
Discovery of a SHP2 Degrader with In Vivo Anti-Tumor Activity

 , , , , ,
, , , , ,
Abstract
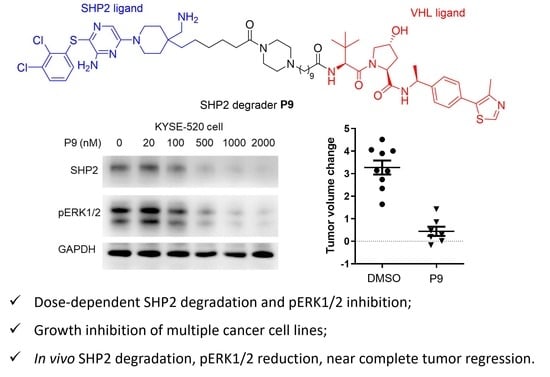
:
1. Introduction
2. Results and Discussion
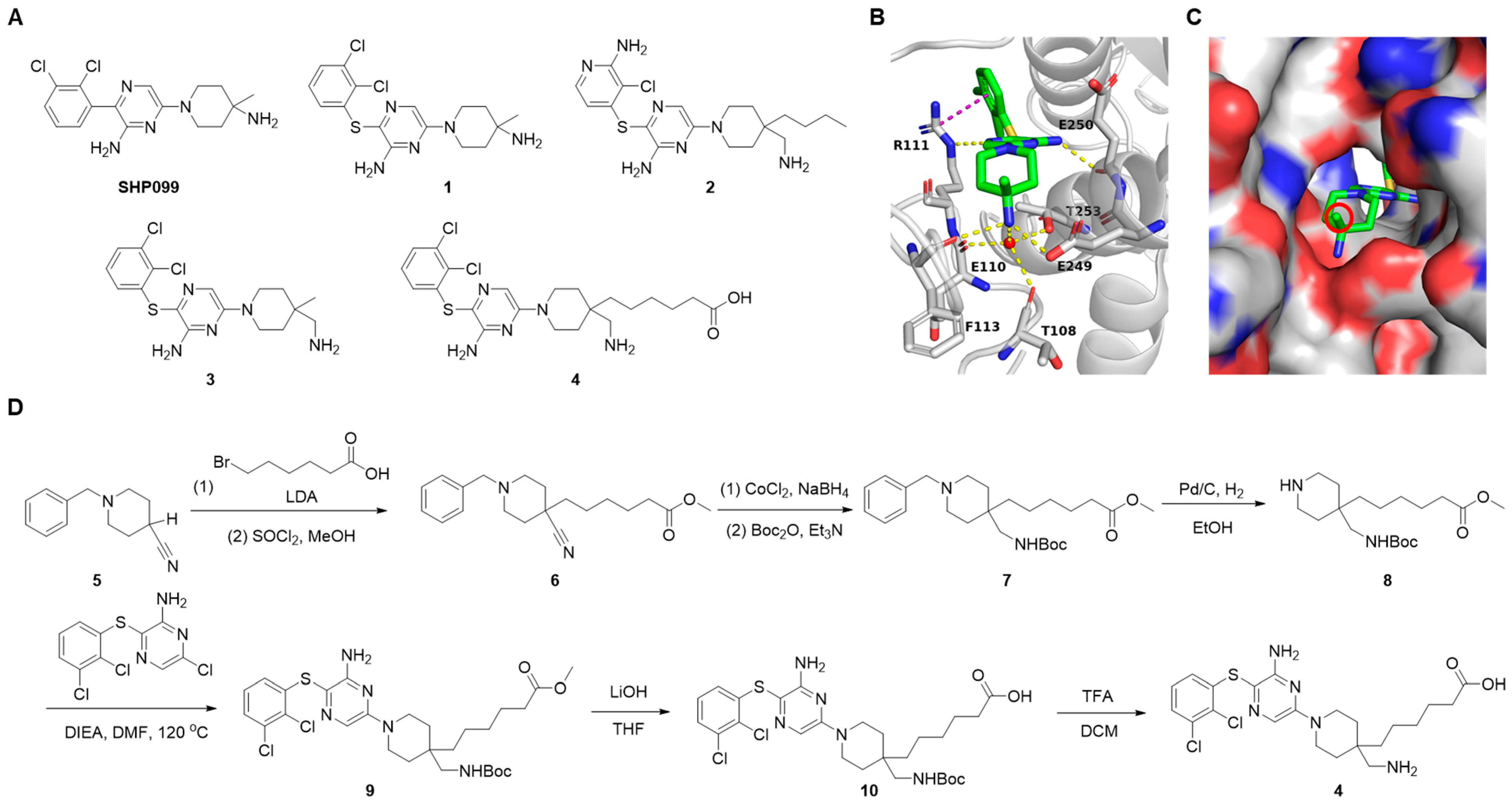
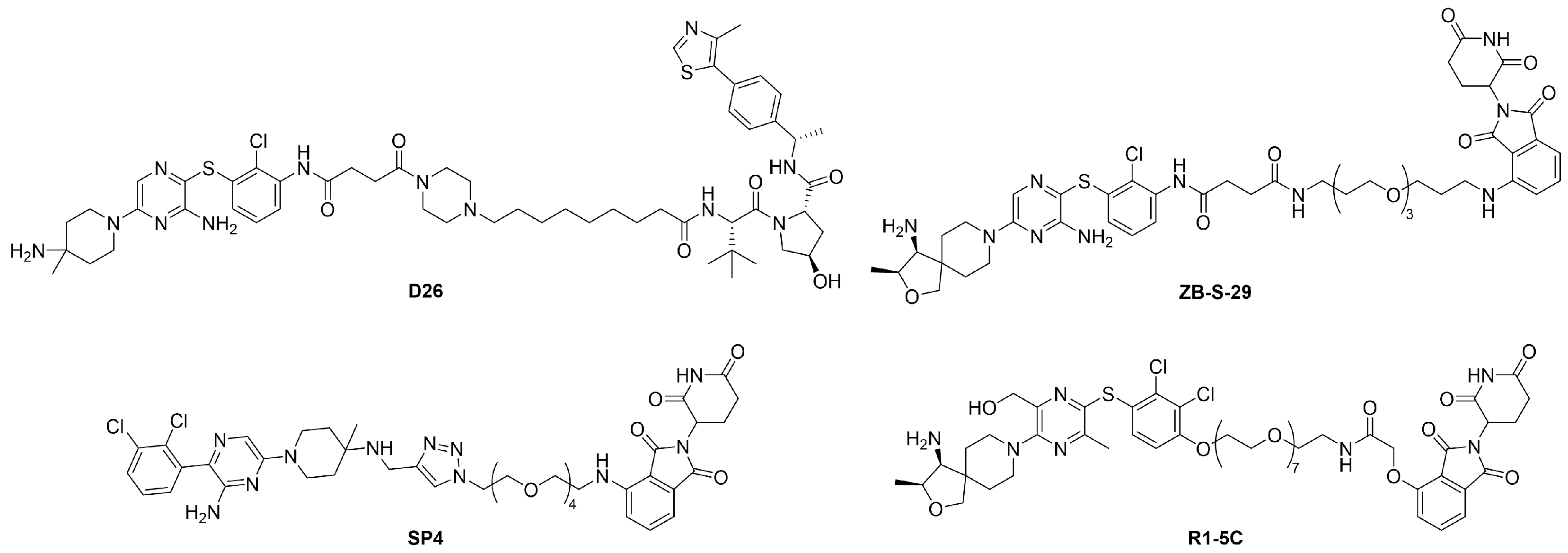
2.1. Design of SHP2 PROTACs
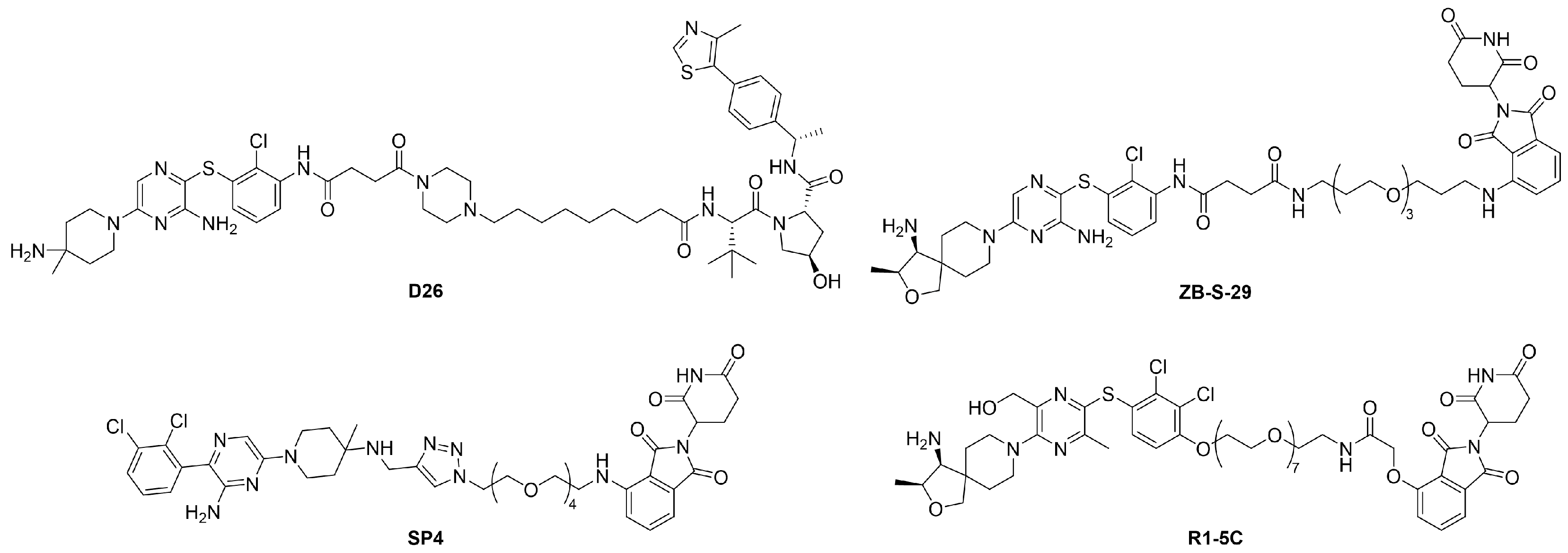
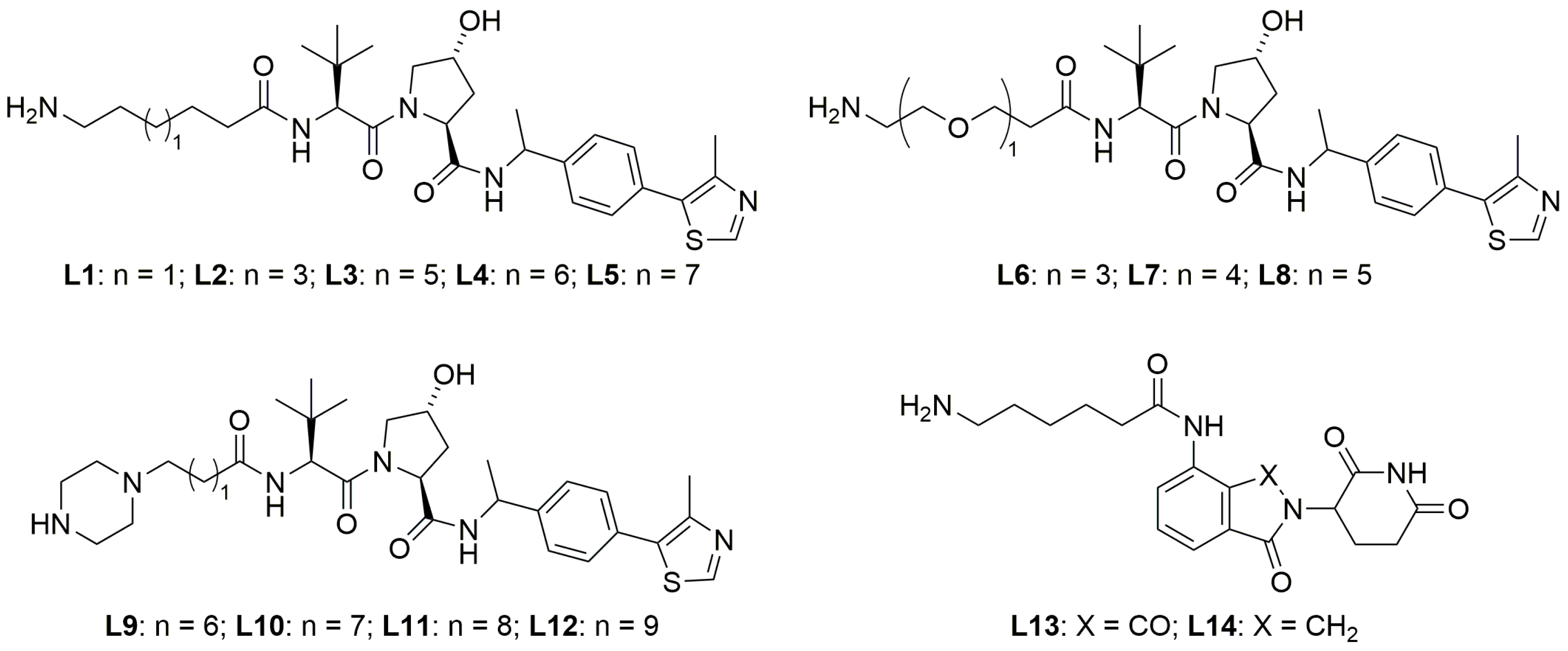
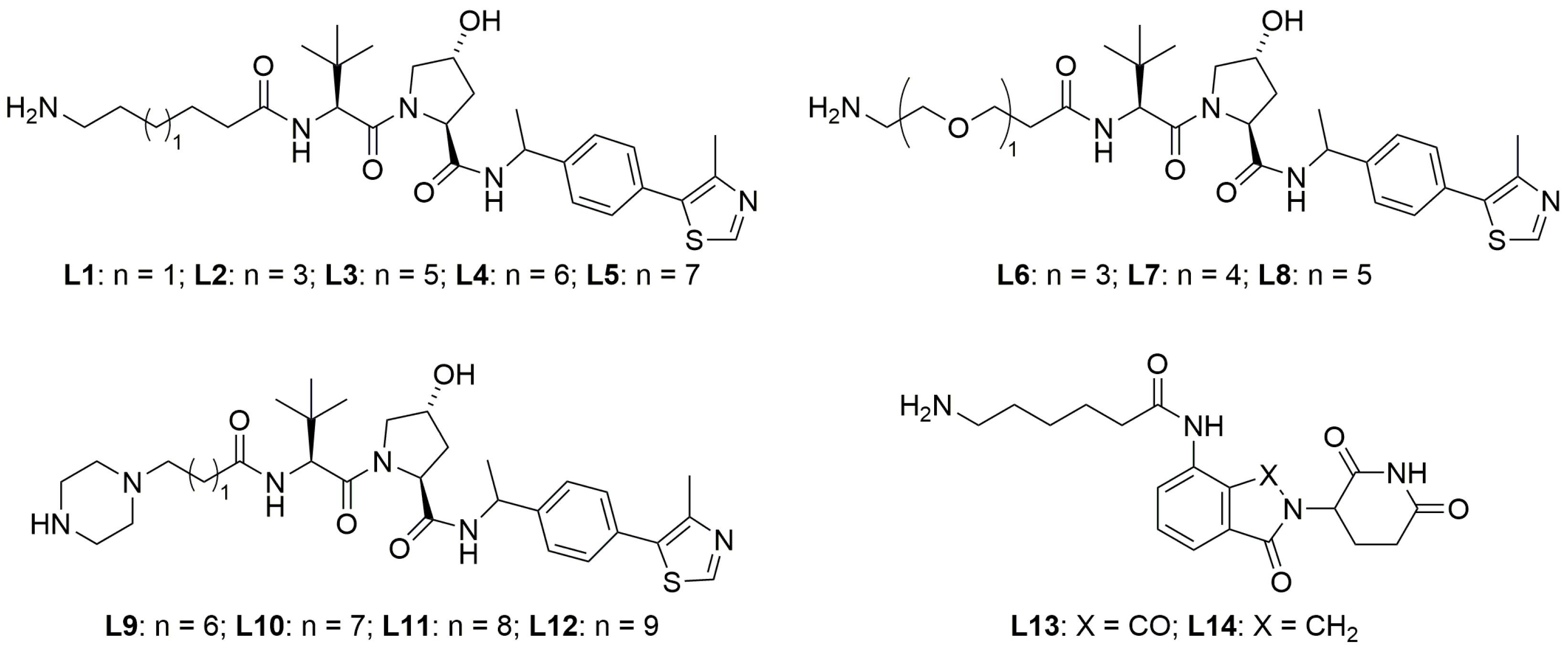
2.2. Exploration of E3 Ligands and Linkers
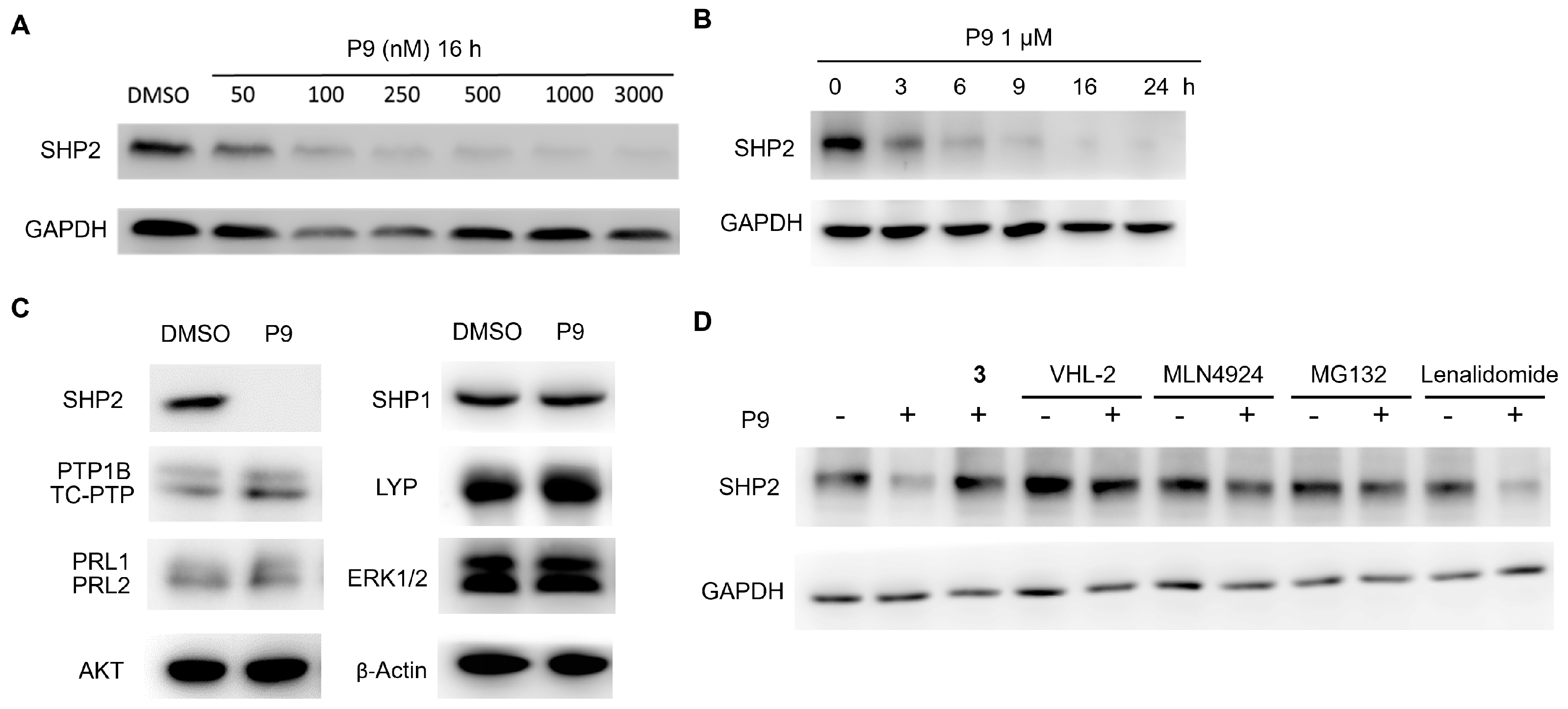
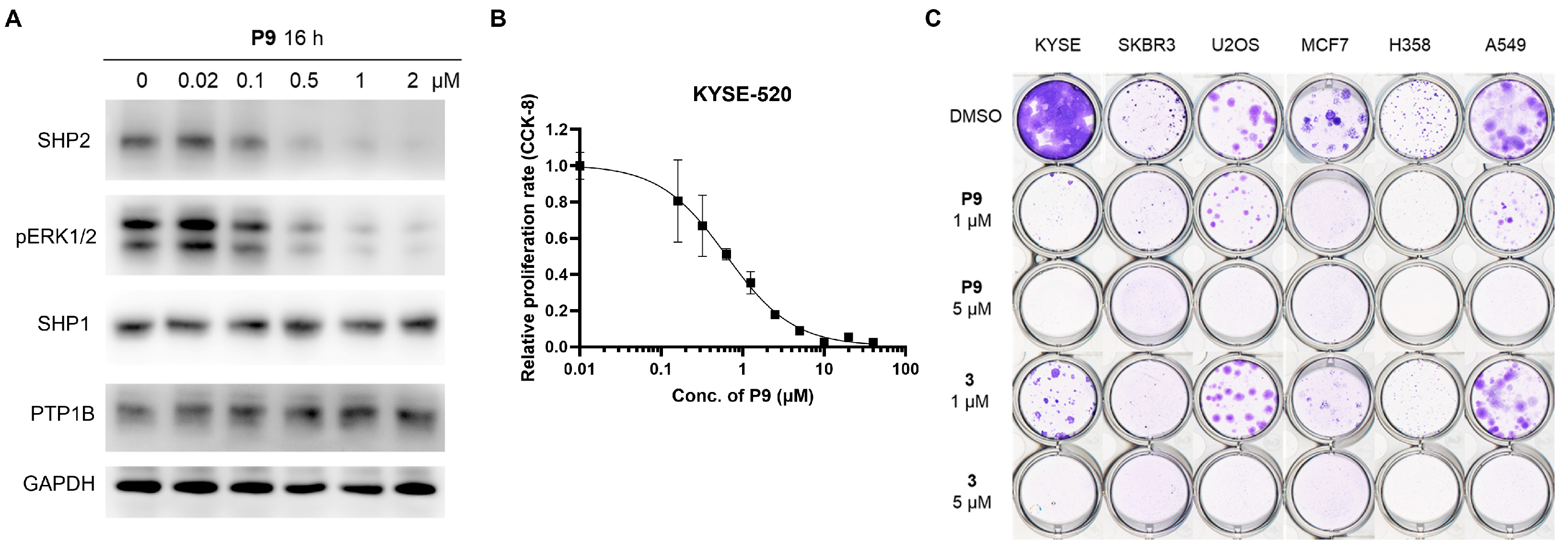
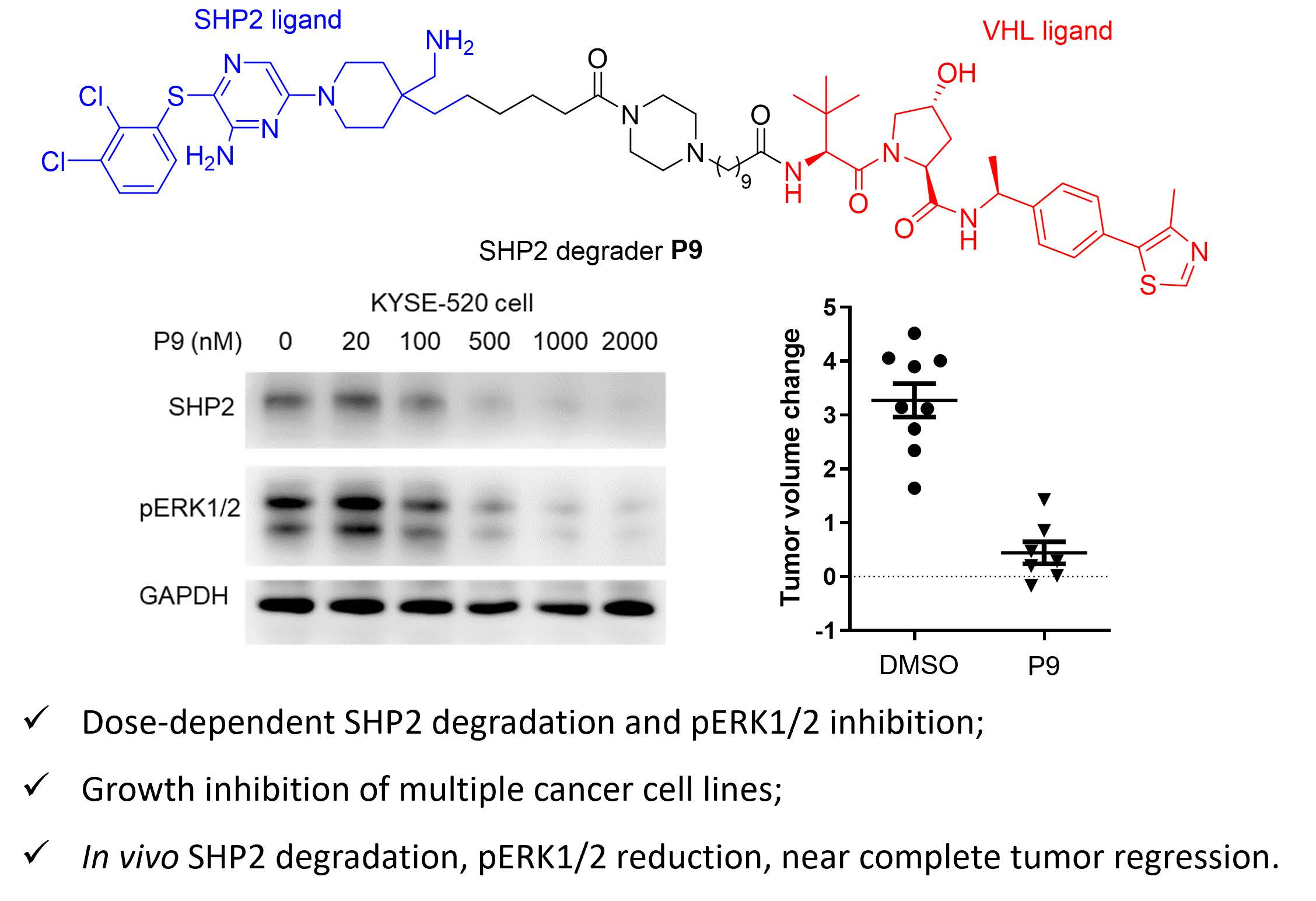
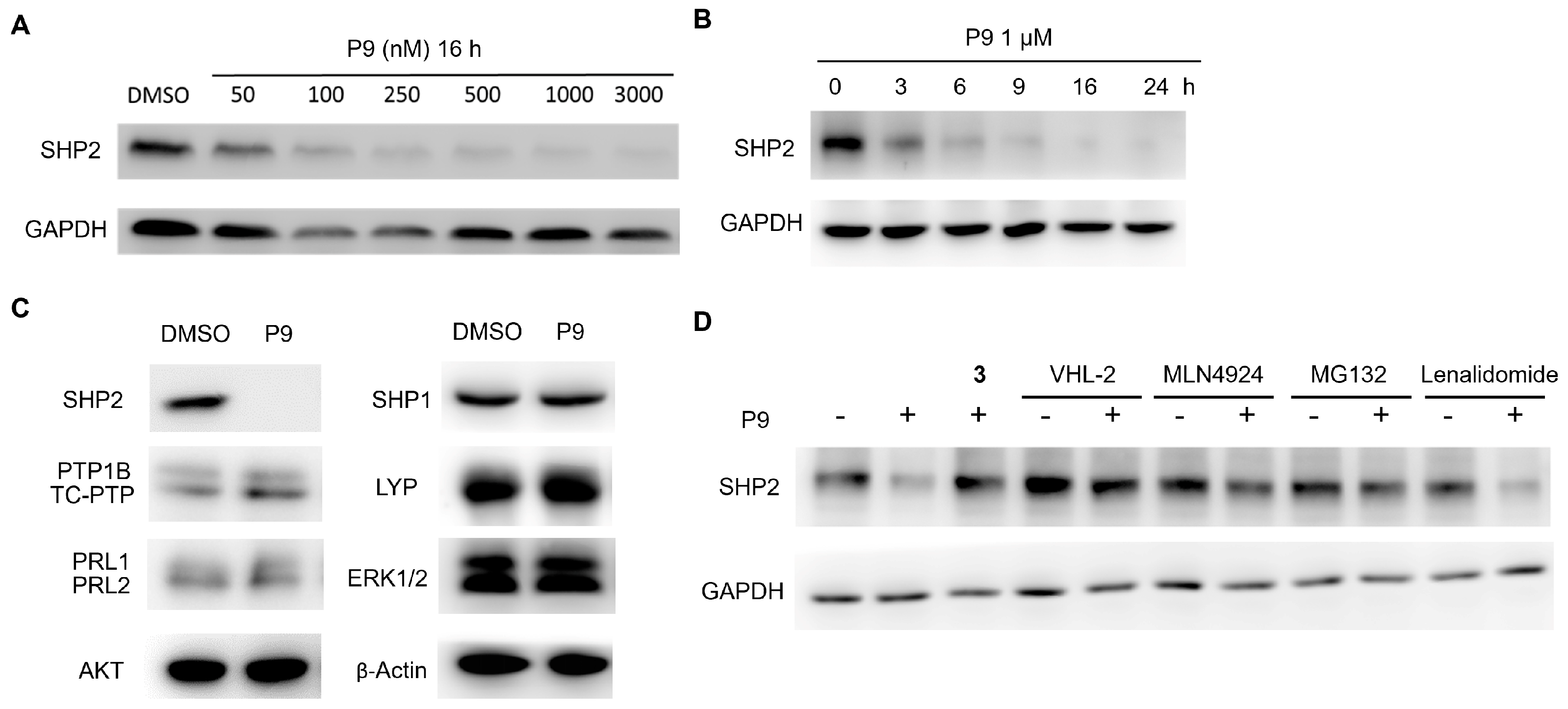
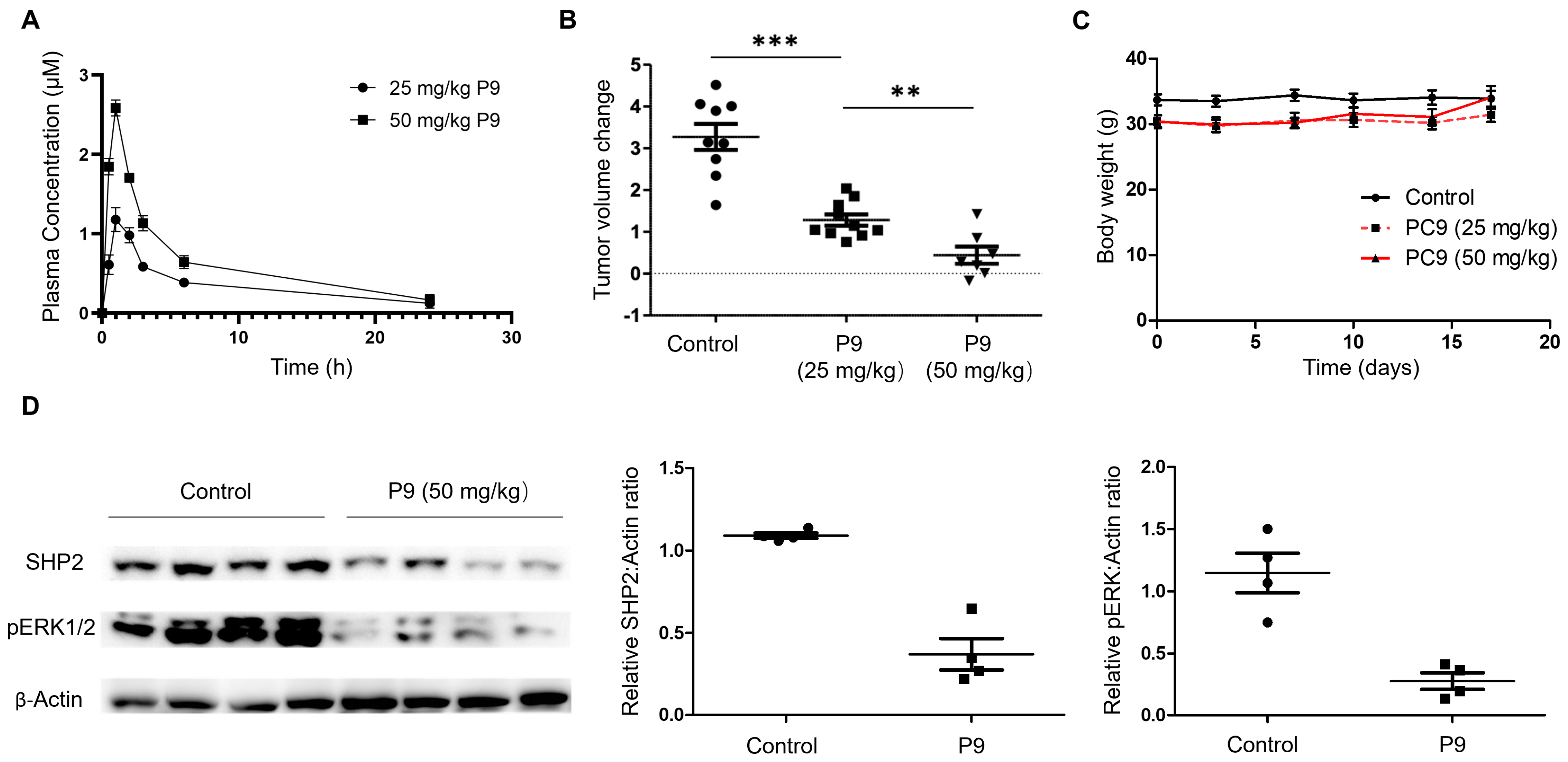
2.3. Dose- and Time-Dependency Study of P9
2.4. Selectivity Evaluation and Mechanism of Action Study of P9
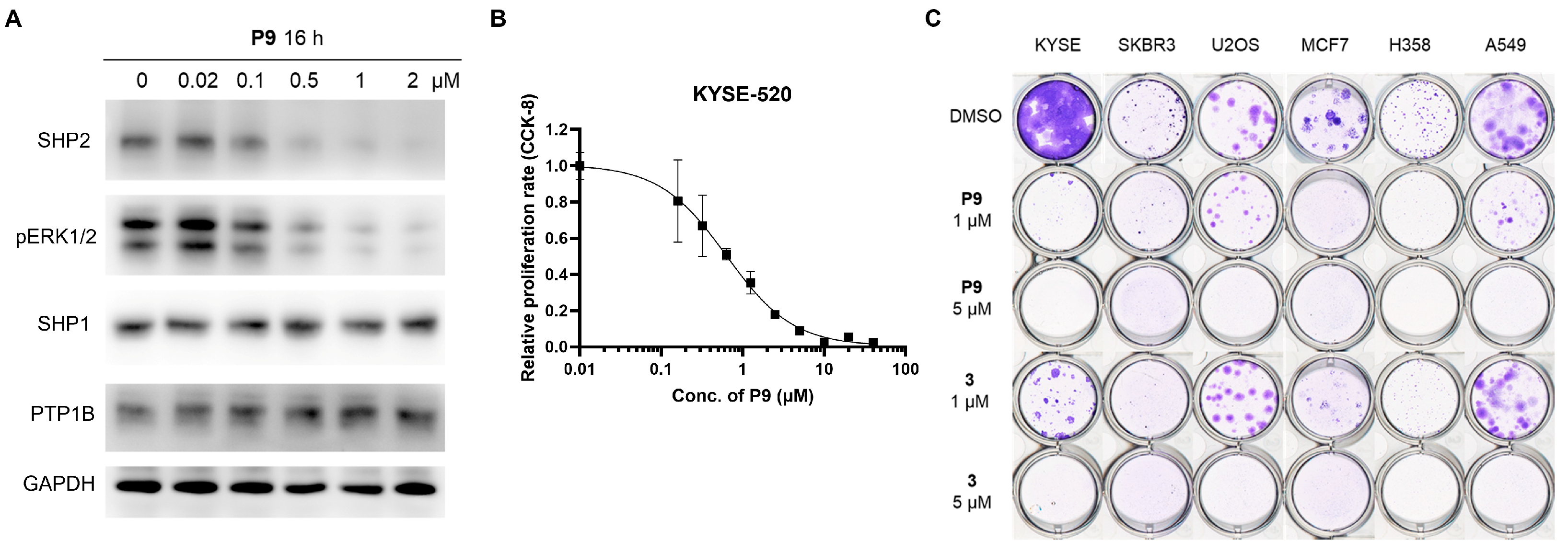
2.5. Tumor Cell Growth Inhibition Study
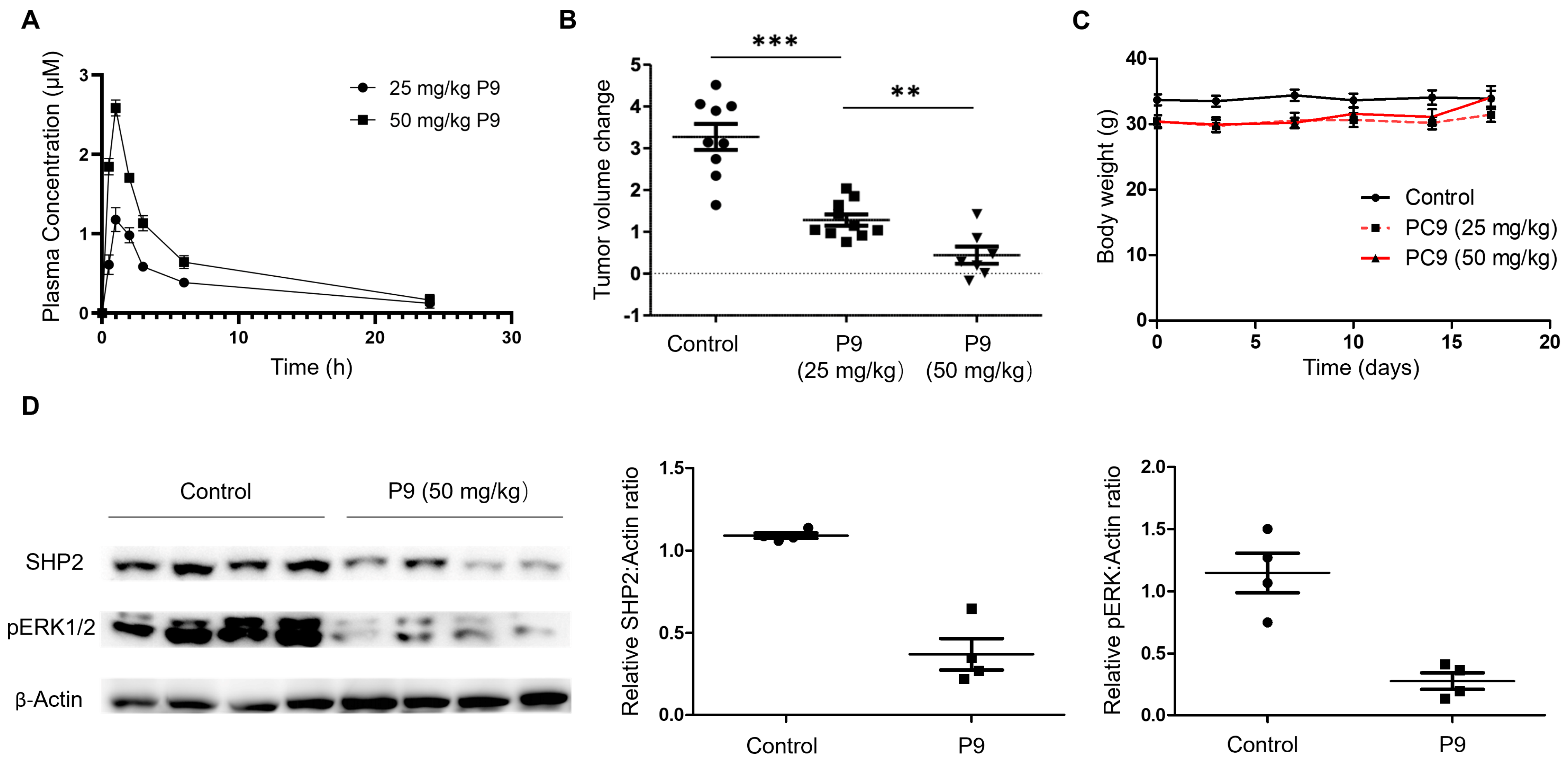
2.6. Evaluation of P9 in a Mouse Xenograft Model
3. Materials and Methods
3.1. General Information
3.2. Synthetic Procedures
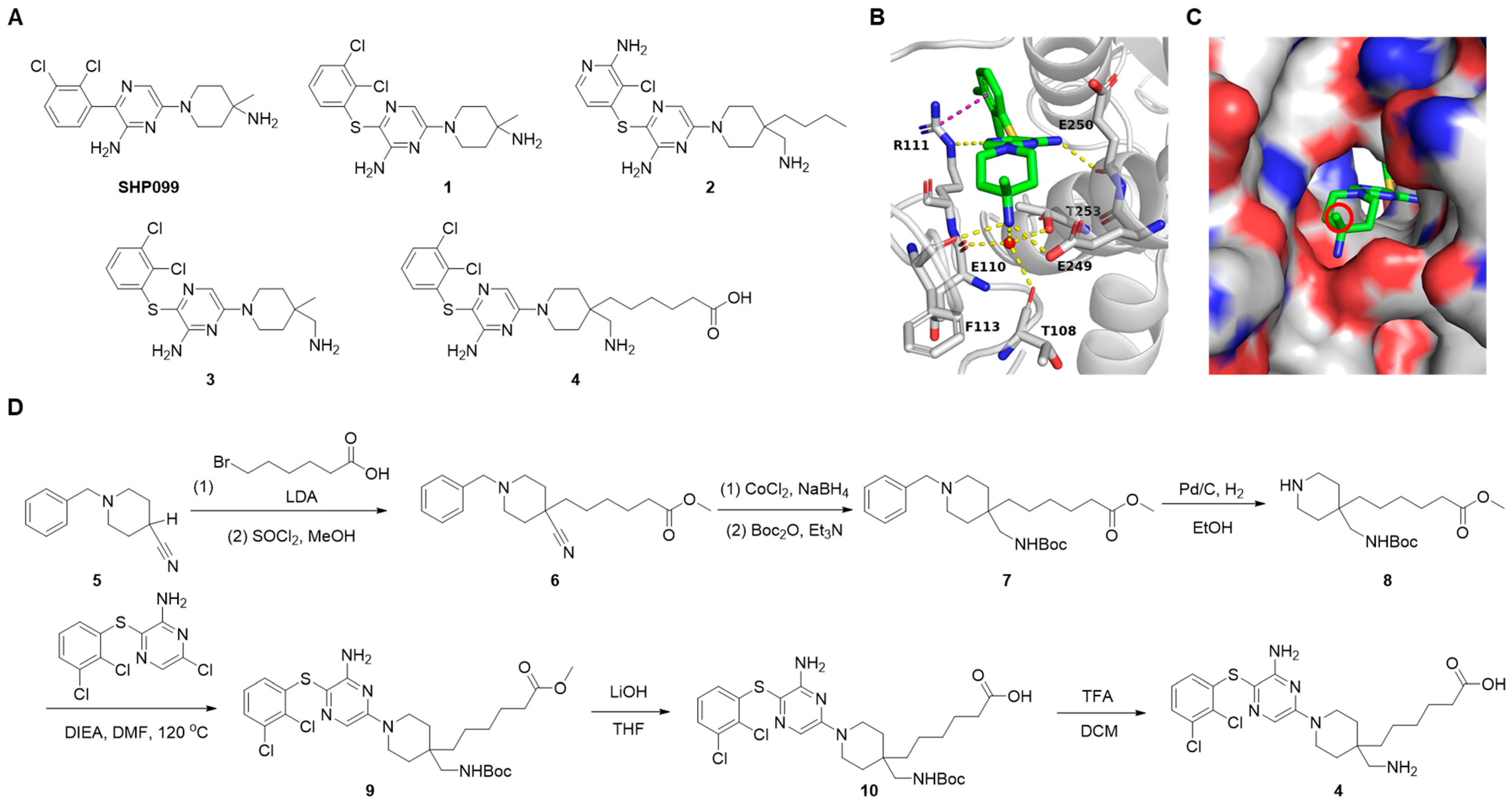
3.2.1. Synthesis of SHP2 Ligand Compound 4
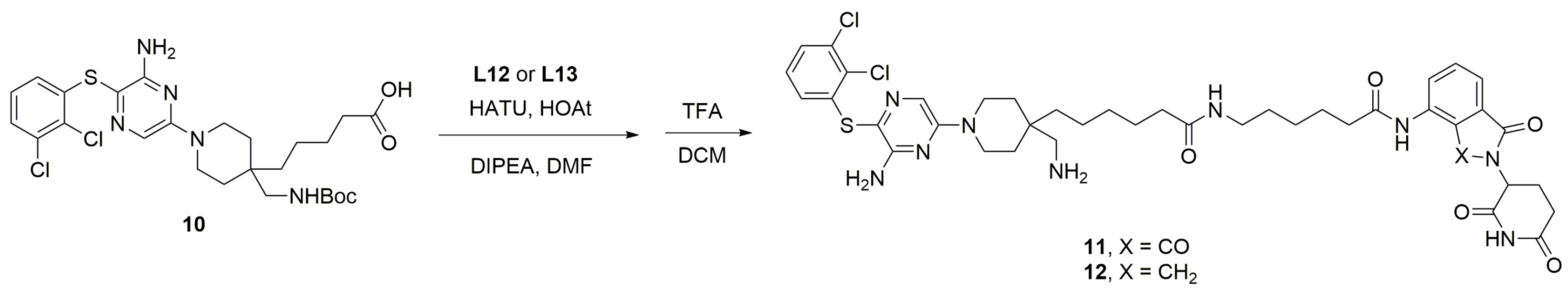
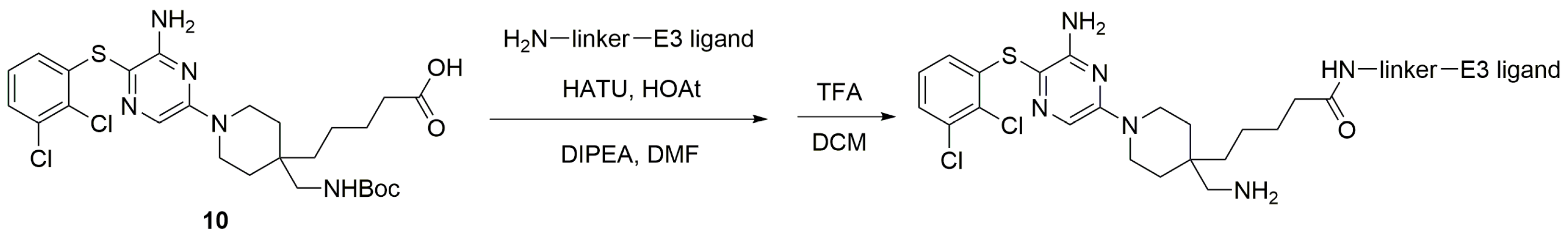
3.2.2. Preparation of SHP2 PROTAC Linkers with E3 Ligands
3.2.3. Synthesis of CRBN-Based SHP2 PROTACs
3.2.4. Synthesis of VHL-Based SHP2 PROTACs
3.3. Cloning, Expression, and Purification of SHP2 Protein
3.4. SHP2 Allosteric Inhibition Assay and Determination of IC50 Values
3.5. PTP Inhibition Assay and Determination of IC50 Values
3.6. Cell Culture, Western-Blot Analysis, and Colony Formation Assay
3.7. Cell Proliferation Assay
3.8. Mouse Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qu, C.K. The SHP-2 Tyrosine Phosphatase: Signaling Mechanisms and Biological Functions. Cell Res. 2000, 10, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Neel, B.G.; Gu, H.; Pao, L. The ‘Shp’ing News: SH2 Domain-Containing Tyrosine Phosphatases in Cell Signaling. Trends Biochem. Sci. 2003, 28, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. Protein Tyrosine Phosphatases: From Genes, to Function, to Disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef]
- Xu, D.; Qu, C.-K. Protein Tyrosine Phosphatases in the JAK/STAT Pathway. Front. Biosci. 2008, 13, 4925. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, M.; Mehler, E.L.; Goldberg, R.; Zampino, G.; Brunner, H.G.; Kremer, H.; Van Der Burgt, I.; Crosby, A.H.; Ion, A.; Jeffery, S.; et al. Mutations in PTPN11, Encoding the Protein Tyrosine Phosphatase SHP-2, Cause Noonan Syndrome. Nat. Genet. 2001, 29, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Kontaridis, M.I.; Swanson, K.D.; David, F.S.; Barford, D.; Neel, B.G. PTPN11 (Shp2) Mutations in LEOPARD Syndrome Have Dominant Negative, Not Activating, Effects. J. Biol. Chem. 2006, 281, 6785–6792. [Google Scholar] [CrossRef]
- Tartaglia, M.; Gelb, B.D. Germ-Line and Somatic PTPN11 Mutations in Human Disease. Eur. J. Med. Genet. 2005, 48, 81–96. [Google Scholar] [CrossRef]
- Tartaglia, M.; Niemeyer, C.M.; Fragale, A.; Song, X.; Buechner, J.; Jung, A.; Hählen, K.; Hasle, H.; Licht, J.D.; Gelb, B.D. Somatic Mutations in PTPN11 in Juvenile Myelomonocytic Leukemia, Myelodysplastic Syndromes and Acute Myeloid Leukemia. Nat. Genet. 2003, 34, 148–150. [Google Scholar] [CrossRef]
- Östman, A.; Hellberg, C.; Böhmer, F.D. Protein-Tyrosine Phosphatases and Cancer. Nat. Rev. Cancer 2006, 6, 307–320. [Google Scholar] [CrossRef]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 Associate with Immunoreceptor Tyrosine-Based Switch Motif of Programmed Death 1 upon Primary Human T Cell Stimulation, but Only Receptor Ligation Prevents T Cell Activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef]
- Li, J.; Jie, H.-B.; Lei, Y.; Gildener-Leapman, N.; Trivedi, S.; Green, T.; Kane, L.P.; Ferris, R.L. PD-1/SHP-2 Inhibits Tc1/Th1 Phenotypic Responses and the Activation of T Cells in the Tumor Microenvironment. Cancer Res. 2015, 75, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-Y. Drugging the Undruggable: Therapeutic Potential of Targeting Protein Tyrosine Phosphatases. Acc. Chem. Res. 2017, 50, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, S.; Overduin, M.; Barr, A.J. Targeting Protein Tyrosine Phosphatase SHP2 for Therapeutic Intervention. Future Med. Chem. 2014, 6, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, S.; Zhao, M.; Yang, X.; Yu, B. Strategies Targeting Protein Tyrosine Phosphatase SHP2 for Cancer Therapy. J. Med. Chem. 2022, 65, 3066–3079. [Google Scholar] [CrossRef]
- Garcia Fortanet, J.; Chen, C.H.-T.; Chen, Y.-N.P.; Chen, Z.; Deng, Z.; Firestone, B.; Fekkes, P.; Fodor, M.; Fortin, P.D.; Fridrich, C.; et al. Allosteric Inhibition of SHP2: Identification of a Potent, Selective, and Orally Efficacious Phosphatase Inhibitor. J. Med. Chem. 2016, 59, 7773–7782. [Google Scholar] [CrossRef]
- Chen, Y.-N.P.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.-T.; Chen, Z.; Cooke, V.G.; et al. Allosteric Inhibition of SHP2 Phosphatase Inhibits Cancers Driven by Receptor Tyrosine Kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef]
- Yuan, X.; Bu, H.; Zhou, J.; Yang, C.-Y.; Zhang, H. Recent Advances of SHP2 Inhibitors in Cancer Therapy: Current Development and Clinical Application. J. Med. Chem. 2020, 63, 11368–11396. [Google Scholar] [CrossRef]
- Lu, H.; Liu, C.; Huynh, H.; Le, T.B.U.; LaMarche, M.J.; Mohseni, M.; Engelman, J.A.; Hammerman, P.S.; Caponigro, G.; Hao, H.-X. Resistance to Allosteric SHP2 Inhibition in FGFR-Driven Cancers through Rapid Feedback Activation of FGFR. Oncotarget 2020, 11, 265–281. [Google Scholar] [CrossRef]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great Opportunities for Academia and Industry. Signal Transduct. Target. Ther. 2019, 4, 64. [Google Scholar] [CrossRef]
- Ruess, D.A.; Heynen, G.J.; Ciecielski, K.J.; Ai, J.; Berninger, A.; Kabacaoglu, D.; Görgülü, K.; Dantes, Z.; Wörmann, S.M.; Diakopoulos, K.N.; et al. Mutant KRAS-Driven Cancers Depend on PTPN11/SHP2 Phosphatase. Nat. Med. 2018, 24, 954–960. [Google Scholar] [CrossRef]
- Guo, W.; Xu, Q. Phosphatase-Independent Functions of SHP2 and Its Regulation by Small Molecule Compounds. J. Pharmacol. Sci. 2020, 144, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Zheng, M.; Liu, Y.; Chen, L.; Li, H. Proteolysis-Targeting Chimera Molecules Targeting SHP2. Future Med. Chem. 2022, 14, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Lu, J.; Wang, M.; Yang, C.-Y.; Wang, S. Discovery of SHP2-D26 as a First, Potent, and Effective PROTAC Degrader of SHP2 Protein. J. Med. Chem. 2020, 63, 7510–7528. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, Z.; Pei, Y.; Song, N.; Xu, L.; Feng, B.; Wang, H.; Luo, X.; Hu, X.; Qiu, X.; et al. Discovery of Thalidomide-Based PROTAC Small Molecules as the Highly Efficient SHP2 Degraders. Eur. J. Med. Chem. 2021, 218, 113341. [Google Scholar] [CrossRef]
- Zheng, M.; Liu, Y.; Wu, C.; Yang, K.; Wang, Q.; Zhou, Y.; Chen, L.; Li, H. Novel PROTACs for Degradation of SHP2 Protein. Bioorganic Chem. 2021, 110, 104788. [Google Scholar] [CrossRef]
- Vemulapalli, V.; Donovan, K.A.; Seegar, T.C.M.; Rogers, J.M.; Bae, M.; Lumpkin, R.J.; Cao, R.; Henke, M.T.; Ray, S.S.; Fischer, E.S.; et al. Targeted Degradation of the Oncogenic Phosphatase SHP2. Biochemistry 2021, 60, 2593–2609. [Google Scholar] [CrossRef]
- Deng, Y.; Ma, G.; Vallega, K.A.; Wang, D.; Wang, M.; Wang, C.; Wang, S.; Ramalingam, S.S.; Sun, S.-Y. Therapeutic Efficacy of the Novel SHP2 Degrader SHP2-D26, Alone or in Combination, against Lung Cancer Is Associated with Modulation of P70S6K/S6, Bim and Mcl-1. Cancer Gene Ther. 2022, 29, 1558–1569. [Google Scholar] [CrossRef]
- LaMarche, M.J.; Acker, M.; Argintaru, A.; Bauer, D.; Boisclair, J.; Chan, H.; Chen, C.H.-T.; Chen, Y.-N.; Chen, Z.; Deng, Z.; et al. Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 13578–13594. [Google Scholar] [CrossRef]
- Pedicone, C.; Fernandes, S.; Dungan, O.M.; Dormann, S.M.; Viernes, D.R.; Adhikari, A.A.; Choi, L.B.; De Jong, E.P.; Chisholm, J.D.; Kerr, W.G. Pan-SHIP1/2 Inhibitors Promote Microglia Effector Functions Essential for CNS Homeostasis. J. Cell Sci. 2019, 133, jcs.238030. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y.; Wu, Y.; Xing, D. Developments of CRBN-Based PROTACs as Potential Therapeutic Agents. Eur. J. Med. Chem. 2021, 225, 113749. [Google Scholar] [CrossRef]
- Bai, L.; Zhang, X.; Ma, N. Water-Promoted Ring-Opening Reactions of N -Substituted Saccharins and Phthalimides by Amines. Chin. J. Chem. 2014, 32, 871–877. [Google Scholar] [CrossRef]
- Diehl, C.J.; Ciulli, A. Discovery of Small Molecule Ligands for the von Hippel-Lindau (VHL) E3 Ligase and Their Use as Inhibitors and PROTAC Degraders. Chem. Soc. Rev. 2022, 51, 8216–8257. [Google Scholar] [CrossRef] [PubMed]
- Bemis, T.A.; La Clair, J.J.; Burkart, M.D. Unraveling the Role of Linker Design in Proteolysis Targeting Chimeras: Miniperspective. J. Med. Chem. 2021, 64, 8042–8052. [Google Scholar] [CrossRef]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.K.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-Induced BET Protein Degradation as a Therapy for Castration-Resistant Prostate Cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef] [PubMed]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS–ERK Signalling in Cancer: Promises and Challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef]
- Ahmed, T.A.; Adamopoulos, C.; Karoulia, Z.; Wu, X.; Sachidanandam, R.; Aaronson, S.A.; Poulikakos, P.I. SHP2 Drives Adaptive Resistance to ERK Signaling Inhibition in Molecularly Defined Subsets of ERK-Dependent Tumors. Cell Rep. 2019, 26, 65–78.e5. [Google Scholar] [CrossRef] [PubMed]
- LaRochelle, J.R.; Fodor, M.; Vemulapalli, V.; Mohseni, M.; Wang, P.; Stams, T.; LaMarche, M.J.; Chopra, R.; Acker, M.G.; Blacklow, S.C. Structural Reorganization of SHP2 by Oncogenic Mutations and Implications for Oncoprotein Resistance to Allosteric Inhibition. Nat. Commun. 2018, 9, 4508. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Gao, C.; Zhao, L.; Yuan, Z.; Chen, Y.; Jiang, Y. Phthalimide Conjugations for the Degradation of Oncogenic PI3K. Eur. J. Med. Chem. 2018, 151, 237–247. [Google Scholar] [CrossRef]
- Kaur, T.; Menon, A.; Garner, A.L. Synthesis of 7-Benzylguanosine Cap-Analogue Conjugates for EIF4E Targeted Degradation. Eur. J. Med. Chem. 2019, 166, 339–350. [Google Scholar] [CrossRef]
- Tominaga, H.; Ishiyama, M.; Ohseto, F.; Sasamoto, K.; Hamamoto, T.; Suzuki, K.; Watanabe, M. A Water-Soluble Tetrazolium Salt Useful for Colorimetric Cell Viability Assay. Anal. Commun. 1999, 36, 47–50. [Google Scholar] [CrossRef]
- Leblanc, A.F.; Sprowl, J.A.; Alberti, P.; Chiorazzi, A.; Arnold, W.D.; Gibson, A.A.; Hong, K.W.; Pioso, M.S.; Chen, M.; Huang, K.M.; et al. OATP1B2 Deficiency Protects against Paclitaxel-Induced Neurotoxicity. J. Clin. Investig. 2018, 128, 816–825. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
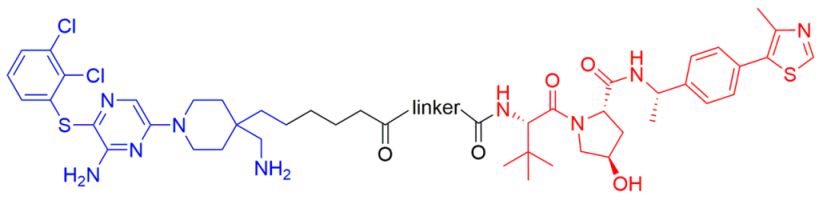 | ||||
|---|---|---|---|---|
| Compound ID | Linker Structure | Linker Length (Atom) | Degradation at 1 μM | Degradation at 10 μM |
| SC5 | 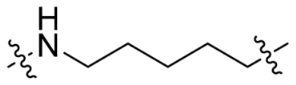 | 6 | <10% | <10% |
| SC7 | 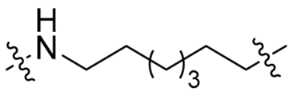 | 8 | <10% | 15% |
| SC9 |  | 10 | N.D. 2 | 25% |
| SC11 | 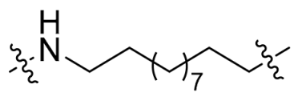 | 12 | 27% | 64% |
| P3 | 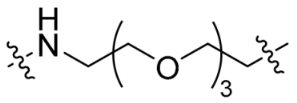 | 12 | <10% | <10% |
| P4 |  | 15 | <10% | <10% |
| P5 | 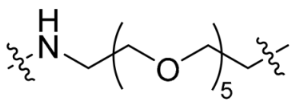 | 18 | <10% | <10% |
| Compound ID | Linker Structure | Linker Length (Atom) | Degradation at 1 μM |
|---|---|---|---|
| SC10 | 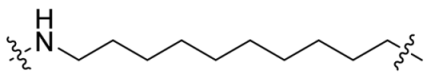 | 11 | 36% |
| P7 | 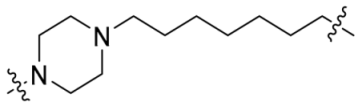 | 11 | 51% |
| P8 |  | 12 | 74% |
| P9 | 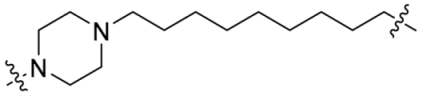 | 13 | >95% |
| P10 |  | 14 | 16% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miao, J.; Bai, Y.; Miao, Y.; Qu, Z.; Dong, J.; Zhang, R.-Y.; Aggarwal, D.; Jassim, B.A.; Nguyen, Q.; Zhang, Z.-Y. Discovery of a SHP2 Degrader with In Vivo Anti-Tumor Activity. Molecules 2023, 28, 6947. https://doi.org/10.3390/molecules28196947
Miao J, Bai Y, Miao Y, Qu Z, Dong J, Zhang R-Y, Aggarwal D, Jassim BA, Nguyen Q, Zhang Z-Y. Discovery of a SHP2 Degrader with In Vivo Anti-Tumor Activity. Molecules. 2023; 28(19):6947. https://doi.org/10.3390/molecules28196947
Chicago/Turabian StyleMiao, Jinmin, Yunpeng Bai, Yiming Miao, Zihan Qu, Jiajun Dong, Ruo-Yu Zhang, Devesh Aggarwal, Brenson A. Jassim, Quyen Nguyen, and Zhong-Yin Zhang. 2023. "Discovery of a SHP2 Degrader with In Vivo Anti-Tumor Activity" Molecules 28, no. 19: 6947. https://doi.org/10.3390/molecules28196947
APA StyleMiao, J., Bai, Y., Miao, Y., Qu, Z., Dong, J., Zhang, R.-Y., Aggarwal, D., Jassim, B. A., Nguyen, Q., & Zhang, Z.-Y. (2023). Discovery of a SHP2 Degrader with In Vivo Anti-Tumor Activity. Molecules, 28(19), 6947. https://doi.org/10.3390/molecules28196947