




ClO2-Mediated Oxidation of the TEMPO Radical: Fundamental Considerations of the Catalytic System for the Oxidation of Cellulose Fibers



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
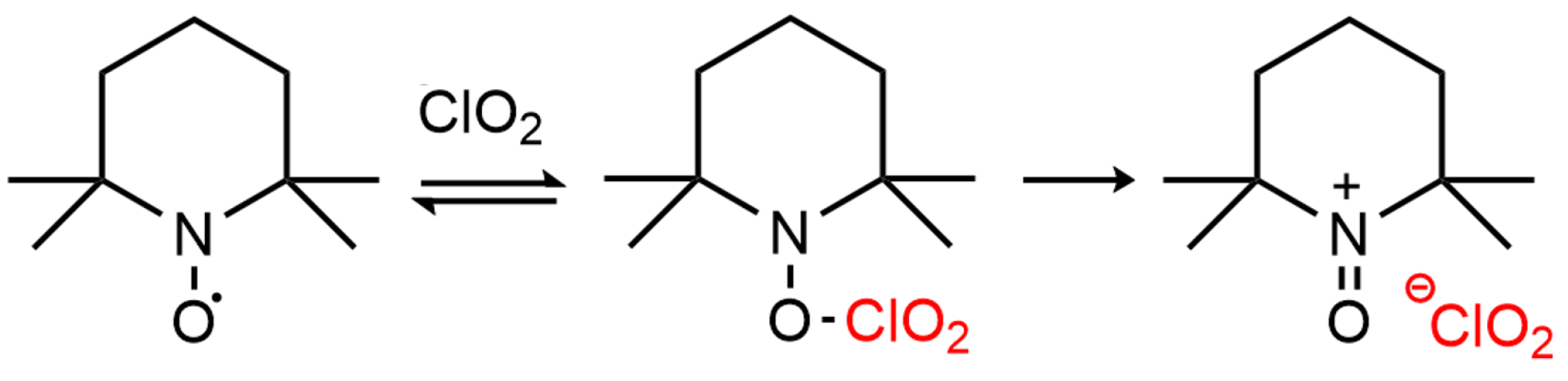
2.1. Investigation of the ClO2-Based TEMPO Activation through EPR Spectroscopy
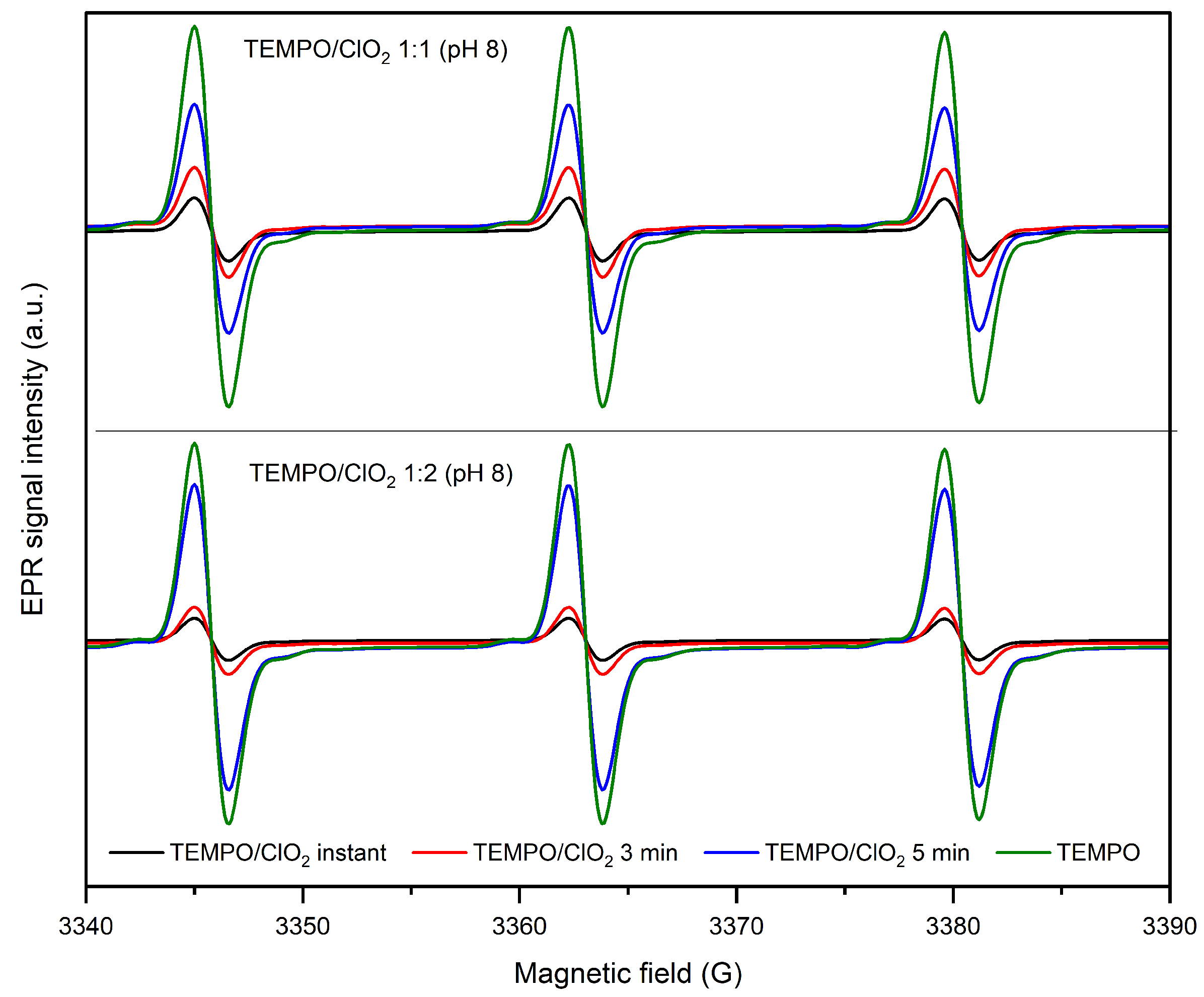
2.1.1. Effect of pH and TEMPO/ClO Molar Ratio
2.1.2. TEMPO Regeneration during ClO Activation

2.1.3. Assessment of a Cellulosic Model Compound during ClO2-Based TEMPO Oxidation
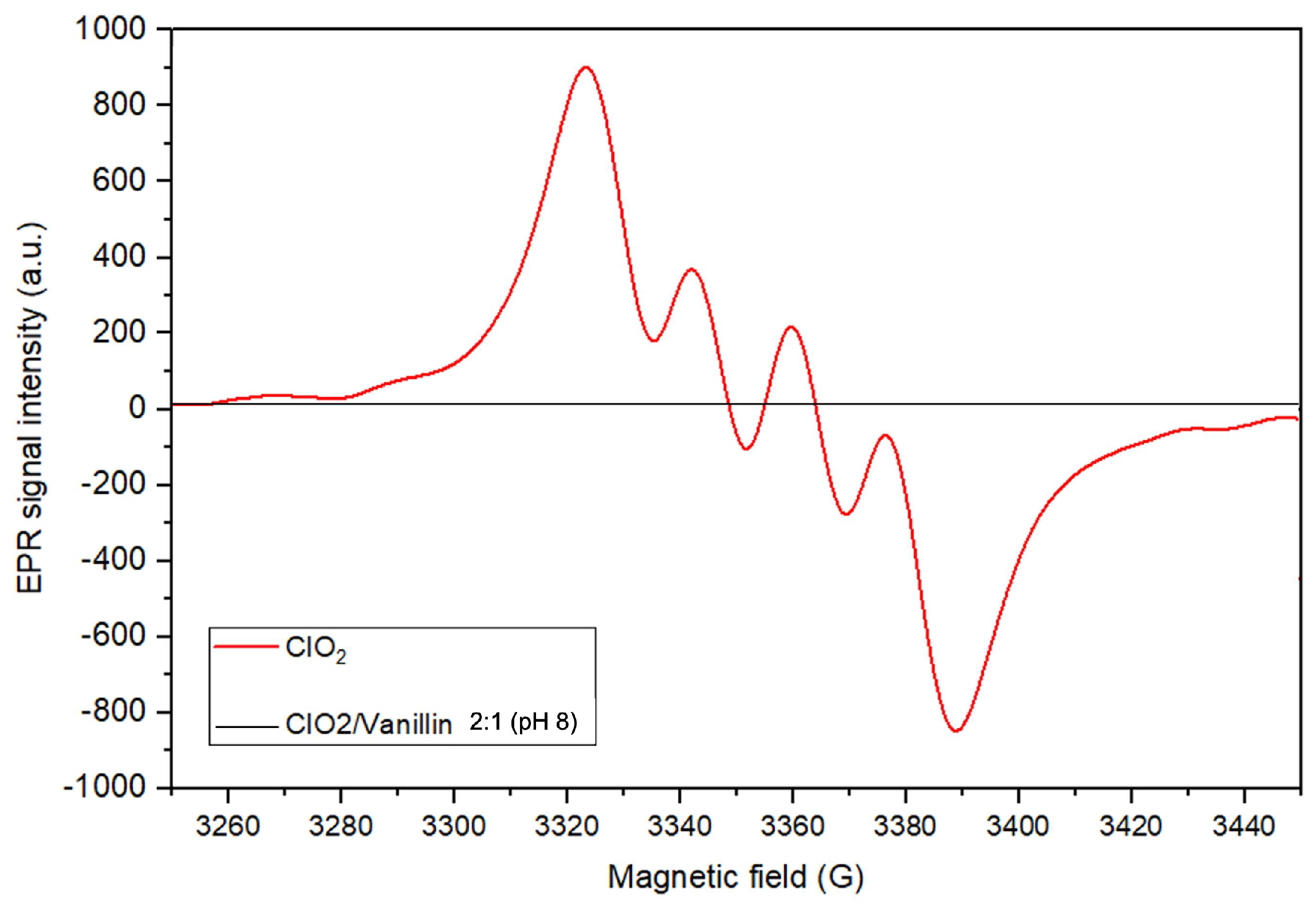
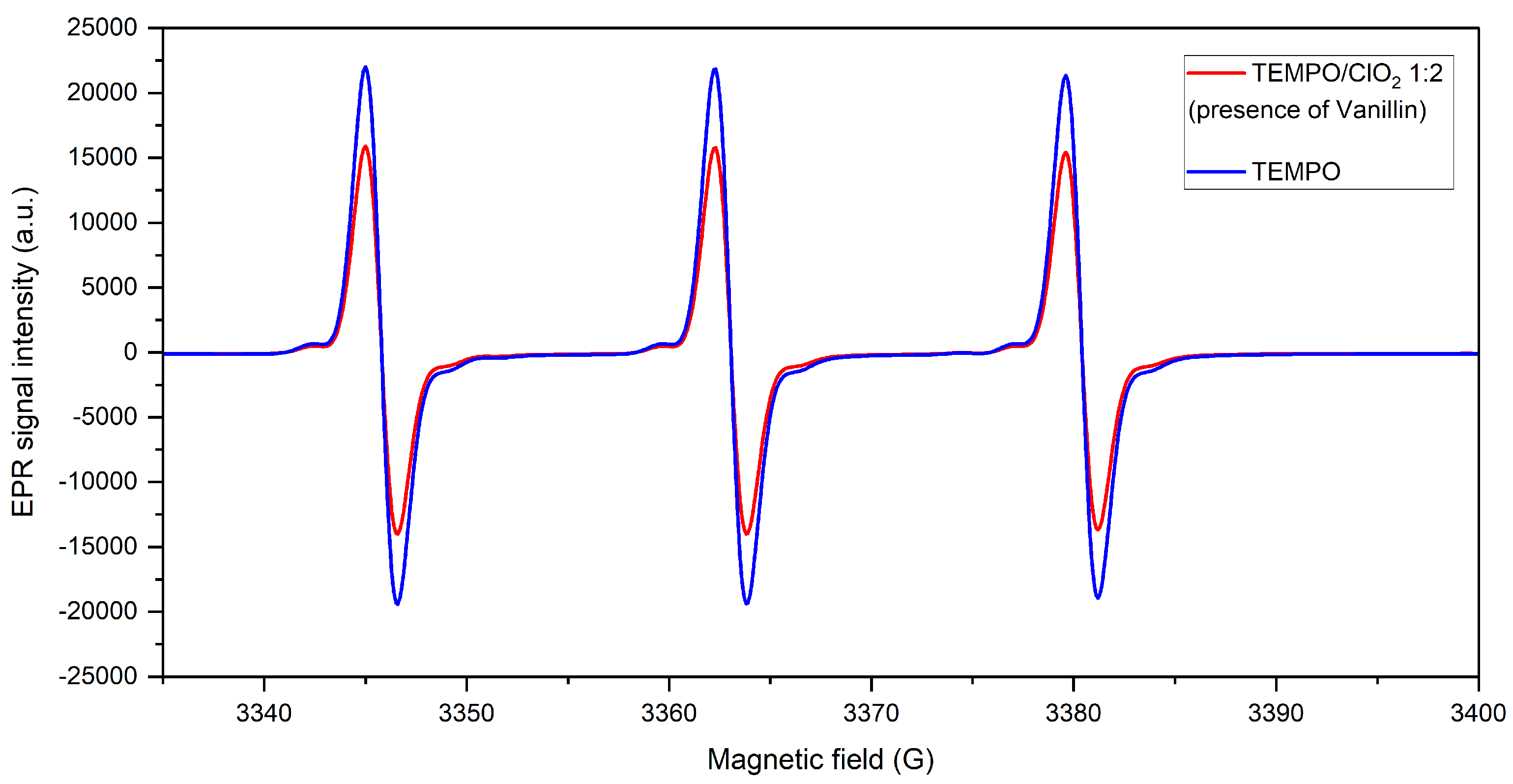
2.1.4. Assessment of a Lignin Model Compound during ClO-Based TEMPO Oxidation
2.1.5. Influence of HOCl as a Co-Oxidant
2.2. UV–Visible Spectroscopy (UV–Vis)
3. Materials and Methods
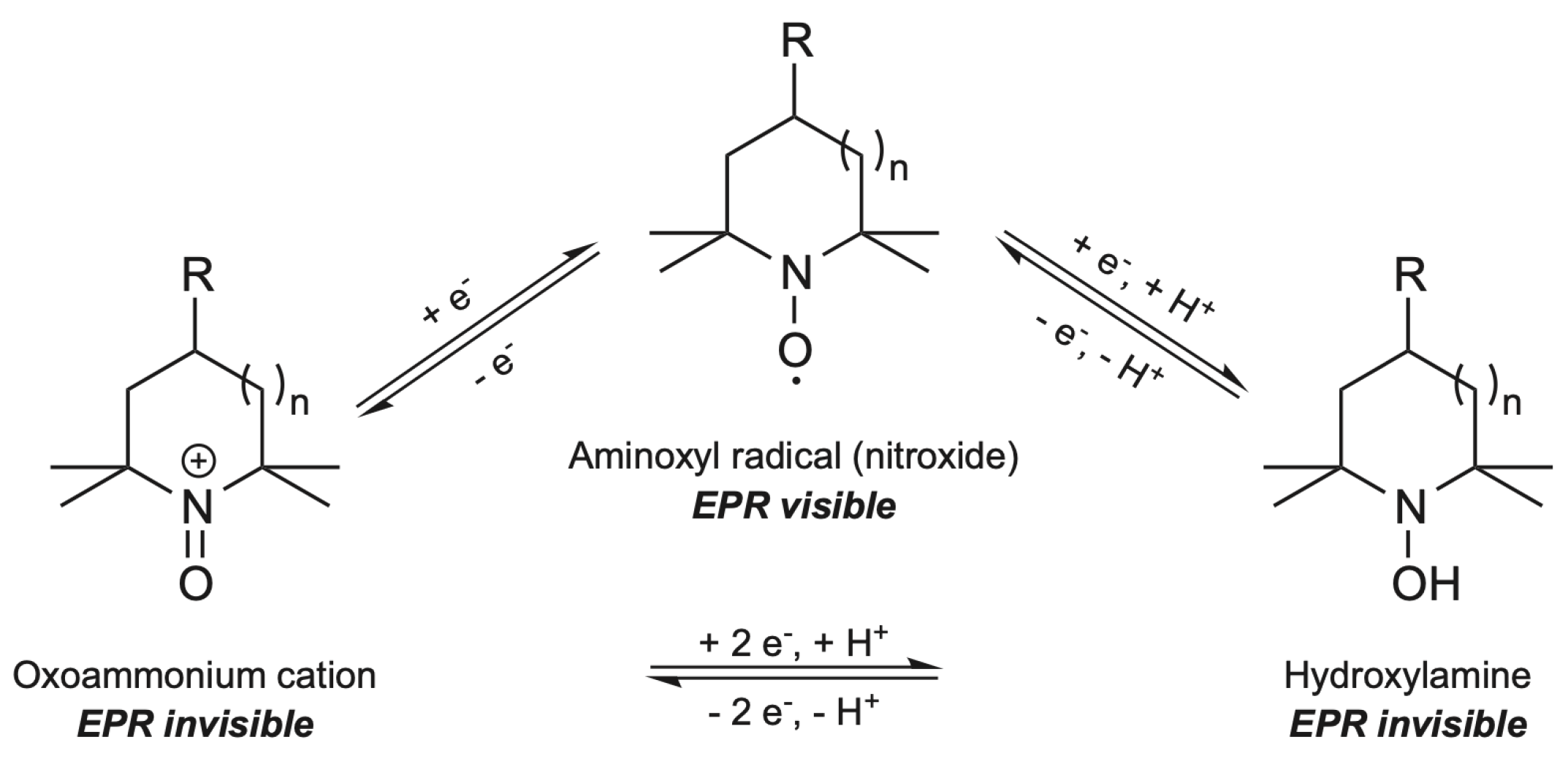
3.1. Electron Paramagnetic Resonance (EPR) Principle for TEMPO Oxidation Detection
3.2. Chemicals
3.3. Technical Parameters for EPR Measurements
3.3.1. Sample Preparation for EPR Analysis
3.3.2. EPR Measurements Procedure
3.4. UV–Visible Spectroscopy (UV–Vis)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Nooy, A.; Besemer, A.; Van Bekkum, H. Highly selective tempo mediated oxidation of primary alcohol groups in polysaccharides. Recl. Trav. Chim. Pays-Bas 1994, 113, 165–166. [Google Scholar] [CrossRef]
- Isogai, A.; Hänninen, T.; Fujisawa, S.; Saito, T. Catalytic oxidation of cellulose with nitroxyl radicals under aqueous conditions. Prog. Polym. Sci. 2018, 86, 122–148. [Google Scholar] [CrossRef]
- Tang, Z.; Li, W.; Lin, X.; Xiao, H.; Miao, Q.; Huang, L.; Chen, L.; Wu, H. TEMPO-oxidized cellulose with high degree of oxidation. Polymers 2017, 9, 421. [Google Scholar] [CrossRef] [PubMed]
- Isogai, A.; Saito, T.; Fukuzumi, H. TEMPO-oxidized cellulose nanofibers. Nanoscale 2011, 3, 71–85. [Google Scholar] [CrossRef]
- Xu, Q.; Li, W.; Cheng, Z.; Yang, G.; Qin, M. TEMPO/NaBr/NaClO-mediated surface oxidation of nanocrystalline cellulose and its microparticulate retention system with cationic polyacrylamide. BioResources 2014, 9, 994–1006. [Google Scholar] [CrossRef]
- Bragd, P.; Van Bekkum, H.; Besemer, A. TEMPO-mediated oxidation of polysaccharides: Survey of methods and applications. Top. Catal. 2004, 27, 49–66. [Google Scholar] [CrossRef]
- Ono, Y.; Takeuchi, M.; Zhou, Y.; Isogai, A. TEMPO/NaBr/NaClO and NaBr/NaClO oxidations of cotton linters and ramie cellulose samples. Cellulose 2021, 28, 6035–6049. [Google Scholar] [CrossRef]
- Saito, T.; Kimura, S.; Nishiyama, Y.; Isogai, A. Cellulose nanofibers prepared by TEMPO-mediated oxidation of native cellulose. Biomacromolecules 2007, 8, 2485–2491. [Google Scholar] [CrossRef]
- Serra, A.; González, I.; Oliver-Ortega, H.; Tarrès, Q.; Delgado-Aguilar, M.; Mutjé, P. Reducing the amount of catalyst in TEMPO-oxidized cellulose nanofibers: Effect on properties and cost. Polymers 2017, 9, 557. [Google Scholar] [CrossRef]
- Shinoda, R.; Saito, T.; Okita, Y.; Isogai, A. Relationship between length and degree of polymerization of TEMPO-oxidized cellulose nanofibrils. Biomacromolecules 2012, 13, 842–849. [Google Scholar] [CrossRef]
- Liu, L.; Chen, Y.Z.; Zhang, Z.J. Preparation of the microfibrillated cellulose and its application in the food packaging paper. Appl. Mech. Mater. 2014, 469, 87–90. [Google Scholar] [CrossRef]
- Gamelas, J.A.; Pedrosa, J.; Lourenço, A.F.; Mutjé, P.; González, I.; Chinga-Carrasco, G.; Singh, G.; Ferreira, P.J. On the morphology of cellulose nanofibrils obtained by TEMPO-mediated oxidation and mechanical treatment. Micron 2015, 72, 28–33. [Google Scholar] [CrossRef]
- Wakabayashi, M.; Fujisawa, S.; Saito, T.; Isogai, A. Nanocellulose film properties tunable by controlling degree of fibrillation of TEMPO-oxidized cellulose. Front. Chem. 2020, 8, 37. [Google Scholar] [CrossRef]
- Lasseuguette, E.; Roux, D.; Nishiyama, Y. Rheological properties of microfibrillar suspension of TEMPO-oxidized pulp. Cellulose 2008, 15, 425–433. [Google Scholar] [CrossRef]
- Islam, M.T.; Alam, M.M.; Patrucco, A.; Montarsolo, A.; Zoccola, M. Preparation of nanocellulose: A review. AATCC J. Res. 2014, 1, 17–23. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Jin, T.; He, H.; Liu, L. Preparation of nanocellulose and its potential in reinforced composites: A review. J. Biomater. Sci. Polym. Ed. 2019, 30, 919–946. [Google Scholar] [CrossRef]
- Bragd, P.L.; Besemer, A.C.; Van Bekkum, H. Bromide-free TEMPO-mediated oxidation of primary alcohol groups in starch and methyl α-D-glucopyranoside. Carbohydr. Res. 2000, 328, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Keasler, V.; De Paula, R.M.; Nilsen, G.; Grunwald, L.; Tidwell, T.J. Biocides overview and applications in petroleum microbiology. Trends Oil Gas Corros. Res. Technol. 2017, 539–562. [Google Scholar] [CrossRef]
- Ganiev, I.; Timerghazin, Q.; Khalizov, A.; Shereshovets, V.; Grigor’ev, A.; Tolstikov, G. Complex of chlorine dioxide with TEMPO and its conversion into oxoammonium salt. J. Phys. Org. Chem. 2001, 14, 38–42. [Google Scholar] [CrossRef]
- Weerawarna, A.S.; Komen, J.L.; Jewell, R.A. Hypochlorite Free Method for Preparation of Stable Carboxylated Carbohydrate Products. U.S. Patent US205/01469A1, 20 January 2005. [Google Scholar]
- Pääkkönen, T.; Pönni, R.; Dou, J.; Nuopponen, M.; Vuorinen, T. Activation of TEMPO by ClO2 for oxidation of cellulose by hypochlorite—Fundamental and practical aspects of the catalytic system. Carbohydr. Polym. 2017, 174, 524–530. [Google Scholar] [CrossRef]
- Tienvieri, T.; Kajanto, I.; Ojala, T.; Saarela, S.; Noupponen, M.; Pääkkönen, T.; Vourinen, T. Method for Catalytic Oxidation of Cellulose and Method for Making a Cellulose Product. U.S. Patent US9410285B2, 19 June 2014. [Google Scholar]
- Dollie, L.; Mortha, G.; Marlin-Dietemann, N. Process for the Catalytic Oxidation of Cellulose Pulp. Finland Patent FR3105224A1, 25 June 2021. [Google Scholar]
- Jefri, U.H.N.M.; Khan, A.; Lim, Y.C.; Lee, K.S.; Liew, K.B.; Kassab, Y.W.; Choo, C.Y.; Al-Worafi, Y.M.; Ming, L.C.; Kalusalingam, A. A systematic review on chlorine dioxide as a disinfectant. J. Med. Life 2022, 15, 313. [Google Scholar] [CrossRef] [PubMed]
- Terhalle, J.; Kaiser, P.; Jütte, M.; Buss, J.; Yasar, S.; Marks, R.; Uhlmann, H.; Schmidt, T.C.; Lutze, H.V. Chlorine dioxide—pollutant transformation and formation of hypochlorous acid as a secondary oxidant. Environ. Sci. Technol. 2018, 52, 9964–9971. [Google Scholar] [CrossRef] [PubMed]
- Pääkkönen, T.; Bertinetto, C.; Pönni, R.; Tummala, G.K.; Nuopponen, M.; Vuorinen, T. Rate-limiting steps in bromide-free TEMPO-mediated oxidation of cellulose—Quantification of the N-Oxoammonium cation by iodometric titration and UV–vis spectroscopy. Appl. Catal. A Gen. 2015, 505, 532–538. [Google Scholar] [CrossRef]
- Medir, M.; Giralt, F. Stability of chlorine dioxide in aqueous solution. Water Res. 1982, 16, 1379–1382. [Google Scholar] [CrossRef]
- Aleksejeva, O.; Barrière, F.; Blum, Z.; Bouffier, L.; Cai, W.R.; Cirovic, S.; Costa, N.L.; Dong, S.; Etienne, M.; Falk, M.; et al. Bioelectrochemistry: Design and Applications of Biomaterials; Walter de Gruyter GmbH & Co., KG: Berlin, Germany, 2019. [Google Scholar]
- Gordon, G.; Kieffer, R.G.; Rosenblatt, D.H. The chemistry of chlorine dioxide. Prog. Inorg. Chem. 1972, 15, 202–286. [Google Scholar]
- Bobbitt, J.M.; BrüCkner, C.; Merbouh, N. Oxoammonium-and Nitroxide-Catalyzed Oxidations of Alcohols. Org. React. 2004, 103–424. [Google Scholar] [CrossRef]
- Bačić, G.; Pavićević, A.; Peyrot, F. In vivo evaluation of different alterations of redox status by studying pharmacokinetics of nitroxides using magnetic resonance techniques. Redox Biol. 2016, 8, 226–242. [Google Scholar] [CrossRef]
- Le, T.P.P.; Opaprakasit, P. Conversion Mechanisms of Nitroxyl Radical (TEMPO), Oxoammonium Cation, and Hydroxylamine in Aqueous Solutions: Two-Dimensional Correlation Ultraviolet–Visible Spectroscopy. Appl. Spectrosc. 2021, 75, 325–335. [Google Scholar] [CrossRef]
- Ma, Z.; Bobbitt, J.M. Organic oxoammonium salts. A new convenient method for the oxidation of alcohols to aldehydes and ketones. J. Org. Chem. 1991, 56, 6110–6114. [Google Scholar] [CrossRef]
- Israeli, A.; Patt, M.; Oron, M.; Samuni, A.; Kohen, R.; Goldstein, S. Kinetics and mechanism of the comproportionation reaction between oxoammonium cation and hydroxylamine derived from cyclic nitroxides. Free Radic. Biol. Med. 2005, 38, 317–324. [Google Scholar] [CrossRef]
- Ryan, M.C.; Whitmire, L.D.; McCann, S.D.; Stahl, S.S. Copper/TEMPO Redox Redux: Analysis of PCET Oxidation of TEMPOH by Copper (II) and the Reaction of TEMPO with Copper (I). Inorg. Chem. 2019, 58, 10194–10200. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Fu, S.; Zhai, H.; Law, K.; Daneault, C. Influence of TEMPO-mediated oxidation on the lignin of thermomechanical pulp. Bioresour. Technol. 2012, 118, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Yuan, Z.; Liu, X.; Qu, J.; Yang, S.; Wang, A.; Wang, C.; Wei, B.; Xu, J.; Ni, Y. Preparation and characterization of lignin-containing cellulose nanofibril from poplar high-yield pulp via TEMPO-mediated oxidation and homogenization. ACS Sustain. Chem. Eng. 2019, 7, 6131–6139. [Google Scholar] [CrossRef]
- Liu, B.; Qin, C.; Zhang, F.; Wang, S.; Liang, C.; Nie, S.; Wang, S.; Yao, S. Reaction mechanism of phenolic lignin and high concentration chlorine dioxide and its application. ACS Omega 2020, 5, 22475–22481. [Google Scholar] [CrossRef]
- Hupperich, K.; Mutke, X.A.; Abdighahroudi, M.S.; Jütte, M.; Schmidt, T.C.; Lutze, H.V. Reaction of chlorine dioxide with organic matter–formation of inorganic products. Environ. Sci. Water Res. Technol. 2020, 6, 2597–2606. [Google Scholar] [CrossRef]
- Vanderkooi, N., Jr.; Poole, T.R. The Electron Paramagnetic Resonance Spectrum of Chlorine Dioxide in Solution. Effect of Temperature and Viscosity on the Line Width. Inorg. Chem. 1966, 5, 1351–1354. [Google Scholar] [CrossRef]
- Ragnar, M. Importance of the structural composition of pulp for the selectivity of ozone and chlorine dioxide bleaching. Nord. Pulp Pap. Res. J. 2001, 16, 72–79. [Google Scholar] [CrossRef]
- Karunakaran, C.; Balamurugan, M. Advances in Electron Paramagnetic Resonance. In Spin Resonance Spectroscopy; Elsevier: Amsterdam, The Netherlands, 2018; pp. 229–280. [Google Scholar]
- Ma, Y.; Loyns, C.; Price, P.; Chechik, V. Thermal decay of TEMPO in acidic media via an N-oxoammonium salt intermediate. Org. Biomol. Chem. 2011, 9, 5573–5578. [Google Scholar] [CrossRef]
- Li, N.; Shi, L.; Wang, X.; Guo, F.; Yan, C. Experimental study of closed system in the chlorine dioxide-iodide-sulfuric acid reaction by UV-vis spectrophotometric method. Int. J. Anal. Chem. 2011, 2011, 130102. [Google Scholar] [CrossRef]
- Wartiovaara, I. The influence of pH on the D1 stage of a D/CED1 bleaching sequence. Paperi ja Puu/Pap. Timber 1982, 9, 534–546. [Google Scholar]
- Wang, L.; Bassiri, M.; Najafi, R.; Najafi, K.; Yang, J.; Khosrovi, B.; Hwong, W.; Barati, E.; Belisle, B.; Celeri, C.; et al. Hypochlorous acid as a potential wound care agent: Part I. Stabilized hypochlorous acid: A component of the inorganic armamentarium of innate immunity. J. Burn. Wounds 2007, 6, e5. [Google Scholar]
- Buffa, J.M.; Grela, M.A.; Aranguren, M.I.; Mucci, V. EPR spectroscopy applied to the study of the TEMPO mediated oxidation of nanocellulose. Carbohydr. Polym. 2016, 136, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Kim, Y.; Moon, J.Y.; Lee, C.W.; Kim, D.; Ha, K.S.; Lee, D.H.; Son, H.; Yoon, S. Investigation of Oxidation Methods of Organic Radical Polymer for Cathode Material in Lithium Ion Batteries. Polymer 2014, 38, 827–831. [Google Scholar]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giraldo Isaza, L.; Mortha, G.; Marlin, N.; Molton, F.; Duboc, C. ClO2-Mediated Oxidation of the TEMPO Radical: Fundamental Considerations of the Catalytic System for the Oxidation of Cellulose Fibers. Molecules 2023, 28, 6631. https://doi.org/10.3390/molecules28186631
Giraldo Isaza L, Mortha G, Marlin N, Molton F, Duboc C. ClO2-Mediated Oxidation of the TEMPO Radical: Fundamental Considerations of the Catalytic System for the Oxidation of Cellulose Fibers. Molecules. 2023; 28(18):6631. https://doi.org/10.3390/molecules28186631
Chicago/Turabian StyleGiraldo Isaza, Laura, Gérard Mortha, Nathalie Marlin, Florian Molton, and Carole Duboc. 2023. "ClO2-Mediated Oxidation of the TEMPO Radical: Fundamental Considerations of the Catalytic System for the Oxidation of Cellulose Fibers" Molecules 28, no. 18: 6631. https://doi.org/10.3390/molecules28186631
APA StyleGiraldo Isaza, L., Mortha, G., Marlin, N., Molton, F., & Duboc, C. (2023). ClO2-Mediated Oxidation of the TEMPO Radical: Fundamental Considerations of the Catalytic System for the Oxidation of Cellulose Fibers. Molecules, 28(18), 6631. https://doi.org/10.3390/molecules28186631