
Development of a Purity Certified Reference Material for Vinyl Acetate


Abstract
:1. Introduction
2. Results and Discussion
2.1. Characterization of the CRM Candidate
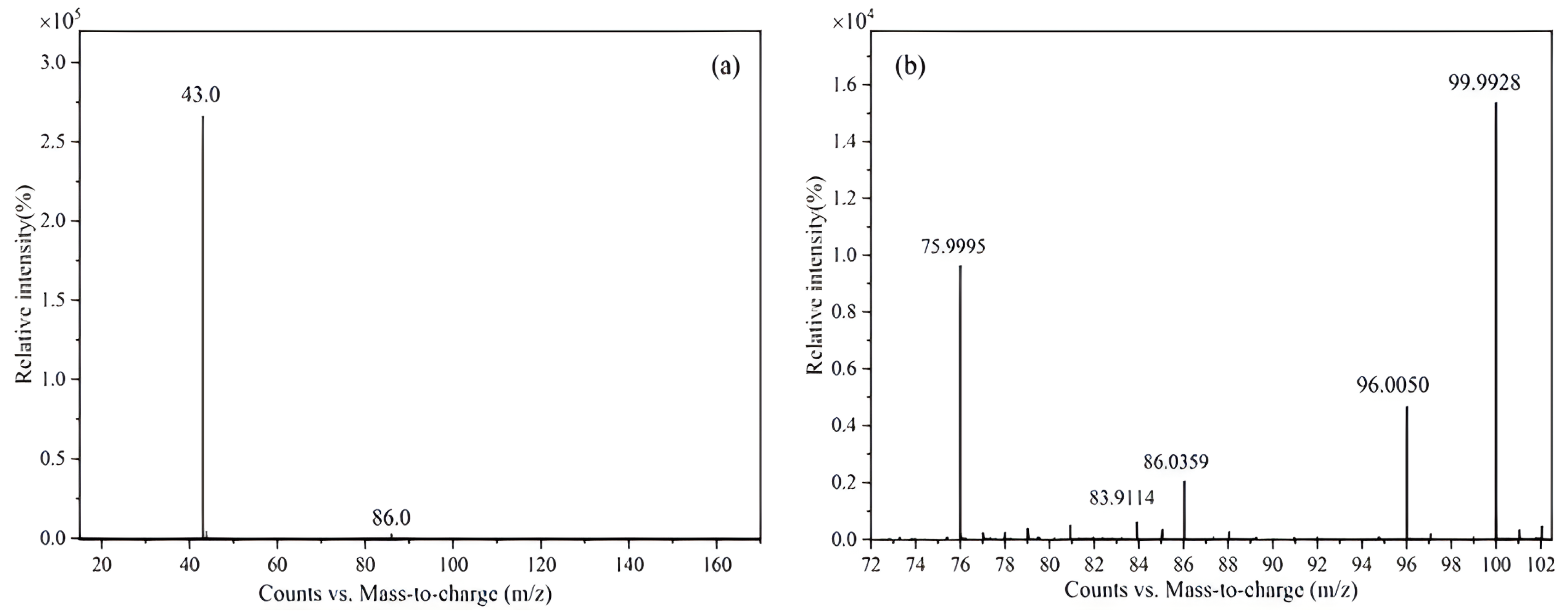
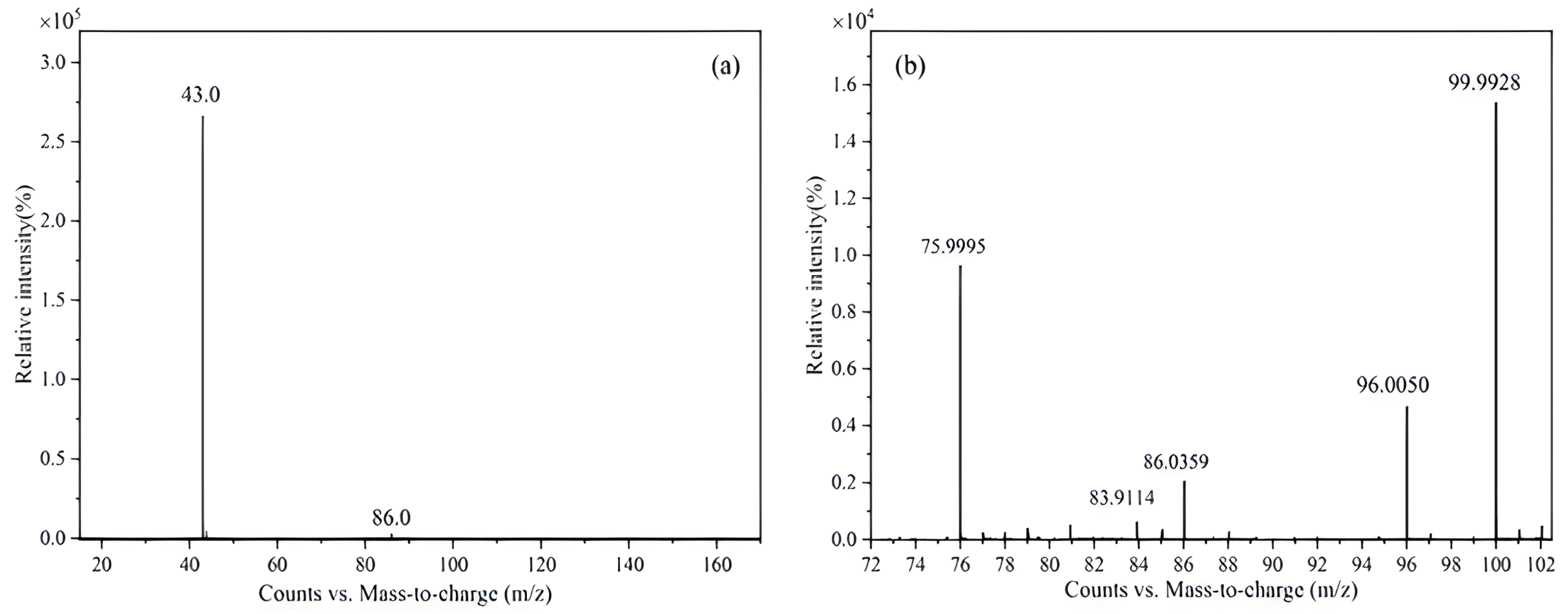
2.1.1. Mass Spectrometry (MS) Analysis
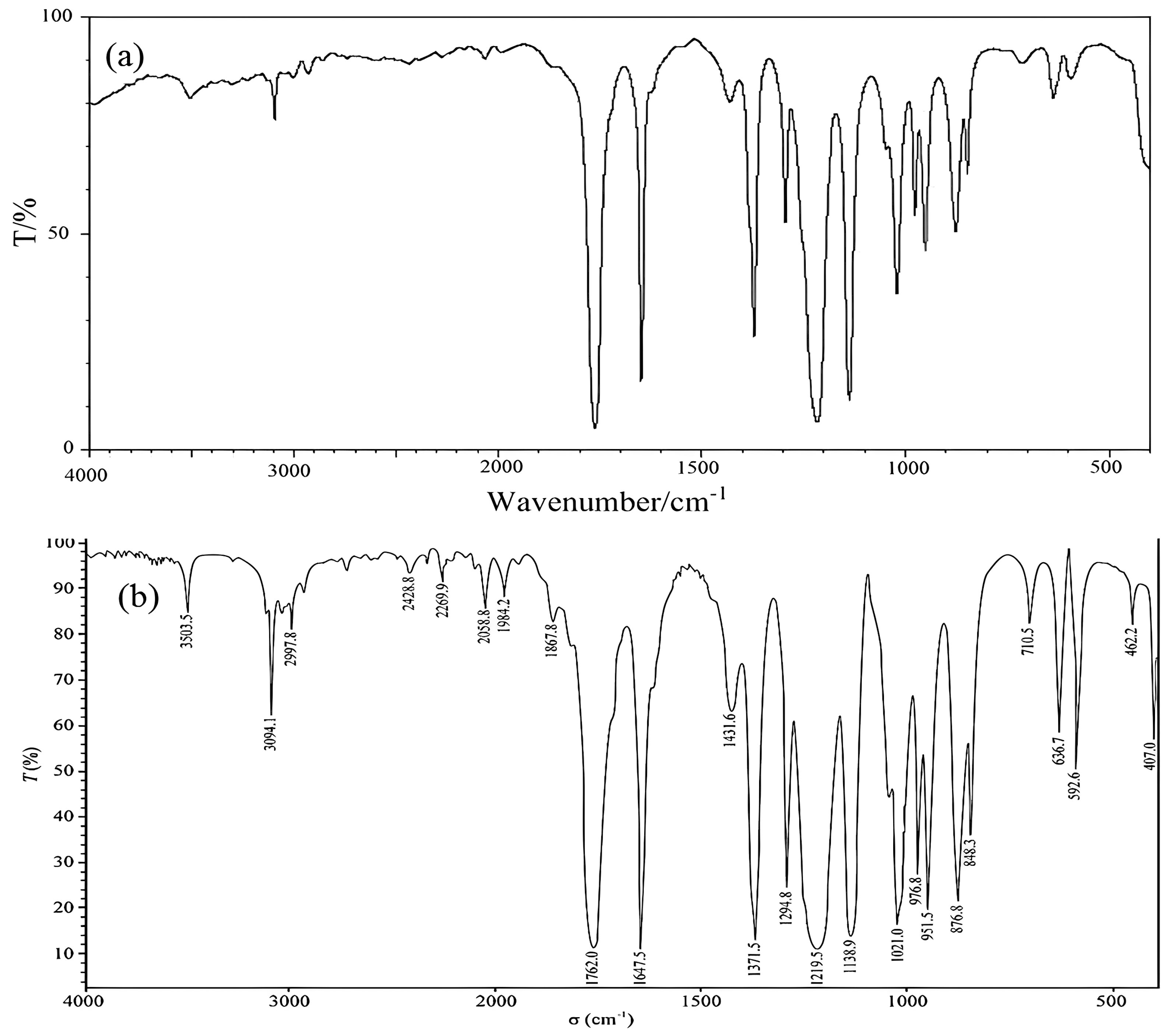
2.1.2. Fourier Transform Infrared Spectroscopy (FT-IR) Analysis
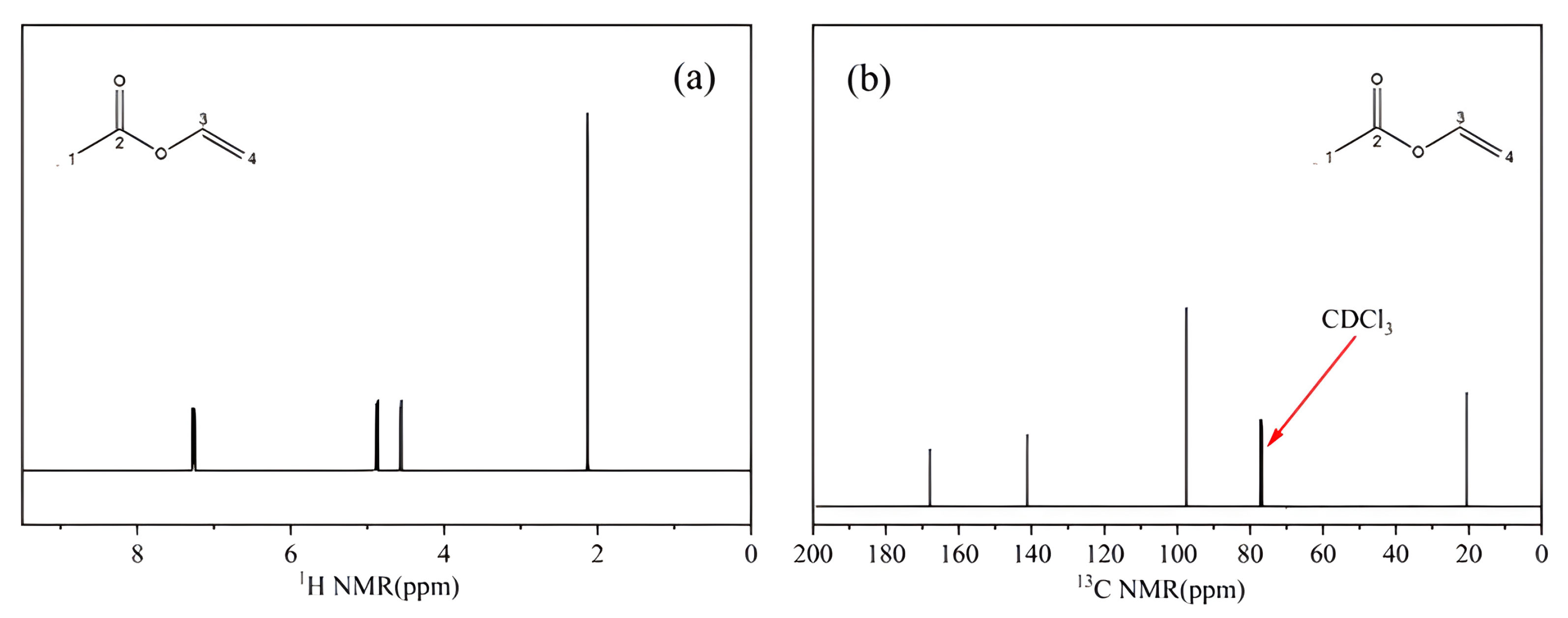
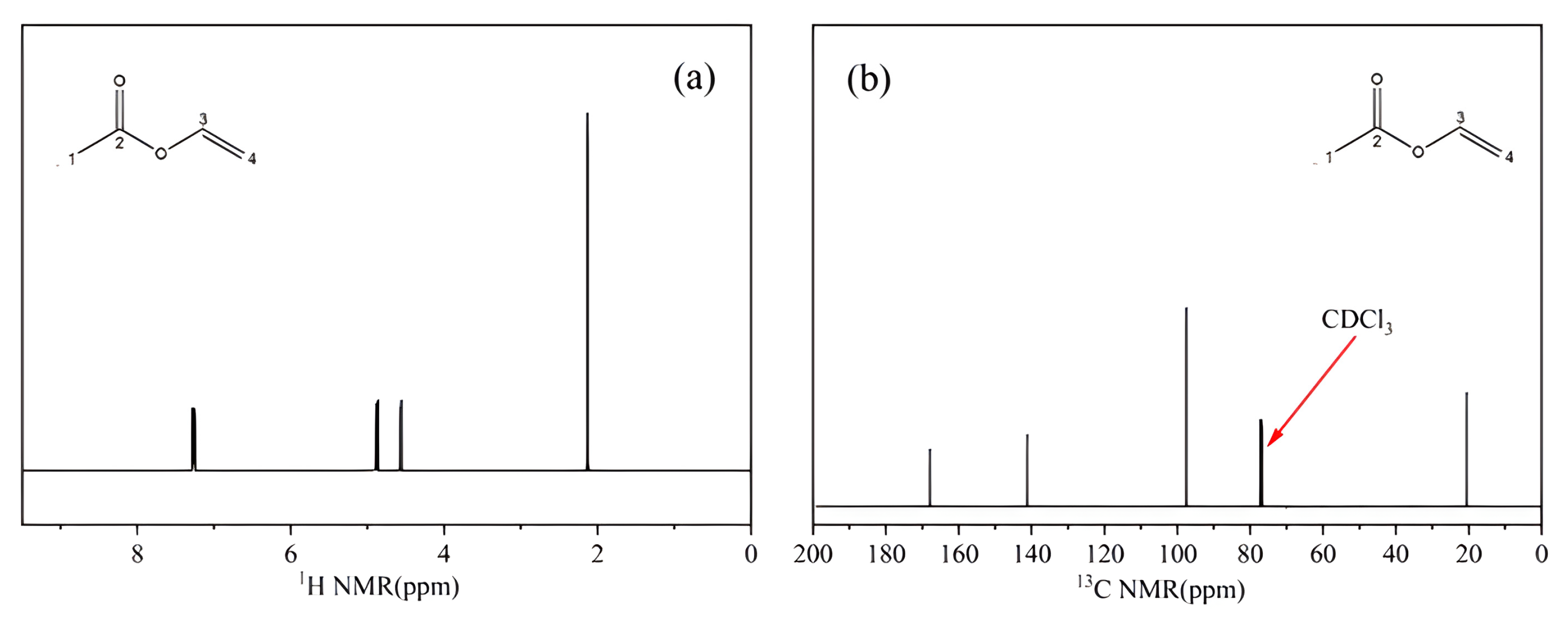
2.1.3. NMR Analysis
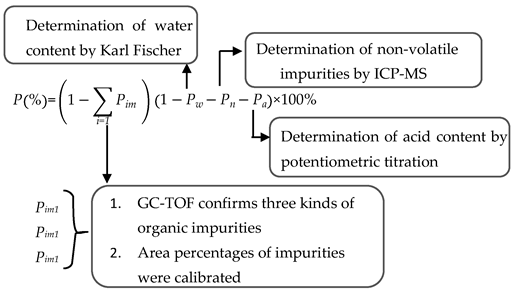
2.2. Purity Determination by the Mass Balance Method
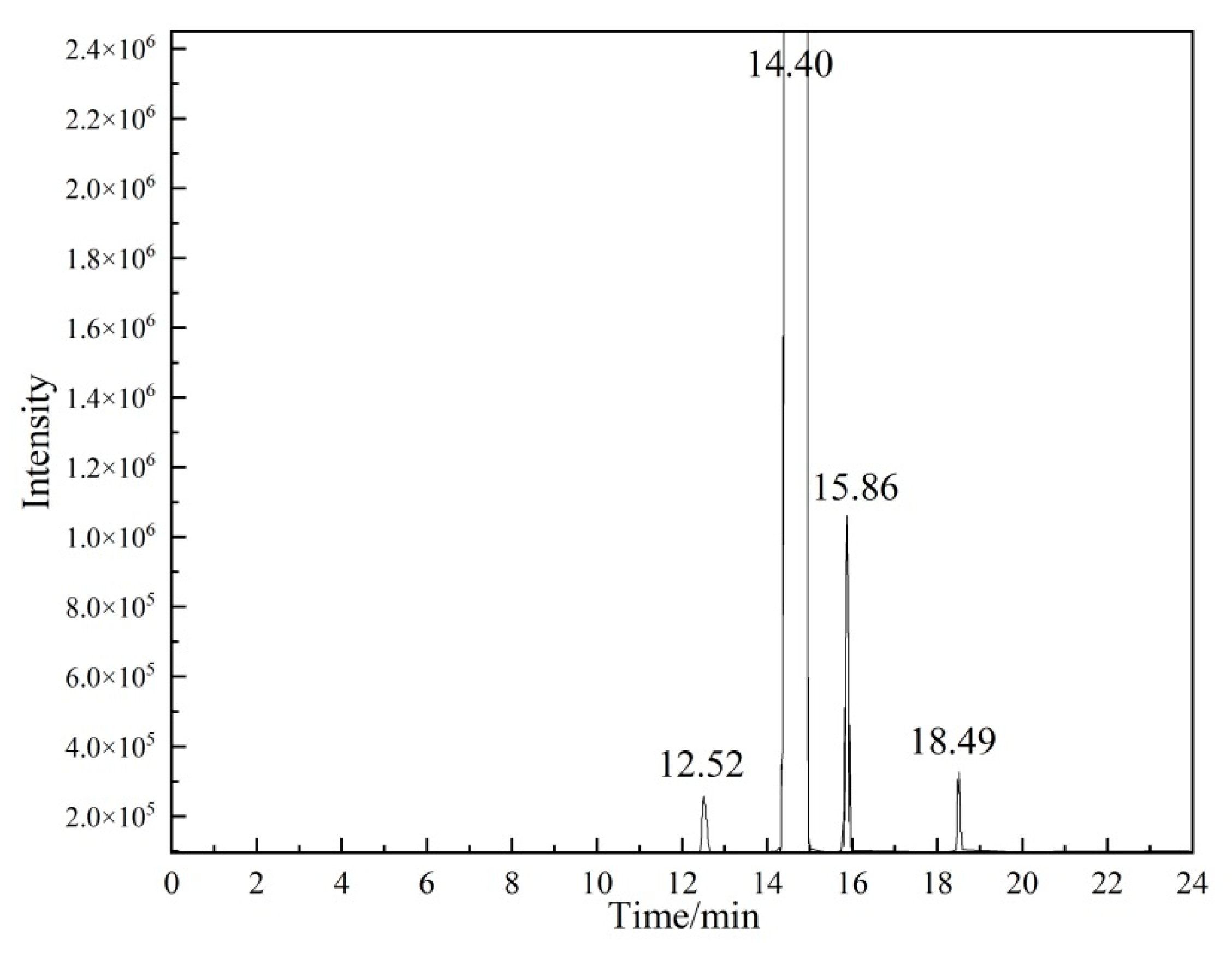
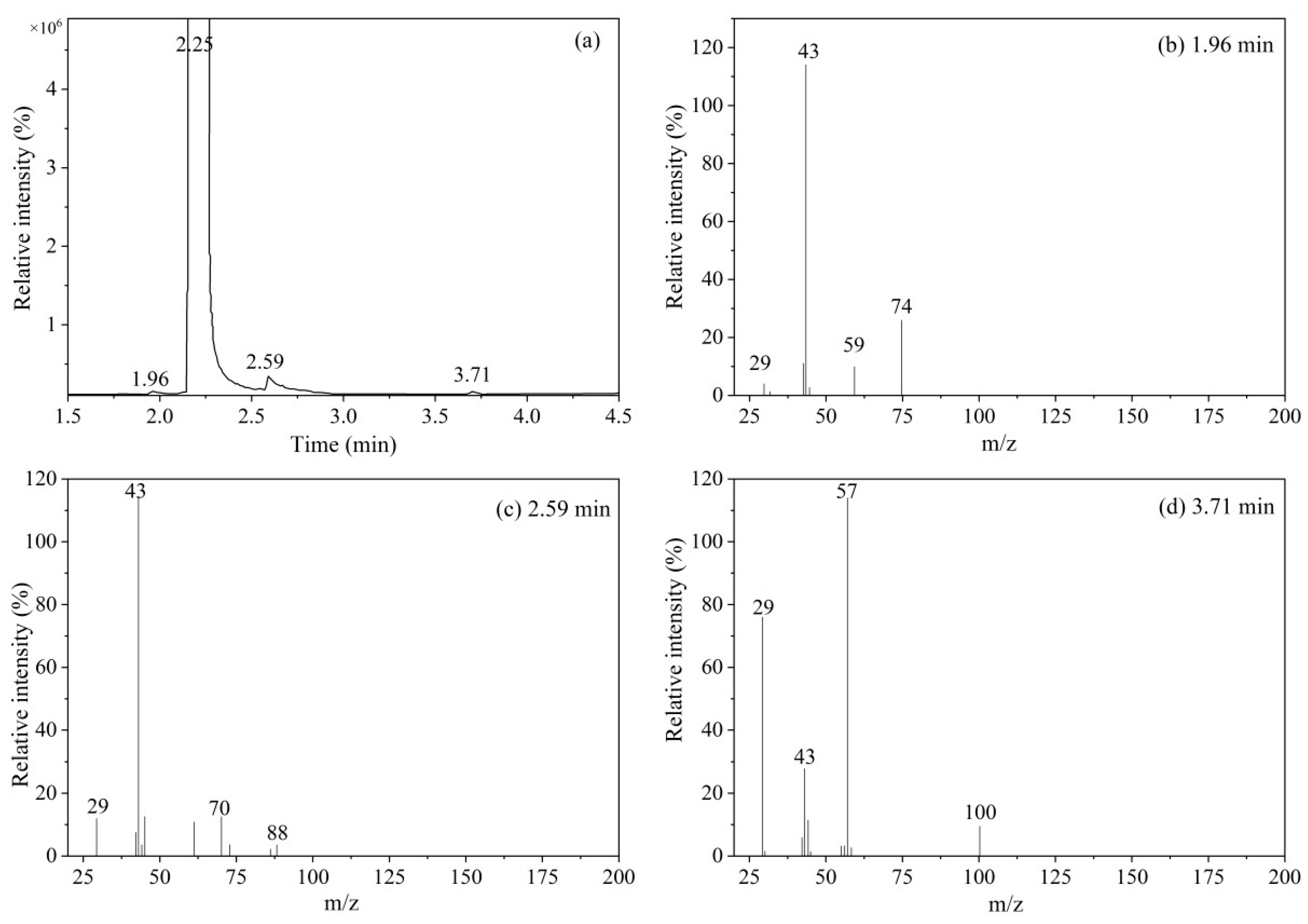
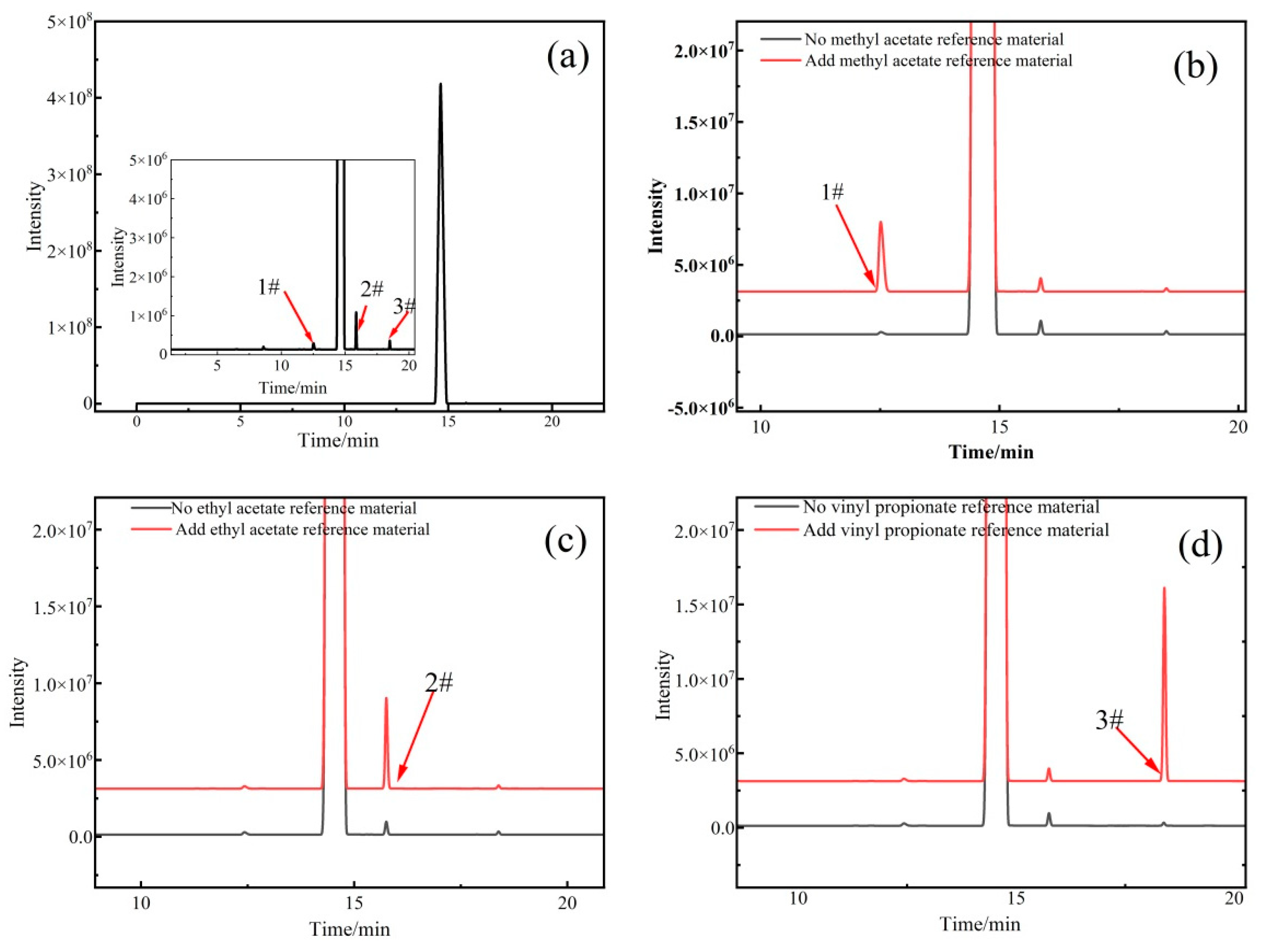
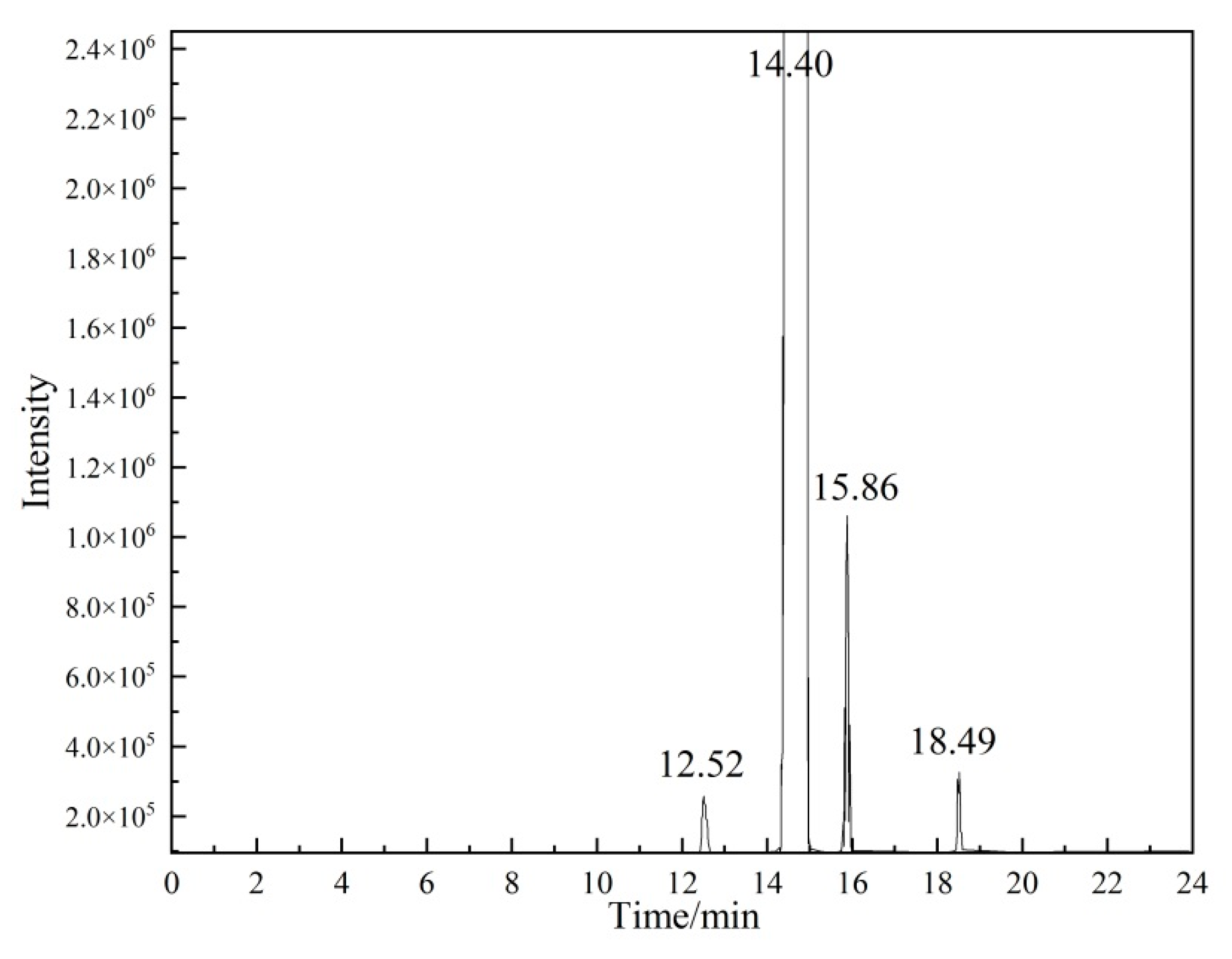
2.2.1. Qualitative Analysis of Organic Impurities
2.2.2. Purity Determined by Mass Balance Method

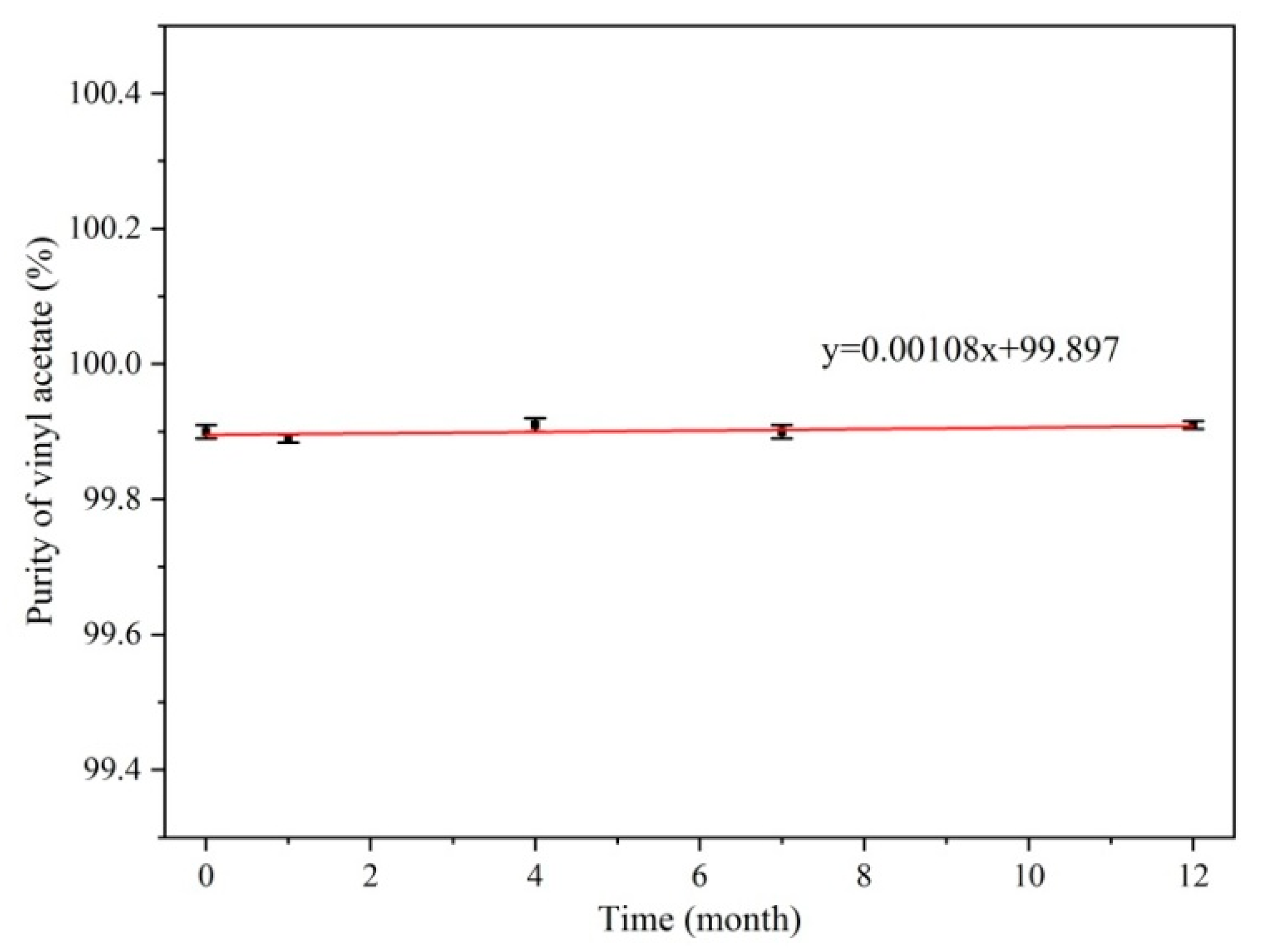
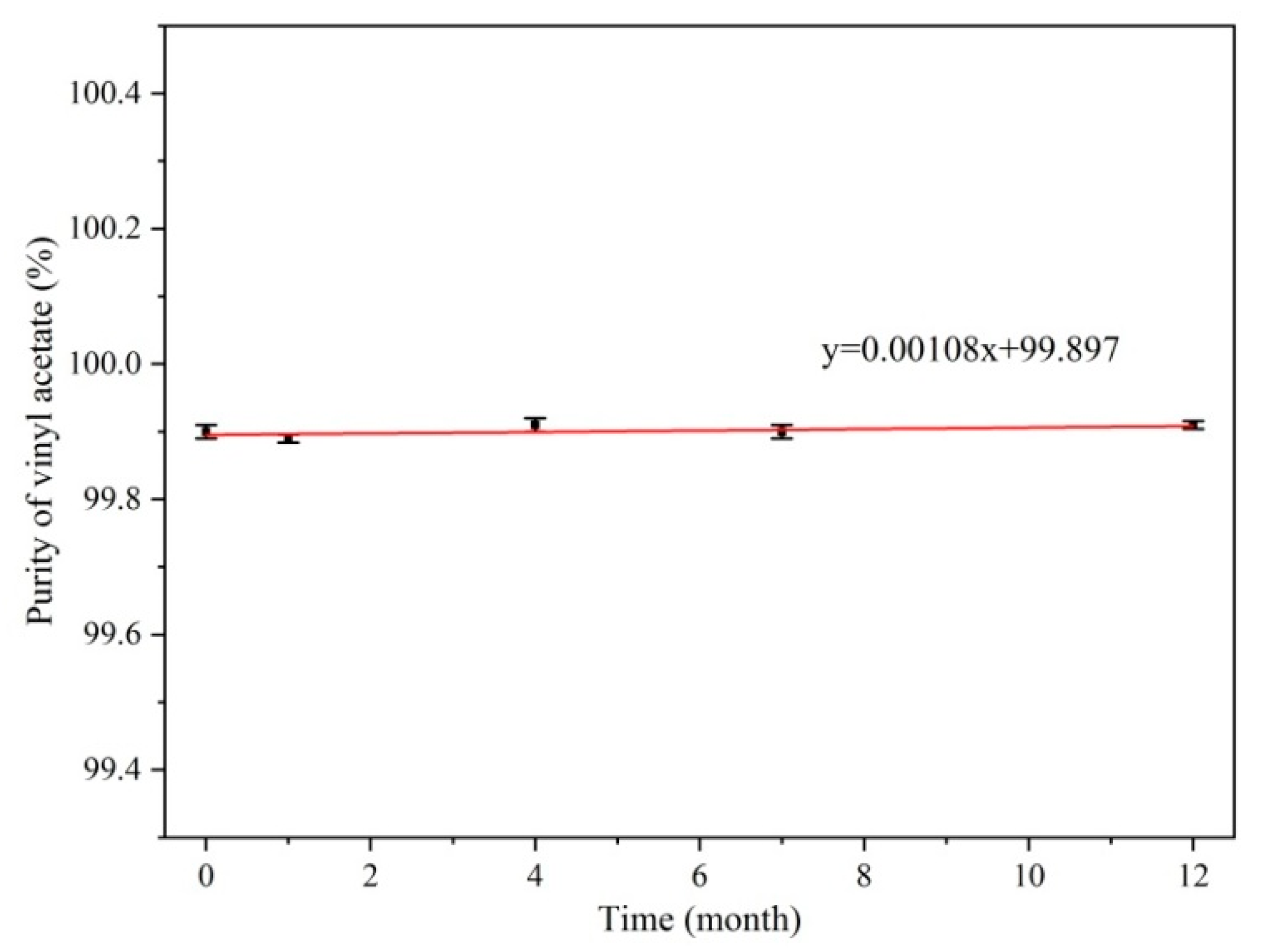
2.3. Homogeneity and Stability Test
2.4. Uncertainty Estimation
2.4.1. Uncertainty of the Mass Balance Method
2.4.2. Combined Uncertainty
3. Materials and Methods
3.1. Apparatus and Materials
3.2. Methods
3.2.1. Preparation of CRM Candidate
3.2.2. Characterization of the CRM Candidate
- (1)
- MS analysis
- (2)
- FT-IR
- (3)
- NMR
3.2.3. Mass Balance Method
- (1)
- Qualitative analysis of organic components
- (2)
- Determination of organic components
- (3)
- Determination of water
- (4)
- Determination of inorganic impurities
- (5)
- Determination of acid
3.3. Homogeneity and Stability Test
Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Neurock, M.; Tysoe, W.T. Mechanistic Insights in the Catalytic Synthesis of Vinyl Acetate on Palladium and Gold/Palladium Alloy Surfaces. Top. Catal. 2013, 56, 19. [Google Scholar] [CrossRef]
- França De Sá, S.; Viana, C.; Ferreira, J.L. Tracing Poly(Vinyl Acetate) Emulsions by Infrared and Raman Spectroscopies: Identification of Spectral Markers. Polymers 2021, 13, 3609. [Google Scholar] [CrossRef] [PubMed]
- Petrocelli, F.P.; Cordeiro, C.F. Continuous Process for the Production of Vinyl Acetate-Ethylene Emulsion Copolymers, Macromolecular Symposia; Wiley Online Library: Hoboken, NJ, USA, 2000; pp. 39–52. [Google Scholar]
- Maes, C.; Luyten, W.; Herremans, G.; Peeters, R.; Carleer, R.; Buntinx, M. Recent Updates on the Barrier Properties of Ethylene Vinyl Alcohol Copolymer (EVOH): A Review. Polym. Rev. 2018, 58, 209–246. [Google Scholar] [CrossRef]
- Zhang, Y.; Pang, B.; Yang, S.; Fang, W.; Yang, S.; Yuan, T.-Q.; Sun, R.-C. Improvement in Wood Bonding Strength of Poly (Vinyl Acetate-Butyl Acrylate) Emulsion by Controlling the Amount of Redox Initiator. Materials 2018, 11, 89. [Google Scholar] [CrossRef] [PubMed]
- Bogdanffy, M.S.; Sarangapani, R.; Plowchalk, D.R.; Jarabek, A.; Andersen, M.E. A biologically based risk assessment for vinyl acetate-induced cancer and noncancer inhalation toxicity. Toxicol. Sci. 1999, 51, 19–35. [Google Scholar] [CrossRef]
- Bogdanffy, M.S.; Valentine, R. Differentiating between local cytotoxicity, mitogenesis, and genotoxicity in carcinogen risk assessments: The case of vinyl acetate. Toxicol. Lett. 2003, 140–141, 83–98. [Google Scholar] [CrossRef]
- Liu, A.-F.; Zhao, J.-Y.; Liu, L.; Shen, M.-J.; Lei, P.-N. Headspace-GC Determination of Amounts of 1-Hexene,1-Octene and Vinyl Acetate Migrated from Polyethylene Food-Contacting Materials into Food Simulants. Phys. Test. Chem. Anal. (Part B Chem. Anal.) 2020, 56, 1073–1078. [Google Scholar]
- Bianchi, O.; Oliveira, R.V.B.; Fiorio, R.; Martins, J.D.N.; Zattera, A.J.; Canto, L.B. Assessment of Avrami, Ozawa and Avrami–Ozawa equations for determination of EVA crosslinking kinetics from DSC measurements. Polym. Test. 2008, 27, 722–729. [Google Scholar] [CrossRef]
- Wenwei, Z.; Xiaoguang, Z.; Li, Y.; Yuefang, Z.; Jiazhen, S. Determination of the vinyl acetate content in ethylene-vinyl acetate copolymers by thermogravimetric analysis. Polymer 1994, 35, 3348–3350. [Google Scholar] [CrossRef]
- Beshah, K. Microstructural analysis of ethylene-vinyl acetate copolymer by 2D NMR spectroscopy. Macromolecules 1992, 25, 5597–5600. [Google Scholar] [CrossRef]
- Meszlényi, G.; Körtvélyessy, G. Direct determination of vinyl acetate content of ethylene-vinyl acetate copolymers in thick films by infrared spectroscopy. Polym. Test. 1999, 18, 551–557. [Google Scholar] [CrossRef]
- Koopmans, R.J.; van der Linden, R.; Vansant, E.F. Quantitative determination of the vinylacetate content in ethylene vinyl-acetate copolymers—A critical review. Polym. Eng. Sci. 1982, 22, 878–882. [Google Scholar] [CrossRef]
- Sun, D.-Z.; Lu, C.-Q.; Zuo, Y.; Qin, Z.-M. Head-Space GC Determination of Formamide in Children’s Articles Made of Plastic of Copolymer of Ethylene and Vinyl Acetate. Phys. Test. Chem. Anal. (Part B Chem. Anal.) 2014, 50, 199–201. [Google Scholar]
- Pang, A.-Q.; Chen, C.-W. Pyrolytic Gas Chromatography Analyzes Vinyl Acetate Content in EVA. Tech. Text. 2000, 7, 36–39. [Google Scholar]
- Shi, M.-J.; Zhang, J. Introduction and Evaluation for the Determination Methods of Vinyl Acetate Content. Plastics 2008, 37, 59+108–110. [Google Scholar]
- Wise, S.A. What is novel about certified reference materials? Anal. Bioanal. Chem. 2018, 410, 2045–2049. [Google Scholar] [CrossRef]
- Gong, H.; Huang, T.; Yang, Y.; Wang, H. Purity determination and uncertainty evaluation of folic acid by mass balance method. Talanta 2012, 101, 96–103. [Google Scholar] [CrossRef]
- Ulberth, F. Certified reference materials for inorganic and organic contaminants in environmental matrices. Anal. Bioanal. Chem. 2006, 386, 1121–1136. [Google Scholar] [CrossRef]
- Hyung, S.-W.; Lee, C.-H.; Kim, B. Development of certified reference materials for accurate determination of fluoroquinolone antibiotics in chicken meat. Food Chem. 2017, 229, 472–478. [Google Scholar] [CrossRef]
- Zhou, J.; Zhao, Y.; Wang, M.; Yang, M.; Wang, T. Production of matrix certified reference material for analysis of salbutamol residue in mutton. Microchem. J. 2022, 175, 107151. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Fang, H.; Chen, H.; Chen, X.; Zhang, Y.; Hong, Z. Development of a new taurine purity certified reference material. Microchem. J. 2022, 181, 107761. [Google Scholar] [CrossRef]
- Davies, S.R.; Jones, K.; Goldys, A.; Alamgir, M.; Chan, B.K.H.; Elgindy, C.; Mitchell, P.S.R.; Tarrant, G.J.; Krishnaswami, M.R.; Luo, Y.; et al. Purity assessment of organic calibration standards using a combination of quantitative NMR and mass balance. Anal. Bioanal. Chem. 2015, 407, 3103–3113. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.-M.; Ding, L.-X.; Hu, C.-Q. A comparative uncertainty study of the purity assessment of chemical reference substances using differential scanning calorimetry (DSC) and mass balance method. Thermochim. Acta 2011, 525, 1–8. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, Y.; Wu, Q.; Xiao, W.; Zhu, J.; Ding, Y. Purity determination and uncertainty estimation of natural products sourcing chemical reference substances by high-performance liquid chromatography and differential scanning calorimetry. Microchem. J. 2021, 166, 106257. [Google Scholar] [CrossRef]
- Nogueira, R.; Garrido, B.C.; Borges, R.M.; Silva, G.E.B.; Queiroz, S.M.; Cunha, V.S. Development of a new sodium diclofenac certified reference material using the mass balance approach and 1H qNMR to determine the certified property value. Eur. J. Pharm. Sci. 2013, 48, 502–513. [Google Scholar] [CrossRef]
- Davies, S.R.; Alamgir, M.; Chan, B.K.H.; Dang, T.; Jones, K.; Krishnaswami, M.; Luo, Y.; Mitchell, P.S.R.; Moawad, M.; Swan, H.; et al. The development of an efficient mass balance approach for the purity assignment of organic calibration standards. Anal. Bioanal. Chem. 2015, 407, 7983–7993. [Google Scholar] [CrossRef]
- Lee, S.; Kwon, H.-J. Purity Assessment of Monosaccharides using Mass Balance Method. Bulletin of the Korean Chemical Society 2020, 41, 1002–1008. [Google Scholar] [CrossRef]
- Wang, S.; Wu, P.; Li, M.; Huang, T.; Shi, N.; Feng, L.; Li, H. Mass balance method for SI-traceable purity assignment of synthetic oxytocin. J. Pharm. Biomed. Anal. 2022, 207, 114401. [Google Scholar] [CrossRef]
- Westwood, S.; Choteau, T.; Daireaux, A.; Josephs, R.D.; Wielgosz, R.I. Mass Balance Method for the SI Value Assignment of the Purity of Organic Compounds. Anal. Chem. 2013, 85, 3118–3126. [Google Scholar] [CrossRef]
- Liu, H.; Cheow, P.S.; Yong, S.; Chen, Y.; Liu, Q.; Teo, T.L.; Lee, T.K. Determination of purity values of amino acid reference materials by mass balance method: An approach to the quantification of related structure impurities. Anal. Bioanal. Chem. 2020, 412, 8023–8037. [Google Scholar] [CrossRef]
- Zhou, J.; Li, F.; Wang, M.; Yan, C.; Yang, M.; Wang, T.; Zhang, L. Preparation of clorprenaline certified reference material: Purity determination and uncertainty evaluation. Microchem. J. 2022, 179, 107502. [Google Scholar] [CrossRef]
- Ma, K.; Wang, H.; Zhao, M.; Xing, J. Purity determination and uncertainty evaluation of theophylline by mass balance method, high performance liquid chromatography and differential scanning calorimetry. Anal. Chim. Acta 2009, 650, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, W.; Huang, T.; Su, P.; Yang, Y. Quantification of a volatile deuterated compound by the differential scanning calorimetry combined with quantitative nuclear magnetic resonance and its verification by the mass balance method combined with gas chromatography-mass spectrometry. Talanta 2022, 246, 123538. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jin, W.; Zhang, Y.; Fang, H.; Chen, H.; Hong, Z.; Huang, X. Development of certified reference materials for four polyunsaturated fatty acid esters. Food Chem. 2022, 389, 133006. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, S.; Li, P.; Li, L.; Ye, J. Establishment of SI-traceable purity assessment of Fumonisin B1 using a combination of quantitative 1H NMR and mass balance. Microchem. J. 2023, 185, 108282. [Google Scholar] [CrossRef]
- Kumar, A.; Misra, D.K. A Review on the Statistical Methods and Implementation to Homogeneity Assessment of Certified Reference Materials in Relation to Uncertainty. Mapan 2020, 35, 457–470. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, M.; Li, F.; Wang, M.; Zhang, Y.; Wei, M.; Li, X.; Qi, X.; Bai, X.; Chai, Y. Development of matrix certified reference material for accurate determination of docosahexaenoic acid in milk powder. Food Chem. 2023, 406, 135012. [Google Scholar] [CrossRef]
- Huang, T.; Li, H.; Zhang, W. Metrological technical specification for purity assessment of organic pure substance certified reference materials in China. Accredit. Qual. Assur. 2021, 26, 279–284. [Google Scholar] [CrossRef]
- Quan, C. Establishment of the purity values of carbohydrate certified reference materials using quantitative nuclear magnetic resonance and mass balance approach. Food Chem. 2014, 153, 378–386. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Impurity No. | Retention Time | Measured Value (m/z) | Theoretical Value (m/z) | Elemental Composition |
|---|---|---|---|---|
| Vinyl acetate | 2.25 min | 86.0366 | 86.0362 | C4H6O2 |
| 43.0177 | 43.0178 | C2H3O | ||
| Impurity #1 | 1.96 min | 74.0361 | 74.0362 | C3H6O2 |
| 59.0124 | 59.0128 | C2H3O2 | ||
| 43.0177 | 43.0178 | C2H3O | ||
| Impurity #2 | 2.59 min | 88.0520 | 88.0519 | C4H8O2 |
| 70.0410 | 70.0143 | C4H6O | ||
| 61.0281 | 61.0284 | C2H5O2 | ||
| 43.0174 | 43.0178 | C2H3O | ||
| Impurity #3 | 3.71 min | 100.02 | 100.02 | C5H8O2 |
| 57.0337 | 57.0335 | C3H5O | ||
| 43.0176 | 43.0178 | C2H3O | ||
| 29.0382 | 29.0386 | C2H5 |
| Measurement | Retention Time (min) | Ai | Concentration (%) | fi (%) | Concentration (Calibration %) | RSD (%) |
|---|---|---|---|---|---|---|
| Methyl acetate | 12.52 | 9,315,686 | 0.02 | 0.86 | -- | -- |
| Ethyl acetate | 15.86 | 33,948,215 | 0.05 | 0.83 | -- | -- |
| Vinyl propionate | 18.49 | 7,934,083 | 0.01 | 1.23 | -- | -- |
| Vinyl acetate (P0) | 14.40 | 63,973,339,424 | 99.92 | -- | 99.93 | 0.0029 |
| 0.030 | 0.0015 | |||||
| 0.0012 | 0.00011 | |||||
| PMB | 99.0 | 0.018 | ||||
| Number | 1 | 2 | 3 | Means |
|---|---|---|---|---|
| 1 | 99.89 | 99.88 | 99.90 | 99.89 |
| 2 | 99.89 | 99.87 | 99.91 | 99.89 |
| 3 | 99.87 | 99.90 | 99.89 | 99.89 |
| 4 | 99.87 | 99.93 | 99.90 | 99.90 |
| 5 | 99.90 | 99.91 | 99.93 | 99.91 |
| 6 | 99.91 | 99.89 | 99.88 | 99.89 |
| 7 | 99.91 | 99.90 | 99.92 | 99.91 |
| 8 | 99.90 | 99.87 | 99.88 | 99.88 |
| 9 | 99.93 | 99.91 | 99.93 | 99.92 |
| 10 | 99.91 | 99.88 | 99.90 | 99.90 |
| 11 | 99.90 | 99.88 | 99.89 | 99.89 |
| 12 | 99.89 | 99.90 | 99.90 | 99.90 |
| 13 | 99.88 | 99.89 | 99.89 | 99.89 |
| 14 | 99.92 | 99.90 | 99.92 | 99.91 |
| 15 | 99.90 | 99.92 | 99.90 | 99.91 |
| Overall mean | 99.90 | |||
| Standard deviation | 0.017 | |||
| Parameters | Values |
|---|---|
| Mean square between groups | = 0.00043 |
| Mean square within groups | = 0.00022 |
| F | F = / = 2.01 |
| F0.05(14, 30) | 2.04 |
| Conclusion |
| Uncertainty Symbols | ||||||
|---|---|---|---|---|---|---|
| Results | 0.0015% | 0.00011% | 0.009% | 0.105% | 0.105% | 0.105% |
| Uncertainty Symbols | Uncertainty Sources | Results |
|---|---|---|
| Homogeneity test | 0.00849% | |
| Long-term stability study | 0.00930% | |
| Mass balance method | 0.105% | |
| GC-FID | 0.105% | |
| Combined uncertainty | 0.11% | |
| Expanded combined uncertainty | 0.3% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, C.; Gao, Q.; Ye, C.; Yang, G.; Zhang, P.; Yang, R.; Zhang, Q.; Ma, K. Development of a Purity Certified Reference Material for Vinyl Acetate. Molecules 2023, 28, 6245. https://doi.org/10.3390/molecules28176245
He C, Gao Q, Ye C, Yang G, Zhang P, Yang R, Zhang Q, Ma K. Development of a Purity Certified Reference Material for Vinyl Acetate. Molecules. 2023; 28(17):6245. https://doi.org/10.3390/molecules28176245
Chicago/Turabian StyleHe, Chen, Qin Gao, Changwen Ye, Guotao Yang, Pengfei Zhang, Rongchao Yang, Qing Zhang, and Kang Ma. 2023. "Development of a Purity Certified Reference Material for Vinyl Acetate" Molecules 28, no. 17: 6245. https://doi.org/10.3390/molecules28176245
APA StyleHe, C., Gao, Q., Ye, C., Yang, G., Zhang, P., Yang, R., Zhang, Q., & Ma, K. (2023). Development of a Purity Certified Reference Material for Vinyl Acetate. Molecules, 28(17), 6245. https://doi.org/10.3390/molecules28176245