
Single-Atom Platinum Catalyst for Efficient CO2 Conversion via Reverse Water Gas Shift Reaction
Abstract
:
1. Introduction
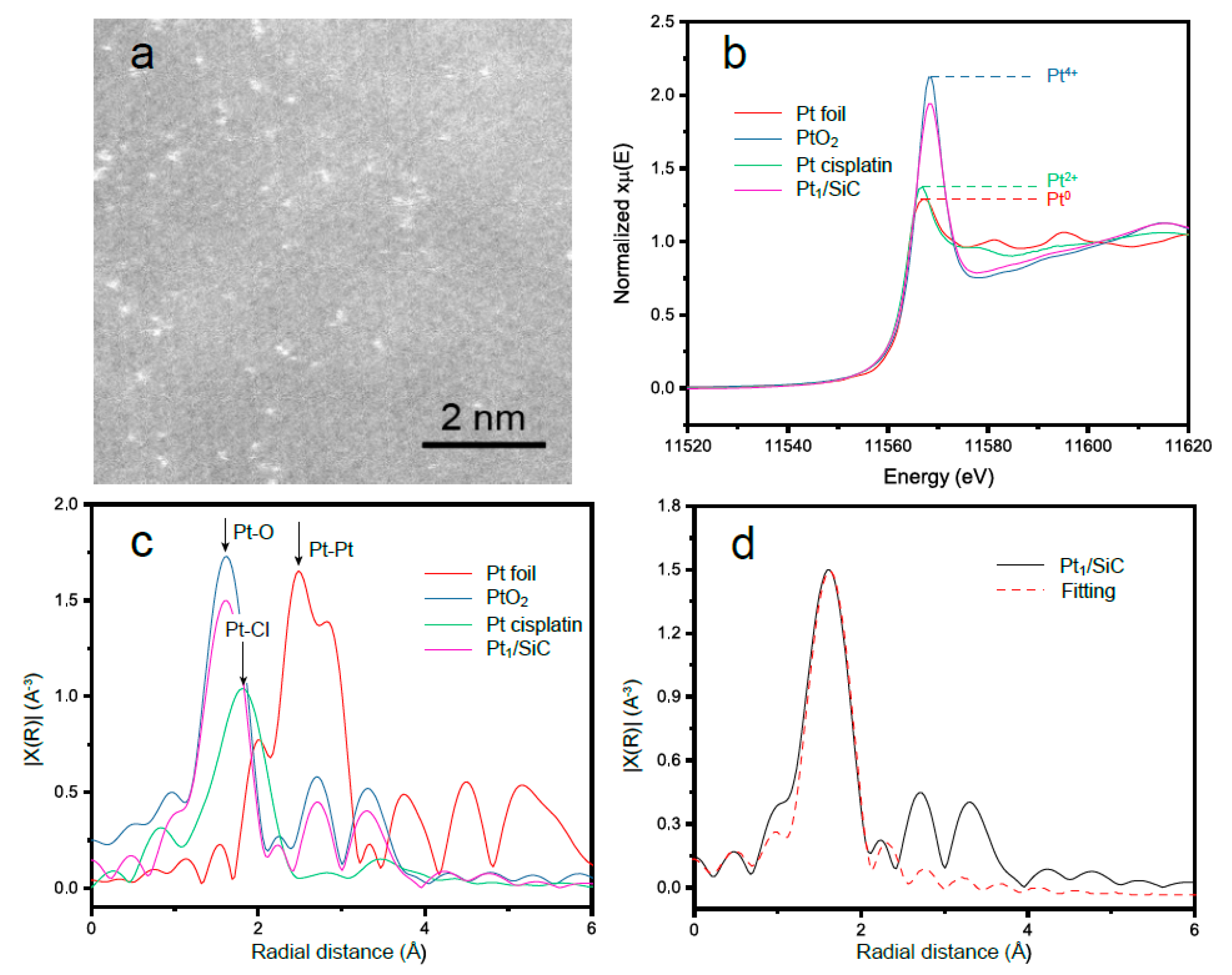
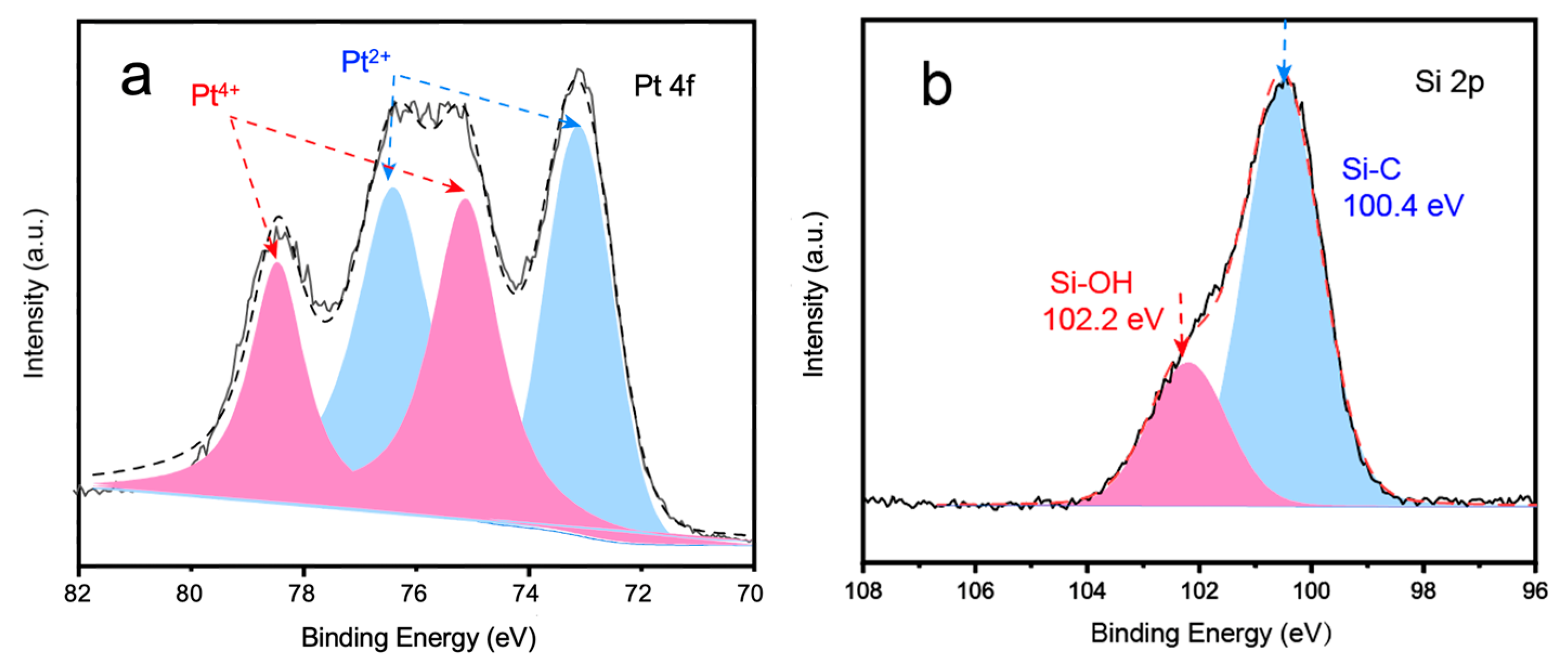
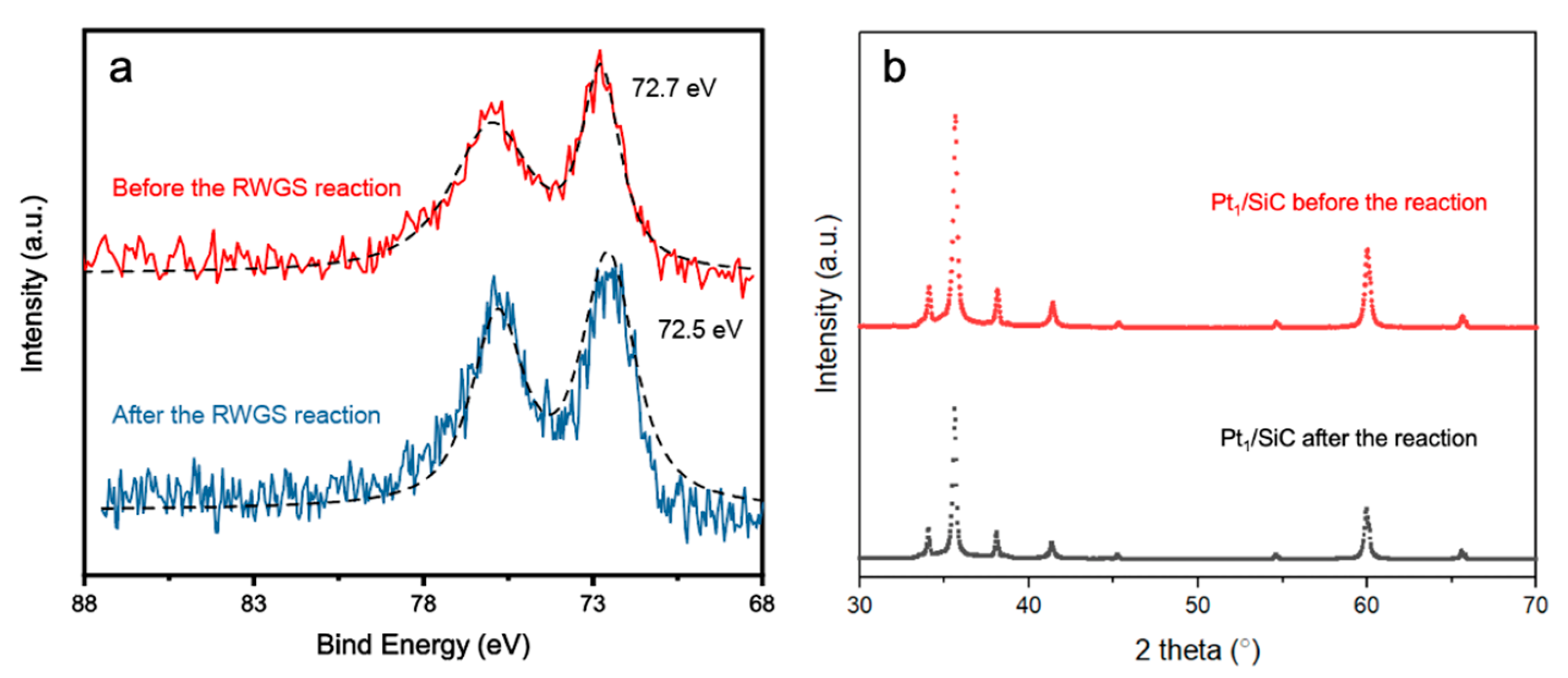
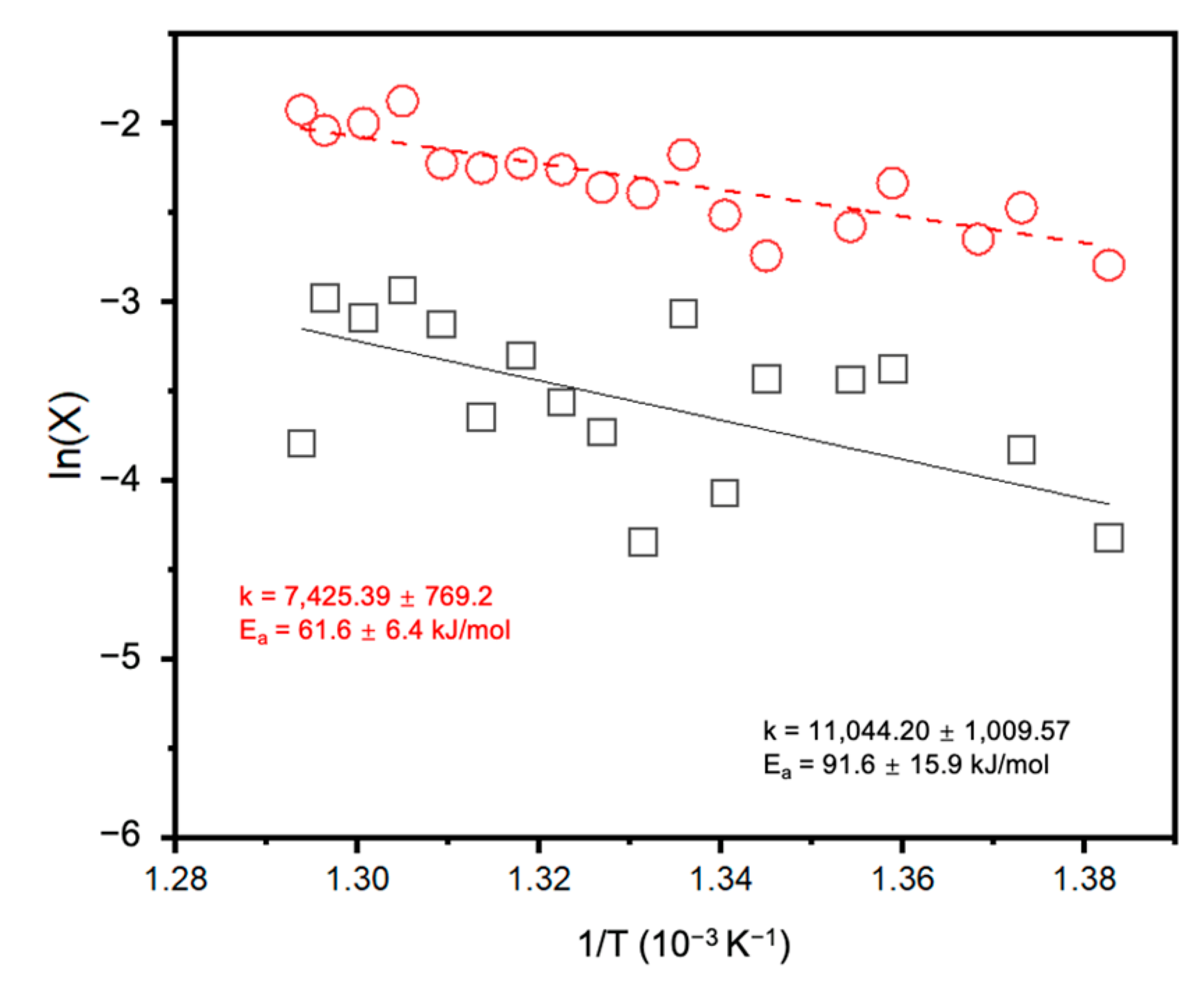
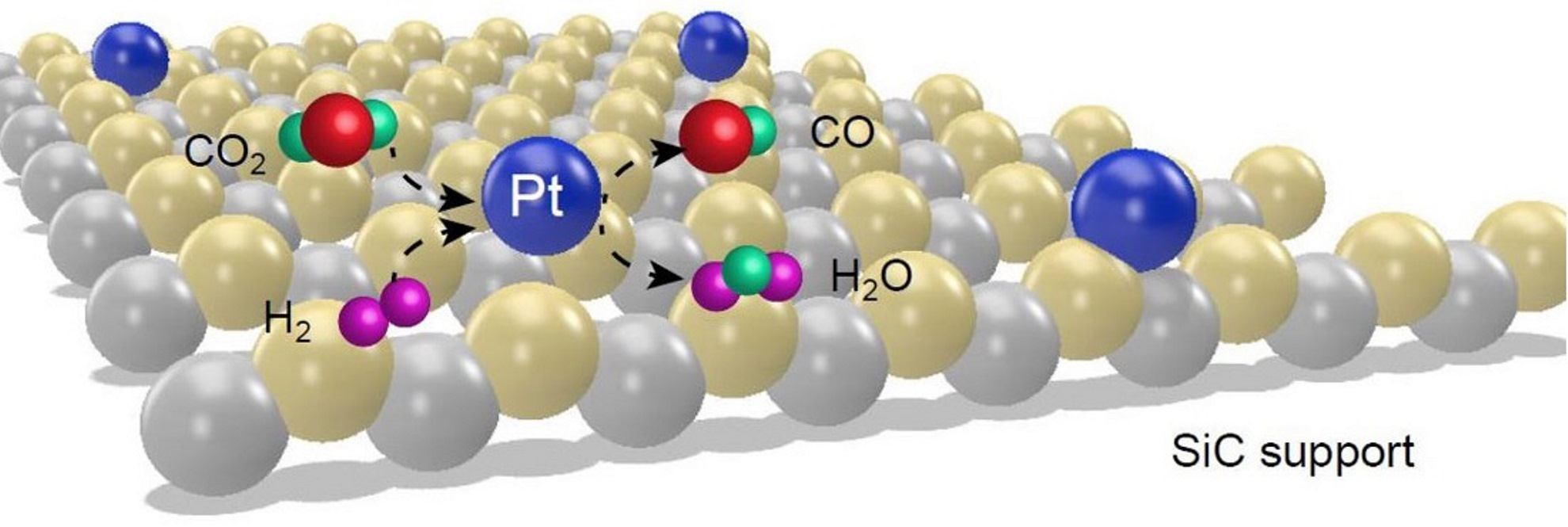
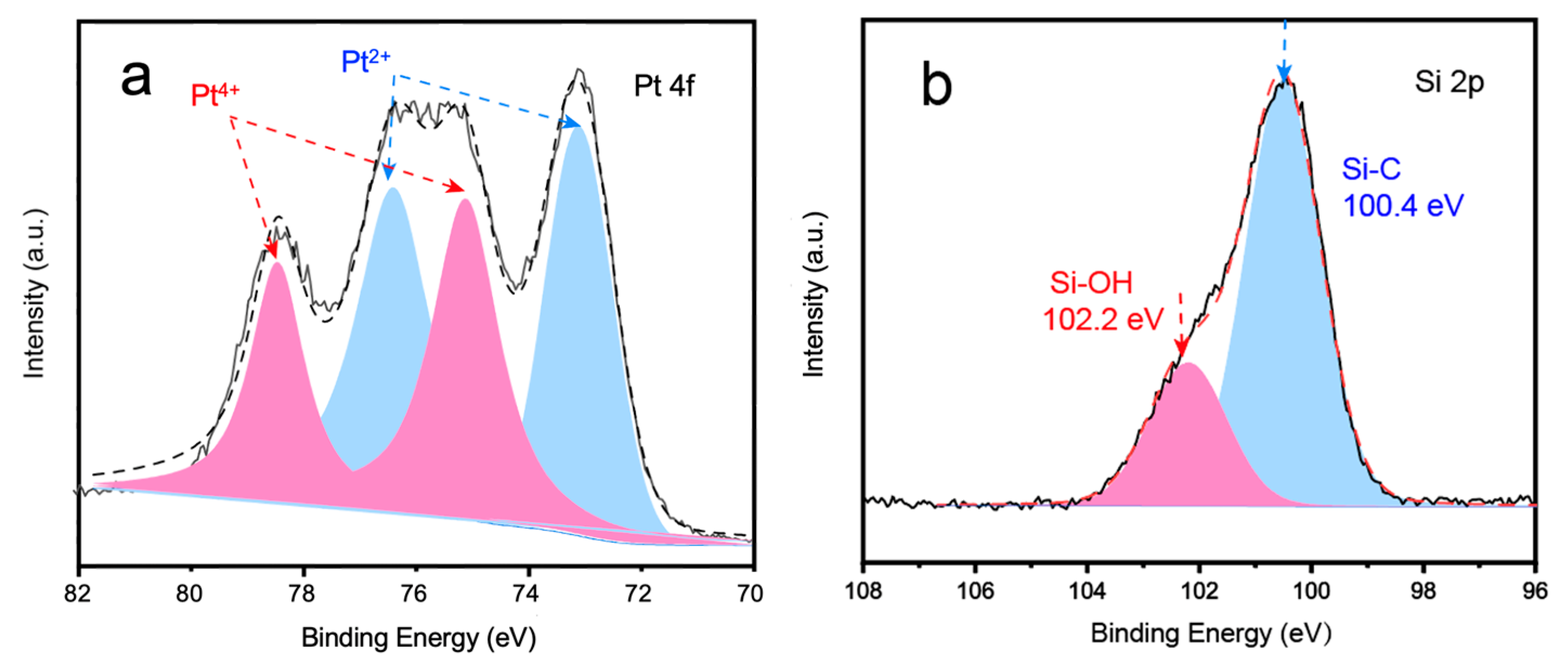
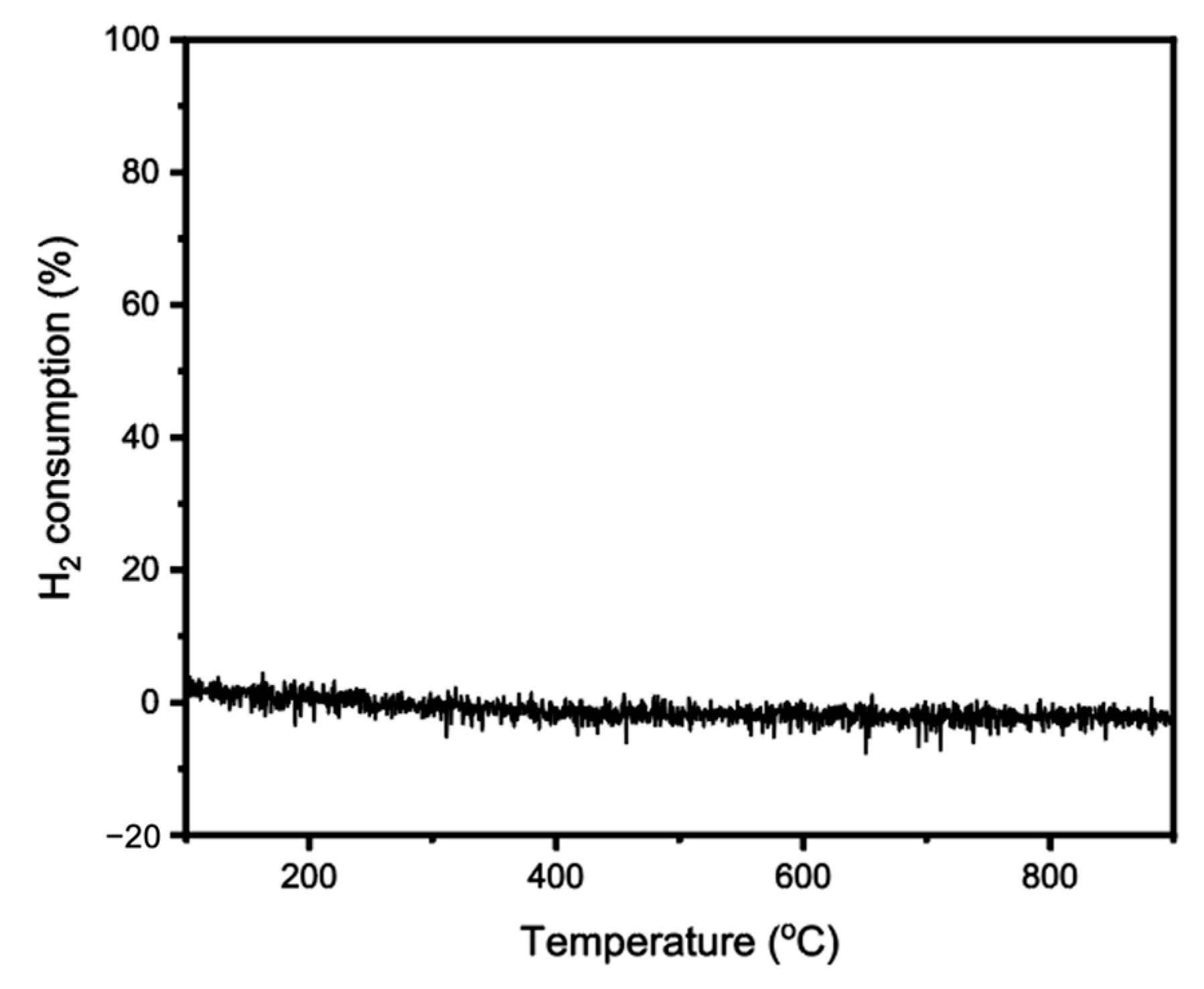
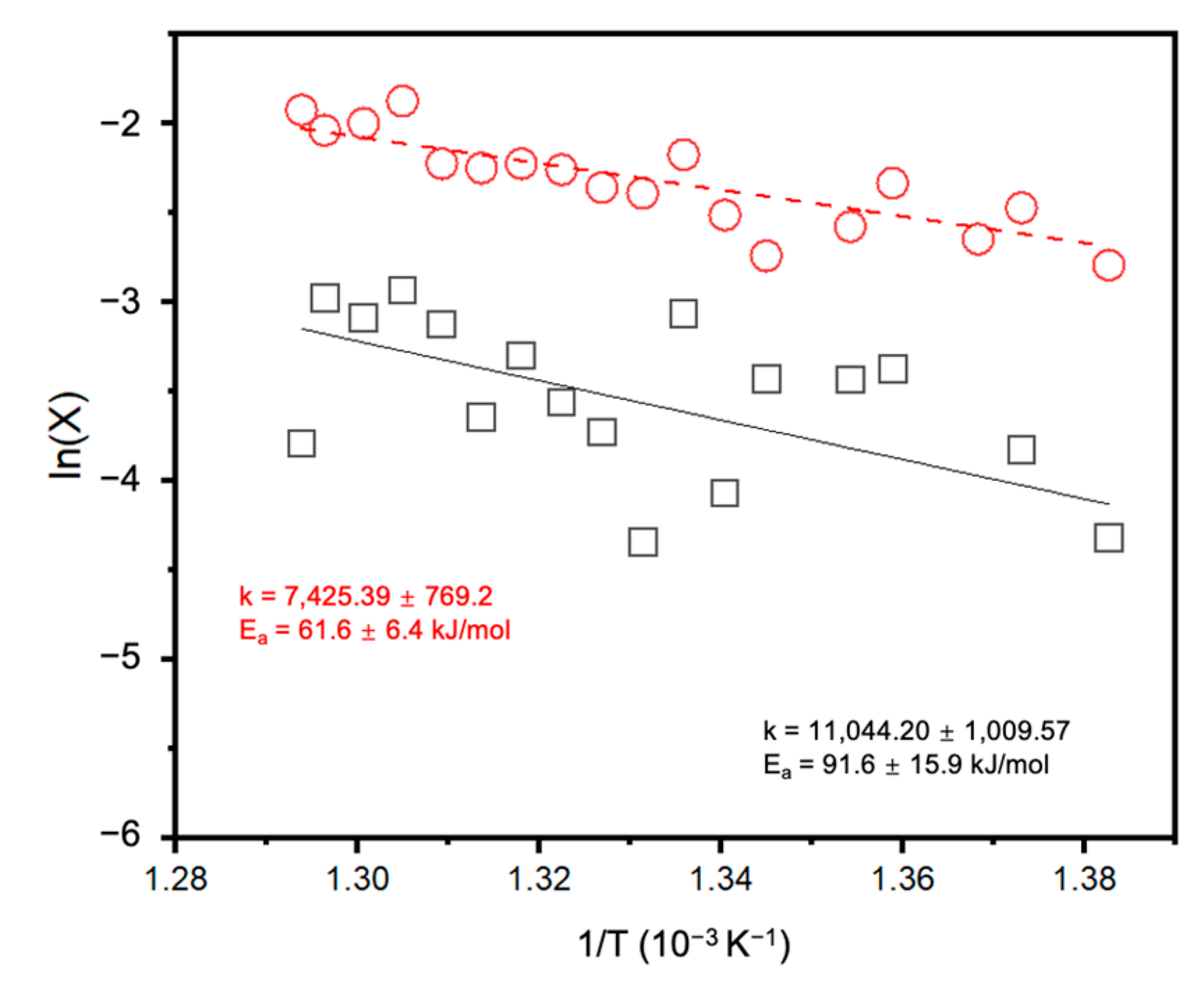
2. Results and Discussion
3. Materials and Methods
3.1. Reagents and Materials
3.2. Preparation of Pt1/SiC Catalysts
3.3. Preparation of Pt/SiC Catalysts
3.4. X-ray Absorption Fine Structure (XAFS)
3.5. X-ray Photoelectron Spectroscopy (XPS)
3.6. Inductively Coupled Plasma Mass Spectrometry (ICP-MS) Experiments
3.7. Temperature-Programmed Reduction (TPR)
3.8. RWGS Reactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yin, H.; Zhao, S.; Zhao, K.; Muqsit, A.; Tang, H.; Chang, L.; Zhao, H.; Gao, Y.; Tang, Z. Ultrathin platinum nanowires grown on single-layered nickel hydroxide with high hydrogen evolution activity. Nat. Commun. 2015, 6, 6430. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.-Y.; Lang, Z.-L.; Yin, L.-Y.; Feng, K.; Xia, Y.-J.; Tan, H.-Q.; Zhu, H.-T.; Zhong, J.; Kang, Z.-H.; Li, Y.-G. Pt-O bond as an active site superior to Pt0 in hydrogen evolution reaction. Nat. Commun. 2020, 11, 490. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; He, N.; Zhu, Q.; Chu, C.; Weon, S.; Rigby, K.; Zhou, X.; Xu, L.; Niu, J.; Stavitski, E. Conflicting roles of coordination number on catalytic performance of single-atom Pt catalysts. ACS Catal. 2021, 11, 5586–5592. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, J.; Wang, J.; Wang, Q.; Wang, Y.; Wang, K.; Wang, Z.; Gu, M.; Tang, Z.; Lim, J. Single-atom catalyst for high-performance methanol oxidation. Nat. Commun. 2021, 12, 5235. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Liu, F.; Tang, Y.; Li, L.; Miao, S.; Su, Y.; Zhang, J.; Huang, J.; Sun, H.; Haruta, M. Maximizing the number of interfacial sites in single-atom catalysts for the highly selective, solvent-free oxidation of primary alcohols. Angew. Chem. Int. Ed. 2018, 57, 7795–7799. [Google Scholar] [CrossRef]
- Banerjee, B.; Amoli, V.; Maurya, A.; Sinha, A.K.; Bhaumik, A. Green synthesis of Pt-doped TiO2 nanocrystals with exposed (001) facets and mesoscopic void space for photo-splitting of water under solar irradiation. Nanoscale 2015, 7, 10504–10512. [Google Scholar] [CrossRef]
- Cai, J.; Yu, Z.; Fan, X.; Li, J. Effect of TiO2 calcination pretreatment on the performance of Pt/TiO2 catalyst for CO oxidation. Molecules 2022, 27, 3875. [Google Scholar] [CrossRef]
- Gawande, M.B.; Fornasiero, P.; Zbořil, R. Carbon-Based Single-Atom Catalysts for Advanced Applications. ACS Catal. 2020, 10, 2231–2259. [Google Scholar] [CrossRef]
- Li, Z.; Hong, R.; Zhang, Z.; Wang, H.; Wu, X.; Wu, Z. Single-Atom Catalysts in Environmental Engineering: Progress, Outlook and Challenges. Molecules 2023, 28, 3865. [Google Scholar] [CrossRef]
- Tiburcio, E.; Greco, R.; Mon, M.; Ballesteros-Soberanas, J.; Ferrando-Soria, J.; López-Haro, M.; Hernández-Garrido, J.C.; Oliver-Meseguer, J.; Marini, C.; Boronat, M. Soluble/MOF-supported palladium single atoms catalyze the ligand-, additive-, and solvent-free aerobic oxidation of benzyl alcohols to benzoic acids. J. Am. Chem. Soc. 2021, 143, 2581–2592. [Google Scholar] [CrossRef]
- Yang, X.-F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-atom catalysts: A new frontier in heterogeneous catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. [Google Scholar] [CrossRef]
- Rivero-Crespo, M.A.; Mon, M.; Ferrando-Soria, J.; Lopes, C.W.; Boronat, M.; Leyva-Pérez, A.; Corma, A.; Hernández-Garrido, J.C.; López-Haro, M.; Calvino, J.J. Confined Pt11+ Water Clusters in a MOF Catalyze the Low-Temperature Water–Gas Shift Reaction with both CO2 Oxygen Atoms Coming from Water. Angew. Chem. Int. Ed. 2018, 57, 17094–17099. [Google Scholar] [CrossRef] [PubMed]
- Daza, Y.A.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Figueiredo, W.T.; Escudero, C.; Perez-Dieste, V.; Ospina, C.A.; Bernardi, F. Determining the Surface Atomic Population of Cu × Ni1–x/CeO2 (0< x ≤ 1) Nanoparticles during the Reverse Water–Gas Shift (RWGS) Reaction. J. Phys. Chem. C 2020, 124, 16868–16878. [Google Scholar]
- Wolf, M.; Gibson, E.K.; Olivier, E.J.; Neethling, J.H.; Catlow, C.R.A.; Fischer, N.; Claeys, M. Water-induced formation of cobalt-support compounds under simulated high conversion Fischer–Tropsch environment. ACS Catal 2019, 9, 4902–4918. [Google Scholar] [CrossRef]
- Cheng, Q.; Tian, Y.; Lyu, S.; Zhao, N.; Ma, K.; Ding, T.; Jiang, Z.; Wang, L.; Zhang, J.; Zheng, L. Confined small-sized cobalt catalysts stimulate carbon-chain growth reversely by modifying ASF law of Fischer–Tropsch synthesis. Nat. Commun. 2018, 9, 3250. [Google Scholar] [CrossRef]
- Wu, H.; Chang, Y.; Wu, J.; Lin, J.; Lin, I.; Chen, C. Methanation of CO2 and reverse water gas shift reactions on Ni/SiO2 catalysts: The influence of particle size on selectivity and reaction pathway. Catal. Sci. Technol. 2015, 5, 4154–4163. [Google Scholar] [CrossRef]
- Xu, J.; Su, X.; Liu, X.; Pan, X.; Pei, G.; Huang, Y.; Wang, X.; Zhang, T.; Geng, H. Methanol synthesis from CO2 and H2 over Pd/ZnO/Al2O3: Catalyst structure dependence of methanol selectivity. Appl. Catal. A Gen. 2016, 514, 51–59. [Google Scholar] [CrossRef]
- Chen, L.; Kovarik, L.; Szanyi, J. Temperature-Dependent Communication between Pt/Al2O3 Catalysts and Anatase TiO2 Dilutant: The Effects of Metal Migration and Carbon Transfer on the Reverse Water–Gas Shift Reaction. ACS Catal. 2021, 11, 12058–12067. [Google Scholar] [CrossRef]
- Chen, L.; Unocic, R.R.; Hoffman, A.S.; Hong, J.; Braga, A.H.; Bao, Z.; Bare, S.R.; Szanyi, J. Unlocking the catalytic potential of TiO2-supported Pt single atoms for the reverse water–gas shift reaction by altering their chemical environment. JACS Au 2021, 1, 977–986. [Google Scholar] [CrossRef]
- Chambers, S.; Droubay, T.; Jennison, D.R.; Mattsson, T. Laminar Growth of Ultrathin Metal Films on Metal Oxides: Co on Hydroxylated α-Al2O3 (0001). Science 2002, 297, 827–831. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Wang, L.; Luo, Q.; Cao, Y.; Dai, Y.; Li, Z.; Li, H.; Zheng, X.; Yan, W.; Yang, J. Molecular-level Insight into How Hydroxyl Groups Boost Catalytic Activity in CO2 Hydrogenation into Methanol. Chem 2018, 4, 613–625. [Google Scholar] [CrossRef]
- Schryer, D.R.; Upchurch, B.T.; Van Norman, J.D.; Brown, K.G.; Schryer, J. Effects of pretreatment conditions on a Pt/SnO2 catalyst for the oxidation of CO in CO2 lasers. J. Catal. 1990, 122, 193–197. [Google Scholar] [CrossRef]
- Fishman, Z.S.; He, Y.; Yang, K.R.; Lounsbury, A.W.; Zhu, J.; Tran, T.M.; Zimmerman, J.B.; Batista, V.S.; Pfefferle, L.D. Hard templating ultrathin polycrystalline hematite nanosheets: Effect of nano-dimension on CO2 to CO conversion via the reverse water-gas shift reaction. Nanoscale 2017, 9, 12984–12995. [Google Scholar] [CrossRef]
- He, Y.; Yang, K.R.; Yu, Z.; Fishman, Z.S.; Achola, L.A.; Tobin, Z.M.; Heinlein, J.A.; Hu, S.; Suib, S.L.; Batista, V.S.; et al. Catalytic manganese oxide nanostructures for the reverse water gas shift reaction. Nanoscale 2019, 11, 16677–16688. [Google Scholar] [CrossRef]
- Ro, I.; Sener, C.; Stadelman, T.M.; Ball, M.R.; Venegas, J.M.; Burt, S.P.; Hermans, I.; Dumesic, J.A.; Huber, G.W. Measurement of intrinsic catalytic activity of Pt monometallic and Pt-MoOx interfacial sites over visible light enhanced PtMoOx/SiO2 catalyst in reverse water gas shift reaction. J. Catal. 2016, 344, 784–794. [Google Scholar] [CrossRef]
- Lu, B.; Kawamoto, K. Preparation of monodispersed NiO particles in SBA-15, and its enhanced selectivity for reverse water gas shift reaction. J. Environ. Chem. Eng. 2013, 1, 300–309. [Google Scholar] [CrossRef]
- Jones, J.; Xiong, H.; DeLaRiva, A.T.; Peterson, E.J.; Pham, H.; Challa, S.R.; Qi, G.; Oh, S.; Wiebenga, M.H.; Hernández, X.I.P. Thermally stable single-atom platinum-on-ceria catalysts via atom trapping. Science 2016, 353, 150–154. [Google Scholar] [CrossRef]
- Lang, R.; Xi, W.; Liu, J.-C.; Cui, Y.-T.; Li, T.; Lee, A.F.; Chen, F.; Chen, Y.; Li, L.; Li, L. Non defect-stabilized thermally stable single-atom catalyst. Nat. Commun. 2019, 10, 234. [Google Scholar] [CrossRef]
- Kobayashi, D.; Kobayashi, H.; Kusada, K.; Yamamoto, T.; Toriyama, T.; Matsumura, S.; Kawaguchi, S.; Kubota, Y.; Haneda, M.; Aspera, S.M.; et al. Boosting reverse water-gas shift reaction activity of Pt nanoparticles through light doping of W. J. Mater. Chem. A 2021, 9, 15613–15617. [Google Scholar] [CrossRef]
- Goguet, A.; Meunier, F.C.; Tibiletti, D.; Breen, A.J.; Burch, R. Spectrokinetic Investigation of Reverse Water-Gas-Shift Reaction Intermediates over a Pt/CeO2 Catalyst. J. Phys. Chem. B 2004, 108, 20240–20246. [Google Scholar] [CrossRef]
- Dao, V.-D.; Hoa, N.T.Q.; Larina, L.L.; Lee, J.-K.; Choi, H.-S. Graphene–platinum nanohybrid as a robust and low-cost counter electrode for dye-sensitized solar cells. Nanoscale 2013, 5, 12237–12244. [Google Scholar] [CrossRef] [PubMed]
- Spencer, M. On the activation energies of the forward and reverse water-gas shift reaction. Catal. Lett. 1995, 32, 9–13. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. Athena, Artemis, Hephaestus: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Su, X.; Liang, B.; Yang, X.; Ren, X.; Duan, H.; Huang, Y.; Zhang, T. Identification of relevant active sites and a mechanism study for reverse water gas shift reaction over Pt/CeO2 catalysts. J. Energy Chem. 2016, 25, 1051–1057. [Google Scholar] [CrossRef]
- Yang, X.; Su, X.; Chen, X.; Duan, H.; Liang, B.; Liu, Q.; Liu, X.; Ren, Y.; Huang, Y.; Zhang, T. Promotion effects of potassium on the activity and selectivity of Pt/zeolite catalysts for reverse water gas shift reaction. Appl. Catal. B Environ. 2017, 216, 95–105. [Google Scholar] [CrossRef]
- Lu, B.; Kawamoto, K. Preparation of mesoporous CeO2 and monodispersed NiO particles in CeO2, and enhanced selectivity of NiO/CeO2 for reverse water gas shift reaction. Mat. Res. Bull. 2014, 53, 70–78. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, D. Study of bimetallic Cu–Ni/γ-Al2O3 catalysts for carbon dioxide hydrogenation. Int. J. Hydrogen Energy 1999, 24, 351–354. [Google Scholar] [CrossRef]
- Chen, C.-S.; Cheng, W.-H.; Lin, S.-S. Study of reverse water gas shift reaction by TPD, TPR and CO2 hydrogenation over potassium-promoted Cu/SiO2 catalyst. Appl. Catal. A Gen. 2003, 238, 55–67. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, X.; Lin, L.; Yao, S.; Zhang, M.; Liu, X.; Wang, X.; Li, Y.-W.; Shi, C.; Ma, D. Highly dispersed copper over β-Mo2C as an efficient and stable catalyst for the reverse water gas shift (RWGS) reaction. ACS Catal. 2017, 7, 912–918. [Google Scholar] [CrossRef]
- Liu, X.; de la Piscina, P.R.; Toyir, J.; Homs, N. CO2 reduction over Cu-ZnGaMO (M = Al, Zr) catalysts prepared by a sol-gel method: Unique performance for the RWGS reaction. Catal. Today 2017, 296, 181–186. [Google Scholar] [CrossRef]
- Zhao, K.; Bkour, Q.; Hou, X.; Kang, S.W.; Park, J.C.; Norton, M.G.; Yang, J.-I.; Ha, S. Reverse water gas shift reaction over CuFe/Al2O3 catalyst in solid oxide electrolysis cell. Chem. Eng. J. 2018, 336, 20–27. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Shell | CN | R/Å | σ2/Å2 |
|---|---|---|---|---|
| PtO2 | Pt-O | 6 | 2.06 | 0.0010 ± 0.0016 |
| Pt foil | Pt-Pt | 12 | 2.80 | 0.0013 ± 0.0014 |
| Pt1/SiC | Pt-O | 5.2 ± 0.7 | 2.05 ± 0.01 | 0.0051 ± 0.0011 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Huang, D. Single-Atom Platinum Catalyst for Efficient CO2 Conversion via Reverse Water Gas Shift Reaction. Molecules 2023, 28, 6630. https://doi.org/10.3390/molecules28186630
He Y, Huang D. Single-Atom Platinum Catalyst for Efficient CO2 Conversion via Reverse Water Gas Shift Reaction. Molecules. 2023; 28(18):6630. https://doi.org/10.3390/molecules28186630
Chicago/Turabian StyleHe, Yulian, and Dahong Huang. 2023. "Single-Atom Platinum Catalyst for Efficient CO2 Conversion via Reverse Water Gas Shift Reaction" Molecules 28, no. 18: 6630. https://doi.org/10.3390/molecules28186630
APA StyleHe, Y., & Huang, D. (2023). Single-Atom Platinum Catalyst for Efficient CO2 Conversion via Reverse Water Gas Shift Reaction. Molecules, 28(18), 6630. https://doi.org/10.3390/molecules28186630