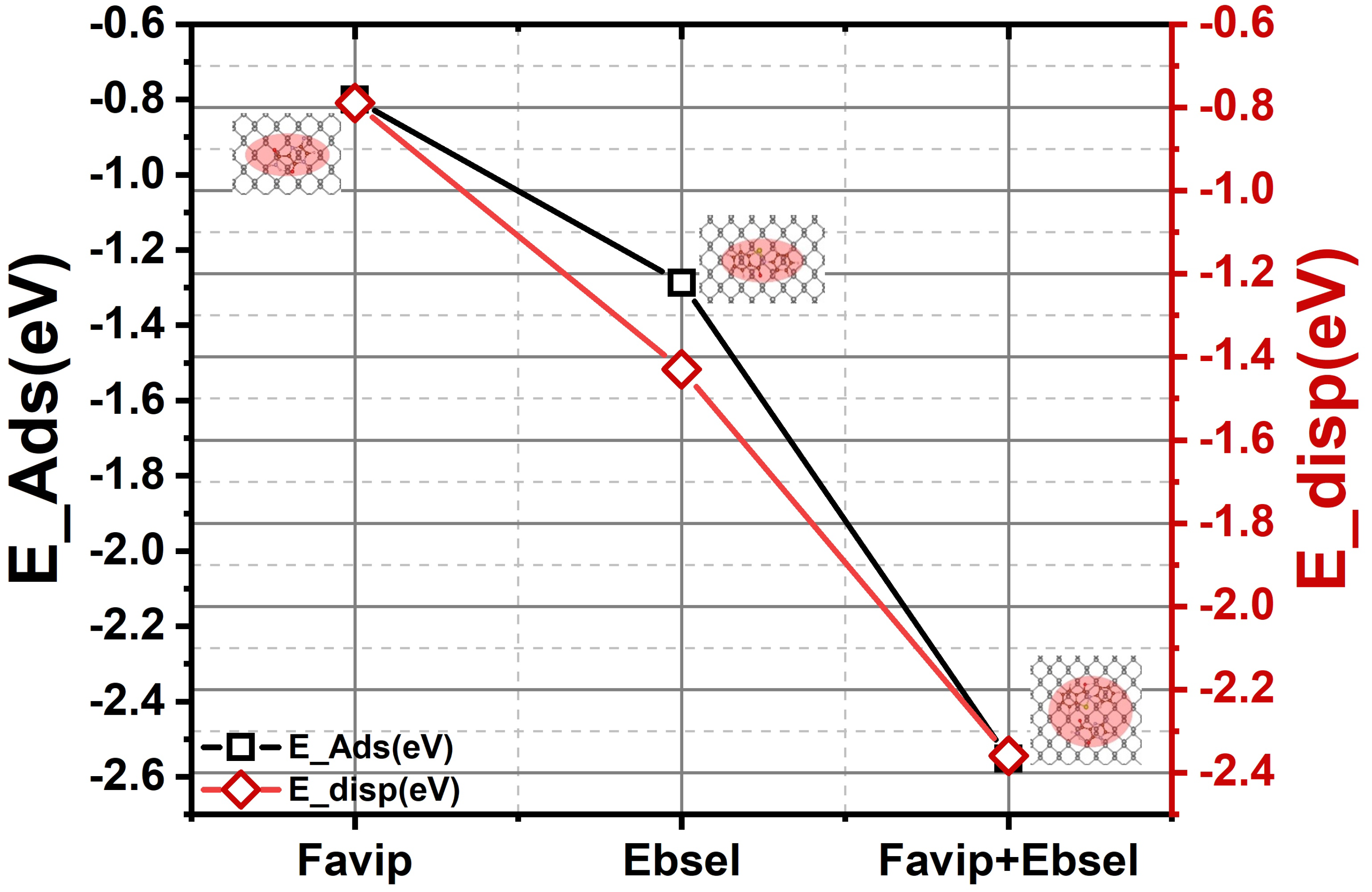
| d–d | EvdW | EBin | EInt | ECov | |
|---|---|---|---|---|---|
| FP | 3.18 | −0.79 | −0.80 | −0.82 | −0.01 |
| EB | 3.28 | −1.43 | −1.29 | −1.31 | 0.14 |
| FP + EB | 3.24 | −2.36 | −2.55 | −2.15 | −0.19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laref, S.; Wang, B.; Gao, X.; Gojobori, T. Computational Studies of Auto-Active van der Waals Interaction Molecules on Ultra-Thin Black-Phosphorus Film. Molecules 2023, 28, 681. https://doi.org/10.3390/molecules28020681
Laref S, Wang B, Gao X, Gojobori T. Computational Studies of Auto-Active van der Waals Interaction Molecules on Ultra-Thin Black-Phosphorus Film. Molecules. 2023; 28(2):681. https://doi.org/10.3390/molecules28020681
Chicago/Turabian StyleLaref, Slimane, Bin Wang, Xin Gao, and Takashi Gojobori. 2023. "Computational Studies of Auto-Active van der Waals Interaction Molecules on Ultra-Thin Black-Phosphorus Film" Molecules 28, no. 2: 681. https://doi.org/10.3390/molecules28020681
APA StyleLaref, S., Wang, B., Gao, X., & Gojobori, T. (2023). Computational Studies of Auto-Active van der Waals Interaction Molecules on Ultra-Thin Black-Phosphorus Film. Molecules, 28(2), 681. https://doi.org/10.3390/molecules28020681