
Resonant Inner-Shell Photofragmentation of Adamantane (C10H16)

 ,
,  , , , , ,
, , , , ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
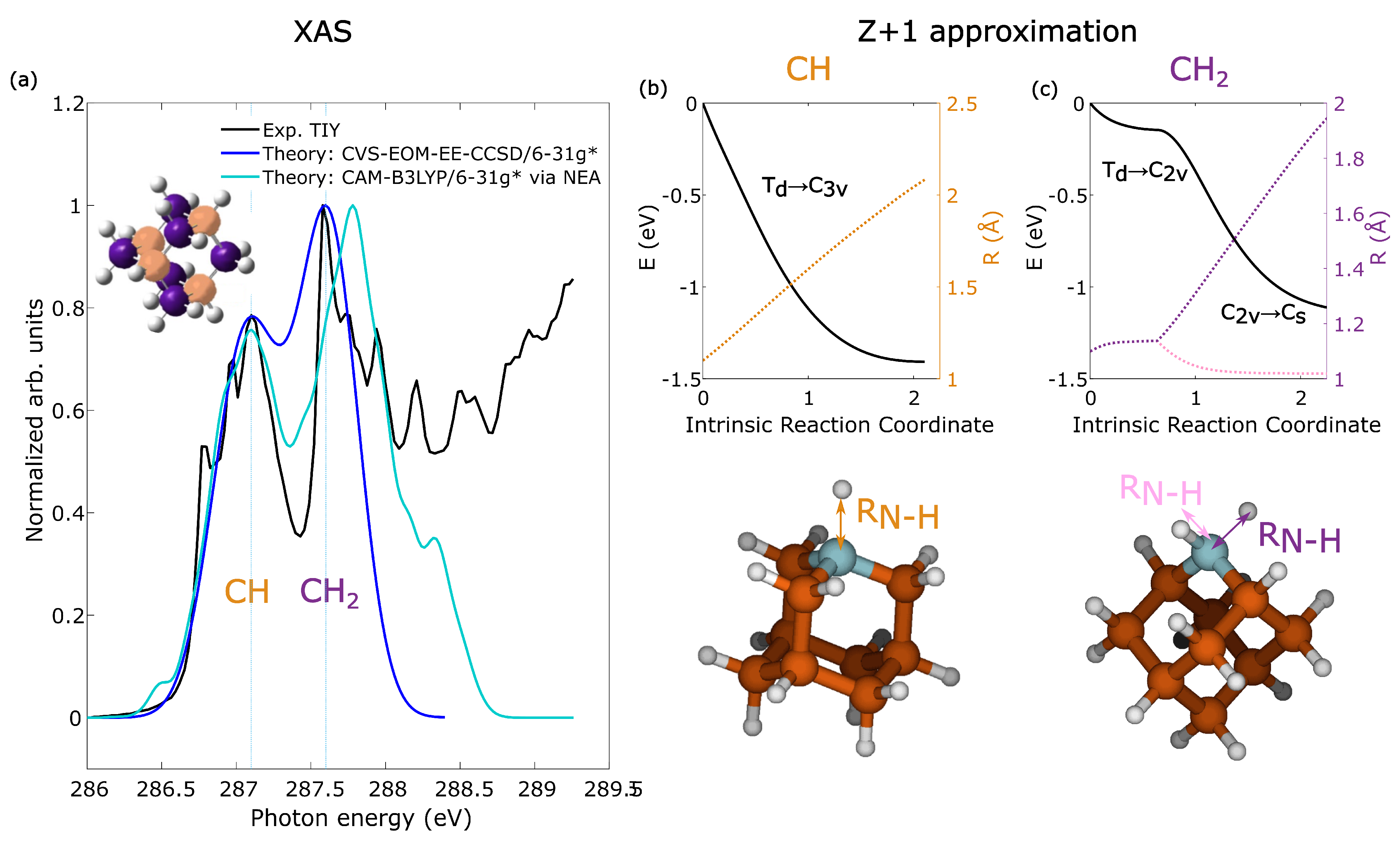
2.1. Resonant Excitation of Adamantane
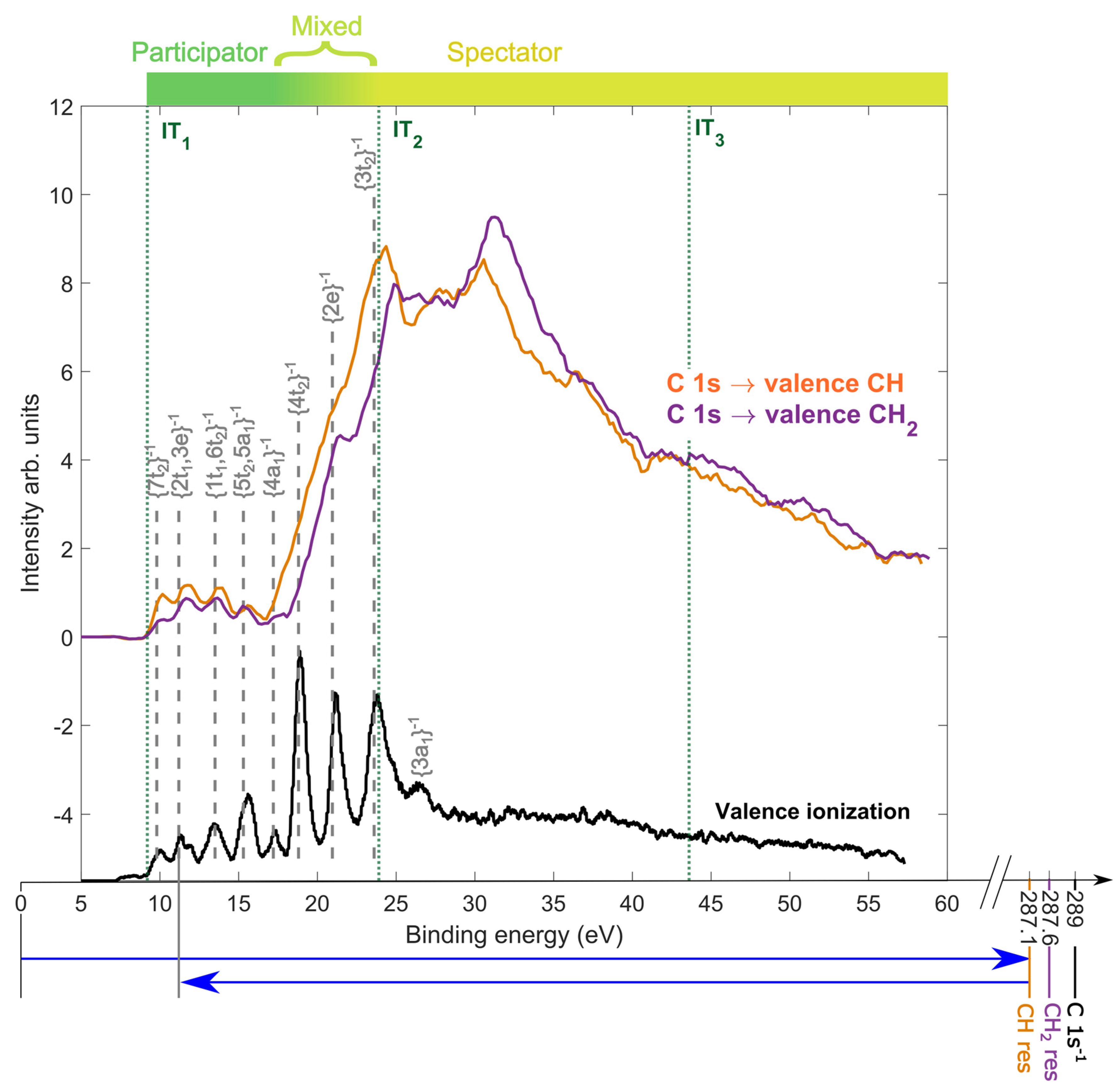
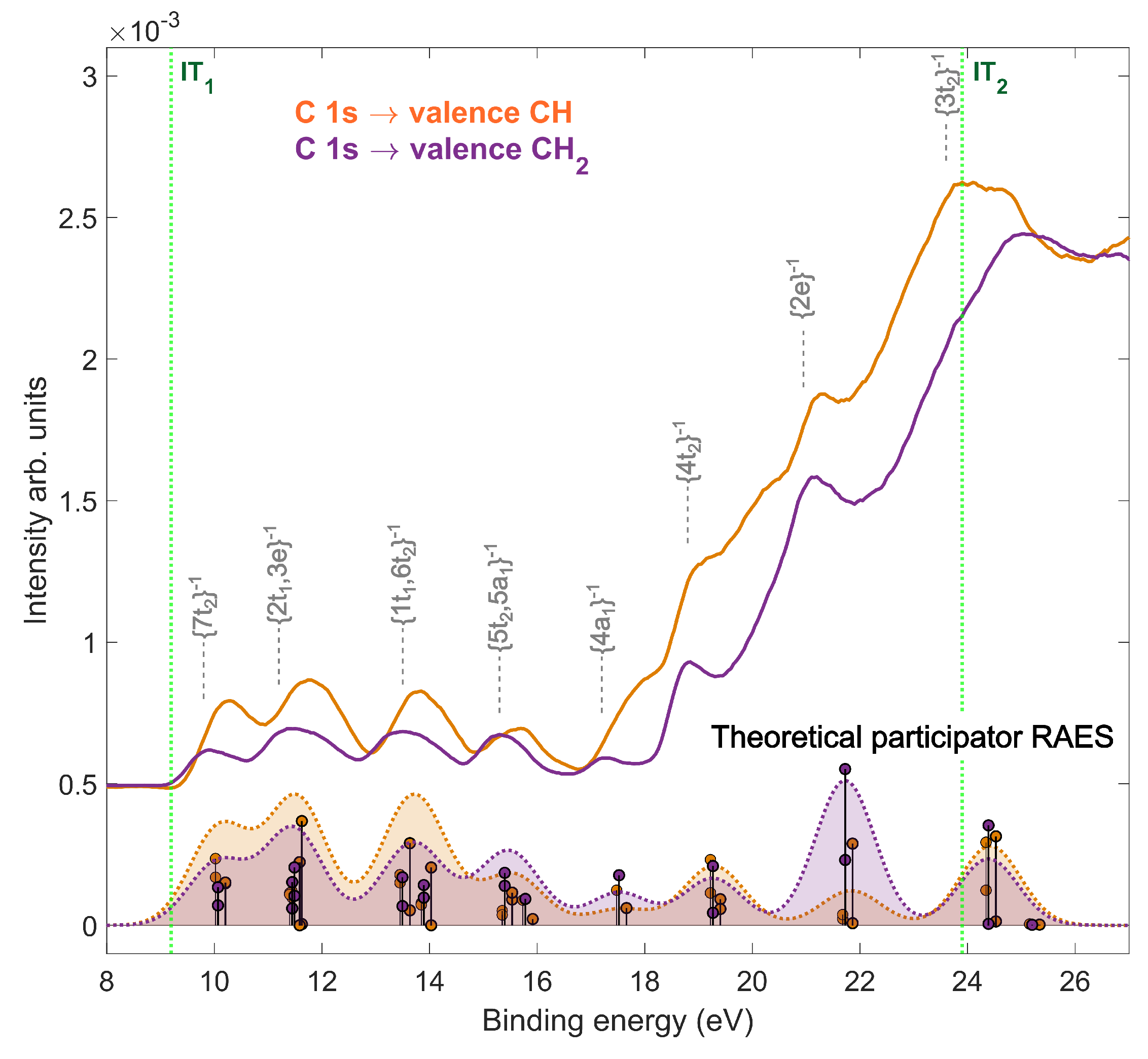
2.2. Resonant Auger Decay of Adamantane
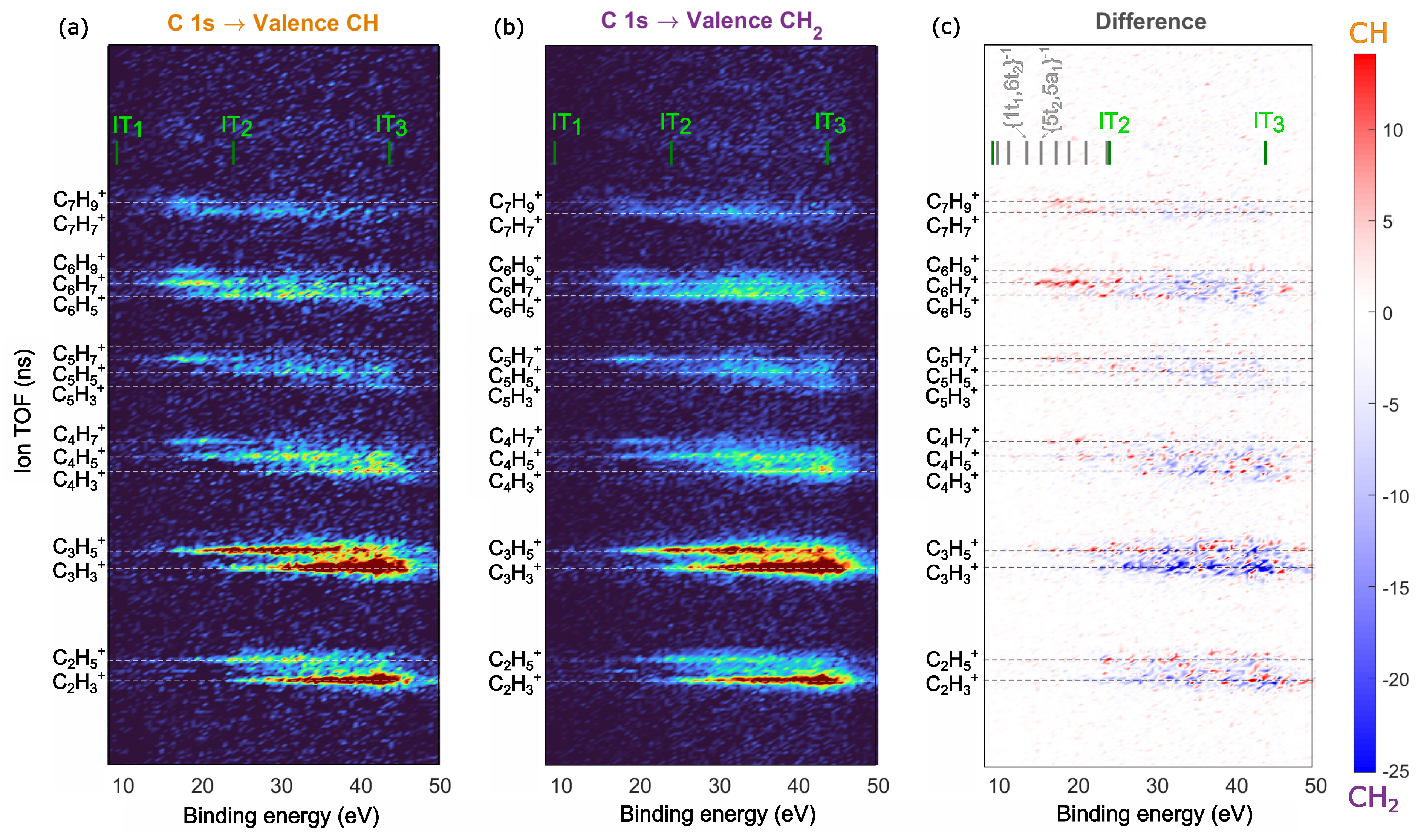
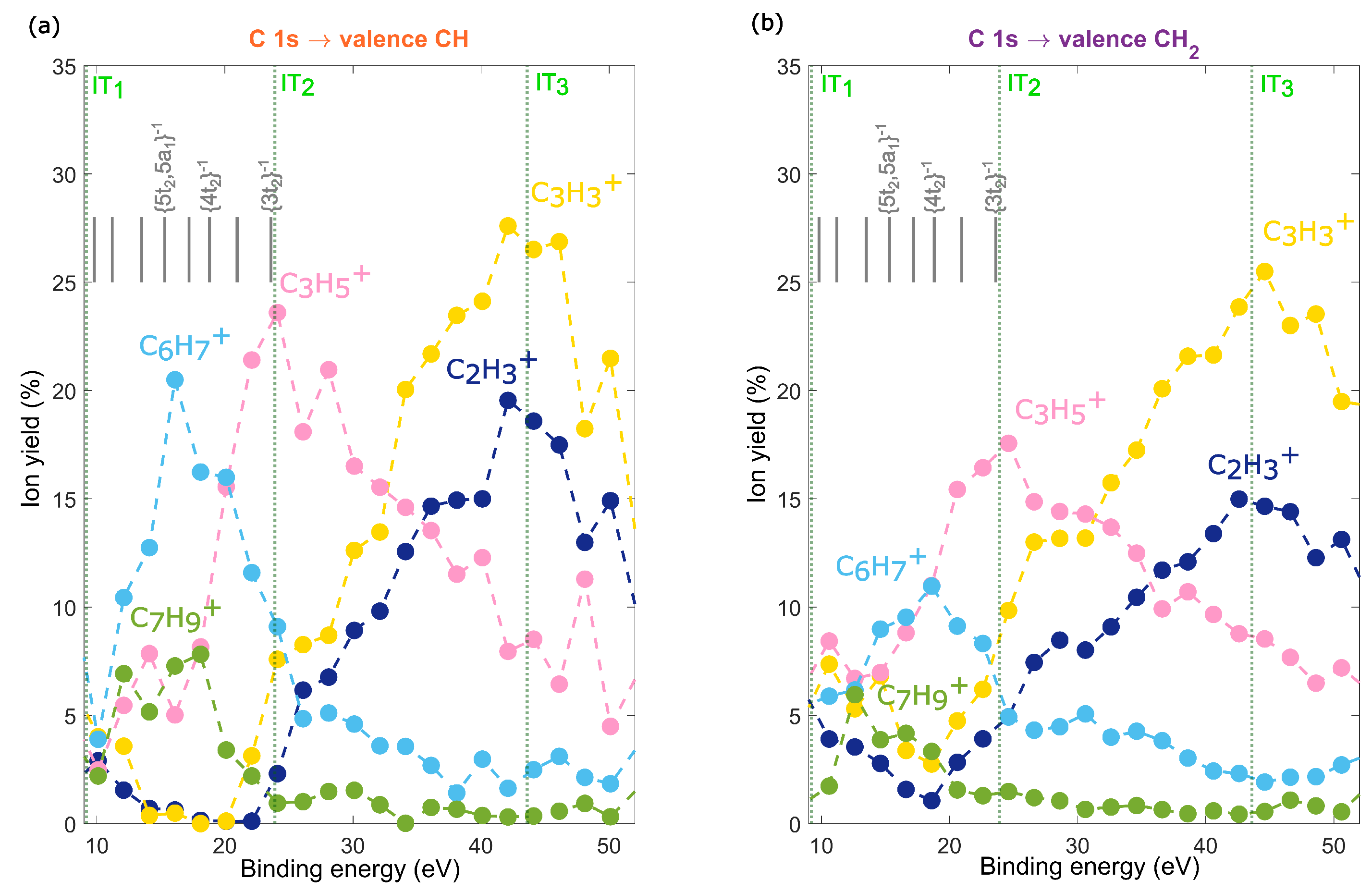
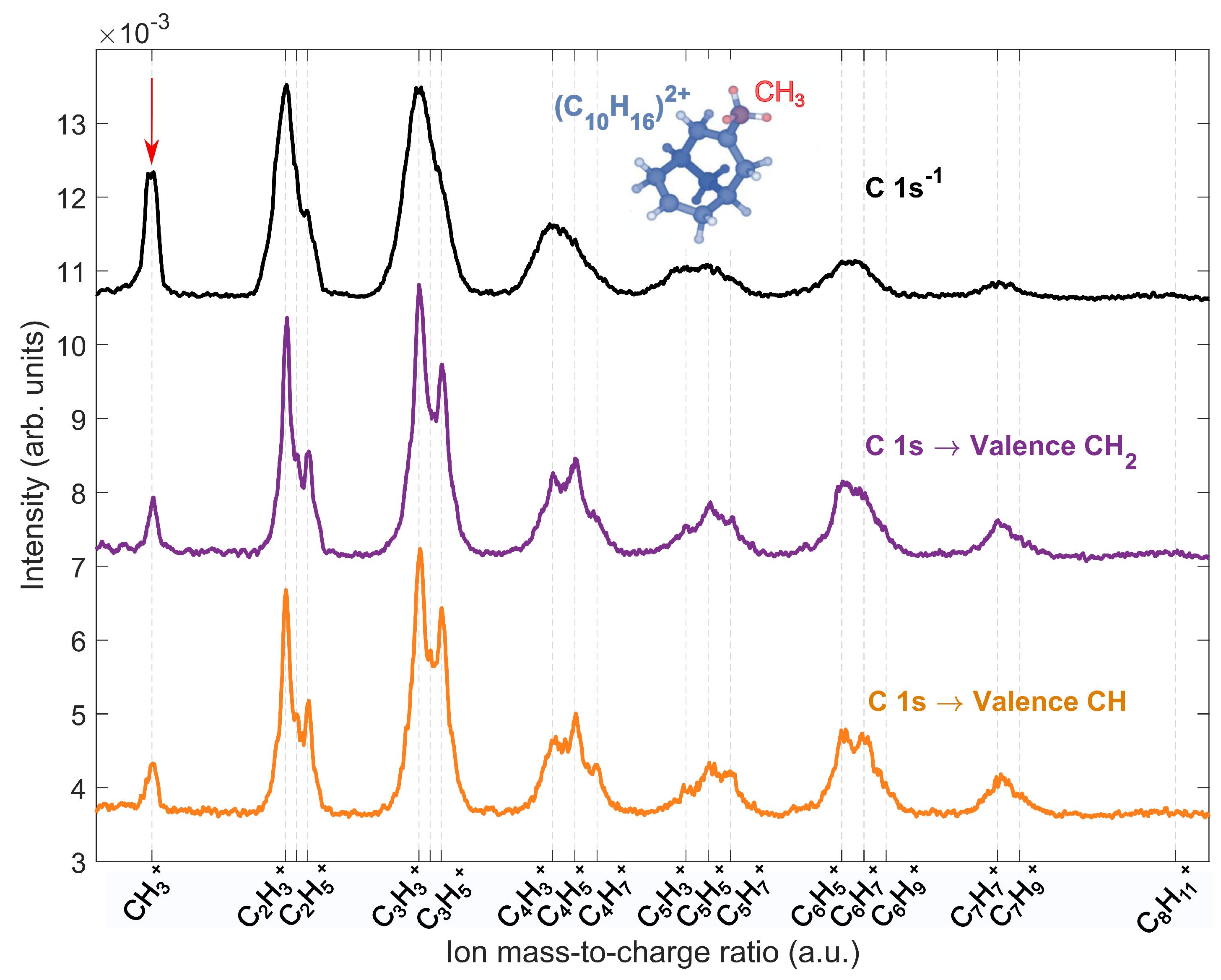
2.3. Site-Selective Fragmentation of Adamantane
2.4. Comparision to Core-Ionized Adamantane
3. Methods
3.1. Experimental
3.2. Theoretical
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Eberhardt, W.; Sham, T.; Carr, R.; Krummacher, S.; Strongin, M.; Weng, S.; Wesner, D. Site-specific fragmentation of small molecules following soft-X-ray excitation. Phys. Rev. Lett. 1983, 50, 1038. [Google Scholar] [CrossRef]
- Nenner, I.; Reynaud, C.; Schmelz, H.; Ferrand-Tanaka, L.; Simon, M.; Morin, P. Site selective fragmentation with soft X-rays: From gaseous polyatomic molecules, free clusters, polymers, adsorbates to biological macromolecules. Z. Phys. Chem. 1996, 195, 43–63. [Google Scholar] [CrossRef]
- Hatherly, P.; Codling, K.; Stankiewicz, M.; Roper, M. Symmetry effects in site-specific fragmentation of carbon dioxide. J. Electron Spectrosc. Relat. Phenom. 1996, 79, 407–410. [Google Scholar] [CrossRef]
- Hitchcock, A.P.; Neville, J.J.; Ju, A.; Cavell, R.G. Ionic fragmentation of inner-shell excited molecules. J. Electron Spectrosc. Relat. Phenom. 1998, 88, 71–75. [Google Scholar] [CrossRef]
- Ibuki, T.; Okada, K.; Saito, K.; Gejo, T. Molecular size effect on the site-specific fragmentation of N and OK shell excited CH3OCOCN and CH3OCOCH2CN molecules. J. Electron Spectrosc. Relat. Phenom. 2000, 107, 39–47. [Google Scholar] [CrossRef]
- Liu, X.; Prümper, G.; Kukk, E.; Sankari, R.; Hoshino, M.; Makochekanwa, C.; Kitajima, M.; Tanaka, H.; Yoshida, H.; Tamenori, Y.; et al. Site-selective ion production of the core-excited CH3 F molecule probed by auger-electron–ion coincidence measurements. Phys. Rev. A 2005, 72, 042704. [Google Scholar] [CrossRef]
- Erben, M.F.; Geronés, M.; Romano, R.M.; Della Védova, C.O. Dissociative photoionization of methoxycarbonylsulfenyl chloride, CH3OC(O)SCl, following sulfur 2p, chlorine 2p, and oxygen 1s excitations. J. Phys. Chem. A 2007, 111, 8062–8071. [Google Scholar] [CrossRef]
- Salén, P.; Kamińska, M.; Squibb, R.J.; Richter, R.; Alagia, M.; Stranges, S.; van der Meulen, P.; Eland, J.H.; Feifel, R.; Zhaunerchyk, V. Selectivity in fragmentation of N-methylacetamide after resonant K-shell excitation. Phys. Chem. Chem. Phys. 2014, 16, 15231–15240. [Google Scholar] [CrossRef]
- de Castilho, R.; Nunez, C.; Lago, A.F.; Santos, A.C.F.; Coutinho, L.H.; Lucas, C.A.; Pilling, S.; Silva-Moraes, M.; de Souza, G. Excitation and ionic fragmentation of the carvone molecule (C10H14O) around the O 1s edge. J. Electron Spectrosc. Relat. Phenom. 2014, 192, 61–68. [Google Scholar] [CrossRef]
- Chagas, G.R.; Satyanarayana, V.S.V.; Kessler, F.; Belmonte, G.K.; Gonsalves, K.E.; Weibel, D.E. Selective fragmentation of radiation-sensitive novel polymeric resist materials by inner-shell irradiation. ACS Appl. Mater. Interfaces 2015, 7, 16348–16356. [Google Scholar] [CrossRef]
- Habenicht, W.; Baiter, H.; Mueller-Dethlefs, K.; Schlag, E.W. Memory effects in molecular fragmentation induced by site-specific core excitation using a reflection time-of-flight mass spectrometer. J. Phys. Chem. 1991, 95, 6774–6780. [Google Scholar] [CrossRef]
- Nagaoka, S.i.; Fujibuchi, T.; Ohshita, J.; Ishikawa, M.; Koyano, I. Fragmentation of F3SiCH2CH2Si(CH3)3 vapour following Si: 2p core-level photoexcitation. A search for a site-specific process in complex molecules. Int. J. Mass Spectrom. Ion Process. 1997, 171, 95–103. [Google Scholar] [CrossRef]
- Schmelz, H.; Reynaud, C.; Simon, M.; Nenner, I. Site-selective fragmentation in core-excited bromo-chloro-alkanes [Br(CH2)nCl]. J. Chem. Phys. 1994, 101, 3742–3749. [Google Scholar] [CrossRef]
- Bolognesi, P.; Kettunen, J.; Cartoni, A.; Richter, R.; Tosic, S.; Maclot, S.; Rousseau, P.; Delaunay, R.; Avaldi, L. Site-and state-selected photofragmentation of 2Br-pyrimidine. Phys. Chem. Chem. Phys. 2015, 17, 24063–24069. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, M.; Fantuzzi, F.; Wohlfart, L.; Kopp, K.; Engels, B.; Bozek, J.; Nicolas, C.; Mayer, D.; Gühr, M.; Holzmeier, F.; et al. Fragmentation of isocyanic acid, HNCO, following core excitation and ionization. J. Chem. Phys. 2021, 154, 114302. [Google Scholar] [CrossRef] [PubMed]
- Oghbaiee, S.; Gisselbrecht, M.; Walsh, N.; Oostenrijk, B.; Laksman, J.; Månsson, E.P.; Sankari, A.; Eland, J.H.; Sorensen, S.L. Site-dependent nuclear dynamics in core-excited butadiene. Phys. Chem. Chem. Physics 2022, 24, 28825–28830. [Google Scholar] [CrossRef]
- Levola, H.; Itälä, E.; Schlesier, K.; Kooser, K.; Laine, S.; Laksman, J.; Ha, D.T.; Rachlew, E.; Tarkanovskaja, M.; Tanzer, K.; et al. Ionization-site effects on the photofragmentation of chloro-and bromoacetic acid molecules. Phys. Rev. A 2015, 92, 063409. [Google Scholar] [CrossRef]
- Candian, A.; Bouwman, J.; Hemberger, P.; Bodi, A.; Tielens, A.G. Dissociative ionisation of adamantane: A combined theoretical and experimental study. Phys. Chem. Chem. Phys. 2018, 20, 5399–5406. [Google Scholar] [CrossRef]
- Boyer, A.; Hervé, M.; Scognamiglio, A.; Loriot, V.; Lépine, F. Time-resolved relaxation and cage opening in diamondoids following XUV ultrafast ionization. Phys. Chem. Chem. Phys. 2021, 23, 27477–27483. [Google Scholar] [CrossRef]
- Maclot, S.; Lahl, J.; Peschel, J.; Wikmark, H.; Rudawski, P.; Brunner, F.; Coudert-Alteirac, H.; Indrajith, S.; Huber, B.A.; Díaz-Tendero, S.; et al. Dissociation dynamics of the diamondoid adamantane upon photoionization by XUV femtosecond pulses. Sci. Rep. 2020, 10, 2884. [Google Scholar] [CrossRef]
- Bouwman, J.; Horst, S.; Oomens, J. Spectroscopic characterization of the product ions formed by electron ionization of adamantane. ChemPhysChem 2018, 19, 3211–3218. [Google Scholar] [CrossRef] [PubMed]
- Waltman, R.J.; Ling, A.C. Mass spectrometry of diamantane and some adamantane derivatives. Can. J. Chem. 1980, 58, 2189–2195. [Google Scholar] [CrossRef]
- Crandall, P.B.; Müller, D.; Leroux, J.; Förstel, M.; Dopfer, O. Optical spectrum of the adamantane radical cation. Astrophys. J. Lett. 2020, 900, L20. [Google Scholar] [CrossRef]
- Richter, R.; Wolter, D.; Zimmermann, T.; Landt, L.; Knecht, A.; Heidrich, C.; Merli, A.; Dopfer, O.; Reiß, P.; Ehresmann, A.; et al. Size and shape dependent photoluminescence and excited state decay rates of diamondoids. Phys. Chem. Chem. Phys. 2014, 16, 3070–3076. [Google Scholar] [CrossRef]
- Ganguly, S.; Gisselbrecht, M.; Eng-Johnsson, P.; Feifel, R.; Hervieux, P.A.; Alfaytarouni, Z.; Fink, R.F.; Díaz-Tendero, S.; Milosavljević, A.R.; Rousseau, P.; et al. Coincidence study of core-ionized adamantane: Site-sensitivity within a carbon cage? Phys. Chem. Chem. Phys. 2022, 24, 28994–29003. [Google Scholar] [CrossRef]
- Willey, T.; Bostedt, C.; Van Buuren, T.; Dahl, J.; Liu, S.; Carlson, R.; Terminello, L.; Möller, T. Molecular limits to the quantum confinement model in diamond clusters. Phys. Rev. Lett. 2005, 95, 113401. [Google Scholar] [CrossRef]
- Willey, T.M.; Lee, J.R.; Brehmer, D.; Paredes Mellone, O.A.; Landt, L.; Schreiner, P.R.; Fokin, A.A.; Tkachenko, B.A.; de Meijere, A.; Kozhushkov, S.; et al. X-ray spectroscopic identification of strain and structure-based resonances in a series of saturated carbon-cage molecules: Adamantane, twistane, octahedrane, and cubane. J. Vac. Sci. Technol. A Vacuum Surf. Film. 2021, 39, 053208. [Google Scholar] [CrossRef]
- Kryzhevoi, N.V.; Dobrodey, N.V.; Cederbaum, L.S. Equivalent core model: Extended theory and applications. J. Chem. Phys. 2003, 118, 2081–2091. [Google Scholar] [CrossRef]
- Morini, F.; Watanabe, N.; Kojima, M.; Deleuze, M.S.; Takahashi, M. Influence of molecular vibrations on the valence electron momentum distributions of adamantane. J. Chem. Phys. 2017, 146, 094307. [Google Scholar] [CrossRef]
- Boschi, R.; Schmidt, W.; Suffolk, R.; Wilkins, B.; Lempka, H.; Ridyard, J. Complete valence shell electronic structure of adamantane from He I and He II photoelectron spectroscopy. J. Electron Spectrosc. Relat. Phenom. 1973, 2, 377–380. [Google Scholar] [CrossRef]
- Sałek, P.; Fink, R.; Gel’mukhanov, F.; Piancastelli, M.; Feifer, R.; Bässler, M.; Sorensen, S.; Miron, C.; Wang, H.; Hjelte, I.; et al. Resonant X-ray Raman Scattering involving avoided crossings in the final-state potential-energy curves. Phys. Rev. A 2000, 62, 062506. [Google Scholar] [CrossRef]
- Piancastelli, M.; Fink, R.; Feifel, R.; Bässler, M.; Sorensen, S.; Miron, C.; Wang, H.; Hjelte, I.; Björneholm, O.; Ausmees, A.; et al. Bond-distance-dependent decay probability of the N 1s→ π* core-excited state in N2. J. Phys. B At. Mol. Opt. Phys. 2000, 33, 1819. [Google Scholar] [CrossRef]
- Feifel, R.; Piancastelli, M.N. Core-level spectroscopy and dynamics of free molecules. J. Electron Spectrosc. Relat. Phenom. 2011, 183, 10–28. [Google Scholar] [CrossRef]
- Patzer, A.; Schütz, M.; Möller, T.; Dopfer, O. Infrared spectrum and structure of the adamantane cation: Direct evidence for Jahn–Teller distortion. Angew. Chem. 2012, 124, 5009–5013. [Google Scholar] [CrossRef]
- Miron, C.; Simon, M.; Morin, P.; Nanbu, S.; Kosugi, N.; Sorensen, S.; Naves de Brito, A.; Piancastelli, M.; Björneholm, O.; Feifel, R.; et al. Nuclear motion driven by the Renner–Teller effect as observed in the resonant Auger decay to the X2Π electronic ground state of N2O+. J. Chem. Phys. 2001, 115, 864–869. [Google Scholar] [CrossRef]
- Antonsson, E.; Patanen, M.; Nicolas, C.; Benkoula, S.; Neville, J.; Sukhorukov, V.; Bozek, J.; Demekhin, P.V.; Miron, C. Dynamics of the C 1 s→ π* excitation and decay in CO 2 probed by vibrationally and angularly resolved Auger spectroscopy. Phys. Rev. A 2015, 92, 042506. [Google Scholar] [CrossRef]
- Travnikova, O.; Miron, C.; Bässler, M.; Feifel, R.; Piancastelli, M.; Sorensen, S.; Svensson, S. Resonant Auger decay study of C1s→ π* core-excited OCS. J. Electron Spectrosc. Relat. Phenom. 2009, 174, 100–106. [Google Scholar] [CrossRef]
- PLEIADES Beamline Website. 2019. Available online: https://www.synchrotron-soleil.fr/fr/lignes-de-lumiere/pleiades (accessed on 1 June 2021).
- Adachi, J.i.; Kosugi, N.; Shigemasa, E.; Yagishita, A. Vibronic Couplings in the C 1s→ n sσg Rydberg Excited States of CO2. J. Phys. Chem. 1996, 100, 19783–19788. [Google Scholar] [CrossRef]
- Danilovic, D.; Bozanic, D.K.; Dojcilovic, R.; Vukmirovic, N.; Sapkota, P.; Vukasinovic, I.; Djokovic, V.; Bozek, J.; Nicolas, C.; Ptasinska, S.; et al. Aerosol Synthesis and Gas-Phase Photoelectron Spectroscopy of Ag-Bi-I Nanosystems. J. Phys. Chem. C 2020, 124, 23930–23937. [Google Scholar] [CrossRef]
- Moddeman, W.; Carlson, T.A.; Krause, M.O.; Pullen, B.; Bull, W.; Schweitzer, G. Determination of the K—LL auger spectra of N2, O2, CO, NO, H2O, and CO2. J. Chem. Phys. 1971, 55, 2317–2336. [Google Scholar] [CrossRef]
- Liu, X.J.; Nicolas, C.; Robert, E.; Miron, C. EPICEA: Probing high-energy electron emission in the molecular frame. J. Phys. Conf. Ser. 2014, 488, 142005. [Google Scholar] [CrossRef]
- Miron, C.; Simon, M.; Leclercq, N.; Morin, P. New high luminosity “double toroidal” electron spectrometer. Rev. Sci. Instruments 1997, 68, 3728–3737. [Google Scholar] [CrossRef]
- Liu, X.J.; Nicolas, C.; Miron, C. Design of a lens table for a double toroidal electron spectrometer. Rev. Sci. Instruments 2013, 84, 033105. [Google Scholar] [CrossRef] [PubMed]
- Prümper, G.; Ueda, K. Electron–ion–ion coincidence experiments for photofragmentation of polyatomic molecules using pulsed electric fields: Treatment of random coincidences. Nucl. Instruments Methods Phys. Res. Sect. A Accel. Spectrometers Detect. Assoc. Equip. 2007, 574, 350–362. [Google Scholar] [CrossRef]
- Skomorowski, W.; Krylov, A.I. Feshbach-Fano approach for calculation of Auger decay rates using equation-of-motion coupled-cluster wave functions. I. Theory and implementation. J. Chem. Phys. 2021, 154, 084124. [Google Scholar] [CrossRef]
- Skomorowski, W.; Krylov, A.I. Feshbach–Fano approach for calculation of Auger decay rates using equation-of-motion coupled-cluster wave functions. II. Numerical examples and benchmarks. J. Chem. Phys. 2021, 154, 084125. [Google Scholar] [CrossRef]
- Epifanovsky, E.; Gilbert, A.T.; Feng, X.; Lee, J.; Mao, Y.; Mardirossian, N.; Pokhilko, P.; White, A.F.; Coons, M.P.; Dempwolff, A.L.; et al. Software for the frontiers of quantum chemistry: An overview of developments in the Q-Chem 5 package. J. Chem. Phys. 2021, 155, 084801. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian~16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H.; et al. Gaussian 09, revision a.02; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Ceriotti, M.; Bussi, G.; Parrinello, M. Nuclear quantum effects in solids using a colorednoise thermostat. Phys. Rev. Lett. 2009, 103, 030603. [Google Scholar] [CrossRef]
- Ceriotti, M.; Bussi, G.; Parrinello, M. Colored-noise thermostats à la carte. J. Chem. Theory Comput. 2010, 6, 1170. [Google Scholar] [CrossRef]
- Crespo-Otero, R.; Barbatti, M. Spectrum simulation and decomposition with nuclear ensemble: Formal derivation and application to benzene, furan and 2-phenylfuran. Theor. Chem. Accounts 2012, 131, 1. [Google Scholar] [CrossRef]
- Prlj, A.; Marsili, E.; Hutton, L.; Hollas, D.; Shchepanovska, D.; Glowacki, D.R.; Slavıcek, P.; Curchod, B.F. Calculating photoabsorption cross-sections for atmospheric volatile organic compounds. ACS Earth Space Chem. 2022, 6, 207. [Google Scholar] [CrossRef] [PubMed]
- Sala, F.D.; Rousseau, R.; Görling, A.; Marx, D. Quantum and thermal fluctuation effects on the photoabsorption spectra of clusters. Phys. Rev. Lett. 2004, 92, 183401. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Brown, R.C.; Heller, E.J. Multidimensional reflection approximation: Application to the photodissociation of polyatomics. J. Phys. Chem. 1983, 87, 2045. [Google Scholar] [CrossRef]
- Lee, S.Y. Semiclassical theory of radiation interacting with a molecule. J. Chem. Phys. 1998, 76, 3064. [Google Scholar] [CrossRef]
- Couto, P.C.D.; Hollas, D.; Slavıcek, P. On the performance of optimally tuned rangeseparated hybrid functionals for X-ray absorption modeling. J. Chem. Theory Comput. 2015, 11, 3234. [Google Scholar] [CrossRef]
- Zeng, W.; Gong, S.; Zhong, C.; Yang, C. Prediction of oscillator strength and transition dipole moments with the nuclear ensemble approach for thermally activated delayed fluorescence emitters. J. Phys. Chem. 2019, 123, 10081. [Google Scholar] [CrossRef]
- Oncak, M.; Sistık, L.; Slavıcek, P. Can theory quantitatively model stratospheric photolysis? ab initio estimate of absolute absorption cross sections of cloocl. J. Chem. Phys. 2010, 133, 174303. [Google Scholar] [CrossRef]
- Slavıcek, P.; Oncak, M.; Hollas, D.; Svoboda, O. Abin, version 1.0. Available online: https://github.com/PHOTOX/ABIN (accessed on 16 July 2023).
- Seritan, S.; Bannwarth, C.; Fales, B.S.; Hohenstein, E.G.; Isborn, C.M.; Kokkila- Schumacher, S.I.; Li, X.; Liu, F.; Luehr, N.; Snyder, J.W.; et al. Terachem: A graphical processing unit-accelerated electronic structure package for large-scale ab initio molecular dynamics. Wiley Interdiscip. Comput. Mol. Sci. 2021, 11, e1494. [Google Scholar] [CrossRef]
- Ufimtsev, I.S.; Mart´ınez, T.J. Quantum chemistry on graphical processing units. 1. strategies for two-electron integral evaluation. J. Chem. Theory Comput. 2008, 4, 222. [Google Scholar] [CrossRef]
- Ufimtsev, I.S.; Martinez, T.J. Quantum chemistry on graphical processing units. 3. analytical energy gradients, geometry optimization, and first principles molecular dynamics. J. Chem. Theory Comput. 2009, 5, 2619. [Google Scholar] [CrossRef]
- Ufimtsev, I.S.; Martinez, T.J. Quantum chemistry on graphical processing units. 2. direct self-consistent-field implementation. J. Chem. Theory Comput. 2009, 5, 1004. [Google Scholar] [CrossRef] [PubMed]
- George, S.D.; Petrenko, T.; Neese, F. Time-dependent density functional calculations of ligand K-edge X-ray absorption spectra. Inorganica Chim. Acta 2008, 361, 965. [Google Scholar] [CrossRef]
- Stener, M.; Fronzoni, G.; de Simone, M. Time dependent density functional theory of core electrons excitations. Chem. Phys. Lett. 2003, 373, 115. [Google Scholar] [CrossRef]
- Krylov, A.I. Equation-of-motion coupled-cluster methods for open-shell and electronically excited species: The hitchhiker’s guide to fock space. Annu. Rev. Phys. Chem. 2008, 59, 433. [Google Scholar] [CrossRef] [PubMed]
- Fransson, T.; Brumboiu, I.E.; Vidal, M.L.; Norman, P.; Coriani, S.; Dreuw, A. Xaboom: An X-ray absorption benchmark of organic molecules based on carbon, nitrogen, and oxygen 1s → π* transitions. J. Chem. Theory Comput. 2021, 17, 1618. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ganguly, S.; Gisselbrecht, M.; Eng-Johnsson, P.; Feifel, R.; Díaz-Tendero, S.; Muchová, E.; Milosavljević, A.R.; Rousseau, P.; Maclot, S. Resonant Inner-Shell Photofragmentation of Adamantane (C10H16). Molecules 2023, 28, 5510. https://doi.org/10.3390/molecules28145510
Ganguly S, Gisselbrecht M, Eng-Johnsson P, Feifel R, Díaz-Tendero S, Muchová E, Milosavljević AR, Rousseau P, Maclot S. Resonant Inner-Shell Photofragmentation of Adamantane (C10H16). Molecules. 2023; 28(14):5510. https://doi.org/10.3390/molecules28145510
Chicago/Turabian StyleGanguly, Smita, Mathieu Gisselbrecht, Per Eng-Johnsson, Raimund Feifel, Sergio Díaz-Tendero, Eva Muchová, Aleksandar R. Milosavljević, Patrick Rousseau, and Sylvain Maclot. 2023. "Resonant Inner-Shell Photofragmentation of Adamantane (C10H16)" Molecules 28, no. 14: 5510. https://doi.org/10.3390/molecules28145510
APA StyleGanguly, S., Gisselbrecht, M., Eng-Johnsson, P., Feifel, R., Díaz-Tendero, S., Muchová, E., Milosavljević, A. R., Rousseau, P., & Maclot, S. (2023). Resonant Inner-Shell Photofragmentation of Adamantane (C10H16). Molecules, 28(14), 5510. https://doi.org/10.3390/molecules28145510