

Experimental IR, Raman, and UV-Vis Spectra DFT Structural and Conformational Studies: Bioactivity and Solvent Effect on Molecular Properties of Methyl-Eugenol


Abstract
Highlights
- The ME molecule has 21 stable configurations.
- For all the tops (except =CH2), the barrier heights are of the same order, while the =CH2 top has a barrier height one order of magnitude higher.
- Like estragole and eugenol, ME also has the same Fermi doublets for the following modes: νs(–CH2) and 2 βs(–CH2); νs(CH3) and 2 δs(CH3).
- The ME molecule has three active sites.
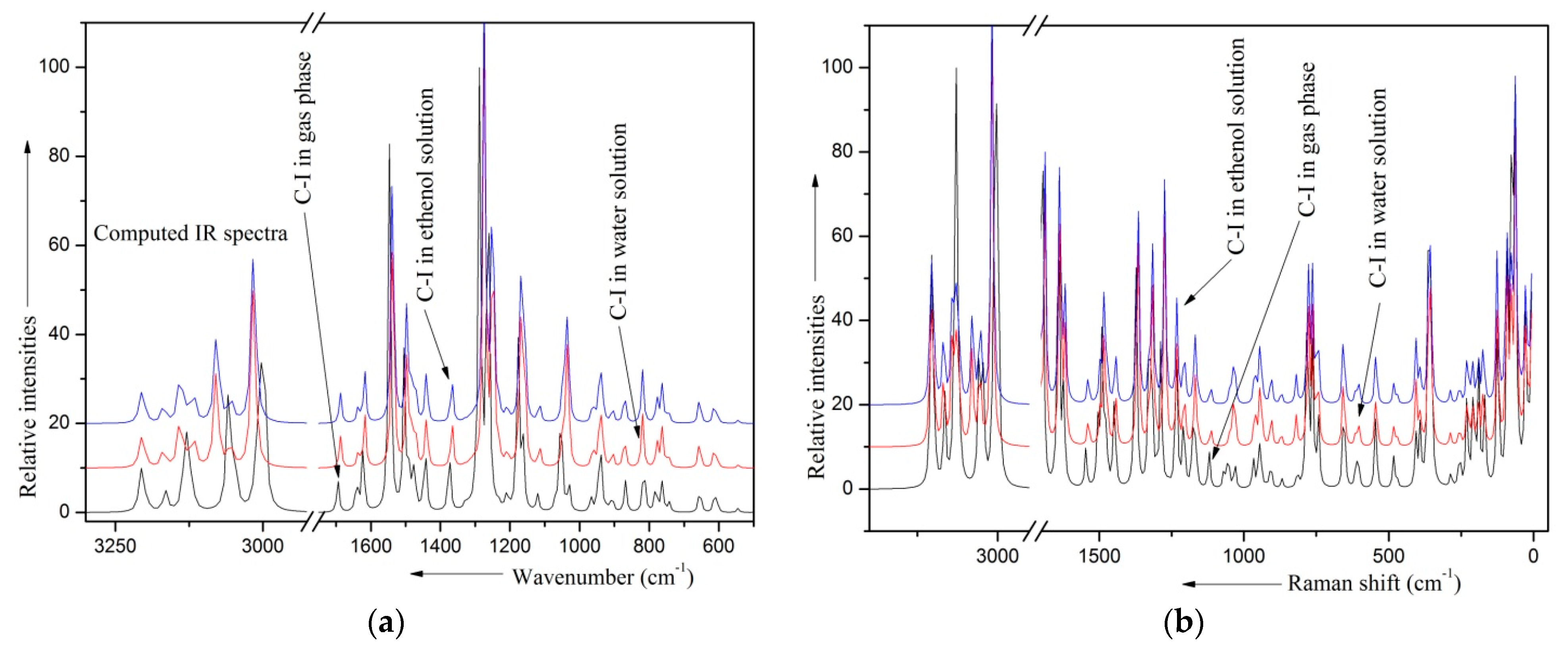
- Vibrational analysis suggests that the solvents affect the internal modes of both OCH3 moieties strongly.
- The methyl-eugenol molecule could be a good choice for the pharmacological applications
- The OCH3 moieties of methyl-eugenol play significant role in interaction with other molecules.
Abstract

1. Introduction
2. Result and Discussion
2.1. Determination of Conformers
2.2. Molecular Geometries
2.3. Barrier Heights
2.4. Bioactive Scores
2.5. APT Charge
2.6. Vibrational Analysis
2.6.1. Methoxy (–OCH3) Group Modes (24)
2.6.2. Allyl (–CH2–CH=CH2) Group Modes (21)
Methylene (–CH2–) Group Modes (6)
Vinyl (–CH=CH2) Group Modes (12)
2.6.3. Phenyl Moiety Modes (30)
Phenyl Ring Modes (12)
C–H Modes (9)
C–O(CH3) and C–C(H2CHCH2) Group Modes (9)
2.7. Conformer Dependent Modes
2.8. Solvent Effects
2.9. MEP Plots
2.10. HOMO-LUMO Plots
3. Experimental Details
4. Computational Detail
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Tan, K.H.; Nishida, R. Methyl eugenol: Its occurrence, distribution, and role in nature, especially in relation to insect behaviour and pollination. J. Insect Sci. 2012, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Kothari, S.K.; Bhattacharya, A.K.; Ramesh, S. Essential oil yield and quality of methyl eugenol rich Ocimum tenuiflorum grown in south India as influenced by method of harvest. J. Chromatogr. A 2004, 1054, 67–72. [Google Scholar] [CrossRef] [PubMed]
- De Vincenzi, M.; Silano, M.; Stacchini, P.; Scazzocchio, B. Constituents of aromatic plants: Methyleugenol. Fitoterapia 2000, 71, 216–221. [Google Scholar] [CrossRef]
- Atkinson, R.G. Phenylpropenes: Occurrence, distribution, and biosynthesis in fruit. J. Agric. Food Chem. 2018, 66, 2259–2272. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, J.; Zhang, J.; Sun, K.; Li, Q.; Kuang, B.; Wang, M.M.Z.; Hou, S.; Gong, N. Methyl eugenol attenuates liver ischemia reperfusion injury via activating PI3K/Akt signalling. Int. Immunopharmacol. 2021, 99, 108023. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Sun, Z.; Ren, Q.; Su, X.; Zhang, D. Methyl eugenol induces potent anticancer effects in RB355 human retinoblastoma cells by inducing autophagy, cell cycle arrest and inhibition of PI3K/Akt signalling pathway. JBUON 2018, 23, 1174–1178. [Google Scholar]
- Tang, F.; Chen, F.; Ling, X.; Huang, Y.; Zheng, X.; Tang, Q. Tan. Inhibitory effect of methyleugenol on IgE-mediated allergic inflammation in RBL-2H3 cells. Mediat. Inflamm. 2015, 2015, 463530. [Google Scholar] [CrossRef]
- Saleh, H.; El-Shorbagy, H.M. Mechanism underlying methyl eugenol attenuation of intestinal ischemia/reperfusion injury. Appl. Physiol. Nutr. Metab. 2017, 42, 1097–1105. [Google Scholar] [CrossRef]
- Carlini, E.A.; Dallmeier, K.; Zelger, J.L. Methyleugenol as a surgical anesthetic in rodents. Experientia 1981, 37, 588–589. [Google Scholar] [CrossRef]
- Shelly, T. Effects of methyl eugenol and raspberry ketone/cue lure on the sexual ehaviour of Bactrocera species (Diptera: Tephritidae). Appl. Entomol. Zool. 2010, 45, 349–361. [Google Scholar] [CrossRef]
- Poucher, W.A. The Raw Materials of Perfumery, Poucher’s Perfumes, Cosmetics and Soaps, 9th ed.; Jouhar, A.J., Ed.; Chapman & Hall: London, UK, 1991; Volume 1, pp. 148–149. [Google Scholar]
- Zhang, X.; Felter, S.P.; Api, A.M.; Joshi, K.; Selechnik, D. A Cautionary tale for using read-across for cancer hazard classification: Case study of isoeugenol and methyl eugenol. Regul. Toxicol. Pharmacol. 2022, 136, 105280. [Google Scholar] [CrossRef] [PubMed]
- Hee, A.K.; Tan, K. Transport of methyl eugenol-derived sex pheromonal components in the male fruit fly, Bactrocera dorsalis. Comp. Biochem. Physiol. C 2006, 143, 422–428. [Google Scholar]
- Gu, X.; Cai, P.; Yang, Y.; Yang, Q.; Yao, M.; Idrees, A.; Ji, Q.; Yang, J.; Chen, J. The response of four braconid parasitoid species to methyl eugenol: Optimization of a biocontrol tactic to suppress Bactrocera dorsalis. Biol. Control 2018, 122, 101–108. [Google Scholar] [CrossRef]
- Norte, M.C.B.; Cosentino, R.M.; Lazarini, C.A. Effects of methyl-eugenol administration on behavioral models related to depression and anxiety, in rats. Phytomedicine 2005, 12, 294–298. [Google Scholar] [CrossRef]
- Rietjens, I.M.; Cohen, S.M.; Fukushima, S.; Gooderham, N.J.; Hecht, S.; Marnett, L.J.; Smith, R.L.; Adams, T.B.; Bastaki, M.; Harman, C.G.; et al. Impact of structural and metabolic variations on the toxicity and carcinogenicity of hydroxy-and alkoxy-substituted allyl- and propenylbenzenes. Chem. Res. Toxicol. 2014, 27, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Chowdhry, B.Z.; Ryall, J.P.; Dines, T.J.; Mendham, A.P. Infrared and Raman spectroscopy of Eugenol, Isoeugenol and Methyl Eugenol: Conformational analysis and Vibrational Assignment from DFT Calculatons of the anhormonic Fundamentals. J. Phys. Chem. A 2015, 112, 5691–5699. [Google Scholar]
- Meijers, S.; Ponec, V.; Finocchio, E.; Busca, G. FTIR Study of the Adsorption and Trasformation of Allylbenzene over Oxide Catalysis. J. Chem. Soc. Faraday Trans. 1995, 91, 1861–1869. [Google Scholar] [CrossRef]
- Owen, N.L.; Hester, R.E. Vibrational Spectra and Torsional Barriers of Anisole and some monohalogenic derivatives. J. Spectrochim. Acta. A 1969, 25, 343–354. [Google Scholar] [CrossRef]
- Yadav, R.K.; Yadav, B.; Srivastav, G.; Jha, O.; Yadav, R.A. Confomational, Structural and Vibrational studies of estragole. Vib. Spect. 2021, 117, 103317. [Google Scholar] [CrossRef]
- Yadav, R.K.; Yadav, B.; Yadav, R.A. Spectroscopic, structural and conformational investigations of the major constituent of the clove oil: Eugenol. J. Comp. Theor. Chem. 2023. [Google Scholar]
- Gupta, V.P. Electron Density Analysis and Electrostatic Potential. In Principles and Applications of Quantum Chemistry; Elsevier: Amsterdam, The Netherlands, 2016; p. 195. [Google Scholar]
- Yadav, B.; Yadav, R.K.; Srivastav, G.; Yadav, R.A. Experimental Raman, FTIR and UV-vis spectra, DFT studies of molecular structures and barrier heights, thermodynamic functions and bioactivity of kaempferol. J. Mol. Struct. 2022, 1258, 132637. [Google Scholar] [CrossRef]
- Srivastav, G.; Yadav, R.K.; Yadav, B.; Yadav, R.A. Vibrational spectra and molecular structure of sulfanilic acid: IR and low temperature Raman studies and DFT investigation of monomeric and dimeric forms. J. Mol. Struct. 2023, 1272, 134143. [Google Scholar] [CrossRef]
- Koopmans, T. Uber die Zuordnung von Wellenfunktionen und Eigenwerten zu den Einzelnen Elektronen Eines Atoms. Phisica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Tezcan, F.; Yerlikaya, G.; Mahmood, A.; Kardaş, G. A novel thiophene Schiff base as an efficient corrosion inhibitor for mild steel in 1.0 M HCl: Electrochemical and quantum chemical studies. J. Mol. Liq. 2018, 269, 398–406. [Google Scholar] [CrossRef]
- Zhang, G.A.; Hou, X.M.; Hou, B.S.; Liu, H.F. Benzimidazole derivatives as novel inhibitors for the corrosion of mild steel in acidic solution: Experimental and theoretical studies. J. Mol. Liq. 2019, 278, 413–427. [Google Scholar] [CrossRef]
- Gupta, N.K.; Verma, C.; Quraishi, M.A.; Mukherjee, A.K. Schiff’s bases derived from l-lysine and aromatic aldehydes as green corrosion inhibitors for mild steel: Experimental and theoretical studies. J. Mol. Liq. 2016, 215, 47–57. [Google Scholar] [CrossRef]
- Heydari, H.; Talebian, M.; Salarvand, Z.; Raeissi, K.; Bagheri, M.; Golozar, M.A. Comparison of two Schiff bases containing O-methyl and nitro substitutes for corrosion inhibiting of mild steel in 1 M HCl solution. J. Mol. Liq. 2018, 254, 177–187. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.1; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Palavarapu, P.L. Ab initio Vibrational Raman and Raman optical activity spectra. J. Phys. Chem. A 1990, 94, 8106–8112. [Google Scholar] [CrossRef]
- Martin, J.M.L.; Valsenoy, V.; Alsenoy, C.V. GAR2PED; University of Antwerp: Antwerp, Belgium, 1995. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conformers | Energy (E) | ΔE Relative to the Conformer I | Relative Abundance | |
|---|---|---|---|---|
| kcal/Mole | kcal/Mole | K | ||
| I | −362,794.265 | 0 | 0 | 32.1 |
| II | −362,793.860 | 0.405 | 204 | 16.2 |
| III | −362,793.644 | 0.622 | 313 | 11.2 |
| IV | −362,793.237 | 1.028 | 518 | 5.7 |
| V | −362,793.212 | 1.054 | 530 | 5.4 |
| VI | −362,792.997 | 1.268 | 638 | 3.8 |
| VII | −362,792.978 | 1.287 | 648 | 3.7 |
| VIII | −362,792.903 | 1.363 | 685 | 3.2 |
| IX | −362,792.881 | 1.384 | 697 | 3.1 |
| X | −362,792.871 | 1.395 | 701 | 3.1 |
| XI | −362,792.859 | 1.408 | 708 | 3.0 |
| XII | −362,792.61 | 1.655 | 833 | 2.0 |
| XIII | −362,792.593 | 1.672 | 842 | 1.9 |
| XIV | −362,792.517 | 1.748 | 880 | 1.7 |
| XV | −362,792.517 | 1.749 | 880 | 1.7 |
| XVI | −362,791.789 | 2.477 | 1246 | 0.5 |
| XVII | −362,791.74 | 2.525 | 1271 | 0.5 |
| XVIII | −362,791.725 | 2.541 | 1279 | 0.4 |
| XIX | −362,791.705 | 2.561 | 1289 | 0.4 |
| XX | −362,791.302 | 2.963 | 1491 | 0.2 |
| XXI | −362,791.301 | 2.964 | 1492 | 0.2 |
| Top | Foldness | * ∆EAB | * ∆EBC | * ∆ECD | * ∆EDE | * ∆EEF | * ∆EFG |
|---|---|---|---|---|---|---|---|
| CHCH2 | 3 | 2.36 | 1.27 | 2.00 | 1.98 | 3.62 | 4.73 |
| CH2CHCH2 | 2 | 2.02 | 1.60 | 1.52 | 1.94 | - | - |
| CH3 | 3 | 3.34 | 3.34 | 3.34 | 3.34 | 3.34 | 3.34 |
| CH3 | 3 | 3.34 | 3.34 | 3.34 | 3.34 | 3.34 | 3.34 |
| 8OCH3 | 3 | 1.13 | 1.54 | 7.15 | 7.15 | 1.54 | 1.13 |
| 9OCH3 | 3 | 2.37 | 1.18 | 2.73 | 2.71 | 1.15 | 2.36 |
| =CH2 | 2 | 93.13 | 93.13 | 93.13 | 93.13 | - | - |
| Bioactive Acceptors | M-Eugenol |
|---|---|
| GPCR ligand | −0.81 |
| Ion channel modulator | −0.38 |
| Kinase inhibitor | −1.06 |
| Nuclear receptor ligand | −0.80 |
| Protease inhibitor | −1.14 |
| Enzyme inhibitor | −0.43 |
| Atoms | C-I | C-II | C-III | C-I In Solvent Effect | |
|---|---|---|---|---|---|
| Water | Ethanol | ||||
| C1 | 0.018 | 0.012 | −0.004 | 0.040 | 0.039 |
| C2 | −0.159 | −0.119 | −0.117 | −0.200 | −0.198 |
| C3 | 0.515 | 0.516 | 0.503 | 0.688 | 0.680 |
| C4 | 0.518 | 0.510 | 0.513 | 0.668 | 0.661 |
| C5 | −0.096 | −0.072 | −0.088 | −0.131 | −0.129 |
| C6 | −0.086 | −0.120 | −0.080 | −0.114 | −0.112 |
| H7 | 0.062 | 0.050 | 0.050 | 0.087 | 0.085 |
| O8 | −0.863 | −0.891 | −0.869 | −1.135 | −1.121 |
| O9 | −0.922 | −0.919 | −0.907 | −1.173 | −1.161 |
| H10 | 0.045 | 0.045 | 0.046 | 0.075 | 0.073 |
| H11 | 0.027 | 0.042 | 0.027 | 0.040 | 0.039 |
| C12 | 0.133 | 0.131 | 0.133 | 0.154 | 0.153 |
| H13 | −0.051 | −0.053 | −0.041 | −0.058 | −0.058 |
| H14 | −0.028 | −0.032 | −0.035 | −0.039 | −0.038 |
| C15 | 0.048 | 0.061 | 0.064 | 0.084 | 0.082 |
| H16 | 0.018 | 0.016 | −0.021 | 0.018 | 0.018 |
| C17 | −0.141 | −0.142 | −0.149 | −0.200 | −0.196 |
| H18 | 0.029 | 0.028 | 0.023 | 0.039 | 0.038 |
| H19 | 0.029 | 0.029 | 0.046 | 0.038 | 0.038 |
| C20 | 0.552 | 0.542 | 0.545 | 0.653 | 0.648 |
| H21 | −0.043 | −0.040 | −0.041 | −0.043 | −0.043 |
| H22 | −0.002 | −0.002 | −0.001 | −0.003 | −0.003 |
| H23 | −0.042 | −0.041 | −0.041 | −0.042 | −0.042 |
| C24 | 0.511 | 0.529 | 0.520 | 0.631 | 0.625 |
| H25 | −0.035 | −0.039 | −0.038 | −0.039 | −0.039 |
| H26 | 0.000 | −0.001 | 0.000 | 0.000 | 0.000 |
| H27 | −0.038 | −0.040 | −0.039 | −0.040 | −0.039 |
| Group/Moiety | Modes in Symbolic Form * | Total | |
|---|---|---|---|
| Phenyl | Ring | 6ν(R) + 3α(R) + 3Φ(R) | 12 |
| (Ph)C–O(CH3) | 2ν + 2β + 2γ | 6 | |
| (Ph)C–C(H2CHCH2) | ν + β + γ | 3 | |
| (Ph)C–H | 3ν + 3β + 3γ | 9 | |
| OCH3 | CH3 | 2νs + 4νas + 2δs + 4δas + 2ρ║ + 2ρ┴ + 2τ | 18 |
| O–C(H3) | 2ν + 2α + 2τ | 6 | |
| Allyl | 2CH2 | 2νs + 2νas + 2βs + 2ρ + 2ω + t + τ | 12 |
| C–C | ν + α + τ | 3 | |
| C–H | ν + 2δ | 3 | |
| C=C | ν + α + τ | 3 | |
| Gas | Solvent | # Mode | |||
|---|---|---|---|---|---|
| Water | Ethanol | ||||
| (1) | (2) | (3) | (1)–(2) | (1)–(3) | |
| 28 (0.29) 0.74 | 27 (0.16) 0.75 | 27 (0.21) 0.75 | 1 | 1 | τ(C1–C12) |
| 64 (1.95) 0.69 | 61 (1.53) 0.75 | 62 (1.63) 0.75 | 3 | 2 | τ(C4–OCH3) |
| 76 (2.48) 0.71 | 77 (2.24) 0.75 | 78 (2.29) 0.75 | −1 | −2 | τ(C12–C15) |
| 89 (0.40) 0.74 | 91 (1.31) 0.75 | 91 (1.39) 0.75 | −2 | −2 | τ(C3–OCH3) |
| 121 (0.36) 0.75 | 122 (0.25) 0.75 | 123 (0.31) 0.75 | −1 | −2 | γ(C1–C12) |
| 165 (0.17) 0.73 | 169 (0.11) 0.73 | 169 (0.13) 0.73 | −4 | −4 | γ(C3–O8) |
| 186 (2.23) 0.50 | 187 (2.9) 0.37 | 187 (2.11) 0.37 | −2 | −2 | β(C3–O8) |
| 204 (0.21) 0.62 | 207 (0.6) 0.68 | 207 (0.8) 0.68 | −3 | −3 | γ(C4–O9) |
| 225 (0.18) 0.69 | 224 (0.9) 0.75 | 224 (0.12) 0.75 | 1 | 1 | τ(OCH3) |
| 250 (0.7) 0.68 | 251 (0.3) 0.67 | 251 (0.4) 0.66 | −1 | −1 | τ(OCH3) |
| 279 (0.3) 0.33 | 282 (0.2) 0.60 | 282 (0.3) 0.60 | −3 | −3 | α(C1–C12–C15) |
| 345 (0.18) 0.25 | 344 (0.5) 0.58 | 344 (0.7) 0.55 | 2 | 2 | β(C1–C12) |
| 354 (0.74) 0.11 | 353 (0.44) 0.10 | 353 (0.49) 0.10 | 1 | 1 | β(C1–O10) |
| 384 (1.12) 0.22 | 383 (1.5) 0.43 | 383 (1.7) 0.42 | 1 | 1 | α(C–O–C)ipc |
| 398 (2.10) 0.72 | 397 (1.12) 0.73 | 397 (1.14) 0.73 | 1 | 1 | α(C14–C15=C17) |
| 462 (1.2) 0.69 | 462 (1.2) 0.68 | 462 (1.2) 0.69 | 1 | 1 | Φ(R) 16b |
| 474 (0.6) 0.61 | 473 (0.3) 0.71 | 473 (0.4) 0.71 | 0 | 0 | α(R) 6b |
| 534 (1.15) 0.52 | 533 (1.11) 0.46 | 533 (1.13) 0.47 | 1 | 1 | α(R) 6a |
| 592 (1.5) 0.54 | 590 (1.3) 0.51 | 591 (1.4) 0.52 | 3 | 2 | α(C–O–C)opc |
| 600 (4.6) 0.72 | 601 (5.3) 0.71 | 601 (5.4) 0.71 | −1 | −1 | τ(C=C) |
| 642 (6.22) 0.49 | 643 (7.15) 0.46 | 643 (6.19) 0.46 | −1 | −1 | α(R) 12 |
| 728 (2.15) 0.17 | 730 (3.15) 0.25 | 730 (3.18) 0.24 | −2 | −2 | Φ(R) 4 |
| 748 (7.30) 0.13 | 748 (9.25) 0.14 | 748 (9.31) 0.14 | 0 | 0 | Φ(R) 16a |
| 766 (6.47) 0.07 | 762 (6.27) 0.07 | 763 (6.34) 0.07 | 3 | 3 | ν(R) 1 |
| 799 (12.3) 0.24 | 803 (12.6) 0.57 | 803 (12.7) 0.56 | −5 | −4 | γ(C–H) |
| 851 (7.2) 0.13 | 854 (8.3) 0.20 | 854 (8.3) 0.19 | −2 | −2 | γ(C2–H7) |
| 888 (4.6) 0.14 | 887 (4.7) 0.13 | 888 (4.8) 0.13 | 2 | 1 | t(C12–H) |
| 903 (1.1) 0.44 | 912 (2.0) 0.41 | 911 (2.0) 0.48 | −9 | −9 | γ(C–H) |
| 920 (12.3) 0.47 | 919 (10.5) 0.67 | 919 (10.5) 0.65 | 1 | 1 | ω(=CH2) |
| 928 (6.11) 0.48 | 927 (5.10) 0.67 | 927 (6.13) 0.66 | 2 | 1 | ν(C12–C15) |
| 948 (3.7) 0.05 | 943 (5.9) 0.05 | 943 (5.10) 0.05 | 5 | 5 | ν(C1–C12) 20a |
| 1005 (6.6) 0.63 | 1004 (5.5) 0.66 | 1004 (5.6) 0.66 | 2 | 1 | δ(=C–H) |
| 1027 (28.9) 0.22 | 1011 (29.8) 0.13 | 1012 (29.10) 0.13 | 17 | 16 | ν(O9–CH3) |
| 1042 (3.5) 0.74 | 1023 (6.2) 0.70 | 1023 (6.3) 0.71 | 19 | 19 | ν(O8–CH3) |
| 1092 (4.8) 0.17 | 1087 (5.4) 0.21 | 1088 (5.5) 0.21 | 6 | 6 | ρ(=CH2) |
| 1134 (16.2) 0.68 | 1130 (31.3) 0.44 | 1130 (29.3) 0.46 | 4 | 4 | β(C–H) 8a |
| 1139 (0.6) 0.75 | 1138 (1.2) 0.61 | 1138 (1.3) 0.64 | 1 | 1 | ρ(CH3) a″ |
| 1139 (0.4) 0.75 | 1139 (1.2) 0.72 | 1139 (1.2) 0.73 | 1 | 1 | ρ(CH3′) a″ |
| 1148 (41.14) 0.06 | 1141 (30.11) 0.05 | 1141 (31.14) 0.05 | 7 | 7 | β(C–H) |
| 1175 (1.6) 0.25 | 1174 (1.6) 0.17 | 1174 (1.7) 0.18 | 1 | 1 | ρ(CH3′) a′ |
| 1180 (2.12) 0.40 | 1179 (2.7) 0.36 | 1179 (2.8) 0.37 | 1 | 1 | ρ(CH3) a′ |
| 1203 (1.38) 0.39 | 1201 (3.22) 0.43 | 1201 (3.27) 0.43 | 2 | 2 | ρ(–CH2) |
| 1231 (91.1) 0.63 | 1218 (64.1) 0.41 | 1219 (65.2) 0.42 | 14 | 13 | [ν(C2–O12)+ ν (C1–O10)]op |
| 1255 (100.36) 0.06 | 1242 (100.50) 0.03 | 1242 (100.60) 0.03 | 13 | 13 | [ν(C2–O12)+ ν(C1–O10)]ip7a |
| 1269 (1.3) 0.57 | 1272 (1.3) 0.47 | 1272 (1.3) 0.47 | −3 | −3 | β(C–H) 3 |
| 1287 (1.39) 0.26 | 1283 (1.34) 0.27 | 1283 (1.41) 0.27 | 4 | 4 | δ(=C–H) |
| 1296 (1.25) 0.39 | 1294 (0.8) 0.41 | 1295 (0.11) 0.41 | 2 | 2 | ω(–CH2) |
| 1340 (14.71) 0.09 | 1332 (10.52) 0.07 | 1333 (11.64) 0.07 | 8 | 7 | ν(R) 14 |
| 1407 (12.5) 0.58 | 1404 (11.4) 0.20 | 1405 (11.4) 0.22 | 3 | 2 | ν(R) 19b |
| 1413 (3.20) 0.39 | 1408 (1.9) 0.44 | 1408 (1.12) 0.43 | 6 | 6 | βs(=CH2) |
| 1438 (9.8) 0.61 | 1435 (5.5) 0.75 | 1436 (5.6) 0.75 | 3 | 3 | δs(CH3′) a′ |
| 1439 (1.6) 0.75 | 1437 (5.4) 0.75 | 1437 (4.5) 0.75 | 2 | 2 | βs(–CH2) |
| 1447 (2.8) 0.49 | 1444 (1.7) 0.54 | 1444 (1.9) 0.54 | 3 | 3 | δs(CH3) a′ |
| 1454 (2.20) 0.75 | 1447 (3.10) 0.75 | 1447 (2.13) 0.75 | 7 | 6 | δas(CH3) a″ |
| 1454 (6.17) 0.75 | 1447 (4.9) 0.75 | 1448 (4.11) 0.75 | 8 | 7 | δas(CH3′) a″ |
| 1467 (20.7) 0.44 | 1459 (22.2) 0.23 | 1459 (22.2) 0.21 | 8 | 7 | δas(CH3) a′ |
| 1467 (15.10) 0.66 | 1460 (6.6) 0.75 | 1460 (6.8) 0.75 | 8 | 7 | δas(CH3′) a′ |
| 1508 (82.10) 0.28 | 1499 (66.7) 0.17 | 1500 (67.8) 0.18 | 8 | 8 | ν(R) 19a |
| 1582 (15.28) 0.75 | 1577 (12.27) 0.74 | 1578 (12.33) 0.74 | 5 | 5 | ν(R) 8a |
| 1600 (7.92) 0.62 | 1595 (4.68) 0.66 | 1595 (4.84) 0.66 | 5 | 5 | ν(R) 8b |
| 1653 (7.90) 0.13 | 1645 (7.64) 0.11 | 1646 (7.78) 0.12 | 7 | 7 | ν(C=C) |
| 2867 (35.60) 0.05 | 2883 (24.22) 0.04 | 2882 (25.29) 0.03 | −15 | −14 | νs(CH3) a′ |
| 2870 (13.100) 0.02 | 2885 (11.25) 0.05 | 2884 (11.51) 0.03 | −14 | −14 | νs(CH3) a′ |
| 2878 (11.83) 0.03 | 2885 (8.100) 0.01 | 2885 (9.100) 0.02 | −7 | −6 | νs(–CH2) |
| 2911 (5.38) 0.73 | 2921 (6.32) 0.71 | 2920 (6.38) 0.71 | −10 | −10 | νas(–CH2) |
| 2921 (15.35) 0.75 | 2943 (12.19) 0.75 | 2942 (12.23) 0.75 | −21 | −20 | νas(CH3) a″ |
| 2924 (16.24) 0.75 | 2945 (12.17) 0.75 | 2944 (12.20) 0.75 | −20 | −19 | νas(CH3) a″ |
| 2983 (6.26) 0.21 | 2980 (7.9) 0.32 | 2980 (7.11) 0.31 | 4 | 3 | νs(=CH2) |
| 2991 (2.63) 0.20 | 2989 (2.47) 0.20 | 2989 (2.56) 0.20 | 3 | 2 | ν(=C–H) |
| 2993 (8.57) 0.48 | 3002 (6.24) 0.53 | 3001 (6.30) 0.53 | −9 | −8 | νas(CH3) a′ |
| 2994 (11.68) 0.41 | 3003 (7.26) 0.50 | 3002 (7.33) 0.50 | −9 | −8 | νas(CH3) a′ |
| 3026 (5.38) 0.36 | 3029 (4.28) 0.35 | 3029 (4.34) 0.35 | −3 | −3 | ν(C–H) 7b |
| 3056 (2.13) 0.20 | 3057 (1.17) 0.23 | 3057 (2.20) 0.23 | −1 | −1 | ν(C–H) 20b |
| 3063 (4.58) 0.21 | 3064 (6.20) 0.67 | 3064 (6.25) 0.67 | −1 | −1 | ν(C–H) 2 |
| 3066 (7.42) 0.59 | 3068 (2.38) 0.22 | 3067 (2.47) 0.22 | −2 | −2 | νas(=CH2) |
| Conformers | Energy in Unit (eV) | Electronegativity (χ) | Chemical Hardness (η) | ||
|---|---|---|---|---|---|
| HOMO | LUMO | ΔE | |||
| I | −5.6844 | −0.3170 | 5.3674 | 3.0007 | 2.6837 |
| II | −5.6447 | −0.3474 | 5.2973 | 2.9961 | 2.6486 |
| III | −5.6776 | −0.3132 | 5.3644 | 2.9954 | 2.6822 |
| Absorption Bands | Excitation Energies (eV) | Oscillator Strength (f) | * Contributions | |
|---|---|---|---|---|
| λexp (nm) | λcal (nm) | |||
| 265 | 247.7 | 5.0047 | 0.0723 | H – 1 → L + 2 (3.4%) H – 1 → L + 4 (3.9%) H – 1 → L + 7 (2.57%) H − 1 → L + 10 (2.87) H → L (2.9%) H → L + 1 (21.98%) H →L + 2 (10.25%) H → L + 3 (45.41%) H → L + 4 (2.64%) H → L + 5 (4.14%) |
| 219 * | 223.4 | 5.5503 | 0.0902 | H − 1 → L + 3 (3.50%) H → L (54.89%) H → L + 1 (5.63%) H → L + 2 (3.99%) H → L + (14.50%) H → L + 6 (7.42%) H → L + 8 (7.78%) H → L + 10 (2.29%) |
| 219 * | 220.7 | 5.6169 | 0.1474 | H − 1 → L + 1 (1.82%) H − 1 → L + 3 (2.85%) H → L (7.84%) H → L + 2 (15.89%) H → L + 3 (6.77%) H → L + 4 (4.97%) H → L + 5 (1.30%) H → L + 7 (6.04%) H → L + 8 (2.60%) H → L + 9 (2.43%) H → L + 10 (47.49%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yadav, R.K.; Yadav, B.; Yadav, R.A.; Kostova, I. Experimental IR, Raman, and UV-Vis Spectra DFT Structural and Conformational Studies: Bioactivity and Solvent Effect on Molecular Properties of Methyl-Eugenol. Molecules 2023, 28, 5409. https://doi.org/10.3390/molecules28145409
Yadav RK, Yadav B, Yadav RA, Kostova I. Experimental IR, Raman, and UV-Vis Spectra DFT Structural and Conformational Studies: Bioactivity and Solvent Effect on Molecular Properties of Methyl-Eugenol. Molecules. 2023; 28(14):5409. https://doi.org/10.3390/molecules28145409
Chicago/Turabian StyleYadav, Rohit Kumar, Bhoopendra Yadav, R. A. Yadav, and Irena Kostova. 2023. "Experimental IR, Raman, and UV-Vis Spectra DFT Structural and Conformational Studies: Bioactivity and Solvent Effect on Molecular Properties of Methyl-Eugenol" Molecules 28, no. 14: 5409. https://doi.org/10.3390/molecules28145409
APA StyleYadav, R. K., Yadav, B., Yadav, R. A., & Kostova, I. (2023). Experimental IR, Raman, and UV-Vis Spectra DFT Structural and Conformational Studies: Bioactivity and Solvent Effect on Molecular Properties of Methyl-Eugenol. Molecules, 28(14), 5409. https://doi.org/10.3390/molecules28145409