Abstract
The present work describes the design and development of seventeen pyrimidine-clubbed benzimidazole derivatives as potential dihydrofolate reductase (DHFR) inhibitors. These compounds were filtered by using ADMET, drug-likeness characteristics calculations, and molecular docking experiments. Compounds 27, 29, 30, 33, 37, 38, and 41 were chosen for the synthesis based on the results of the in silico screening. Each of the synthesized compounds was tested for its in vitro antibacterial and antifungal activities using a variety of strains. All the compounds showed antibacterial properties against Gram-positive bacteria (Staphylococcus aureus and Staphylococcus pyogenes) as well as Gram-negative bacteria (Escherichia coli and Pseudomonas aeruginosa). Most of the compounds either had a higher potency than chloramphenicol or an equivalent potency to ciprofloxacin. Compounds 29 and 33 were effective against all the bacterial and fungal strains. Finally, the 1,2,3,4-tetrahydropyrimidine-2-thiol derivatives with a 6-chloro-2-(chloromethyl)-1H-benzo[d]imidazole moiety are potent enough to be considered a promising lead for the discovery of an effective antibacterial agent.
Keywords:
DHFR; antifungal; antibacterial; pyrimidines; benzimidazoles; ADMETlab 2.0; molecular docking 1. Introduction
The treatment of nosocomial infections poses a significant global risk to public health due to drug-resistant bacteria, such as methicillin-resistant Staphylococcus aureus (MRSA) and multidrug-resistant Escherichia coli [1,2,3]. Research commissioned by the United Kingdom Government estimates that “the cost in terms of lost global production between now and 2050 would be an astounding 100 trillion USD” if no action is taken. Infections caused by fungi may significantly threaten human health, especially for immunocompromised patients. When it comes to clinical care, invasive fungal infections (IFIs) pose a significant challenge on a global scale [4,5,6]. It is imperative that attempts to discover new antibiotic agents be stepped up to keep pace with the worrisome increase in cases of antibiotic resistance being demonstrated by disease-causing microbes [7,8]. One crucial step is the identification of potent inhibitors of receptors that are critical to the bacteria’s survival.
One of these targets, known as dihydrofolate reductase (DHFR), may be found in the Gram-negative bacterium Escherichia coli and several microorganisms [9]. This enzyme is essential to the continued existence of the microbe. Since the middle of the 20th century, it has been shown that the DHFR enzyme may be used as a therapeutic target for the treatment of infections. DHFR plays a role in the production of raw materials for cell proliferation in both prokaryotic and eukaryotic cells. This is accomplished by catalyzing the reduction of dihydrofolate to tetrahydrofolate via the use of NADPH in the process [10]. Inhibitors of DHFR are employed extensively in the treatment of fungal infections, bacterial diseases, and mycobacterial diseases, as well as in the fight against malaria [11,12,13]. When it comes to finding novel and effective inhibitors for this enzyme, the traditional drug-discovery methods only depend on the rational drug design-based elaboration of core scaffolds.
The vast majority of DHFR inhibitors, which are either already in use or are the subject of research, are derivatives of folic acid that have a 2,4-diamino substitution in the pyrimidine ring. Structurally, these compounds belong to a variety of distinct classes (pyrimidines, pteridines, quinazolines, and pyrido-pyrimidines) [14,15]. It has been shown that several derivatives based on amino pyrimidine can inhibit DHFR, which in turn results in antibacterial activity [16,17]. Because of this, the pyrimidine scaffold was chosen for the current work to design and develop new DHFR inhibitors as possible antibacterial and antifungal medications. As an innovative approach, we have tried to merge the benzimidazole and pyrimidine nuclei in the hope of obtaining novel derivatives with synergistic effects. We aimed to obtain derivatives with a lower toxicity profile and improved biological potential. First, these new potential inhibitors were filtered by ADMET and drug-likeness calculations. Successively, molecular docking experiments were carried out. The compounds that showed substantial promise for DHFR inhibition were placed through a wet lab synthesis, which was then followed by a biological assessment.
2. Results and Discussion
2.1. In Silico ADMET Profile of Designed Molecules
The ability for researchers to explore the biological effects of potential pharmacological candidates is made feasible by pharmacokinetic features, making them an essential part of the drug development process [18]. We designed a set of new pyrimidine-clubbed benzimidazole derivatives and performed ADMET, drug-like, and molecular docking calculations before starting the synthesis (Figure 1, 2D structures of the compounds). The physicochemical properties of the designed molecules 25–41 are tabulated in Table 1. In the physicochemical analysis, the values of all the molecules are displayed within the acceptable range, i.e., molecular weights, nHA (H-bond acceptors), nHD (H-bond donors), nRot (rotatable bonds), van der Waals volume, and TPSA (total polar surface area). The drug’s lipophilicity, which is essential for solubility, absorption, membrane penetration, plasma protein binding, distribution, and tissue penetration, is directly connected to the logP and logS values. The significance of the drug’s lipophilicity necessitated the inclusion of logP and logS as elements of Lipinski’s rule of five. In the present investigation, all these parameters were within the accepted range and displayed optimum oral bioavailability, indicating they can be developed to be delivered through the oral route [19,20].
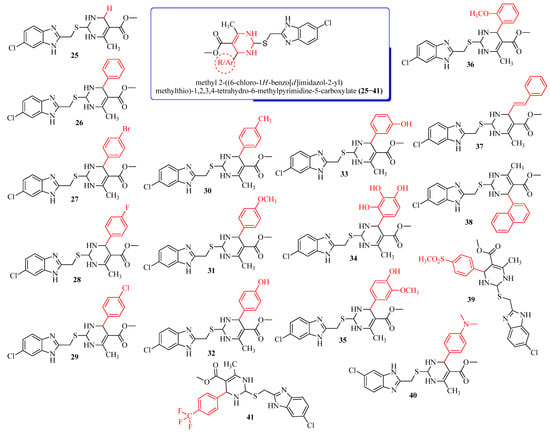
Figure 1.
Parent nucleus and substitutions of methyl 2-((6-chloro-1H-benzo[d]imidazol-2-yl)methylthio)-1,2,3,4-tetrahydro-6-methylpyrimidine-5-carboxylate derivatives (25–41).

Table 1.
Physicochemical properties of methyl 2-((6-chloro-1H-benzo[d]imidazol-2-yl)methylthio)-1,2,3,4-tetrahydro-6-methylpyrimidine-5-carboxylate derivatives (25–41).
The drug-like properties of the molecules are exemplified in Table 2. The different parameters, such as the QED (quantitative estimate of drug-likeness), NP score (natural product-likeness score), Lipinski rule, Pfizer rule, GSK rule, Golden Triangle rule, and Chelator rule, were calculated. The QED is an indicator of drug-likeness that was introduced in 2012 and is an index of drug-likeness that is modeled using the information available on marketed medications. It is frequently used in the present small-molecule drug development process for computational approaches and to assess drug-like features. Most of the designed compounds showed an attractive range of QED [21,22]. Typically, the NP score falls somewhere in the range of −5 to 5. If the score is higher, then there is a greater likelihood that the molecule in question is an NP [23,24]. All the designed molecules displayed NP-like properties except for 30, which showed a −9.932 NP score. The compounds satisfying the GSK rule may have a more favorable ADMET profile, but unfortunately, only 25 accepted the rule. The compounds satisfying the Golden Triangle rule may have a more favorable ADMET profile, and all the molecules accepted under the rule show a favorable ADMET profile.
Table 2.
Drug-likeness properties of methyl 2-((6-chloro-1H-benzo[d]imidazol-2-yl)methylthio)-1,2,3,4-tetrahydro-6-methylpyrimidine-5-carboxylate derivatives (25–41).
The absorption parameters of the molecules are illustrated in Table 3. As a model of how medications are absorbed by the human digestive tract, the human colon epithelial cancer cell line, known as Caco-2, is employed. This model is useful for determining whether a substance is appropriate for oral administration, predicting intestinal permeability, and researching drug efflux. Caco-2 permeability is optimum when the value is higher than −5.15 log units, and fortunately, all the molecules displayed optimum Caco-2 permeability [25]. It is possible to acquire a better knowledge of the process of drug efflux with the aid of MDCK-MDR1 cells, which also draws attention to early potential problems with drug permeability. It has been discovered that the permeability of MDCK-MDR1 may, in addition to intestinal permeability, be used as an accurate predictor of the permeability of the blood–brain barrier [26]. Many of the molecules displayed P-gp-inhibitor and P-gp-substrate activity. All the designed molecules displayed excellent human intestinal absorption (HIA). The molecules’ bioavailability of 20% and 30% were within acceptable limits.
Table 3.
An absorption parameter of methyl 2-((6-chloro-1H-benzo[d]imidazol-2-yl)methylthio)-1,2,3,4-tetrahydro-6-methylpyrimidine-5-carboxylate derivatives (25–41).
The distribution and metabolism profile of the molecules are depicted in Table 4. The plasma protein binding (PPB, <90%) drugs with high protein bound within them may have a low therapeutic index. Many of the molecules displayed a PPB at less than 90%. The volume distribution (VD; optimal 0.04–20 L/kg) of all molecules was within the acceptable limit range. None of the molecules displayed a BBB penetration potential. Cytochrome (CYP) enzymes play an important role in drug metabolism; therefore, their substrate or inhibitor contributes to the drug’s action. None of the molecules in the current study demonstrated CYP inhibitory or substrate potential [27].
Table 4.
Distribution and metabolism profile of methyl 2-((6-chloro-1H-benzo[d]imidazol-2-yl)methylthio)-1,2,3,4-tetrahydro-6-methylpyrimidine-5-carboxylate derivatives (25–41).
An excretion and toxicity profile of the molecules is tabulated in Table 5. Many of the molecules displayed a moderate to low clearance (CL, High: >15 mL/min/kg; moderate: 5–15 mL/min/kg; low: <5 mL/min/kg) rate. All the molecules exhibited a short half-life (T1/2, <3 h). The toxicity profile of the molecules suggested favorable properties, and many of the values were within the range. The physicochemical radar of the developed molecules obtained from the ADMETlab 2.0 web server is reported in Figure 2, which indicates the molecules’ favorable physicochemical parameters to be developed further [27]. Most of the developed molecules displayed physicochemical properties within the upper limit of the acceptable range, as per the radar images. The physicochemical radar contains almost all the properties that are ideal for the development of any lead as a potential therapeutic agent. An environmental toxicity profile (bioconcentration factors, IGC50, LC50FM, and LC50DM) of the designed molecules is shown in Table 6. The environmental toxicity profile of the molecules was optimum and within the acceptable range.
Table 5.
Excretion and toxicity profile of methyl 2-((6-chloro-1H-benzo[d]imidazol-2-yl)methylthio)-1,2,3,4-tetrahydro-6-methylpyrimidine-5-carboxylate derivatives (25–41).
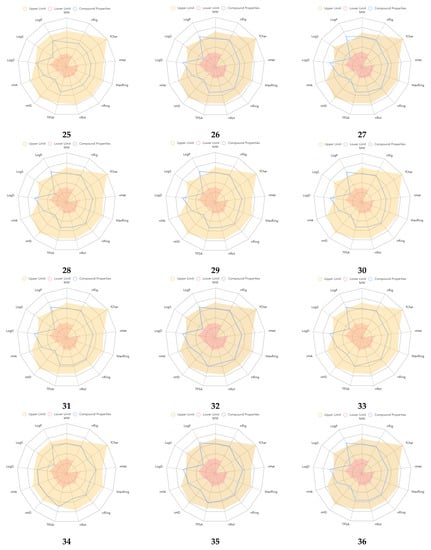
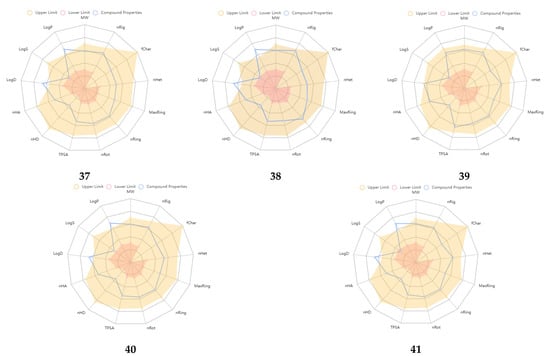
Figure 2.
Physicochemical radar of developed molecules obtained from ADMETlab 2.0 web server.
Table 6.
Environmental toxicity profile of methyl 2-((6-chloro-1H-benzo[d]imidazol-2-yl)methylthio)-1,2,3,4-tetrahydro-6-methylpyrimidine-5-carboxylate derivatives (25–41).
2.2. Molecular Docking
In the docking calculations, comparisons have been made between the binding affinities of the designed derivatives and the binding mode of the native ligand that is found in the crystal structure of DHFR (PDB ID: 5CCC). The molecular interactions of the titled compounds are exemplified in the Supplementary Information; in Table 7, the most potent compounds’ 2D- and 3D-docking poses are described. The native ligand displayed a binding affinity with DHFR of −8.5 kcal/mol, and it established six conventional hydrogen bonds with Asp27, Ala6, Ile5, and Arg57, in addition to one carbon–hydrogen bond with Ile94. It has established many hydrophobic interactions, such as Pi–sigma bonds, Pi–Pi T-shaped bonds, alkyl bonds, and Pi–alkyl bonds with Ile50, Phe31, Ile94, Ile5, and Ala7. Compound 27 exhibited a binding affinity value of -8.6 kcal/mol with the formation of five hydrophobic bonds (pi-sigma, alkyl, pi-alkyl) with Leu28, Lys32, Leu28, Ala7, and Phe31. Compound 29 exhibited a binding affinity value of −9.3 kcal/mol with the formation of one hydrogen bond and several hydrophobic bonds (pi–sigma, pi–pi T-shaped alkyl, pi–alkyl) with Leu28, Phe31, Ile50, Ile5, Ala7, Met20, Trp30, and Phe31. Compound 30 displayed a binding affinity value of −9.6 kcal/mol with the formation of one carbon–hydrogen bond and many hydrophobic bonds (pi–sigma, pi–pi T-shaped alkyl, pi–alkyl) with Leu28, Phe31, Ile50, Ile5, Ala7, Met20, Trp30, and Phe31.
Table 7.
The 2D- and 3D-docking postures of molecules selected for the synthesis.
Compound 33 displayed a binding affinity value of −9.0 kcal/mol with the formation of two conventional hydrogen bonds and one carbon–hydrogen bond with Met20, Ile94, and Asp27. It also displayed many hydrophobic bonds (pi–sigma, pi–pi T-shaped alkyl, pi–alkyl) with Leu28, Leu54 Phe31, Ile50, Ile5, Ala7, Met20, Trp30, and Phe31. Compound 37 displayed a binding affinity value of −8.7 kcal/mol with the formation of one conventional hydrogen bond and one carbon–hydrogen bond with Ala7 and Asp27. It also displayed many hydrophobic bonds (pi–pi T-shaped, alkyl, pi–alkyl) with Tyr100, Ile50, Met20, Leu28, and Ile14. Compound 38 displayed a binding affinity value of −9 kcal/mol with the formation of one conventional hydrogen bond with Ile94. It also displayed many hydrophobic bonds (pi–pi T-shaped, amide-Pi stacked, alkyl, pi–alkyl) with Leu28, Met20, Ile5, Phe31, Ala7, and Phe31. It also displayed electrostatic interactions with Glu17. Compound 41 displayed a binding affinity value of −9 kcal/mol with the formation of one conventional hydrogen bond and one carbon bond with Thr113 and Trp30. It also displayed many hydrophobic bonds (pi–sigma, alkyl, pi–alkyl) with Leu28, Lys32, Ile5, Phe31, Ala7, and Phe31. Therefore, from the above results, the compounds that showed a binding affinity value lower than the native compound in the X-ray complex (27, 29, 30, 33, 37, 38, and 41) were selected for the synthesis and biological evaluation.
2.3. Synthesis of the Selected Compounds
Compounds 27, 29, 30, 33, 37, 38, and 41 were chosen for the synthesis based on the results of the in silico screening and molecular docking investigations.
Target derivatives can be prepared by condensing the suitable building blocks 4–21 with benzimidazole 24. Initially, the 1,2,3,4-tetrahydropyrimidine-2-thiol derivatives 4–21 were synthesized using a modified Biginelli reaction, where a solution of ethyl acetoacetate 1, (1.3 g, 10 mmol), thiourea 3, (1.14 g, 15 mmol), ferric chloride (FeCl3.6H2O, 2.5 mmol), and conc. HCl (1–2 drops) in EtOH (20 mL) was heated with the appropriate aldehydes 2 (10 mmol) under reflux for 4–5 hrs. The yields obtained were in the range of 80–95%. Moreso, 6-Chloro-2-(chloromethyl)-1H-benzo[d]imidazole 24 was synthesized by a microwave-assisted synthesis by refluxing 4-chloro-1,2-phenylenediamine 22 and chloroacetyl chloride 23 in the presence of 4N HCl as the catalyst. The yield was 87%. The yellowish-brown product was recrystallized from dioxane; m.p. 142–144 °C. The detailed procedure of the synthesis and physical characterizations of compounds 4–21 and 24 are described in our previously published paper [28]. In the key step, 24 (1.66 g, 0.01 mol) and 4–21 (0.01 mol) were condensed by heating them with potassium hydroxide (KOH) in H2O: acetone (2:1) at about 50–60 °C for 45 min. The reaction mixture was cooled to room temperature and poured into ice-cold water; the precipitate was separated by filtration, and the products [methyl 2-((6-chloro-1H-benzo[d]imidazol-2-yl)methylthio)-1,2,3,4-tetrahydro-6-methylpyrimidine-5-carboxylate derivatives] 27, 29, 30, 33, 37, 38, and 41 were recrystallized from ethanol. The yield was 80–90%. The reaction scheme for the synthesis is depicted in Figure 3.
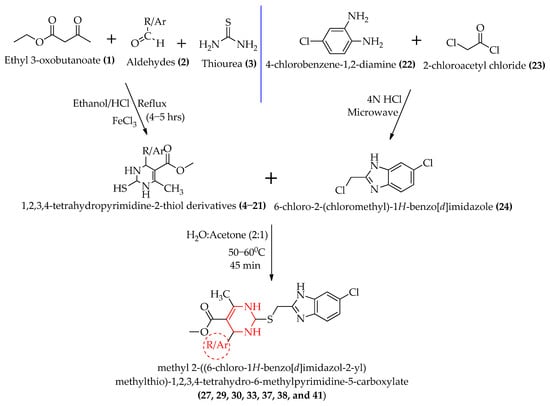
Figure 3.
The reaction scheme for the synthesis of methyl 2-((6-chloro-1H-benzo[d]imidazol-2-yl)methylthio)-1,2,3,4-tetrahydro-6-methylpyrimidine-5-carboxylate derivatives.
2.4. In Vitro Antibacterial Activity
The findings of the synthetic derivatives’ antibacterial and antifungal activities are listed in Table 8, which display the MICs and MFCs, respectively (n = 3).
Table 8.
The antibacterial and antifungal properties shown by the compounds that were synthesized (n = 3).
Xue-Qian Bai et al. reported some pyrimidine derivatives as potential antimicrobial agents, where one compound presented the most potent inhibitory activities against Staphylococcus aureus, Escherichia coli, and Candida albicans, with a MIC of 2.4 μmol/L. Additionally, it was the most potent, with MICs of 2.4 or 4.8 μmol/L against four multidrug-resistant, Gram-positive bacterial strains [29]. Omaima G. Shaaban et al. synthesized and evaluated some 3,4-dihydrothieno[2,3-d]pyrimidine derivatives as potential antimicrobial agents. Many of the derivatives displayed half of the potency of levofloxacin against Pseudomonas aeruginosa and Proteus vulgaris and also half the activity of ampicillin against the Gram-positive bacterium B. subtilis [30]. In the current study, each of the selected and synthetized compounds was tested for its in vitro antibacterial and antifungal activities using a variety of strains. All the compounds produced showed antibacterial activities against Gram-positive bacteria (Staphylococcus aureus and Staphylococcus pyogenes) as well as Gram-negative bacteria (Escherichia coli and Pseudomonas aeruginosa). All the compounds had actions against Gram-positive and Gram-negative bacteria that were much more powerful than that of ampicillin. Most of the compounds either had a higher potency than chloramphenicol or an equivalent potency to ciprofloxacin. Against Escherichia coli, 33 and 41 were sensitive at 25 µg/mL, whereas 29, 37, and 38 were sensitive at 50 µg/mL. It was observed that 27 and 30 were non-sensitive against Escherichia coli.
Infections brought on by the opportunistic bacteria Pseudomonas aeruginosa are often treated with the drug ciprofloxacin. In spite of the widespread administration of ciprofloxacin, the number of Pseudomonas aeruginosa strains that have developed resistance to the drug continue to rise [31]. Infections caused by Pseudomonas aeruginosa are notoriously difficult to treat because of the bacteria’s high levels of inherent and acquired antibiotic resistance. Once Pseudomonas aeruginosa has taken hold in a human host, it quickly creates genetic changes that make it resistant to antibiotics and better able to adapt to the host environment [32]. Therefore, it is not surprising that ciprofloxacin displayed low sensitivity (25 µg/mL) against Pseudomonas aeruginosa. However, all the compounds were sensitive at 50 µg/mL except 38, which was non-sensitive against Pseudomonas aeruginosa. Against Staphylococcus aureus, 27, 33, 37, 38, and 41 were sensitive at 25 µg/mL, whereas 29 and 30 were sensitive at 50 µg/mL. Against Staphylococcus pyogenes, 30 and 33 were sensitive at 25 µg/mL, whereas 29, 37, 38, and 41 were sensitive at 50 µg/mL. Compound 27 was non-sensitive against Staphylococcus pyogenes.
Candida, a yeast, is developing increased resistance to antifungal medications. Candida infections may be challenging to treat because of the possibility of drug resistance. Two patients with oral candidiasis, who did not improve while using nystatin in conjunction with triamcinolone acetonide, are presented. High in vitro resistance to nystatin was seen when triamcinolone acetonide was used in conjunction with the Candida albicans isolates collected from the patients after therapy [33]. This might be a reason why nystatin demonstrated low sensitivity (100 µg/mL) against Candida albicans, whereas all the compounds were sensitive at 100 µg/mL, which is equipotent to nystatin and more potent than griseofulvin except for 33, which was sensitive at 200 µg/mL. Against Aspergillus niger, all the compounds were equipotent with nystatin and griseofulvin (100 µg/mL) except for 30, which was sensitive at 200 µg/mL and 37 was non-sensitive. Against Aspergillus clavatus, all the compounds were sensitive at 100 µg/mL except for 38 and 41, which were non-sensitive. It was observed that 29 and 33 were sensitive against all the bacterial and fungal strains.
From the above results, it was observed that these molecules have enough potential as antimicrobial agents. These molecules displayed an optimum binding affinity for the DHFR enzyme and showed significant inhibition. Therefore, we proposed that these molecules exert antimicrobial activity via the inhibition of DHFR.
3. Materials and Methods
3.1. Pre-ADMET Profile and Drug-Likeness Properties
The in silico ADMET assessment models are a new type of tool that has been created to provide medicinal chemists with extra support in the process of the creation and optimization of leads. ADMETlab 2.0 is a revamped version of the AMDETlab web server, which is commonly used for predicting the pharmacokinetics and toxic characteristics of various compounds (https://admetmesh.scbdd.com/ accessed on 21 August 2022) [27].
3.2. Molecular Docking
Using Autodock vina in PyRx 0.8, the hypothesized derivatives, as well as the native ligand, were docked to the crystal structure of the wild-type E. coli dihydrofolate reductase [34]. The structures of the proposed derivatives and the native ligand were drawn using ChemDraw Ultra 12.0. (Mol File format). By using the open-Babel tool, the ligands were imported into the PyRx software. By using the Universal Force Field (UFF), each of the ligands was optimized in terms of reducing the amount of energy [35]. The ligands were then converted to the PDBQT format and prepared for docking purposes. The crystal structure of wild-type E. coli dihydrofolate reductase was obtained from the RCSB Protein Data Bank (PDB) with PDB ID: 5CCC (https://www.rcsb.org/structure/5CCC, accessed on 28 August 2022). The enzyme structure was refined using Discovery Studio Visualizer (version 19.1.0.18287), and then it was purified and prepared for docking using the same program [36]. The output file of the enzyme was saved in a PDB file format and imported to PyRx to perform the molecular docking studies. In order to aid molecular docking, a three-dimensional grid box (size_x = 41.7862652138Å; size_y = 39.1754565902Å; size_z = 37.1398050256Å) with an exhaustiveness value of eight was developed [34]. The strategy reported in previous papers was used in order to carry out the complete molecular docking method as well as to locate cavities and active amino acid residues [18,28,37,38,39,40,41,42,43,44,45,46]. The exposed cavity of the DHFR is shown with the co-crystallized ligand molecule in Figure 4.
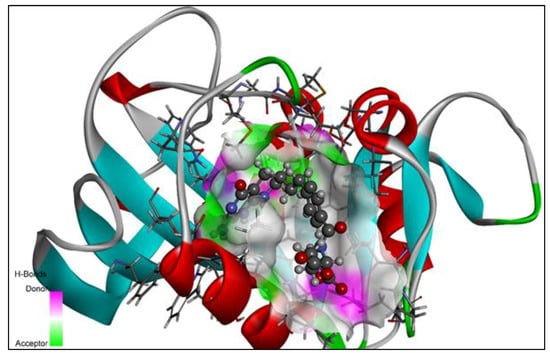
Figure 4.
The native ligand 5,10-dideazatetrahydrofolic acid is shown in the allosteric location of the DHFR in this 3D ribbon image.
3.3. Chemistry
From the Lab Trading Laboratory in Aurangabad, Maharashtra, India, all the essential chemicals and reagents of synthetic quality were obtained. The progression of the reaction was monitored and verified using thin-layer chromatography (TLC, Merck precoated silica GF 254). The melting points were determined using a VEEGO Model VMP-D melting point apparatus. The mass spectra were determined by SAPALA ORGANICS PVT LTD using an LC-MS spectrometer, and the results are reported in the Supplementary Information. 1H NMR and 13C NMR were calculated. CDCl3 was used as the solvent, and TMS was used as the internal standard. The chemical shift values were stated in δ ppm.
Methyl 2-((6-chloro-1H-benzo[d]imidazol-2-yl)methylthio)-4-(4-bromophenyl)-1,2,3,4-tetrahydro-6-methylpyrimidine-5-carboxylate(27)
Molecular formula: C21H20BrClN4O2S, molecular weight: 508.50 g/mol; m.p. (0C): 208–210; Rf Value: 0.85; % Yield: 78. Solubility: ethanol, methanol, DCM, chloroform. Elemental analysis (calc.): C, 49.67; H, 3.97; Br, 15.73; Cl, 6.98; N, 11.03; S, 6.31. 1H NMR δ ppm: 1.72 (s, pyrimidine 6-methyl protons, 3H), 2.01, 2.12 (two-s, pyrimidine ―NH, 2H), 3.72 (s, ―SCH2, 2H), 3.77 (s, acetatemethyl, 3H), 4.60 (s, 4H-pyrimidine, 1H), 4.81 (s, 2H-pyrimidine, 1H), 5.05 (s, imidazole-NH, 1H), 7.12, 7.85 (q, Ar-H, 4H, J = 1.5 and 7.5), 7.14, 7.53, 8.36 (t, Ar-H, 3H, J = 7.5). 13C NMR δ ppm: 14.1 (C10), 26.3 (C19), 52.3 (C30), 58.9 (C3), 81.4 (C1), 106 (C4), 115.8 (C28), 116.6 (C25), 121.4 (C14), 124.1 (C26), 129.2 (C27), 130.1 (C9 and C12), 131.4 (C13 and C15), 137 (C23), 138.5 (C11), 140.3 (C22), 141.5 (C20), 153.9 (C5), 167.2 (C7). MS: m/z 508.34, 509.32 (m + 1), 510.90 (m + 2).
Methyl 2-((6-chloro-1H-benzo[d]imidazol-2-yl) methylthio)-4-(4-chlorophenyl)-1, 2, 3, 4-tetrahydro-6-methylpyrimidine-5-carboxylate(29)
Molecular formula: C21H20Cl2N4O2S, molecular weight: 464.38 gm/mol; m.p. (°C): 213–216; Rf Value: 0.85; % Yield: 76. Solubility: ethanol, methanol, DCM, chloroform. Elemental analysis (calc.): C, 54.43; H, 4.35; Cl, 15.30; N, 12.09; S, 6.92. 1H NMR δ ppm: 1.91, 2.0 (two-s, pyrimidine ―NH, 2H), 3.26 (s, pyrimidine 6-methyl protons, 3H), 3.70 (s, ―SCH2, 2H), 3.77 (s, acetatemethyl, 3H), 4.58 (s, 4H-pyrimidine, 1H), 4.80 (s, 2H-pyrimidine, 1H), 5.0 (s, imidazole-NH, 1H), 7.34, 7.37 (q, Ar-H, 4H, J = 1.5 and 7.5), 7.14, 7.53, 8.36 (t, Ar-H, 3H, J = 7.5). 13C NMR δ ppm: 14.1 (C10), 26.3 (C19), 52.3 (C30), 58.9 (C3), 81.4 (C1), 106 (C4), 115.8 (C28), 116.6 (C25), 121.4 (C14), 124.1 (C26), 129.2 (C27), 130.1 (C9 and C12), 131.4 (C13 and C15), 137 (C23), 138.5 (C11), 140.3 (C22), 141.5 (C20), 153.9 (C5), 167.2 (C7). MS: m/z 464.16, 465.17 (m + 1), 466.17 (m + 2).
Methyl 2-((6-chloro-1H-benzo[d]imidazol-2-yl)methylthio)-1,2,3,4-tetrahydro-6-methyl- 4-p-tolylpyrimidine-5-carboxylate(30)
Molecular formula: C22H23ClN4O2S, molecular weight: 443.22 g/mol; m.p. (°C): 228–238; Rf Value: 0.59; % Yield: 50. Solubility: ethanol, methanol, DCM, chloroform. Elemental analysis (calc.): C, 59.65; H, 5.23; Cl, 8.00; N, 12.65; S, 7.24. 1H NMR δ ppm: 1.91, 2.0 (two-s, pyrimidine ―NH, 2H), 2.26 (s, pyrimidine 6-methyl protons, 3H), 2.34 (s, 4-methyl of phenyl, 3H), 3.70 (s, ―SCH2, 2H), 3.77 (s, acetatemethyl, 3H), 4.59 (s, 4H-pyrimidine, 1H), 4.80 (s, 2H-pyrimidine, 1H), 5.0 (s, imidazole-NH, 1H), 7.11 (q, Ar-H, 4H, J = 7.5), 7.14, 7.53, 8.36 (t, Ar-H, 3H, J = 7.5). 13C NMR δ ppm: 14.1 (C17), 21.3 (C16), 26.3 (C19), 52.3 (C30), 58.9 (C3), 81.4 (C1), 106 (C4), 115.8 (C28), 116.6 (C25), 124.1 (C26), 127.8 (C9 and C11), 128.8 (C14 and C15), 129.2 (C27), 136.5 (C10), 136.7 (C12), 137 (C23), 140.3 (C22), 141.5 (C20), 153.9 (C5), 167.2 (C7). MS: m/z 431.15, 432.15 (m + 1), 433.16 (m + 2).
Methyl 2-((6-chloro-1H-benzo[d]imidazol-2-yl)methylthio)-1,2,3,4-tetrahydro-4-(3-hy-droxyphenyl)-6-methylpyrimidine-5-carboxylate (33)
Molecular formula: C22H23ClN4O2S, molecular weight: 445.42 g/mol; m.p. (°C): 268–286; Rf Value: 0.68; % Yield: 50. Solubility: ethanol, methanol, DCM, chloroform. Elemental analysis (calc.): C, 57.69; H, 4.74; Cl, 7.93; N, 12.60; O, 10.79; S, 7.21. 1H NMR δ ppm: 1.91, 2.0 (two-s, pyrimidine ―NH, 2H), 2.26 (s, pyrimidine 6-methyl protons, 3H), 3.70 (s, ―SCH2, 2H), 3.77 (s, acetatemethyl, 3H), 4.59 (s, 4H-pyrimidine, 1H), 4.80 (s, 2H-pyrimidine, 1H), 5.0 (s, imidazole-NH, 1H), 5.35 (s, ―OH, 1H), 6.76, 6.79, 6.97, 7.16 (q, Ar-H, 4H, J = 1.5 and 7.5), 7.14, 7.53, 8.36 (t, Ar-H, 3H, J = 7.5). 13C NMR δ ppm: 14.1 (C10), 26.3 (C19), 52.3 (C30), 59.2 (C3), 81.4 (C1), 106 (C4), 113.6 (C12), 114.2 (C14), 115.8 (C28), 116.6 (C25), 120.5 (C16), 124.1 (C26), 129.2 (C27), 129.9 (C15), 137 (C23), 139.5 (C11), 140.3 (C22), 141.5 (C20), 153.9 (C5), 156.8 (C13), 167.2 (C7). MS: m/z 445.13, 446.14 (m + 1), 447.14 (m + 2).
Methyl 2-((6-chloro-1H-benzo[d]imidazol-2-yl)methylthio)-1,2,3,4-tetrahydro-6-methyl-4-styrylpyrimidine-5-carboxylate(37)
Molecular formula: C23H23ClN4O2S, molecular weight: 455.97 gm/mol; m.p. (°C): 280–295; Rf Value: 0.90; % Yield: 78. Solubility: ethanol, methanol, DCM, chloroform. Elemental analysis (calc.): C, 60.72; H, 5.10; Cl, 7.79; N, 12.31; S, 7.05. 1H NMR δ ppm: 1.91, 2.0 (two-s, pyrimidine ―NH, 2H), 2.26 (s, pyrimidine 6-methyl protons, 3H), 3.70 (s, ―SCH2, 2H), 3.77 (s, acetatemethyl, 3H), 3.99 (s, 4H-pyrimidine, 1H), 4.80 (s, 2H-pyrimidine, 1H), 5.0 (s, imidazole-NH, 1H), 6.19, 6.56 (d, ethelene protons, 2H, J = 15.1), 7.24, 7.40, 7.33 (t, Ar-H, 5H, J = 7.5), 7.14, 7.53, 8.36 (t, Ar-H, 3H, J = 7.5). 13C NMR δ ppm: 14.2 (C18), 26.3 (C20), 51.0 (C3), 52.3 (C31), 81.9 (C1), 106.0 (C4), 115.8 (C29), 116.6 (C26), 123.3 (C10), 124.1 (C27), 127.9 (C15), 128.5 (C13 and C17), 128.6 (C14 and C16), 129.2 (C28), 136.4 (C12), 137 (C24), 140.3 (C23), 141.5 (C21), 153.9 (C5), 167.2 (C7). MS: m/z 455.13, 456.14 (m + 1), 457.14 (m + 2).
Methyl 2-((6-chloro-1H-benzo[d]imidazol-2-yl)methylthio)-1,2,3,4-tetrahydro-6-methyl-4-(naphthalen-1-yl)pyrimidine-5-carboxylate (38)
Molecular formula: C25H23ClN4O2S, molecular weight: 479 g/mol; m.p. (°C): 280–295; Rf Value: 0.90; % Yield: 70. Solubility: ethanol, methanol, DCM, chloroform. Elemental analysis (calc.): C, 62.69; H, 4.84; Cl, 7.40; N, 11.70; S, 6.69. 1H NMR δ ppm: 1.91, 2.0 (two-s, pyrimidine ―NH, 2H), 2.26 (s, pyrimidine 6-methyl protons, 3H), 3.70 (s, ―SCH2, 2H), 3.77 (s, acetatemethyl, 3H), 4.59 (s, 4H-pyrimidine, 1H), 4.80 (s, 2H-pyrimidine, 1H), 5.0 (s, imidazole-NH, 1H), 7.00, 7.42, 7.52, 7.54, 7.92, 8.05, 8.18 (m, napthyl-H, 7H, J = 1.5 and 7.5), 7.14, 7.53, 8.36 (t, Ar-H, 3H, J = 7.5). 13C NMR δ ppm: 14.1 (C20), 26.3 (C22), 52.3 (C33), 57.2 (C3), 81.4 (C1), 106.0 (C4), 115.8 (C31), 116.6 (C28), 124.1 (C29), 124.2 (C11), 125.5 (C18), 125.6 (C16), 125.8 (C17), 126.5 (C13), 126.9 (C12), 132.6 (C19), 133.5 (C14), 134.0 (C10), 137.0 (C26), 140.3 (C25), 141.5 (C23), 153.9 (C5), 167.2 (C7). MS: m/z 479.13, 480.14 (m + 1), 481.14 (m + 2).
Methyl 2-((6-chloro-1H-benzo[d]imidazol-2-yl)methylthio)-4-(4-(trifluoromethyl) phenyl)-1,2,3,4-tetrahydro-6-methylpyrimidine-5-carboxylate(41)
Molecular formula: C22H20ClF3N4O2S, molecular weight: 497.90 m/mol; m.p. (°C): 259–278; Rf Value: 0.89; % Yield: 76. Solubility: ethanol, methanol, DCM, chloroform. Elemental analysis (calc.): C, 53.17; H, 4.06; Cl, 7.13; F, 11.457; N, 11.27; S, 6.45. 1H NMR δ ppm: 1.91, 2.0 (two-s, pyrimidine ―NH, 2H), 2.26 (s, pyrimidine 6-methyl protons, 3H), 3.70 (s, ―SCH2, 2H), 3.77 (s, acetatemethyl, 3H), 4.59 (s, 4H-pyrimidine, 1H), 4.80 (s, 2H-pyrimidine, 1H), 5.0 (s, imidazole-NH, 1H), 7.16, 7.50 (d, Ar-H, 4H, J = 7.5), 7.14, 7.53, 8.36 (t, Ar-H, 3H, J = 7.5). 13C NMR δ ppm: 14.1 (C10), 26.3 (C22), 52.3 (C33), 58.9 (C3), 81.4 (C1), 106.0 (C4), 115.8 (C31), 116.6 (C28), 124.1 (C29 and C17), 124.9 (C13 and C15), 128.2 (C9 and C12), 129.2 (C30), 129.3 (C14), 137.0 (C26), 140.3 (C25), 141.5 (C23), 153.9 (C5), 167.2 (C7). MS: m/z 497.13, 498.14 (m + 1), 499.14 (m + 2).
3.4. In Vitro Biological Evaluation
By using the broth dilution method, several different doses of derivatives were produced in the DMSO so that their antibacterial and antifungal properties could be evaluated against the reference strains (Gram-positive bacteria (Staphylococcus aureus and Staphylococcus pyogenes), Gram-negative bacteria (Escherichia coli and Pseudomonas aeruginosa), and fungal (Candida albicans, Aspergillus niger, and Aspergillus clavatus)). The bacteria were kept alive in a nutrient-rich Mueller–Hinton broth, and the drugs were diluted. The turbidity of the broth was used to identify the test strains that were used to inoculate the broth with 108 colony-forming units (cfu) per milliliter. For both the primary and secondary screenings, stock solutions of the synthesized derivate were diluted in a step-by-step process to a concentration of 2 mg/mL. The first screen comprised screening of the synthesized derivatives at concentrations of 1000, 500, and 250 μg/mL; further screenings of the active derivatives were performed at concentrations of 200, 100, 50, 25, 12.5, and 6.250 μg/mL. A control that did not contain antibiotics was subcultured (before being inoculated) by distributing one loopful of media uniformly over a fourth of a plate of medium that was adequate for growing the test organisms. This was followed by overnight incubation at 37 °C. The lowest concentrations of derivatives that were able to prevent the development of bacteria or fungi were used as the minimal inhibitory concentrations (MICs). In order to establish the correctness of the MIC, it was compared with the quantity of control growth that occurred before the incubation process (the original inoculum). The antibiotics gentamycin, ampicillin, chloramphenicol, ciprofloxacin, and norfloxacin served as the standards for determining the antibacterial activity, while nystatin and griseofulvin served as the criteria for determining the antifungal activity [28,40,46].
4. Conclusions
We designed and developed some methyl 2-((6-chloro-1H-benzo[d]imidazol-2-yl)methylthio)-1,2,3,4-tetrahydro-6-methylpyrimidine-5-carboxylate derivatives, 25–41, as potential DHFR inhibitors. The ADMET profiles of all the created compounds were positive, and some of them even showed reduced binding affinities (in terms of DHFR) compared to the native ligand. In the present investigation, several bacterial and fungal strains (Gram-positive bacteria (Staphylococcus aureus and Staphylococcus pyogenes), Gram-negative bacteria (Escherichia coli and Pseudomonas aeruginosa), and fungal (Candida albicans, Aspergillus niger, and Aspergillus clavatus)) were used to evaluate the in vitro antibacterial and antifungal properties of the synthesized compounds. It was observed that 29 and 33 were sensitive against all the bacterial and fungal strains. The results showed that the most promising compounds were those derived from 1,2,3,4-tetrahydropyrimidine-2-thiol with a 6-chloro-2-(chloromethyl)-1H-benzo[d]imidazole moiety, possibly because this moiety plays a vital role in boosting the compounds’ antibacterial characteristics. These compounds inhibited the DHFR enzyme with substantial binding affinity. Thus, we reasoned, these compounds may be antimicrobial because they inhibit DHFR. We thus conclude that these compounds have the potential to serve as lead compounds for the creation of further effective antibacterial and antifungal compounds.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28020501/s1, Table S1. The molecular interactions of designed derivatives with DHFR, Figure S1. Mass Spectrum of Synthesized Compound 27, Figure S2. Mass Spectrum of Synthesized Compound 29, Figure S3. Mass Spectrum of Synthesized Compound 30, Figure S4. Mass Spectrum of Synthesized Compound 33, Figure S5. Mass Spectrum of Synthesized Compound 37, Figure S6. Mass Spectrum of Synthesized Compound 38, Figure S7. Mass Spectrum of Synthesized Compound 41.
Author Contributions
Conceptualization, M.A.H. and S.L.K.; methodology, M.A.H. and A.M.; software, M.A.H., R.R. and S.J.A.; validation, M.A.H., V.B.T., F.I. and F.A.S.; formal analysis, M.S.C., F.I. and S.L.K.; investigation, M.A.H. and M.S.C.; resources, M.A.H.; data curation, M.A.H., G.C., A.M.A. and M.T.; writing—original draft preparation, M.A.H., A.M., R.R., S.J.A., V.B.T., A.M.A., M.T. and M.S.C.; writing—review and editing, M.A.H., A.M., R.R., S.J.A., V.B.T., M.S.C., F.I., F.A.S., A.M.A., M.T. and S.L.K.; visualization, F.I. and S.L.K.; supervision, S.L.K.; project administration, S.L.K.; funding acquisition, A.M.A. and M.T. All authors have read and agreed to the published version of the manuscript.
Funding
University of Palermo: FFR-D15-161363.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Loi, V.V.; Huyen, N.T.T.; Busche, T.; Tung, Q.N.; Gruhlke, M.C.H.; Kalinowski, J.; Bernhardt, J.; Slusarenko, A.J.; Antelmann, H. Staphylococcus Aureus Responds to Allicin by Global S-Thioallylation—Role of the Brx/BSH/YpdA Pathway and the Disulfide Reductase MerA to Overcome Allicin Stress. Free Radic. Biol. Med. 2019, 139, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Jouhar, L.; Jaafar, R.F.; Nasreddine, R.; Itani, O.; Haddad, F.; Rizk, N.; Hoballah, J.J. Microbiological Profile and Antimicrobial Resistance among Diabetic Foot Infections in Lebanon. Int. Wound J. 2020, 17, 1764–1773. [Google Scholar] [CrossRef]
- Anwar, K.; Hussein, D.; Salih, J. Antimicrobial Susceptibility Testing and Phenotypic Detection of MRSA Isolated from Diabetic Foot Infection. Int. J. Gen. Med. 2020, 13, 1349–1357. [Google Scholar] [CrossRef]
- Marchese, A.; Barbieri, R.; Sanches-Silva, A.; Daglia, M.; Nabavi, S.F.; Jafari, N.J.; Izadi, M.; Ajami, M.; Nabavi, S.M. Antifungal and Antibacterial Activities of Allicin: A Review. Trends Food Sci. Technol. 2016, 52, 49–56. [Google Scholar] [CrossRef]
- Hasan, M.F.; Das, R.; Khan, A.; Rahman, M. The Determination of Antibacterial and Antifungal Activities of Polygonum Hydropiper (L.) Root Extract. Adv. Biol. Res. 2009, 3, 53–56. [Google Scholar]
- Indora, N.; Kaushik, D. Design, Development and Evaluation of Ethosomal Gel of Fluconazole for Topical Fungal Infection. Int. J. Eng. Sci. Invent. Res. Dev. 2015, I, 280–306. [Google Scholar]
- Murali, T.S.; Kavitha, S.; Spoorthi, J.; Bhat, D.V.; Prasad, A.S.B.; Upton, Z.; Ramachandra, L.; Acharya, R.V.; Satyamoorthy, K. Characteristics of Microbial Drug Resistance and Its Correlates in Chronic Diabetic Foot Ulcer Infections. J. Med. Microbiol. 2014, 63, 1377–1385. [Google Scholar] [CrossRef]
- Sánchez-Sánchez, M.; Cruz-Pulido, W.L.; Bladinieres-Cámara, E.; Alcalá-Durán, R.; Rivera-Sánchez, G.; Bocanegra-García, V. Bacterial Prevalence and Antibiotic Resistance in Clinical Isolates of Diabetic Foot Ulcers in the Northeast of Tamaulipas, Mexico. Int. J. Low. Extrem. Wounds 2017, 16, 129–134. [Google Scholar] [CrossRef]
- Rengarajan, J.; Sassetti, C.M.; Naroditskaya, V.; Sloutsky, A.; Bloom, B.R.; Rubin, E.J. The Folate Pathway Is a Target for Resistance to the Drug Para-Aminosalicylic Acid (PAS) in Mycobacteria. Mol. Microbiol. 2004, 53, 275–282. [Google Scholar] [CrossRef]
- Srinivasan, B.; Tonddast-Navaei, S.; Roy, A.; Zhou, H.; Skolnick, J. Chemical Space of Escherichia Coli Dihydrofolate Reductase Inhibitors: New Approaches for Discovering Novel Drugs for Old Bugs. Med. Res. Rev. 2019, 39, 684–705. [Google Scholar] [CrossRef]
- He, J.; Qiao, W.; An, Q.; Yang, T.; Luo, Y. Dihydrofolate Reductase Inhibitors for Use as Antimicrobial Agents. Eur. J. Med. Chem. 2020, 195, 112268. [Google Scholar] [CrossRef] [PubMed]
- Wróbel, A.; Arciszewska, K.; Maliszewski, D.; Drozdowska, D. Trimethoprim and Other Nonclassical Antifolates an Excellent Template for Searching Modifications of Dihydrofolate Reductase Enzyme Inhibitors. J. Antibiot. 2020, 73, 5–27. [Google Scholar] [CrossRef] [PubMed]
- Songsungthong, W.; Prasopporn, S.; Bohan, L.; Srimanote, P.; Leartsakulpanich, U.; Yongkiettrakul, S. A Novel Bicyclic 2,4-Diaminopyrimidine Inhibitor of Streptococcus Suis Dihydrofolate Reductase. PeerJ 2021, 9, e10743. [Google Scholar] [CrossRef] [PubMed]
- Banjanac, M.; Tatic, I.; Ivezic, Z.; Tomic, S.; Dumic, J. Pyrimido-Pyrimidines: A Novel Class of Dihydrofolate Reductase Inhibitors. Food Technol. Biotechnol. 2009, 47, 236–245. [Google Scholar]
- Gebauer, M.G.; McKinlay, C.; Gready, J.E. Synthesis of Quaternised 2-Aminopyrimido[4,5-d]Pyrimidin-4(3H)-Ones and Their Biological Activity with Dihydrofolate Reductase. Eur. J. Med. Chem. 2003, 38, 719–728. [Google Scholar] [CrossRef]
- Ahmed Elkanzi, N.A. Synthesis and Biological Activities of Some Pyrimidine Derivatives: A Review. Orient. J. Chem. 2020, 36, 1001–1015. [Google Scholar] [CrossRef]
- Bhat, A.R.; Dongre, R.S.; Naikoo, G.A.; Hassan, I.U.; Ara, T. Proficient Synthesis of Bioactive Annulated Pyrimidine Derivatives: A Review. J. Taibah Univ. Sci. 2017, 11, 1047–1069. [Google Scholar] [CrossRef]
- Khan, A.; Unnisa, A.; Sohel, M.; Date, M.; Panpaliya, N.; Saboo, S.G.; Siddiqui, F.; Khan, S. Investigation of Phytoconstituents of Enicostemma Littorale as Potential Glucokinase Activators through Molecular Docking for the Treatment of Type 2 Diabetes Mellitus. Silico Pharmacol. 2022, 10, 1. [Google Scholar] [CrossRef]
- Waring, M.J. Lipophilicity in Drug Discovery. Expert Opin. Drug Discov. 2010, 5, 235–248. [Google Scholar] [CrossRef]
- Lobo, S. Is There Enough Focus on Lipophilicity in Drug Discovery? Expert Opin. Drug Discov. 2020, 15, 261–263. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the Chemical Beauty of Drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef]
- Kosugi, T.; Ohue, M. Quantitative Estimate Index for Early-Stage Screening of Compounds Targeting Protein-Protein Interactions. Int. J. Mol. Sci. 2021, 22, 10925. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Roggo, S.; Schuffenhauer, A. Natural Product-Likeness Score and Its Applications in the Drug Discovery Process. Chem. Cent. J. 2008, 2, S2. [Google Scholar] [CrossRef]
- Menke, J.; Massa, J.; Koch, O. Natural Product Scores and Fingerprints Extracted from Artificial Neural Networks. Comput. Struct. Biotechnol. J. 2021, 19, 4593–4602. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.B.; Zgair, A.; Taha, D.A.; Zang, X.; Kagan, L.; Kim, T.H.; Kim, M.G.; Yun, H.y.; Fischer, P.M.; Gershkovich, P. Quantitative Analysis of Lab-to-Lab Variability in Caco-2 Permeability Assays. Eur. J. Pharm. Biopharm. 2017, 114, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; West, M.; Patel, N.C.; Wager, T.; Hou, X.; Johnson, J.; Tremaine, L.; Liras, J. Validation of Human MDR1-MDCK and BCRP-MDCK Cell Lines to Improve the Prediction of Brain Penetration. J. Pharm. Sci. 2019, 108, 2476–2483. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Kale, M.; Siddiqui, F.; Nema, N. Novel Pyrimidine-Benzimidazole Hybrids with Antibacterial and Antifungal Properties and Potential Inhibition of SARS-CoV-2 Main Protease and Spike Glycoprotein. Digit. Chin. Med. 2021, 4, 102–119. [Google Scholar] [CrossRef]
- Bai, X.Q.; Li, C.S.; Cui, M.Y.; Song, Z.W.; Zhou, X.Y.; Zhang, C.; Zhao, Y.; Zhang, T.Y.; Jiang, T.Y. Synthesis and Molecular Docking Studies of Novel Pyrimidine Derivatives as Potential Antibacterial Agents. Mol. Divers. 2020, 24, 1165–1176. [Google Scholar] [CrossRef] [PubMed]
- Shaaban, O.G.; Issa, D.A.E.; El-Tombary, A.A.; Abd El Wahab, S.M.; Abdel Wahab, A.E.; Abdelwahab, I.A. Synthesis and Molecular Docking Study of Some 3,4-Dihydrothieno[2,3-d]Pyrimidine Derivatives as Potential Antimicrobial Agents. Bioorg. Chem. 2019, 88, 102934. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.; Patrick, W.M.; Lamont, I.L. Mechanisms of Ciprofloxacin Resistance in Pseudomonas Aeruginosa: New Approaches to an Old Problem. J. Med. Microbiol. 2019, 68, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Liu, H.; Pan, X.; Ma, Z.; Wang, D.; Zhang, X.; Zhu, G.; Bai, F.; Cheng, Z.; Wu, W.; et al. Mechanisms for Development of Ciprofloxacin Resistance in a Clinical Isolate of Pseudomonas Aeruginosa. Front. Microbiol. 2021, 11, 598291. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.V.; Dinsdale, R.C.W. Nystatin—Resistance of Candida Albicans Isolates from Two Cases of Oral Candidiasis. Br. J. Oral Surg. 1982, 20, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Discovery Studio Modeling Environment, Release 3.5; Accelrys Software Inc.: San Diego, CA, USA, 2012.
- Almerico, A.M.; Tutone, M.; Lauria, A. Docking and Multivariate Methods to Explore HIV-1 Drug-Resistance: A Comparative Analysis. J. Comput. Aided. Mol. Des. 2008, 22, 287–297. [Google Scholar] [CrossRef]
- Khan, S.L.; Siddiui, F.A. Beta-Sitosterol: As Immunostimulant, Antioxidant and Inhibitor of SARS-CoV-2 Spike Glycoprotein. Arch. Pharmacol. Ther. 2020, 2, 12–16. [Google Scholar] [CrossRef]
- Khan, S.L.; Sonwane, G.M.; Siddiqui, F.A.; Jain, S.P.; Kale, M.A.; Borkar, V.S. Discovery of Naturally Occurring Flavonoids as Human Cytochrome P450 (CYP3A4) Inhibitors with the Aid of Computational Chemistry. Indo Glob. J. Pharm. Sci. 2020, 10, 58–69. [Google Scholar] [CrossRef]
- Shntaif, A.H.; Khan, S.; Tapadiya, G.; Chettupalli, A.; Saboo, S.; Shaikh, M.S.; Siddiqui, F.; Amara, R.R. Rational Drug Design, Synthesis, and Biological Evaluation of Novel N-(2-Arylaminophenyl)-2,3-Diphenylquinoxaline-6-Sulfonamides as Potential Antimalarial, Antifungal, and Antibacterial Agents. Digit. Chin. Med. 2021, 4, 290–304. [Google Scholar] [CrossRef]
- Chaudhari, R.N.; Khan, S.L.; Chaudhary, R.S.; Jain, S.P.; Siddiqui, F.A. Β-Sitosterol: Isolation from Muntingia Calabura Linn Bark Extract, Structural Elucidation and Molecular Docking Studies As Potential Inhibitor of SARS-CoV-2 Mpro (COVID-19). Asian J. Pharm. Clin. Res. 2020, 13, 204–209. [Google Scholar] [CrossRef]
- Unnisa, A.; Khan, S.L.; Sheikh, F.A.H.; Mahefooz, S.; Kazi, A.A.; Siddiqui, F.A.; Gawai, N.; Saboo, S.G. In-Silico Inhibitory Potential of Triphala Constituents Against Cytochrome P450 2E1 for the Prevention of Thioacetamide-Induced Hepatotoxicity. J. Pharm. Res. Int. 2021, 33, 367–375. [Google Scholar] [CrossRef]
- Khan, S.L.; Siddiqui, F.A.; Shaikh, M.S.; Nema, N.V.; Shaikh, A.A. Discovery of Potential Inhibitors of the Receptor-Binding Domain (RBD) of Pandemic Disease-Causing SARS-CoV-2 Spike Glycoprotein from Triphala through Molecular Docking. Curr. Chin. Chem. 2021, 2, 74–84. [Google Scholar] [CrossRef]
- Khan, S.L.; Siddiqui, F.A.; Jain, S.P.; Sonwane, G.M. Discovery of Potential Inhibitors of SARS-CoV-2 (COVID-19) Main Protease (Mpro) from Nigella Sativa (Black Seed) by Molecular Docking Study. Coronaviruses 2020, 2, 384–402. [Google Scholar] [CrossRef]
- Tutone, M.; Virzì, A.; Almerico, A.M. Reverse screening on indicaxanthin from Opuntia ficus-indica as natural chemoactive and chemopreventive agent, J. Theor. Biol. 2018, 455, 147–160. [Google Scholar] [CrossRef]
- Siddiqui, F.A.; Khan, S.L.; Marathe, R.P.; Nema, N.V. Design, Synthesis, and In Silico Studies of Novel N-(2-Aminophenyl)-2,3- Diphenylquinoxaline-6-Sulfonamide Derivatives Targeting Receptor- Binding Domain (RBD) of SARS-CoV-2 Spike Glycoprotein and Their Evaluation as Antimicrobial and Antimalarial Agents. Lett. Drug Des. Discov. 2021, 18, 915–931. [Google Scholar] [CrossRef]
















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).