1. Introduction
A broad definition of drug polymorphism includes molecular polymorphs, amorphous phases, hydrates and solvates, salts, and cocrystals, while a strict definition only includes crystal structures composed of the same molecules, atoms, or ions [
1]. Polymorphism is a fundamental consideration in drug crystallization, and polymorph control is an important factor in drug safety and efficacy. Different crystal structures can confer different physical chemical properties, such as solubility, dissolution rates, and stability, and lead to different biological utilization [
2]. For Biopharmaceutics Classification System (BCS) II and IV drugs, a low effective blood drug concentration is often caused by the low solubility of a drug polymorph, and it is necessary to find polymorphs with better solubility. A metastable drug polymorph can have serious drawbacks due to high solubility and phase transition during storage into a more stable, ineffective form, which may not be clinically suitable. Many technologies have been developed to distinguish drug polymorphs, including powder X-ray diffraction (PXRD), differential scanning calorimetry (DSC), Fourier transform infrared spectroscopy (FT-IR), Raman spectroscopy (Raman), solid–state nuclear magnetic resonance (ss-NMR), ultraviolet–visible spectroscopy (UV–Vis), hot-stage microscopy, small- angle X-ray scattering (SAXS), neutron scattering, and flow-driven spectral chaos (FSC) [
3,
4,
5,
6].
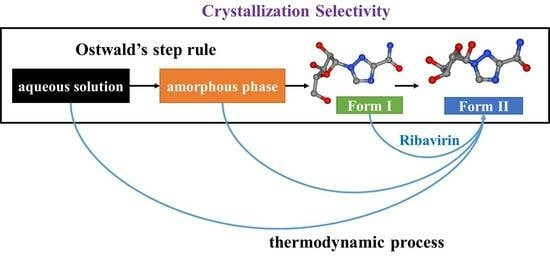
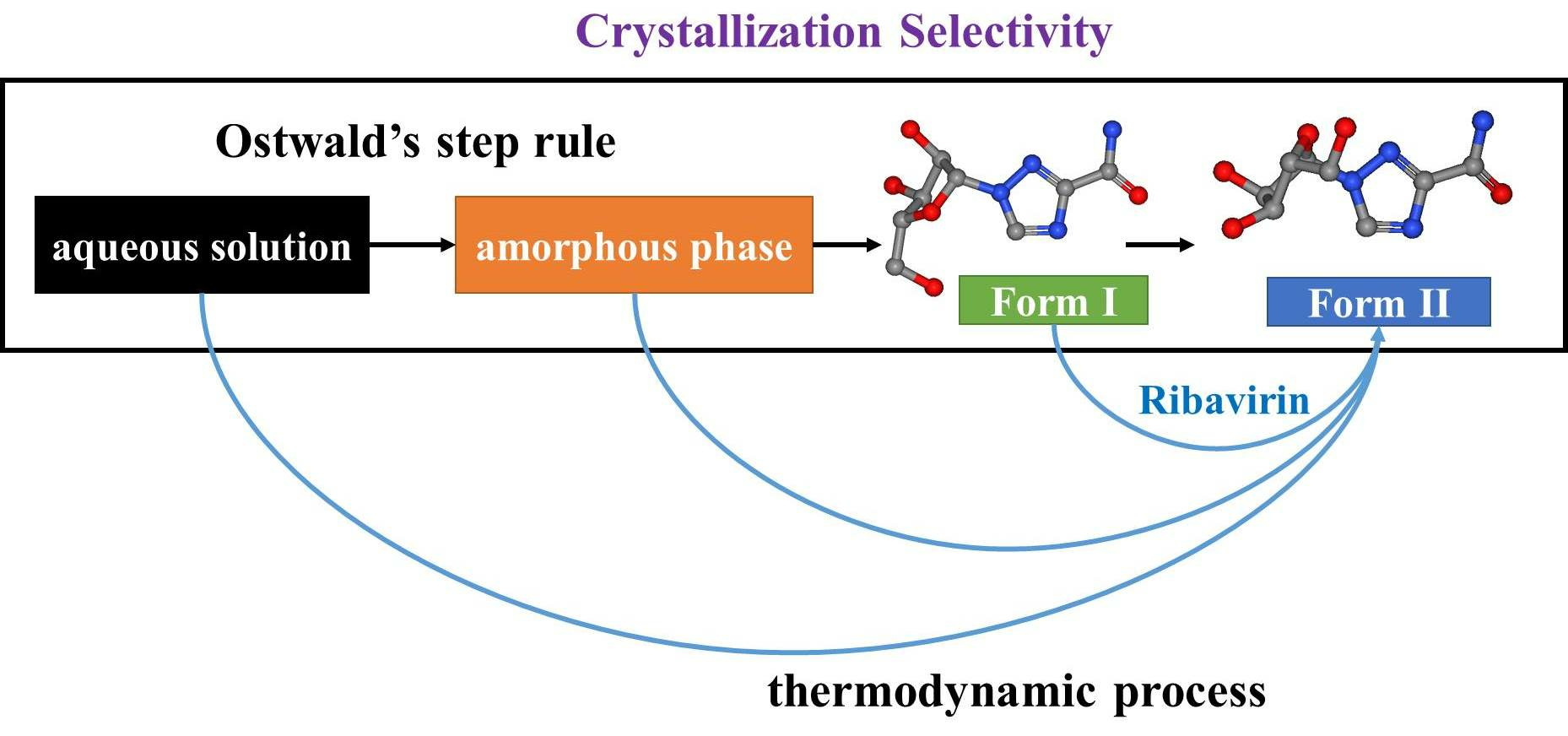
The control of molecular polymorphisms has been studied thoroughly in the laboratory and in industrial production, and selective crystallization processes are observed in many different systems [
7,
8], such as solution crystallization, melt crystallization, amorphous phase recrystallization, solvate desolvation, and evaporative crystallization [
9,
10]. For example, it is easy to obtain the metastable inosine α form from aqueous solution or the stable inosine β form from dimethylsulfoxide (DMSO) solution [
11]. Amorphous guanine has a tendency to recrystallize into the metastable anhydrous guanine β form in biominerals and in the laboratory [
12,
13], and amorphous calcium carbonate can have different short-range orders, such as calcite or vaterite [
14]. The product of guanine monohydrate dehydration is a framework of guanine monohydrate [
15], and the dehydration product of inosine dihydrate is the metastable inosine α form [
16]. However, theoretical explanations of selective crystallization processes are still somewhat inadequate.
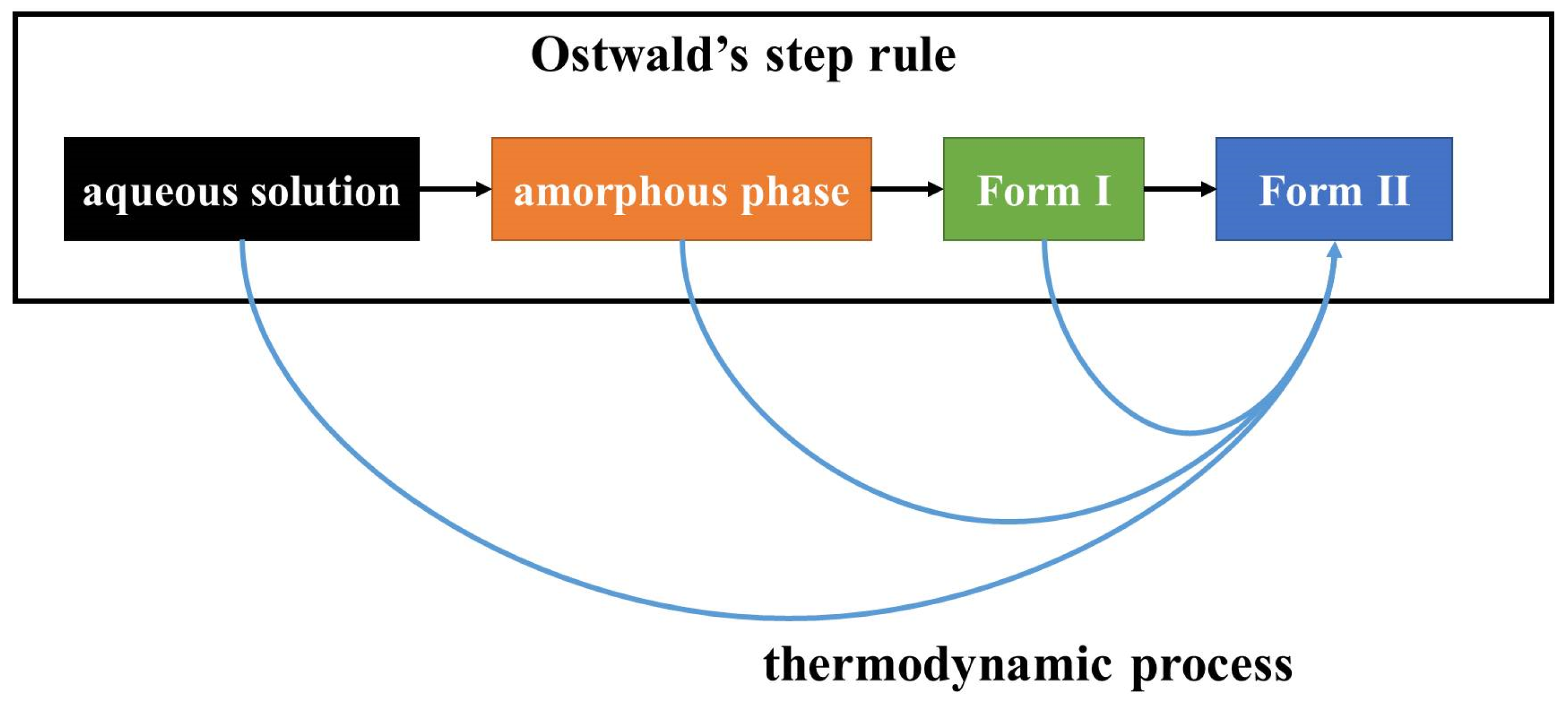
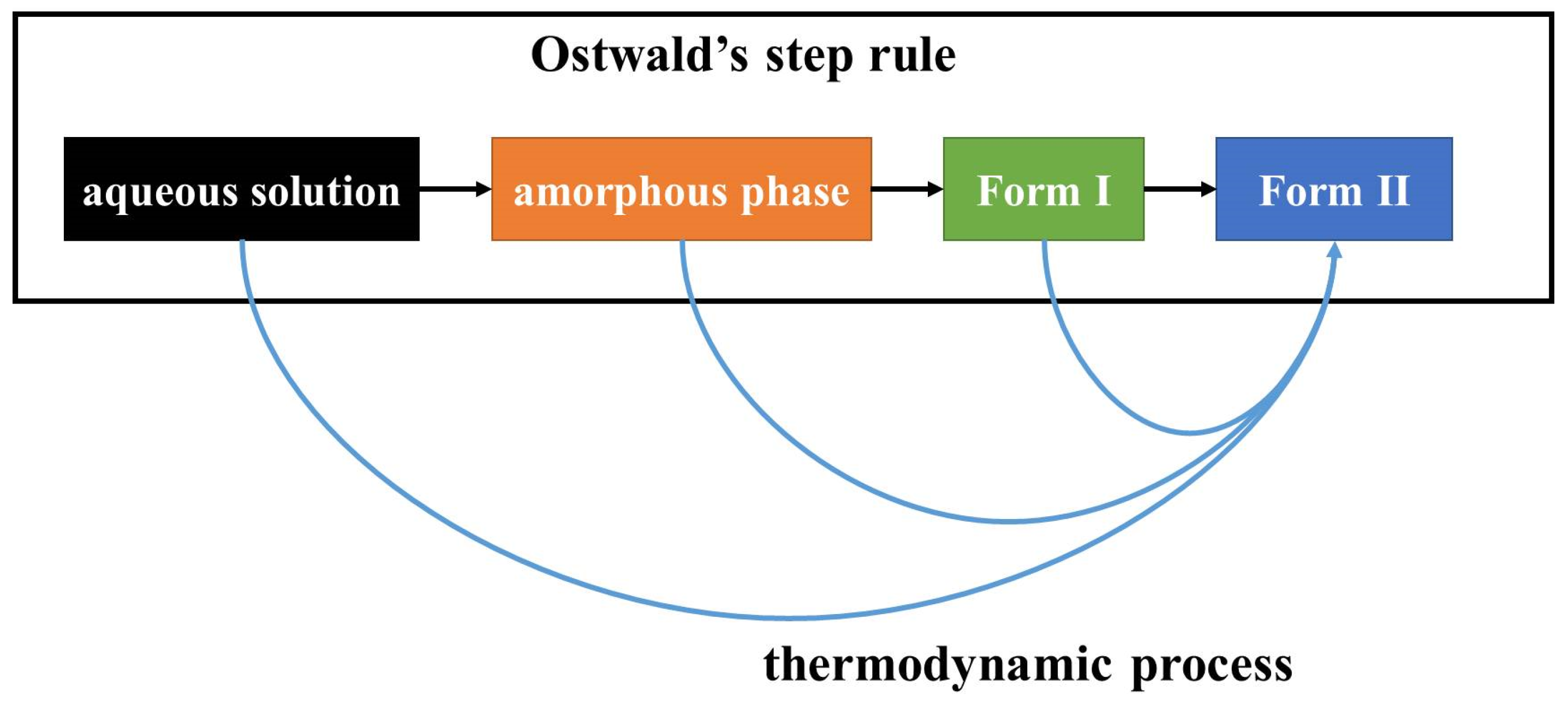
The first step in crystallization is nucleation. In classical nucleation theory (CNT), monomers such as molecules, atoms, or ions in a supersaturated condition coalesce into a nucleation cluster, and then additional monomers attach one by one to form a crystalline phase [
17]. The molecular arrangement in the nucleation cluster determines the polymorph. CNT nucleation is suitable to describe a process via monomer-by-monomer addition.
However, the crystallization process can also apply to the association of multi-ion complexes, oligomeric clusters, crystalline or amorphous nanoparticles, and monomer rich liquid droplets [
18], for example. Non-classical nucleation theory (NCNT) was proposed to describe crystallization processes with these non-monomer additions. In the field of molecular crystals, the hypothesis that a certain molecular assembly in solution induces a polymorph of similar assembly structure was proposed to explain solution crystallization selectivity [
3,
4]. However, this theory cannot explain all crystallization processes, and the correlation between the solution associate and crystal synthon sometimes cannot be established [
3,
4]. We believe that the molecular assembly theory can explain the process from the non-monomer intermediate phase (assemblies) to crystals, but that it fails to explain the relationships between the monomer and crystal or monomer and non-monomer intermediate phase.
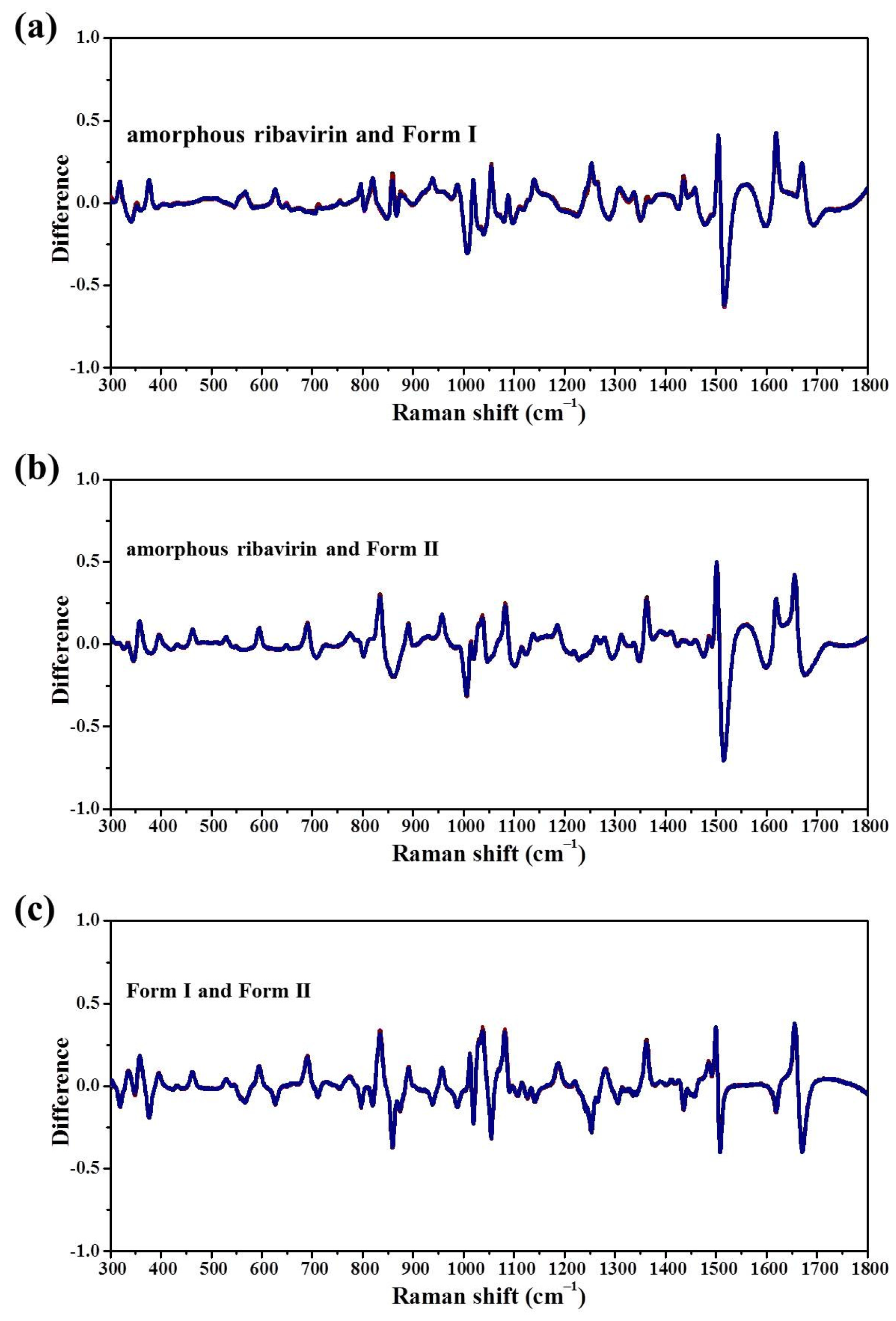
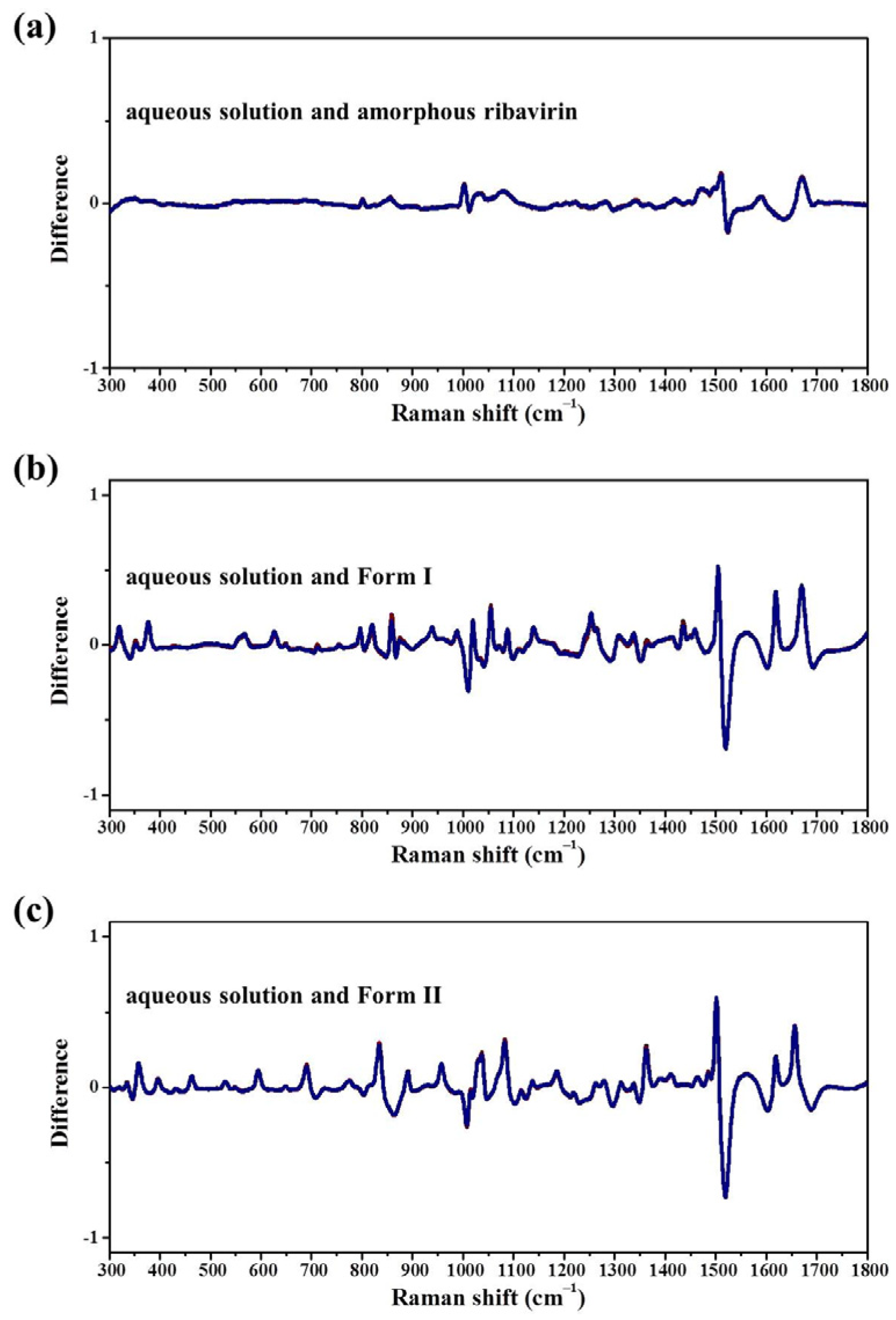
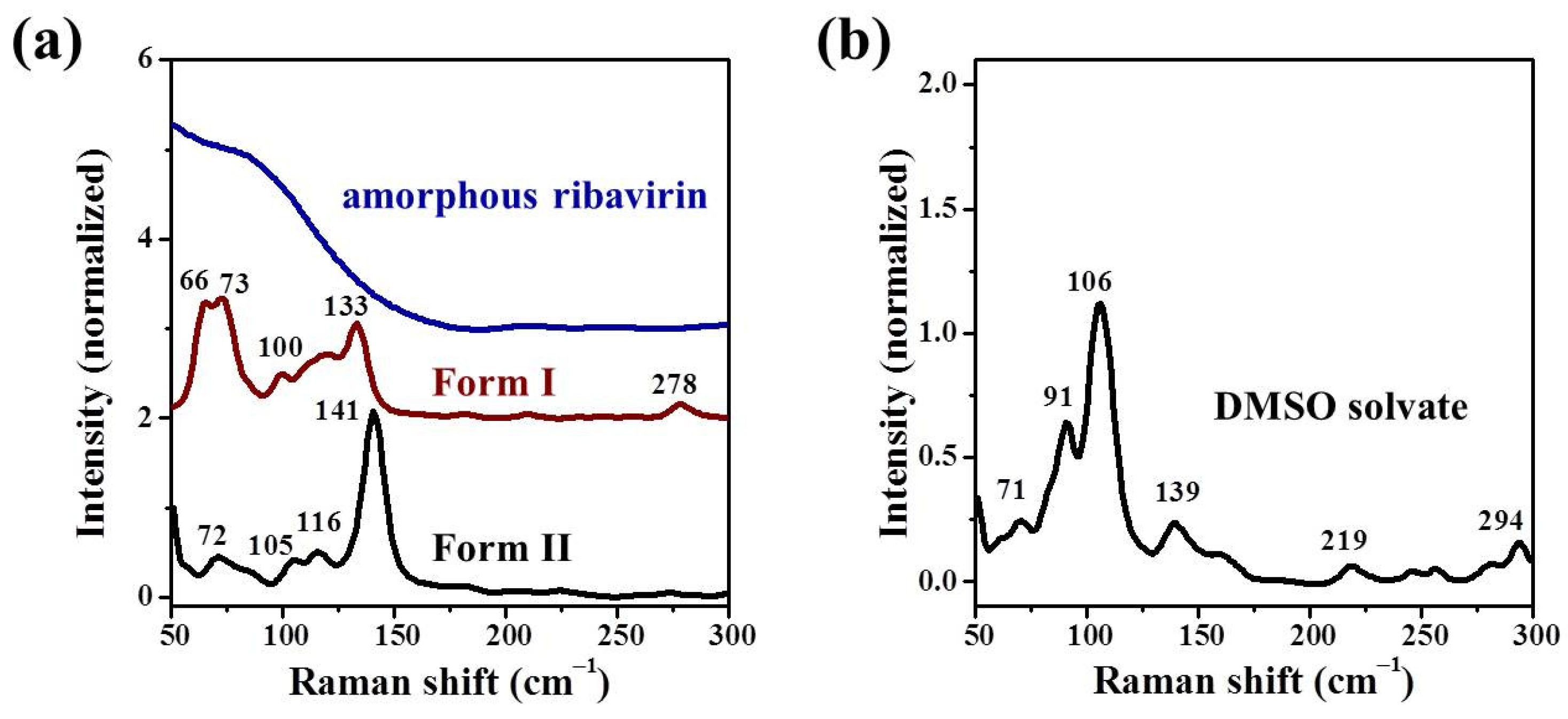
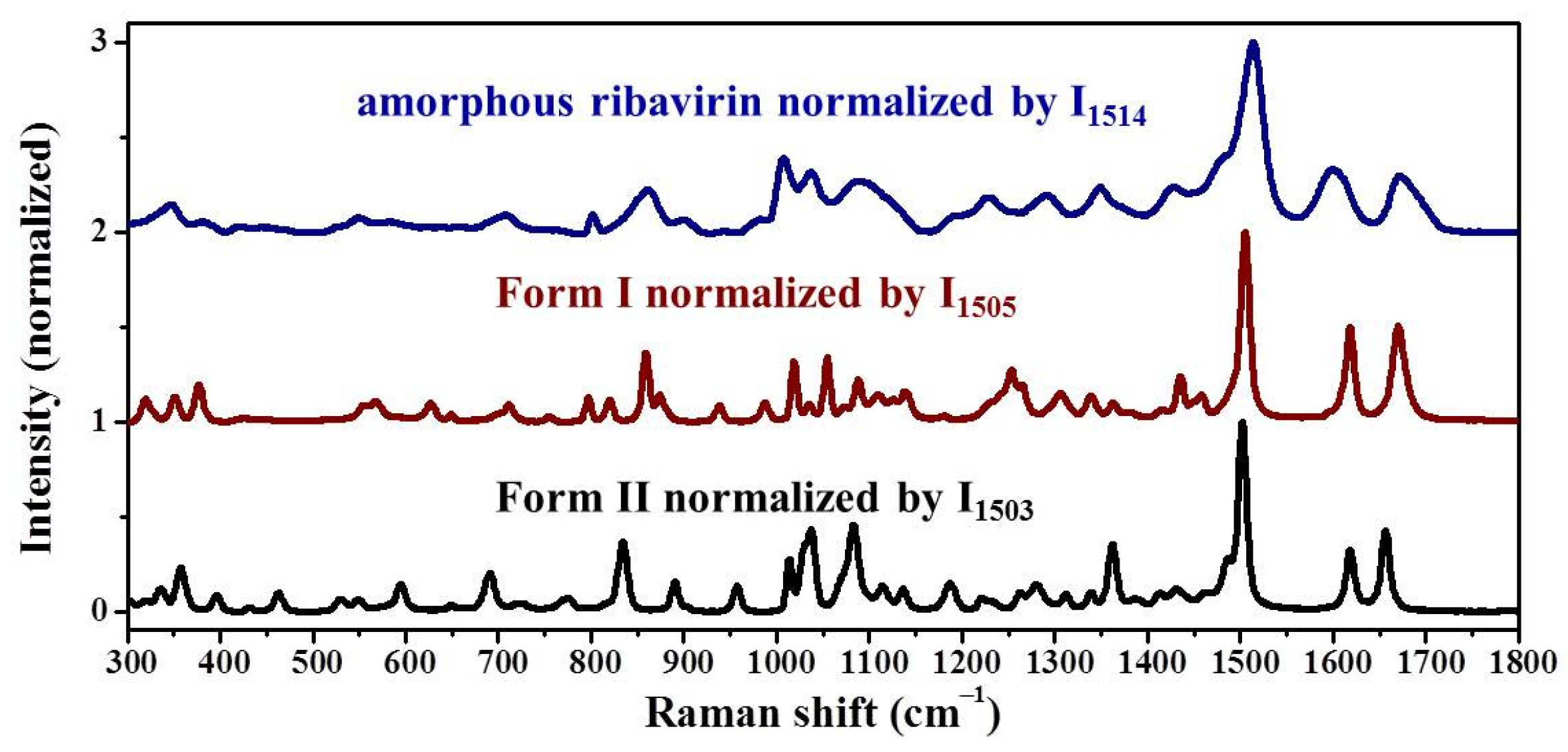
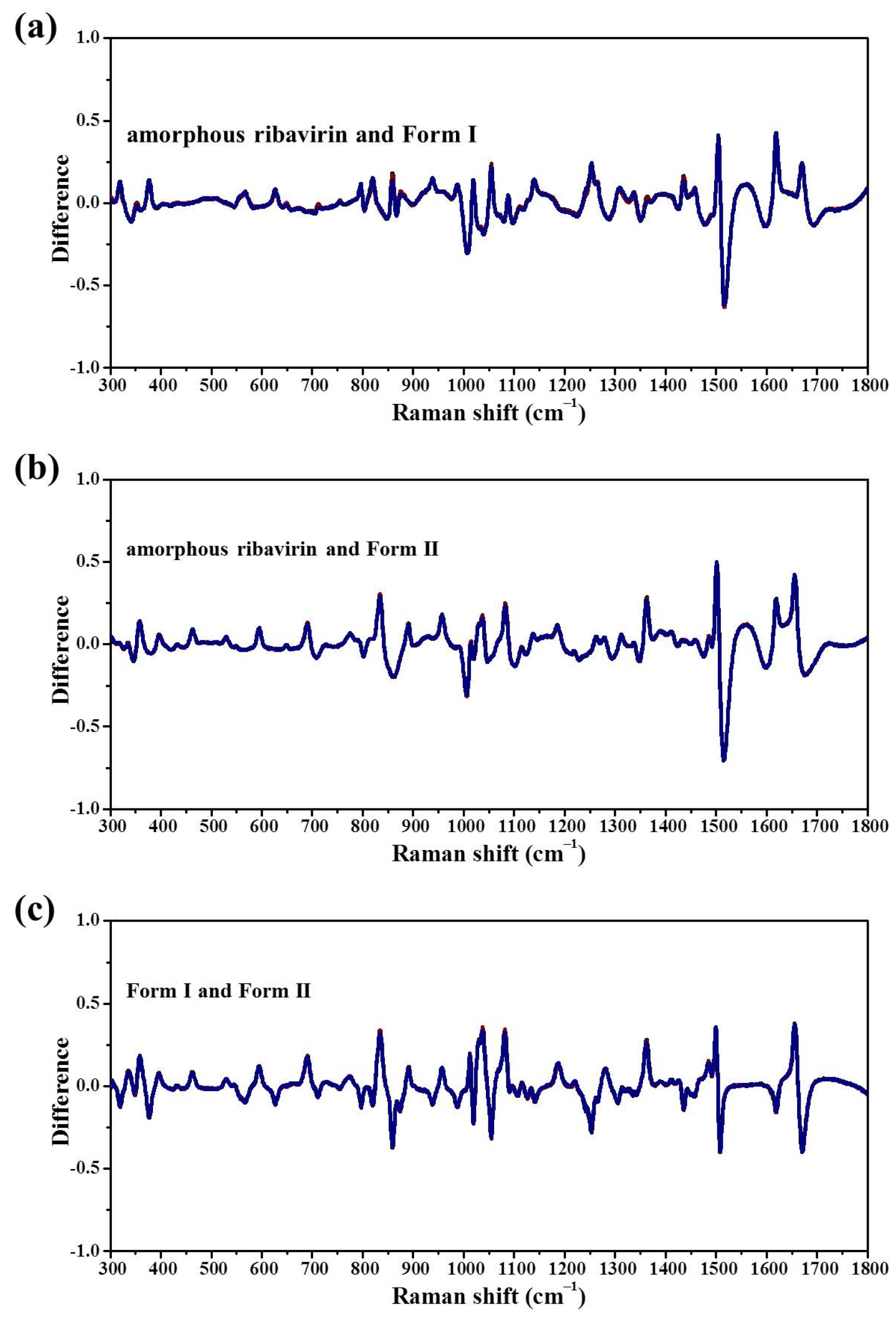
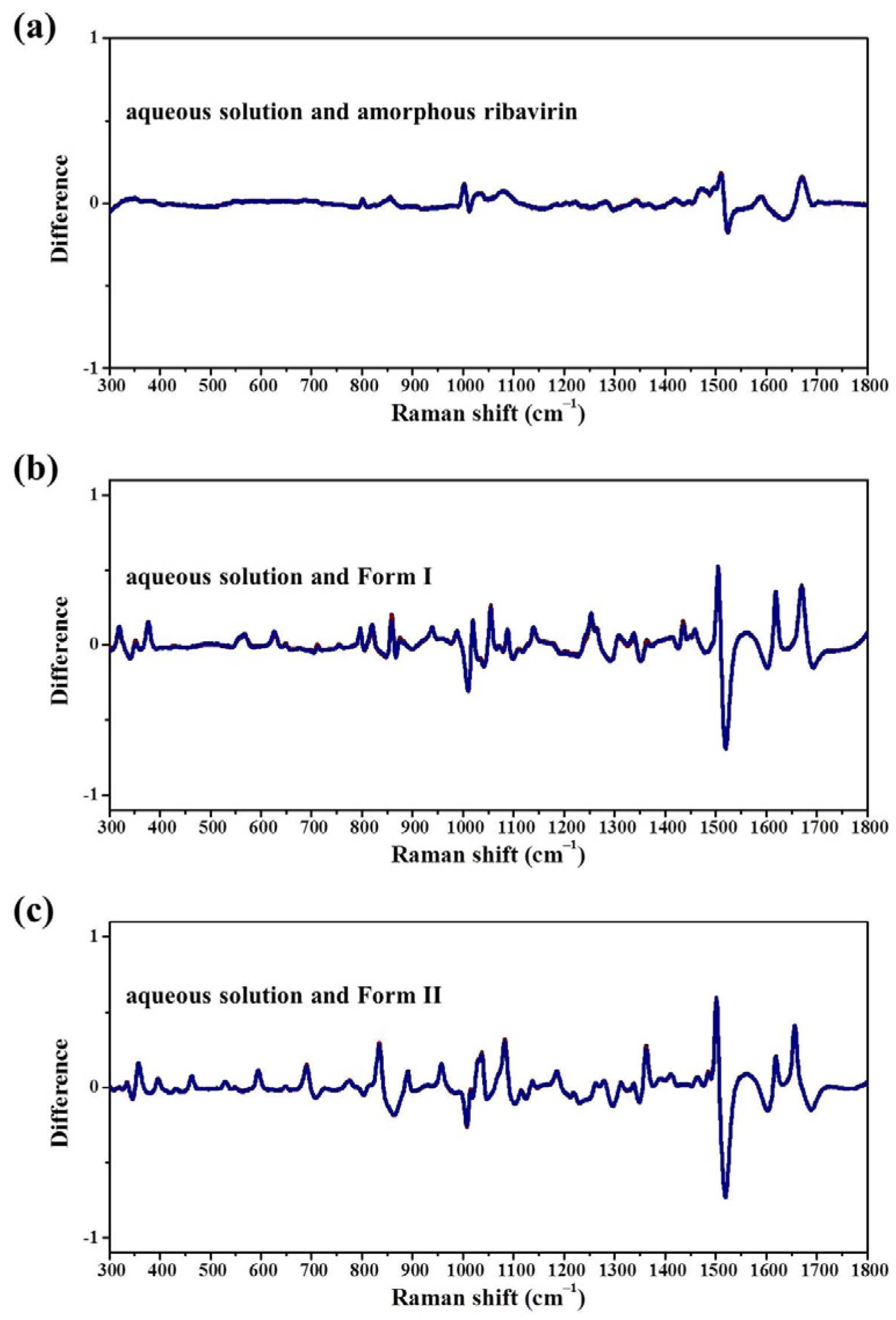
Standard analysis techniques that relate to monomer assembly and crystals include FTIR, Raman, NMR, and neutron scattering. However, they are inadequate in correlating all the related phases at the same time and establishing similarities among these phases. Our group has proposed mid-frequency Raman difference spectroscopy (MFRDS) to solve this problem and has established phase relationships to explain the selective crystallization behaviors of inosine and simvastatin [
11,
19]. It is still necessary, however, to test the power of MFRDS and identify new selective crystallization processes for well-researched molecules based on MFRDS results.
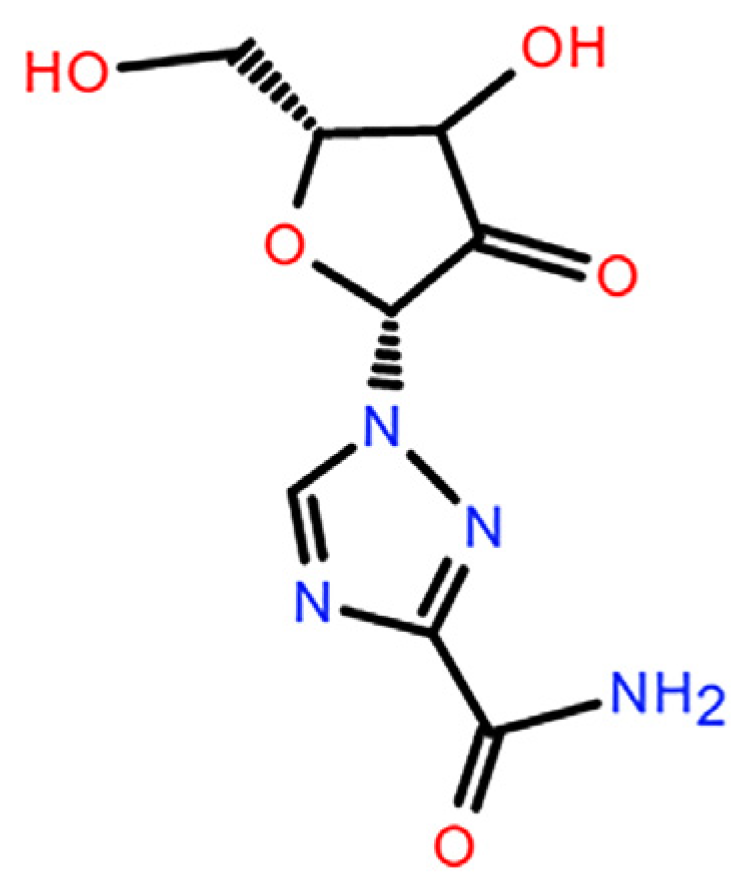
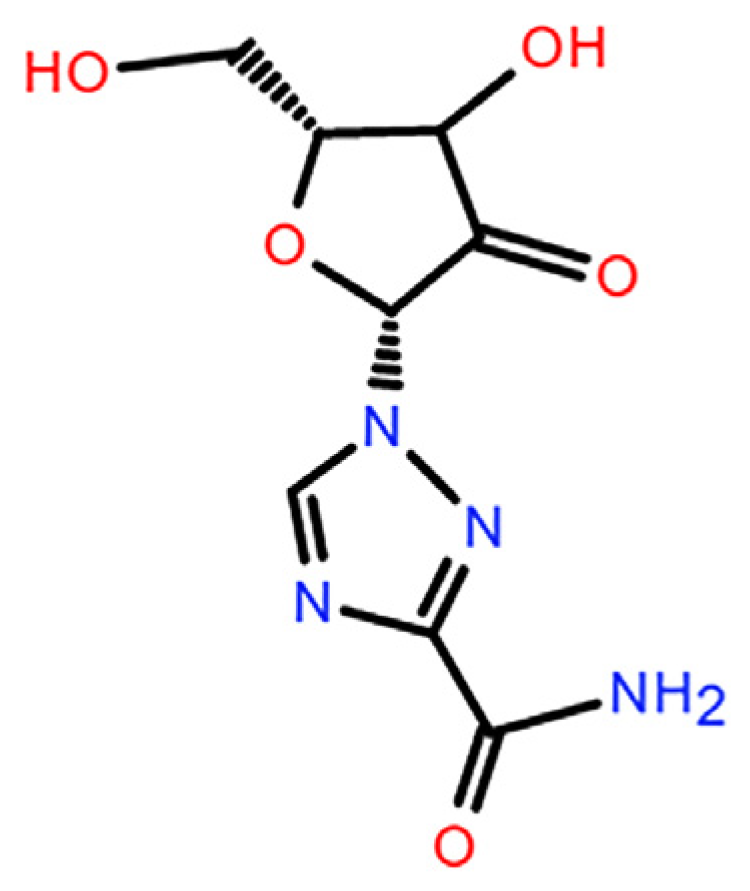
Ribavirin (C
8H
12N
4O
5, 1-β-D-ribofuranosyl-lH-1, 2, 4-triazole-3-carboxamide, CAS: 36791-04-5,
Figure 1) is a broad-spectrum nucleoside antiviral drug and was recommended in the Interim Guidance for Diagnosis and Treatment (Seventh Edition) of COVID-19 [
20]. The molecular structure of ribavirin contains flexible ribofuranosyl and rigid triazole structures. The flexible part endows ribavirin with rich conformational potential and its many hydrogen-bonding units induce complex intermolecular interactions. These factors favor the formation of polymorphs and amorphous phases, as is the case for guanosine and inosine [
11].
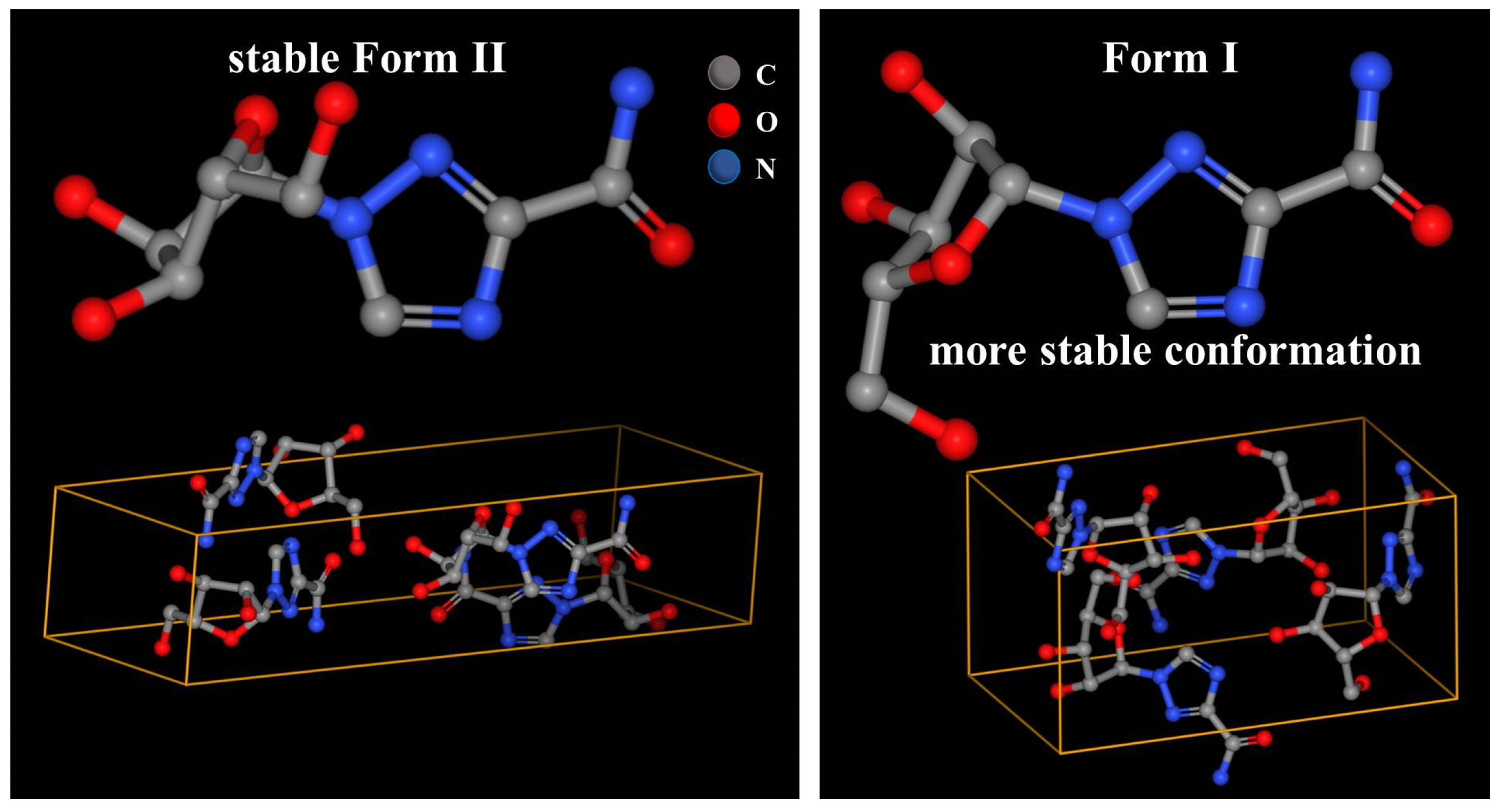
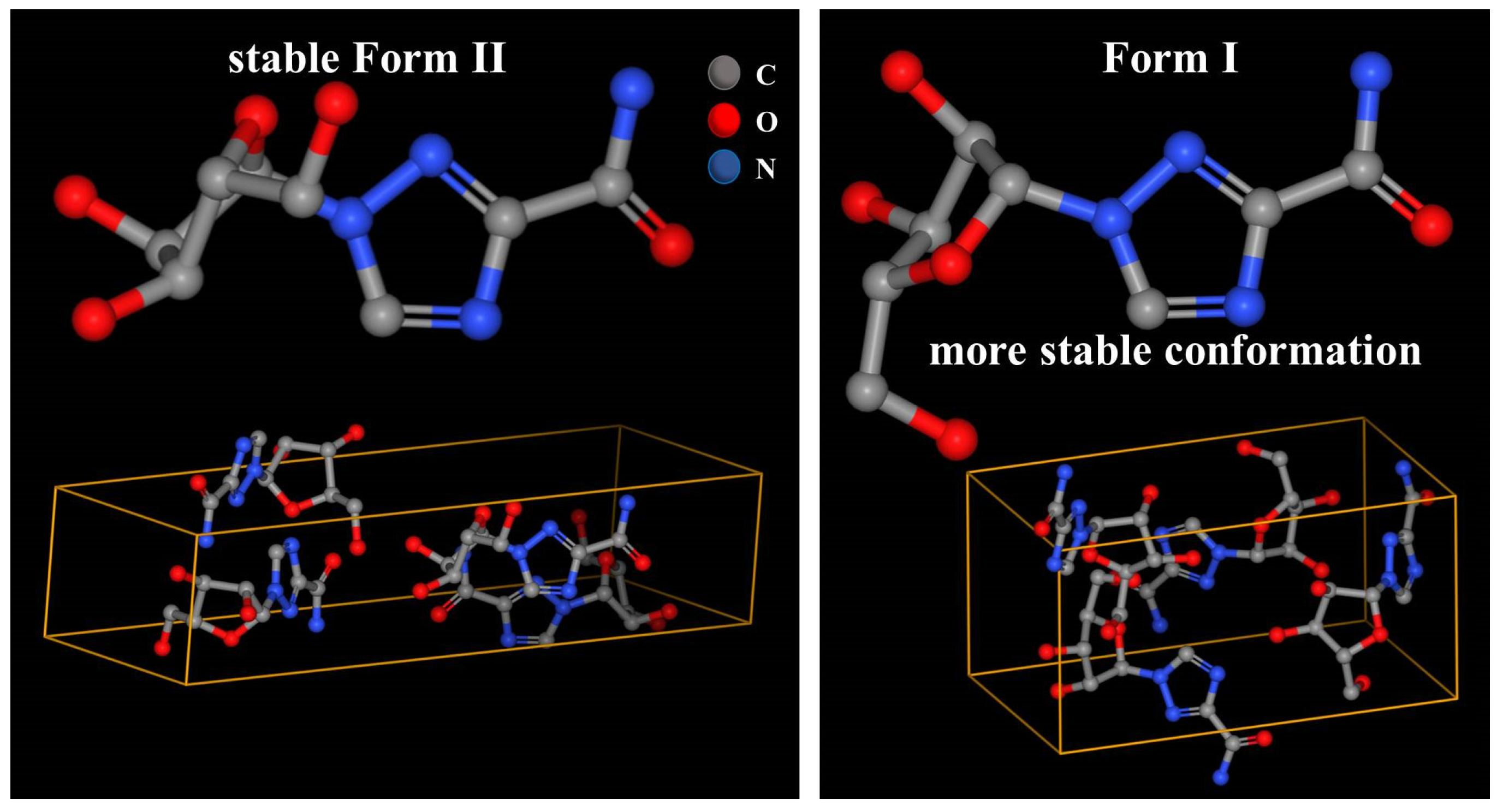
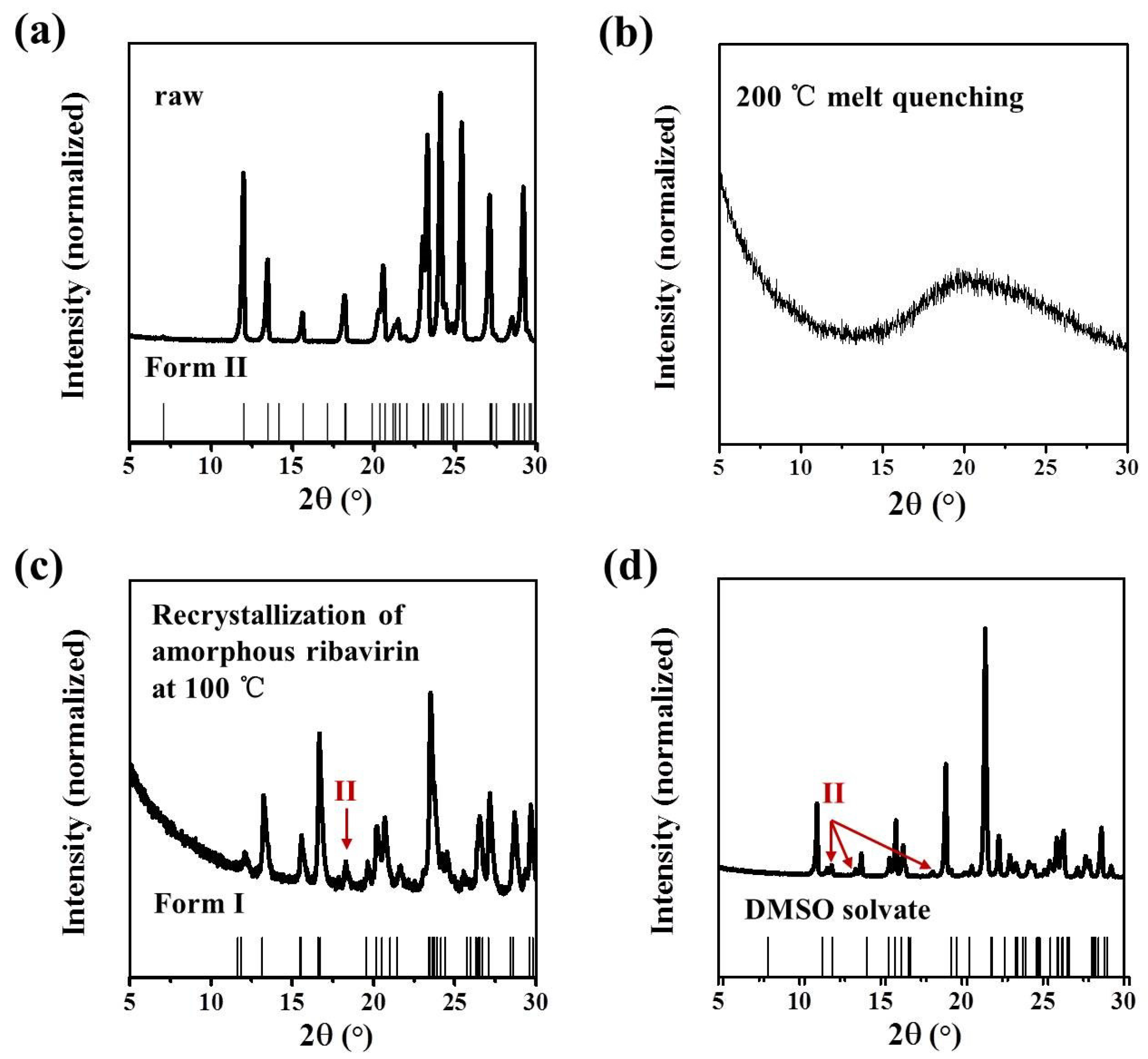
Anhydrous ribavirin has two polymorphs (stable Form II and metastable Form I,
Table 1) and one amorphous phase. The conformations are quite different in Form II and Form I (
Figure 2), and the conformational energy of Form I is significantly lower [
21,
22]. The single crystal of ribavirin DMSO solvate has been reported (
Table 1) [
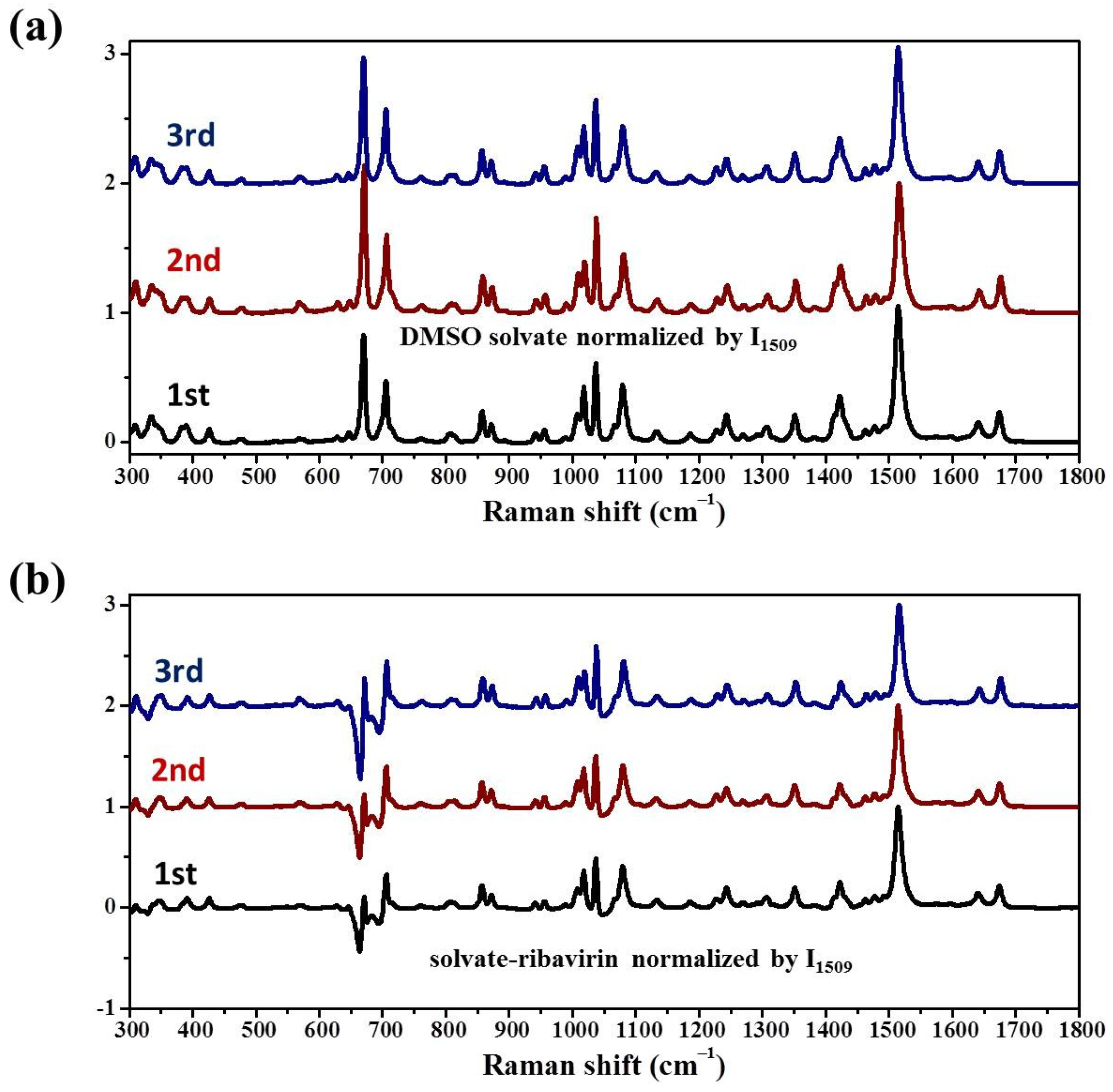
23] but has not been published in the Cambridge Crystallographic Data Centre (CCDC). While the main synthesis methods of ribavirin solid forms are listed (
Table 2), the selective crystallization of ribavirin solution, the recrystallization of amorphous ribavirin, and the desolvation of ribavirin DMSO solvate have not been explained.
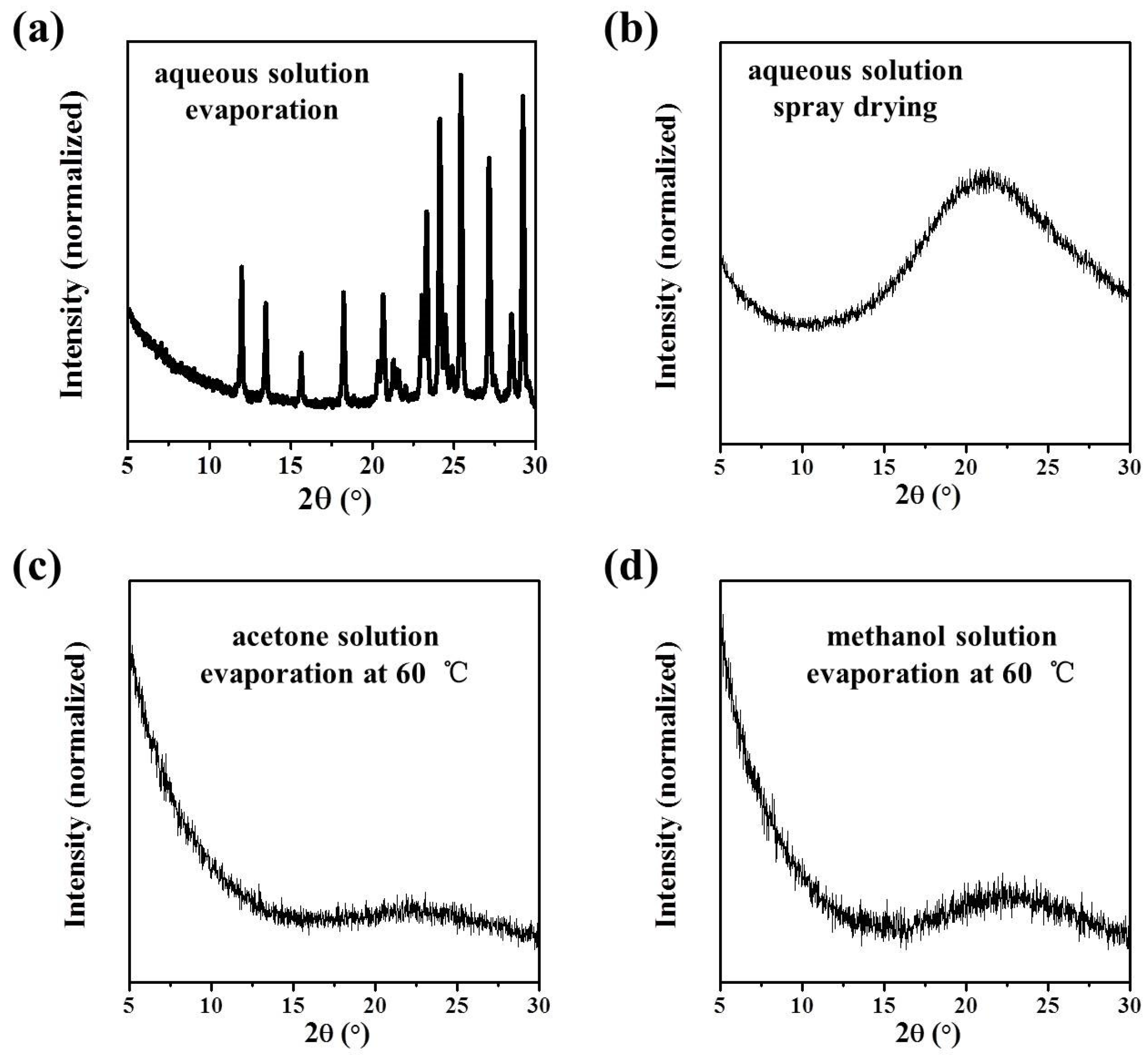
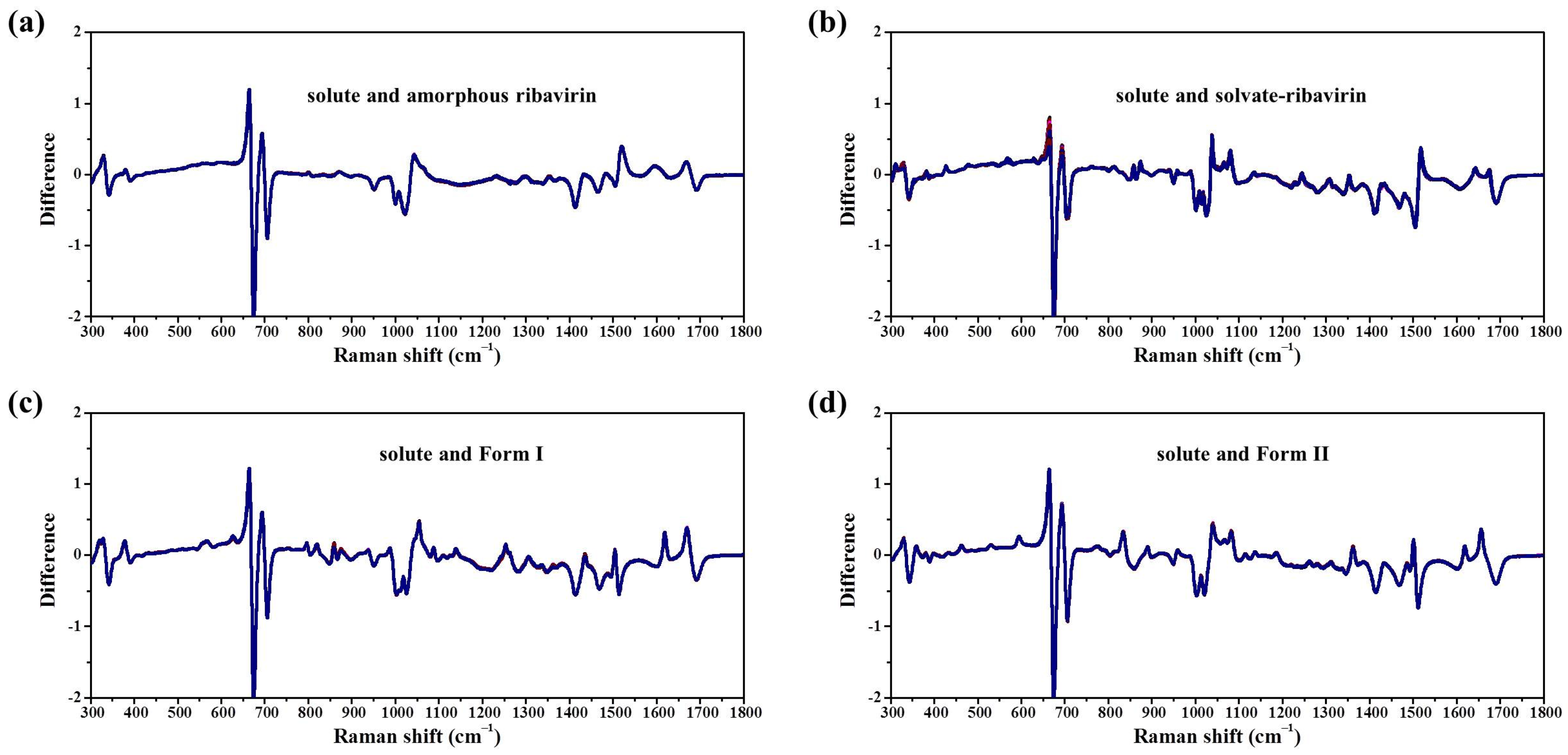
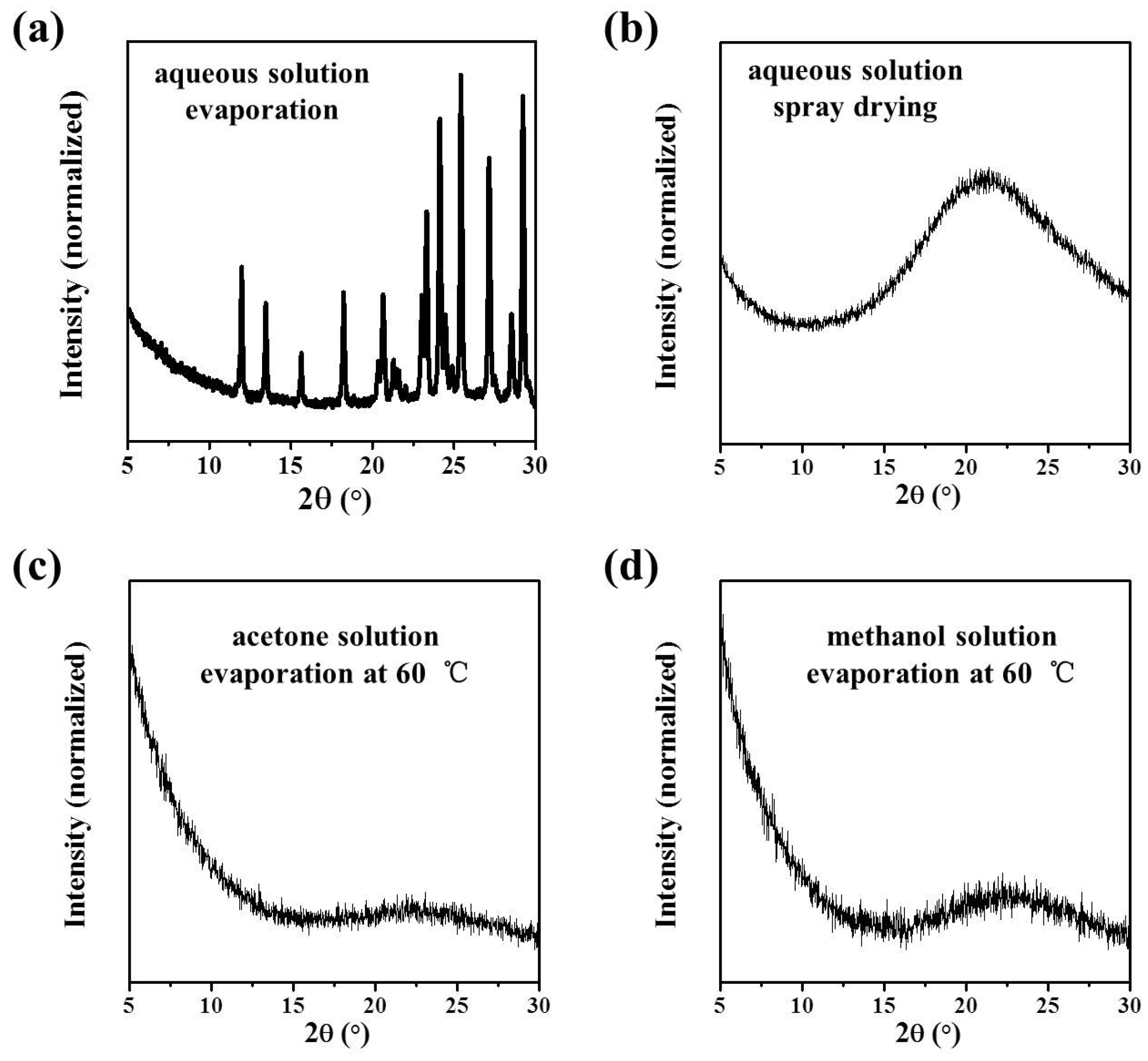
In this work, the crystallization selectivity of ribavirin solution and the amorphous phase are studied by MFRDS. Selective crystallizations from amorphous ribavirin to Form I and from aqueous solution to amorphous ribavirin are effected and explained in depth. Furthermore, reasons for the poor reproducibility of ribavirin DMSO solvate are proposed. In general, MFRDS is further demonstrated as a theoretical foundation for the preparation of amorphous and metastable phases of organic molecules.
4. Experiments
4.1. Materials
Ribavirin (C8H12N4O5, 98%, Form II) and ethyl acetate (C4H8O2, AR) were bought from shanghai Aladdin, China. dimethyl sulfoxide (DMSO, C2H6OS, AR) from Beijing Sinopharm, China. ethanol (C2H6O, AR) from Shanghai Hushi, China.
4.2. Polymorphs
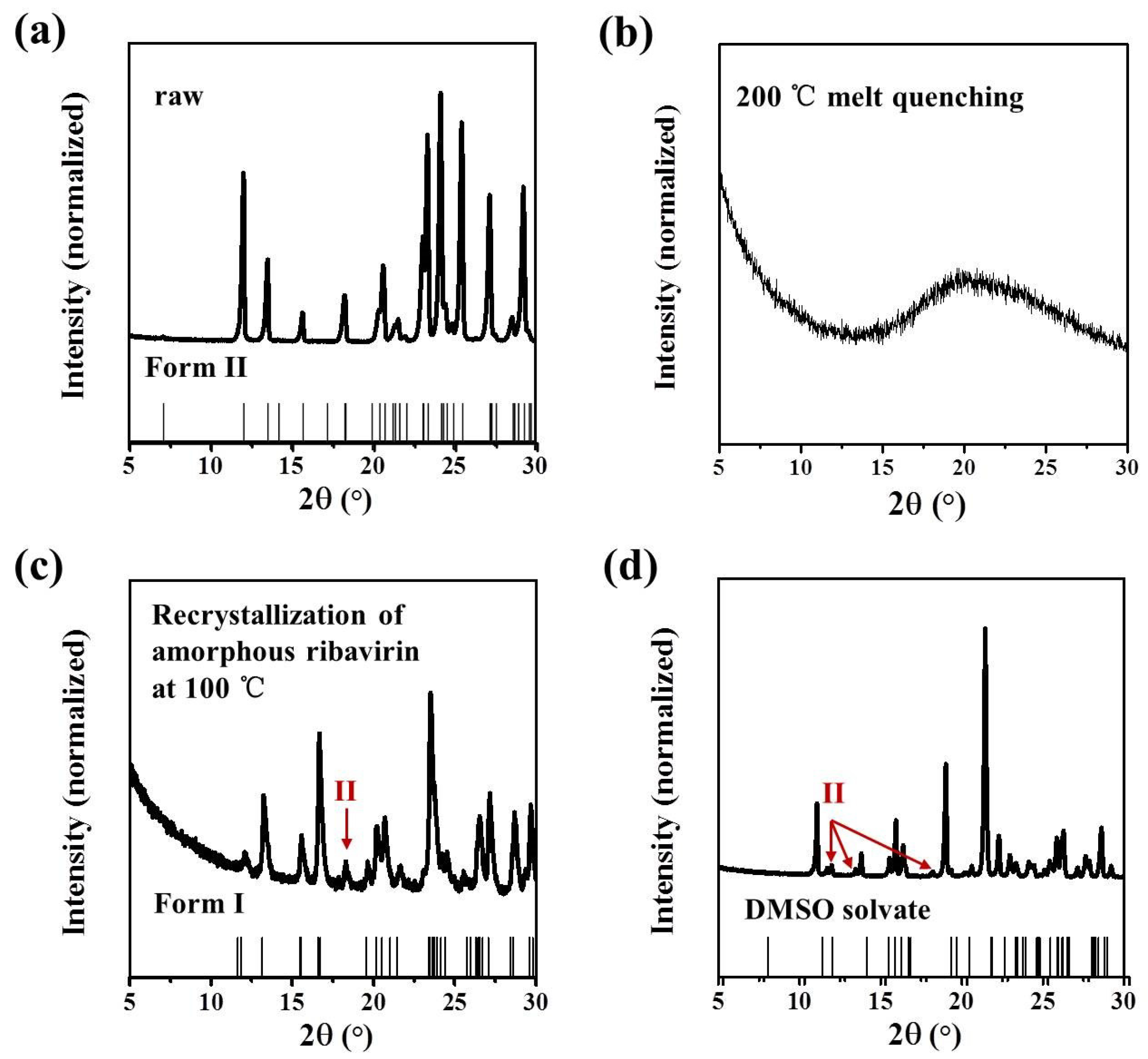
Commercial ribavirin was provided as Form II. Quenched amorphous ribavirin was prepared by melting ribavirin powder on an aluminum substrate at 200 °C (DLAB HP380-Pro) for several seconds until the powder melted completely, and then cooling the liquid under atmospheric conditions. Spray dried amorphous ribavirin was obtained from 1 wt% ribavirin aqueous solution by spray drying (Shanghai Pilotech, Shanghai, China. YC-015, inlet 160 °C, 1.5–2 L·h−1). Ribavirin Form I was synthesized by heating amorphous ribavirin at 100 °C for several hours to ensure complete recrystallization of the amorphous phase. Ribavirin DMSO solvate was obtained by dissolving commercial ribavirin (0.5 g) in DMSO (2.5 mL), adding ethyl acetate (10 mL) and stirring overnight. The obtained DMSO solvate was filtered and washed with ethanol.
4.3. Characterization
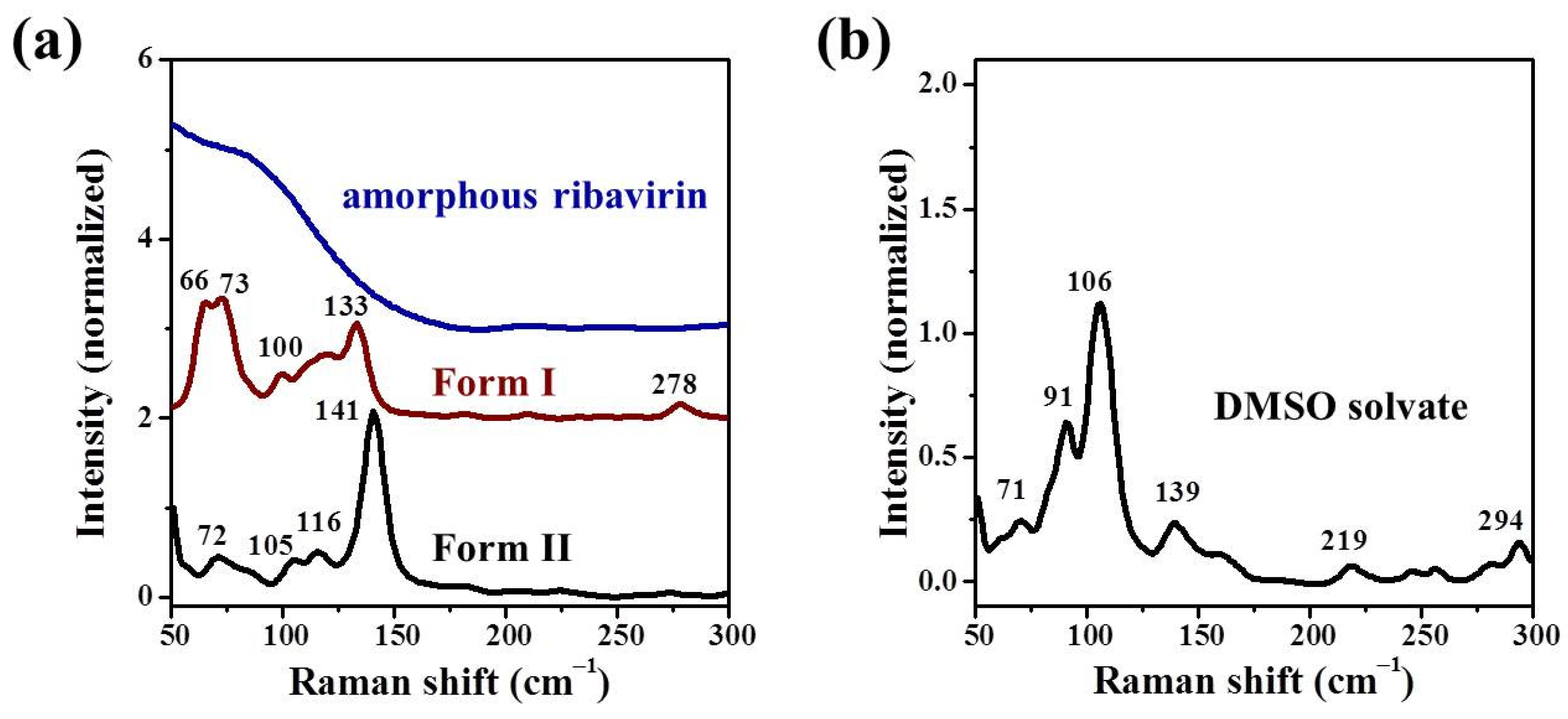
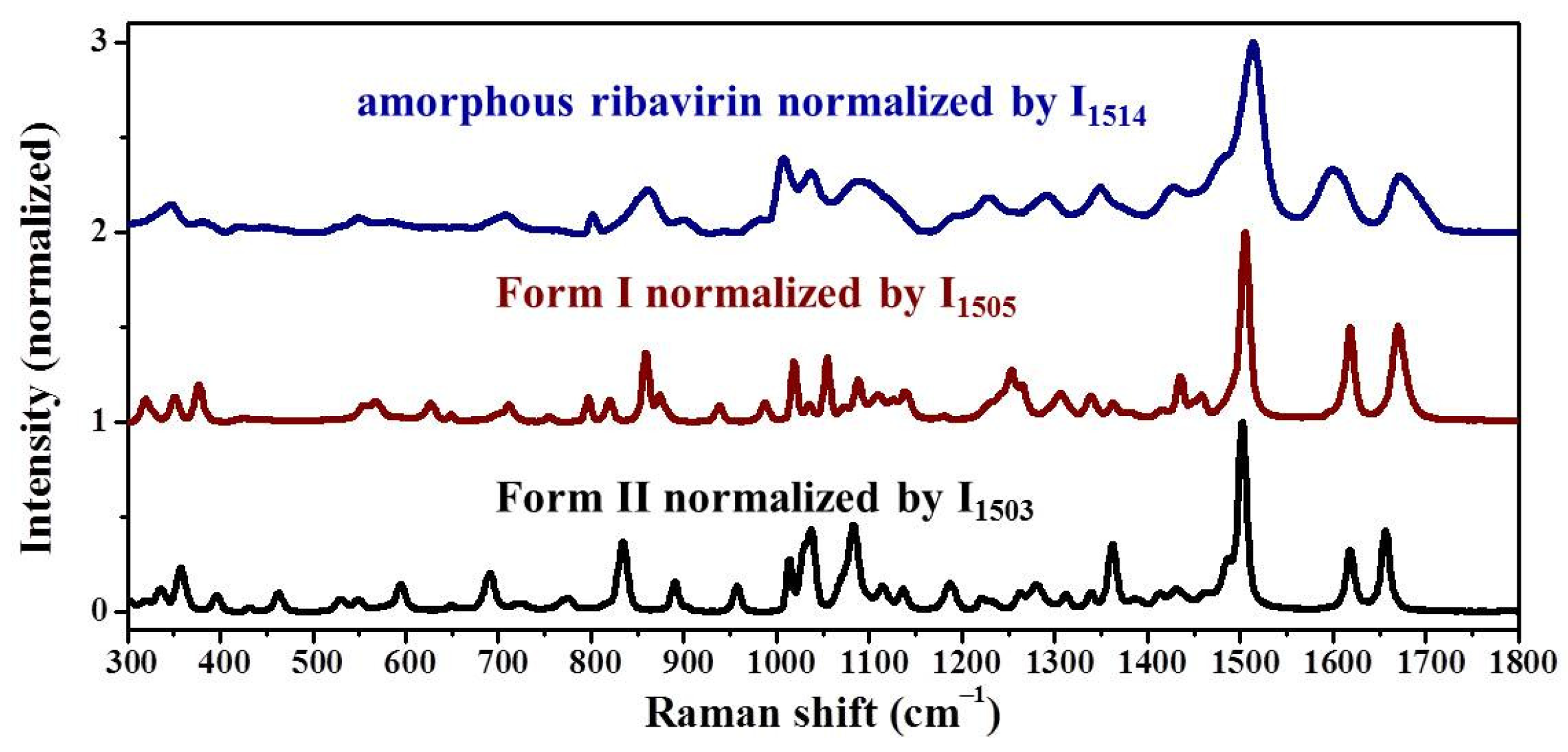
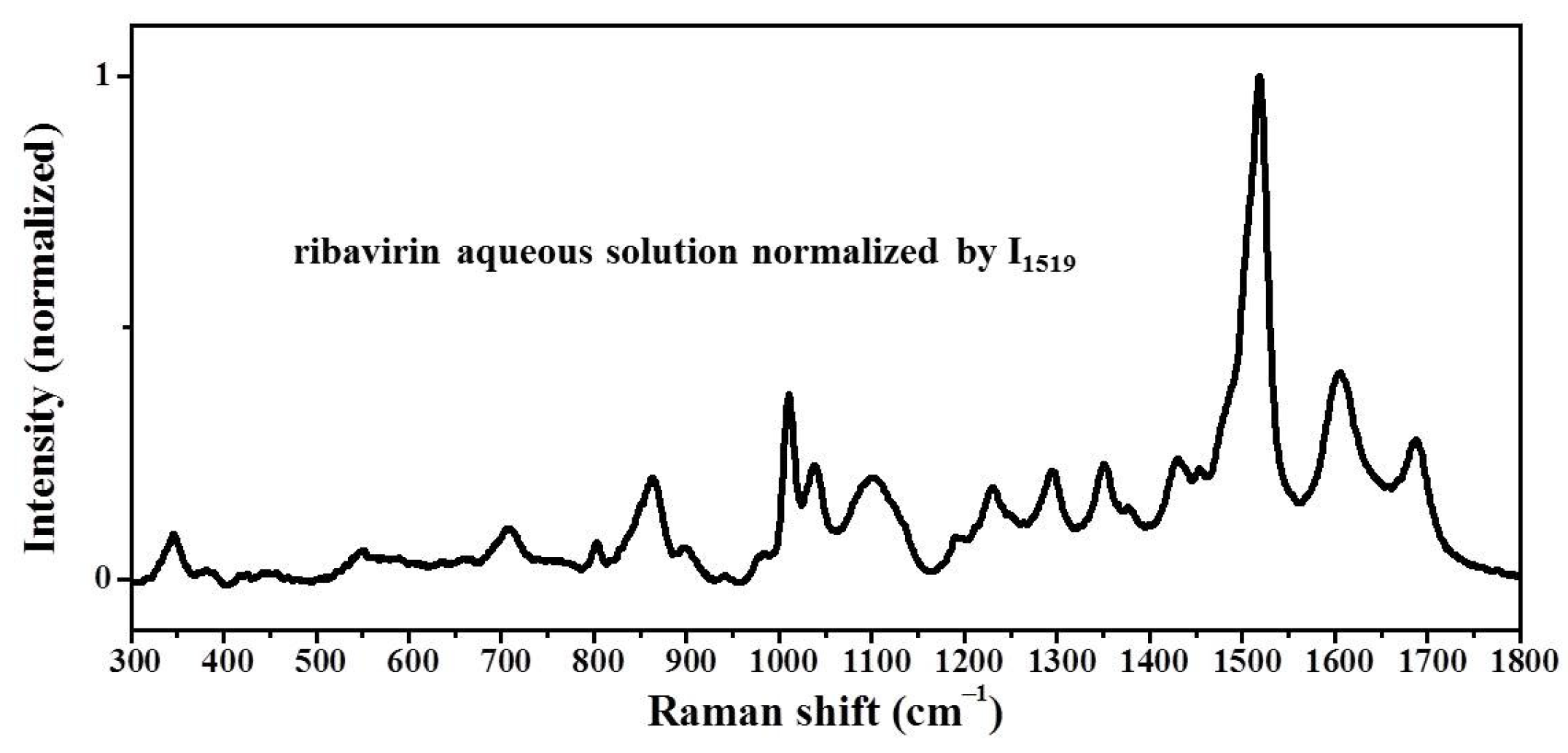
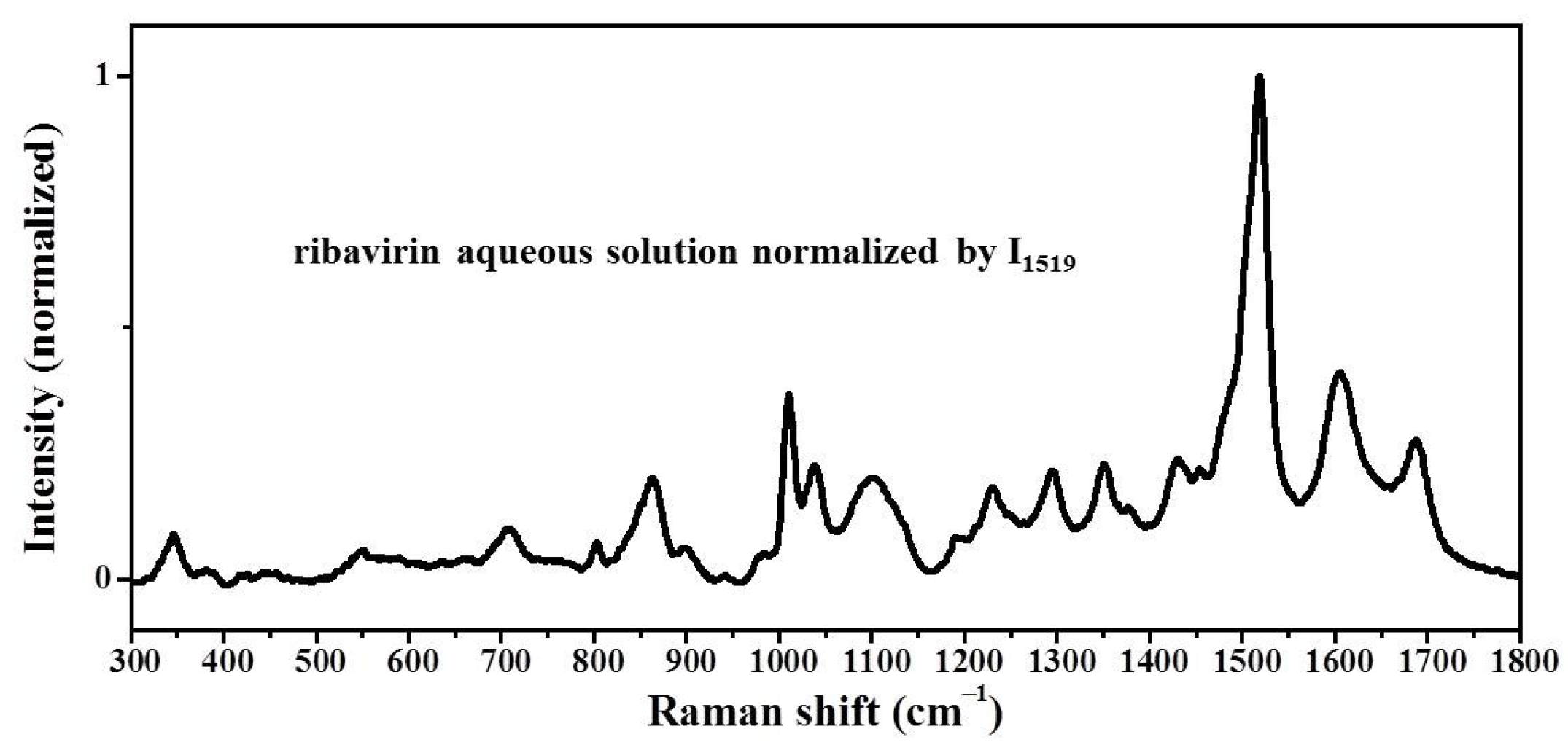
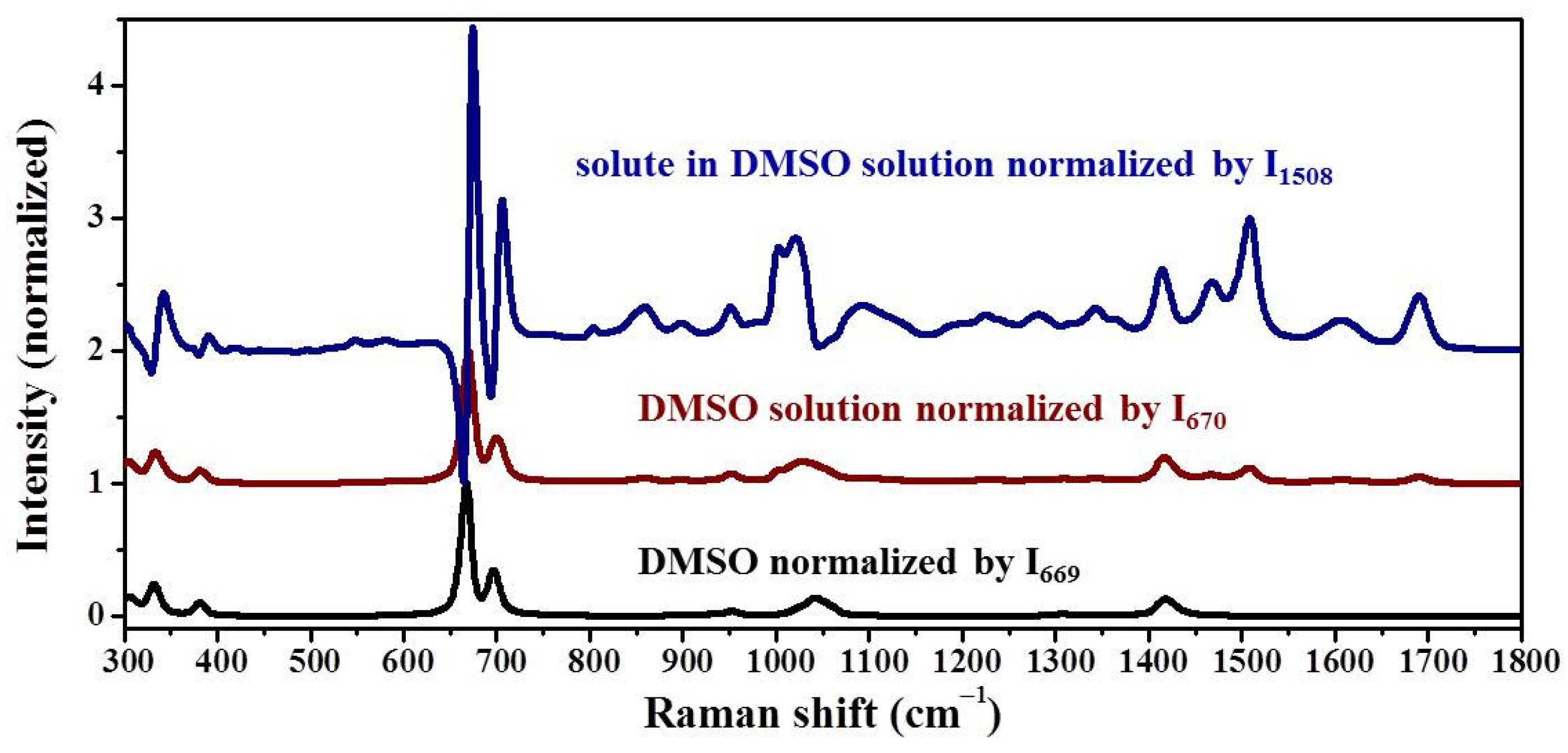
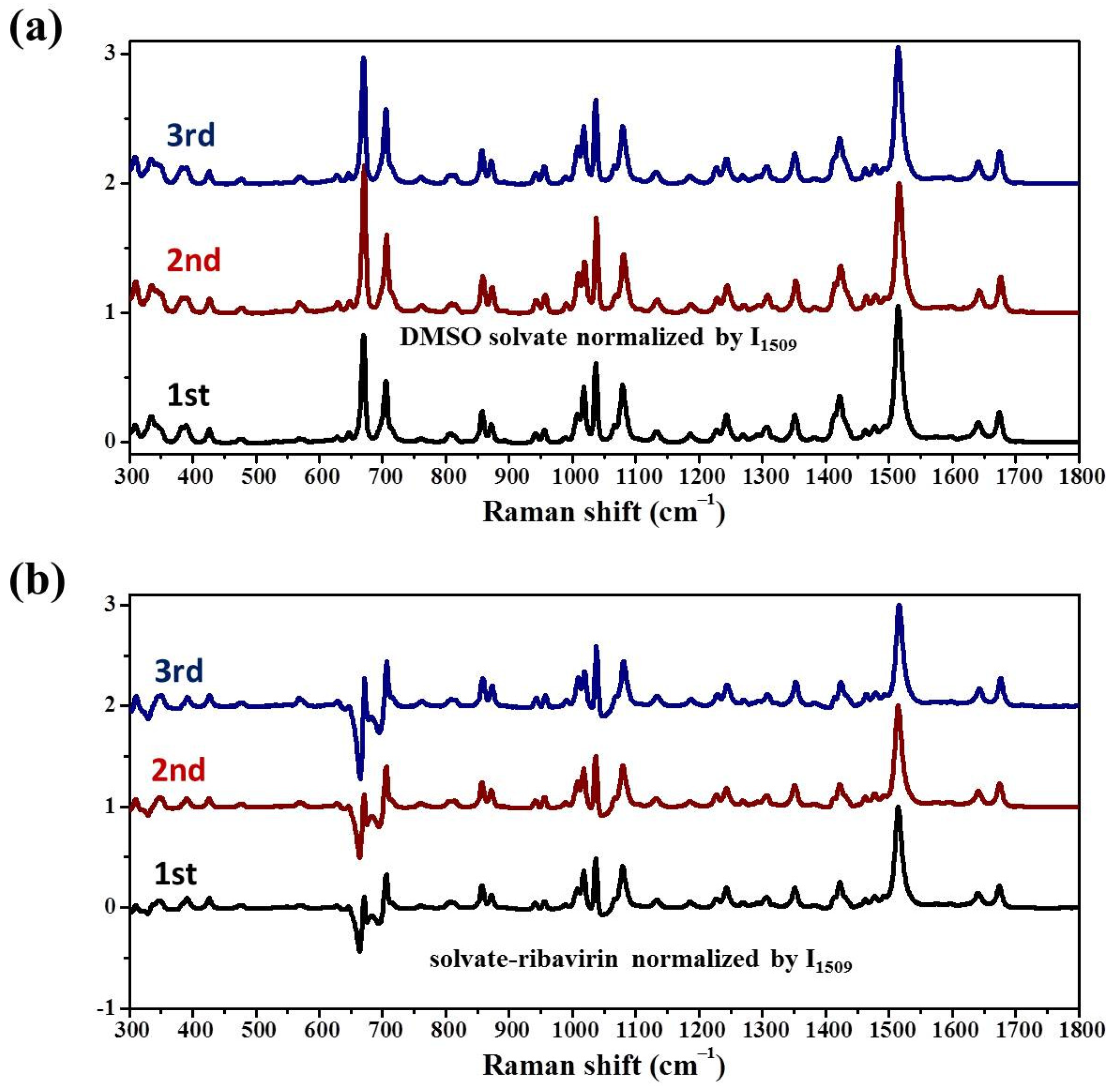
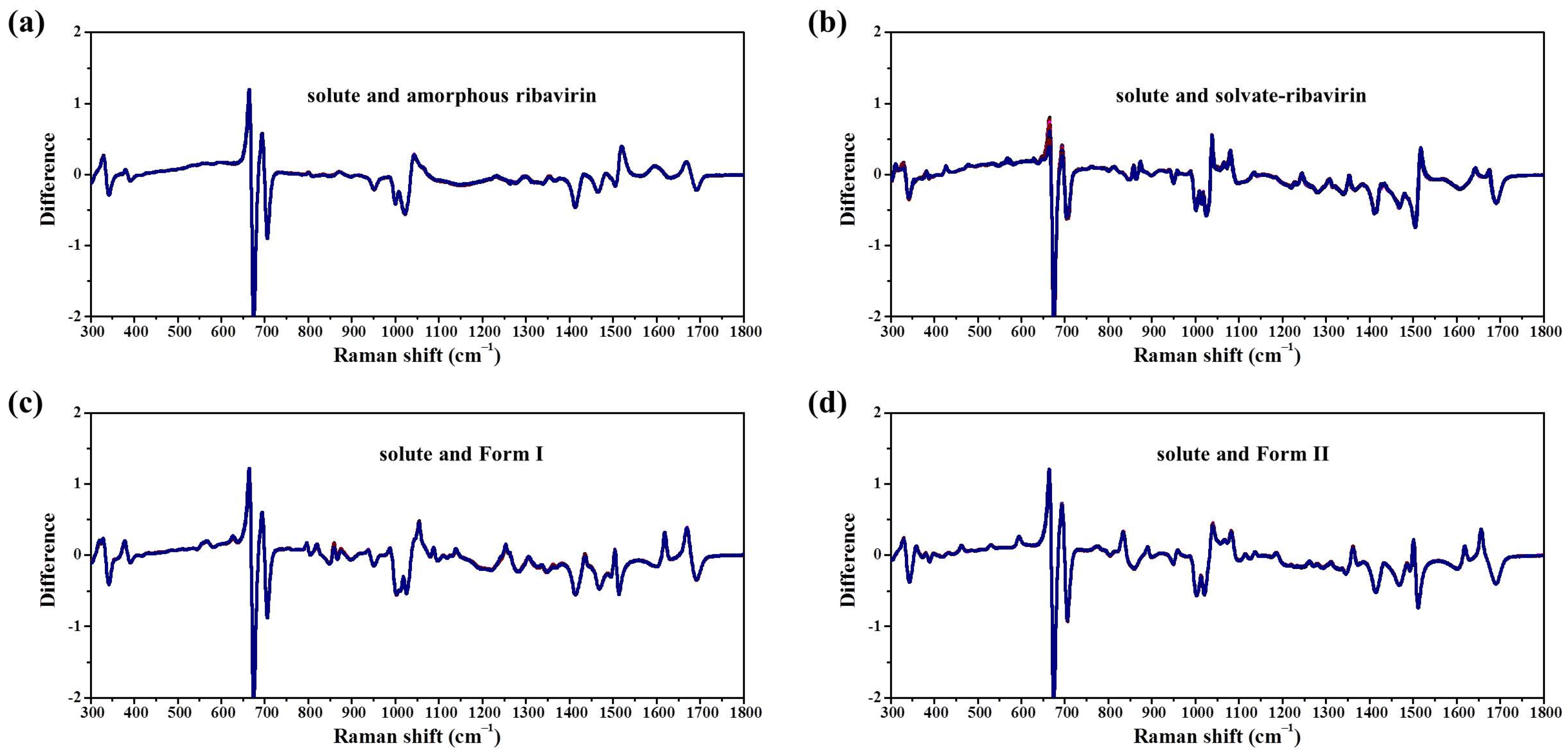
Samples were characterized by powder X-ray diffraction (PXRD, Philips X’Pert Pro, Almelo, The Netherlands, Cu Kα, 40 kV, 30 mA, 5–30°, 4°·min−1) and confocal Raman spectroscopy (DXR3xi, Thermo Fisher Scientific, USA, 532 nm, 40 mW, 0.002–0.02 s, 1000 scans, 50–3400 cm−1, 50× objective lens). All Raman spectra were normalized in the range of 300–1800 cm−1 to the strongest band. MFRDS were carried out by the direct subtraction of two normalized MFRS and a polynomial fitting process to deduce the baseline.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}