

In Silico Studies of Novel Vemurafenib Derivatives as BRAF Kinase Inhibitors



Abstract
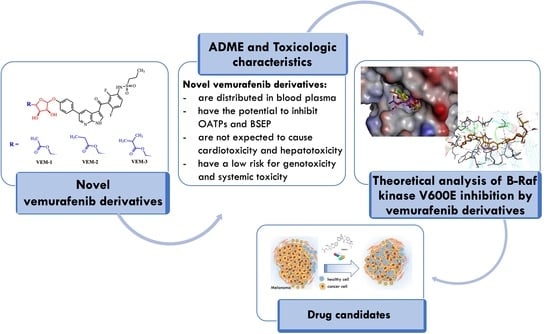
1. Introduction
2. Results and Discussion
2.1. Drug-Likeness Screening, Biotransformation, and Bioavailability
2.2. Simulation of Plasma Concentration–Time Profile in Humans
2.3. Toxicity Assessment
2.4. Binding Site in the BRAFV600E Kinase
2.5. Binding Free Energy and Intermolecular Interactions in the BRAFV600E Binding Pocket
2.6. Stability Assessment of MD Simulations
2.7. Chemical Reactivity of VEM-3
3. Materials and Methods
3.1. Starting Structure Preparation and Modeling
3.2. Preliminary Molecular Docking
3.3. Molecular Dynamic Simulations and Binding Free Energy Calculations
3.4. Computation of Physicochemical, Biopharmaceutical, and Toxicological Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Bonner, T.; O’Brien, S.J.; Nash, W.G.; Rapp, U.R.; Morton, C.C.; Leder, P. The human homologs of the raf (mil) oncogene are located on human chromosomes 3 and 4. Science 1984, 223, 71–74. [Google Scholar] [CrossRef]
- Molina, J.R.; Adjei, A.A. The Ras/Raf/MAPK pathway. J. Thorac. Oncol. 2006, 1, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef]
- Swaika, A.; Crozier, J.A.; Joseph, R.W. Vemurafenib: An evidence-based review of its clinical utility in the treatment of metastatic melanoma. Drug Des. Dev. Ther. 2014, 8, 775–787. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef]
- Halaban, R.; Zhang, W.; Bacchiocchi, A.; Cheng, E.; Parisi, F.; Ariyan, S.; Krauthammer, M.; McCusker, J.P.; Kluger, Y.; Sznol, M. PLX4032, a selective BRAF(V600E) kinase inhibitor, activates the ERK pathway and enhances cell migration and proliferation of BRAF melanoma cells. Pigment. Cell Melanoma Res. 2010, 23, 190–200. [Google Scholar] [CrossRef]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef]
- Smalley, K.S.; Lioni, M.; Dalla Palma, M.; Xiao, M.; Desai, B.; Egyhazi, S.; Hansson, J.; Wu, H.; King, A.J.; Van Belle, P.; et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomas. Mol. Cancer Ther. 2008, 7, 2876–2883. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Menzies, A.M.; Zimmer, L.; Eroglu, Z.; Ye, F.; Zhao, S.; Rizos, H.; Sucker, A.; Scolyer, R.A.; Gutzmer, R.; et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur. J. Cancer 2015, 51, 2792–2799. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Erin Chen, Y.; Kumar, R.; Taylor, M.; Jenny Njauw, C.N.; Miao, B.; Frederick, D.T.; Wargo, J.A.; Flaherty, K.T.; Jonsson, G.; et al. MITF Modulates Therapeutic Resistance through EGFR Signaling. J. Investig. Dermatol. 2015, 135, 1863–1872. [Google Scholar] [CrossRef]
- Hu, W.; Jin, L.; Jiang, C.C.; Long, G.V.; Scolyer, R.A.; Wu, Q.; Zhang, X.D.; Mei, Y.; Wu, M. AEBP1 upregulation confers acquired resistance to BRAF (V600E) inhibition in melanoma. Cell Death Dis. 2013, 4, e914. [Google Scholar] [CrossRef]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef]
- Das Thakur, M.; Salangsang, F.; Landman, A.S.; Sellers, W.R.; Pryer, N.K.; Levesque, M.P.; Dummer, R.; McMahon, M.; Stuart, D.D. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013, 494, 251–255. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef]
- Pelster, M.S.; Amaria, R.N. Combined targeted therapy and immunotherapy in melanoma: A review of the impact on the tumor microenvironment and outcomes of early clinical trials. Ther. Adv. Med. Oncol. 2019, 11, 1758835919830826. [Google Scholar] [CrossRef] [PubMed]
- Rager, T.; Eckburg, A.; Patel, M.; Qiu, R.; Gantiwala, S.; Dovalovsky, K.; Fan, K.; Lam, K.; Roesler, C.; Rastogi, A.; et al. Treatment of Metastatic Melanoma with a Combination of Immunotherapies and Molecularly Targeted Therapies. Cancers 2022, 14, 3779. [Google Scholar] [CrossRef]
- Froelich, W. New BRAF inhibitor shows promise in a variety of cancers. Oncol. Times 2020, 42, 37. [Google Scholar] [CrossRef]
- Tutuka, C.S.A.; Andrews, M.C.; Mariadason, J.M.; Ioannidis, P.; Hudson, C.; Cebon, J.; Behren, A. PLX8394, a new generation BRAF inhibitor, selectively inhibits BRAF in colonic adenocarcinoma cells and prevents paradoxical MAPK pathway activation. Mol. Cancer 2017, 16, 112. [Google Scholar] [CrossRef] [PubMed]
- Sumardika, I.W.; Youyi, C.; Kondo, E.; Inoue, Y.; Ruma, I.M.W.; Murata, H.; Kinoshita, R.; Yamamoto, K.I.; Tomida, S.; Shien, K.; et al. beta-1,3-Galactosyl-O-Glycosyl-Glycoprotein beta-1,6-N-Acetylglucosaminyltransferase 3 Increases MCAM Stability, Which Enhances S100A8/A9-Mediated Cancer Motility. Oncol. Res. 2018, 26, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Very, N.; Lefebvre, T.; El Yazidi-Belkoura, I. Drug resistance related to aberrant glycosylation in colorectal cancer. Oncotarget 2018, 9, 1380–1402. [Google Scholar] [CrossRef]
- Kudo, T.; Nakagawa, H.; Takahashi, M.; Hamaguchi, J.; Kamiyama, N.; Yokoo, H.; Nakanishi, K.; Nakagawa, T.; Kamiyama, T.; Deguchi, K.; et al. N-glycan alterations are associated with drug resistance in human hepatocellular carcinoma. Mol. Cancer 2007, 6, 32. [Google Scholar] [CrossRef]
- De Vellis, C.; Pietrobono, S.; Stecca, B. The Role of Glycosylation in Melanoma Progression. Cells 2021, 10, 2136. [Google Scholar] [CrossRef]
- Nisiewicz, M.K.; Kowalczyk, A.; Sobiepanek, A.; Jagielska, A.; Wagner, B.; Nowakowska, J.; Gniadek, M.; Grudzinski, I.P.; Kobiela, T.; Nowicka, A.M. Tracking of Glycans Structure and Metallomics Profiles in BRAF Mutated Melanoma Cells Treated with Vemurafenib. Int. J. Mol. Sci. 2021, 22, 439. [Google Scholar] [CrossRef]
- Tang, L.; Chen, X.; Zhang, X.; Guo, Y.; Su, J.; Zhang, J.; Peng, C.; Chen, X. N-Glycosylation in progression of skin cancer. Med. Oncol. 2019, 36, 50. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Ntie-Kang, F. An in silico evaluation of the ADMET profile of the StreptomeDB database. Springerplus 2013, 2, 353. [Google Scholar] [CrossRef]
- Durmus, S.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Oral availability and brain penetration of the B-RAFV600E inhibitor vemurafenib can be enhanced by the P-GLYCOprotein (ABCB1) and breast cancer resistance protein (ABCG2) inhibitor elacridar. Mol. Pharm. 2012, 9, 3236–3245. [Google Scholar] [CrossRef] [PubMed]
- Thapar, M.M.; Ashton, M.; Lindegardh, N.; Bergqvist, Y.; Nivelius, S.; Johansson, I.; Bjorkman, A. Time-dependent pharmacokinetics and drug metabolism of atovaquone plus proguanil (Malarone) when taken as chemoprophylaxis. Eur. J. Clin. Pharmacol. 2002, 58, 19–27. [Google Scholar] [CrossRef]
- Goldinger, S.M.; Rinderknecht, J.; Dummer, R.; Kuhn, F.P.; Yang, K.H.; Lee, L.; Ayala, R.C.; Racha, J.; Geng, W.; Moore, D.; et al. A single-dose mass balance and metabolite-profiling study of vemurafenib in patients with metastatic melanoma. Pharmacol. Res. Perspect. 2015, 3, e00113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Heinzmann, D.; Grippo, J.F. Clinical Pharmacokinetics of Vemurafenib. Clin. Pharmacokinet. 2017, 56, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef]
- Sargsyan, K.; Grauffel, C.; Lim, C. How Molecular Size Impacts RMSD Applications in Molecular Dynamics Simulations. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA. Discovery Studio Modeling Environment, Release 2017; Dassault Systèmes: San Diego, CA, USA, 2016. [Google Scholar]
- Halgren, T.A. Merck molecular force field 3. Molecular geometries and vibrational frequencies for MMFF94. J. Comput. Chem. 1996, 17, 553–586. [Google Scholar] [CrossRef]
- Hehre, W.; Ohlinger, S. Spartan 20 for Windows, Machintosh and Linux Tutorial and Users ’Guide; Wavefunction Inc.: Irvine, CA, USA, 2022. [Google Scholar]
- Brose, M.S.; Volpe, P.; Feldman, M.; Kumar, M.; Rishi, I.; Gerrero, R.; Einhorn, E.; Herlyn, M.; Minna, J.; Nicholson, A.; et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002, 62, 6997–7000. [Google Scholar]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Gagnon, J.K.; Law, S.M.; Brooks, C.L., 3rd. Flexible CDOCKER: Development and application of a pseudo-explicit structure-based docking method within CHARMM. J. Comput. Chem. 2016, 37, 753–762. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Vangunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, B.; Abed, S.N.; Al-Attraqchi, O.; Kuche, K.; Tekade, R.K. Computer-Aided Prediction of Pharmacokinetic (ADMET) Properties; Dosage Form Design Parameters; Elsevier: Amsterdam, The Netherlands, 2018; Volume 21. [Google Scholar]
- Martinez-Mayorga, K.; Madariaga-Mazon, A.; Medina-Franco, J.L.; Maggiora, G. The impact of chemoinformatics on drug discovery in the pharmaceutical industry. Expert Opin. Drug Discov. 2020, 15, 293–306. [Google Scholar] [CrossRef]
- ADMET Predictor; Version 10.1; Simulations Plus: Lancaster, CA, USA, 2020.
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Hosea, N.A.; Jones, H.M. Predicting pharmacokinetic profiles using in silico derived parameters. Mol. Pharm. 2013, 10, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Agoram, B.; Woltosz, W.S.; Bolger, M.B. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv. Drug Deliv. Rev. 2001, 50, S41–S67. [Google Scholar] [CrossRef]
- Savoia, P.; Zavattaro, E.; Cremona, O. Clinical Implications of Acquired BRAF Inhibitors Resistance in Melanoma. Int. J. Mol. Sci. 2020, 21, 9730. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Vd | Sw | Peff | MDCK | %Unbnd | RBP | BBB Filter | logBB |
|---|---|---|---|---|---|---|---|---|
| Expected Values | ||||||||
| (≤3.7 L/kg) | (≥0.010 mg/mL) | (≥0.5 cm/s·10−4) | (≥30 cm/s·10−7) | (>10%) | (<1) | (High/Low) | ||
| vemurafenib | 1.54 | 0.0000006 | 1.683 | 135.93 | 2.60 | 0.72 | Low | −0.152 |
| VEM-1 | 1.02 | 0.00013 | 0.337 | 12.62 | 4.77 | 0.69 | Low | −1.157 |
| VEM-2 | 1.05 | 0.00011 | 0.284 | 13.89 | 4.06 | 0.68 | Low | −1.103 |
| VEM-3 | 1.10 | 0.000092 | 0.241 | 14.22 | 3.86 | 0.68 | Low | −1.011 |
| Compound | P-gp Substrate/ Inhibitor | Bcrp Substrate/ Inhibitor | BSEP Inhibitor | BSEP_IC50 | OATP1B1 Inhibitor | OATP1B3 Inhibitor |
|---|---|---|---|---|---|---|
| (≤60 μM) | ||||||
| vemurafenib | No | Yes | Yes | 22.67 | Yes | No |
| VEM-1 | Yes | Yes | Yes | 11.44 | Yes | Yes |
| VEM-2 | Yes | Yes | Yes | 9.80 | Yes | Yes |
| VEM-3 | Yes | Yes | Yes | 10.07 | Yes | Yes |
| Compound | MRTD | hERG Filter | hERG pIC50 | AlkPhos | GGT | LDH | SGOT |
|---|---|---|---|---|---|---|---|
| Expected Values | |||||||
| (>3.16 mg/kg/day) | (Yes/No) | (>5.5) | |||||
| vemurafenib | Below 3.16 | Yes | 5.06 | T | NT | NT | NT |
| VEM-1 | Below 3.16 | No | 4.38 | T | NT | NT | NT |
| VEM-2 | Below 3.16 | No | 4.36 | T | NT | NT | NT |
| VEM-3 | Below 3.16 | No | 4.33 | T | NT | NT | NT |
| Compound | Mouse TD50 | Rat TD50 | Rat Acute LD50 | Chrom_Aberr | MUT97+1537 | MUT(*) |
|---|---|---|---|---|---|---|
| Expected Values | ||||||
| (<25 mg/kg/day) | (<4 mg/kg/day) | (<300 mg/kg) | ||||
| vemurafenib | 623.15 | 21.93 | 44.0 | NT | Yes | No |
| VEM-1 | 628.50 | 1.99 | 135.98 | NT | No | No |
| VEM-2 | 669.95 | 1.70 | 140.63 | NT | Yes | No |
| VEM-3 | 670.56 | 1.38 | 156.29 | NT | Yes | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Żołek, T.; Mazurek, A.; Grudzinski, I.P. In Silico Studies of Novel Vemurafenib Derivatives as BRAF Kinase Inhibitors. Molecules 2023, 28, 5273. https://doi.org/10.3390/molecules28135273
Żołek T, Mazurek A, Grudzinski IP. In Silico Studies of Novel Vemurafenib Derivatives as BRAF Kinase Inhibitors. Molecules. 2023; 28(13):5273. https://doi.org/10.3390/molecules28135273
Chicago/Turabian StyleŻołek, Teresa, Adam Mazurek, and Ireneusz P. Grudzinski. 2023. "In Silico Studies of Novel Vemurafenib Derivatives as BRAF Kinase Inhibitors" Molecules 28, no. 13: 5273. https://doi.org/10.3390/molecules28135273
APA StyleŻołek, T., Mazurek, A., & Grudzinski, I. P. (2023). In Silico Studies of Novel Vemurafenib Derivatives as BRAF Kinase Inhibitors. Molecules, 28(13), 5273. https://doi.org/10.3390/molecules28135273