
Length-Dependent Transition from Extended to Folded Shapes in Short Oligomers of an Azetidine-Based α-Amino Acid: The Critical Role of NH···N H-Bonds

 , , , , , and
, , , , , and
Abstract
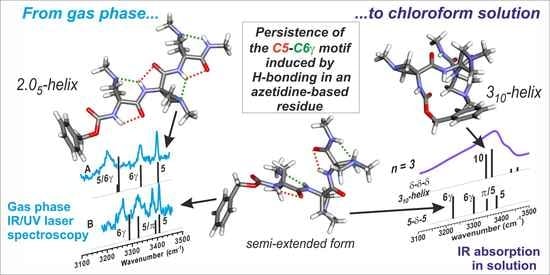
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
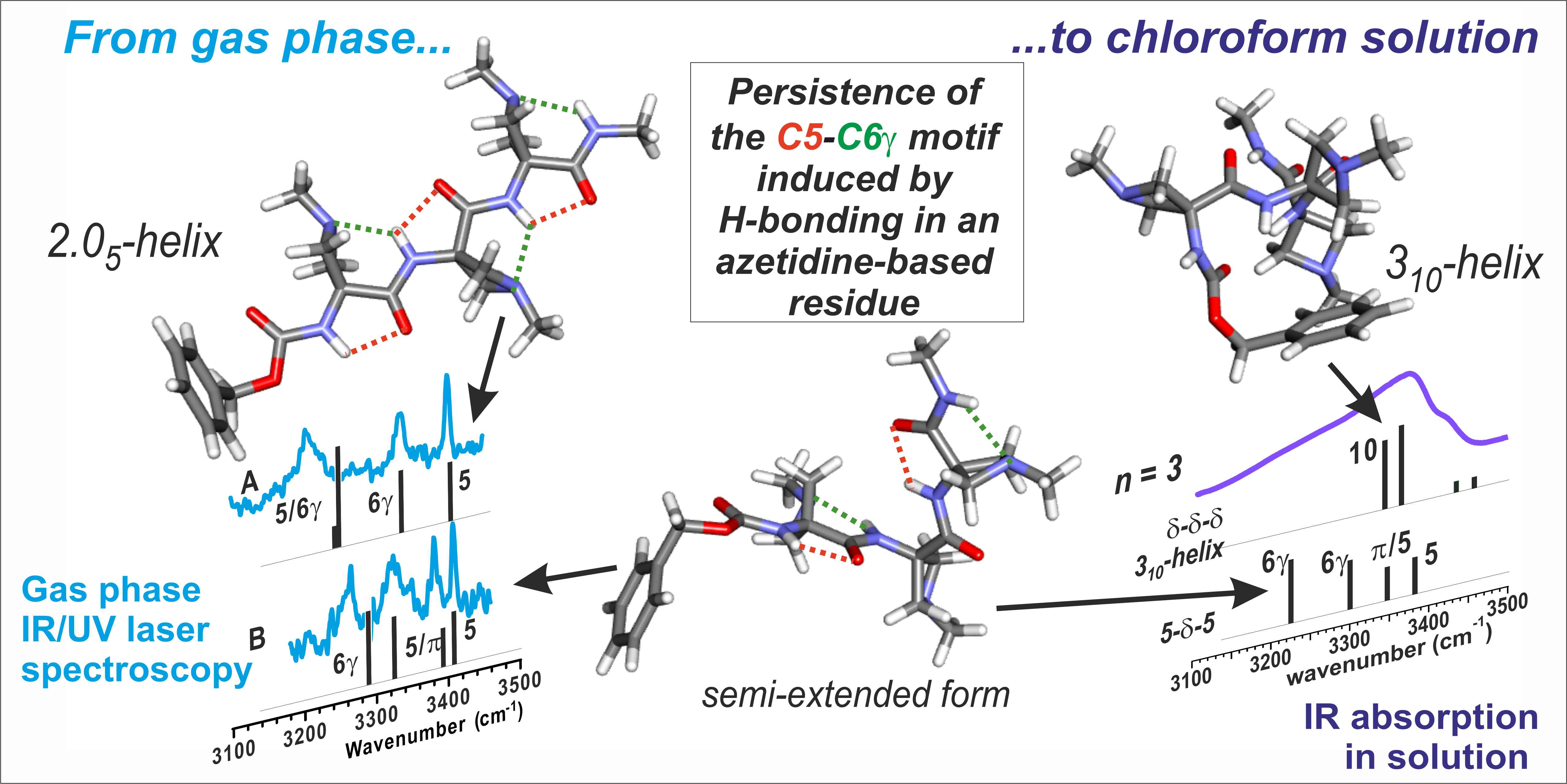
1. Introduction
2. Results
2.1. Gas Phase Conformational Analysis
2.1.1. Theoretical Landscapes
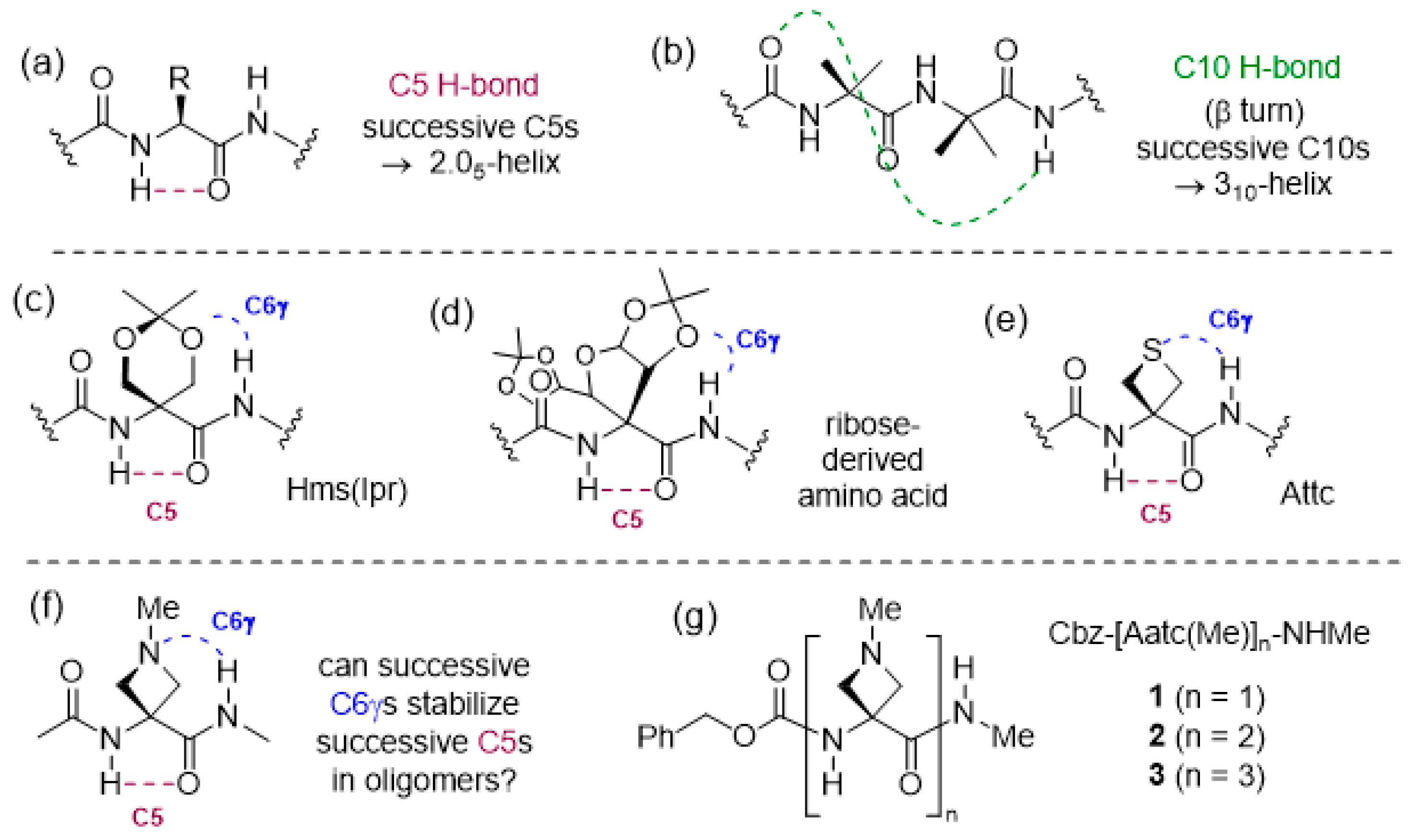
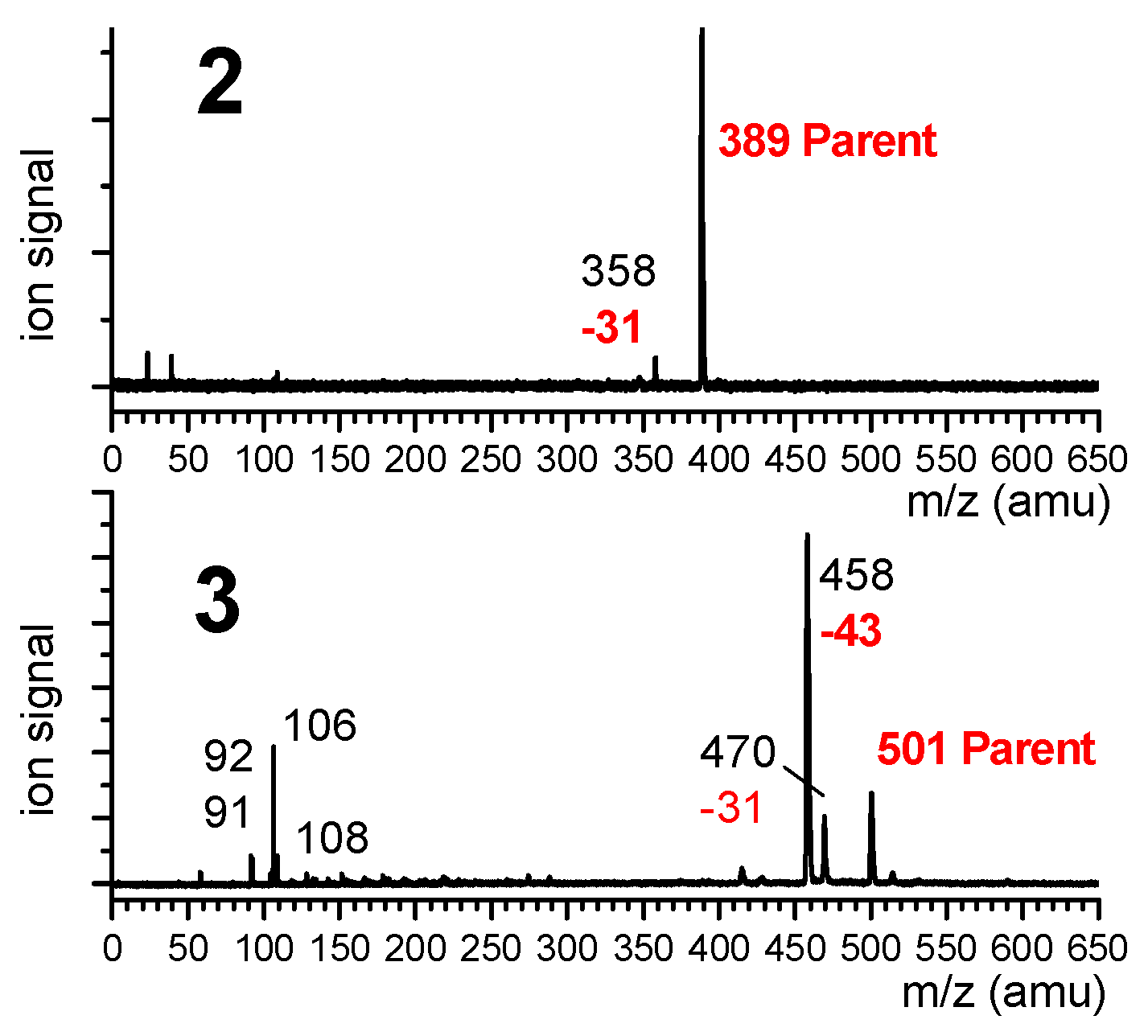
2.1.2. Gas Phase Laser Spectroscopy and Quantum Chemistry Calculations
2.2. Conformational Analysis in Chloroform Solution
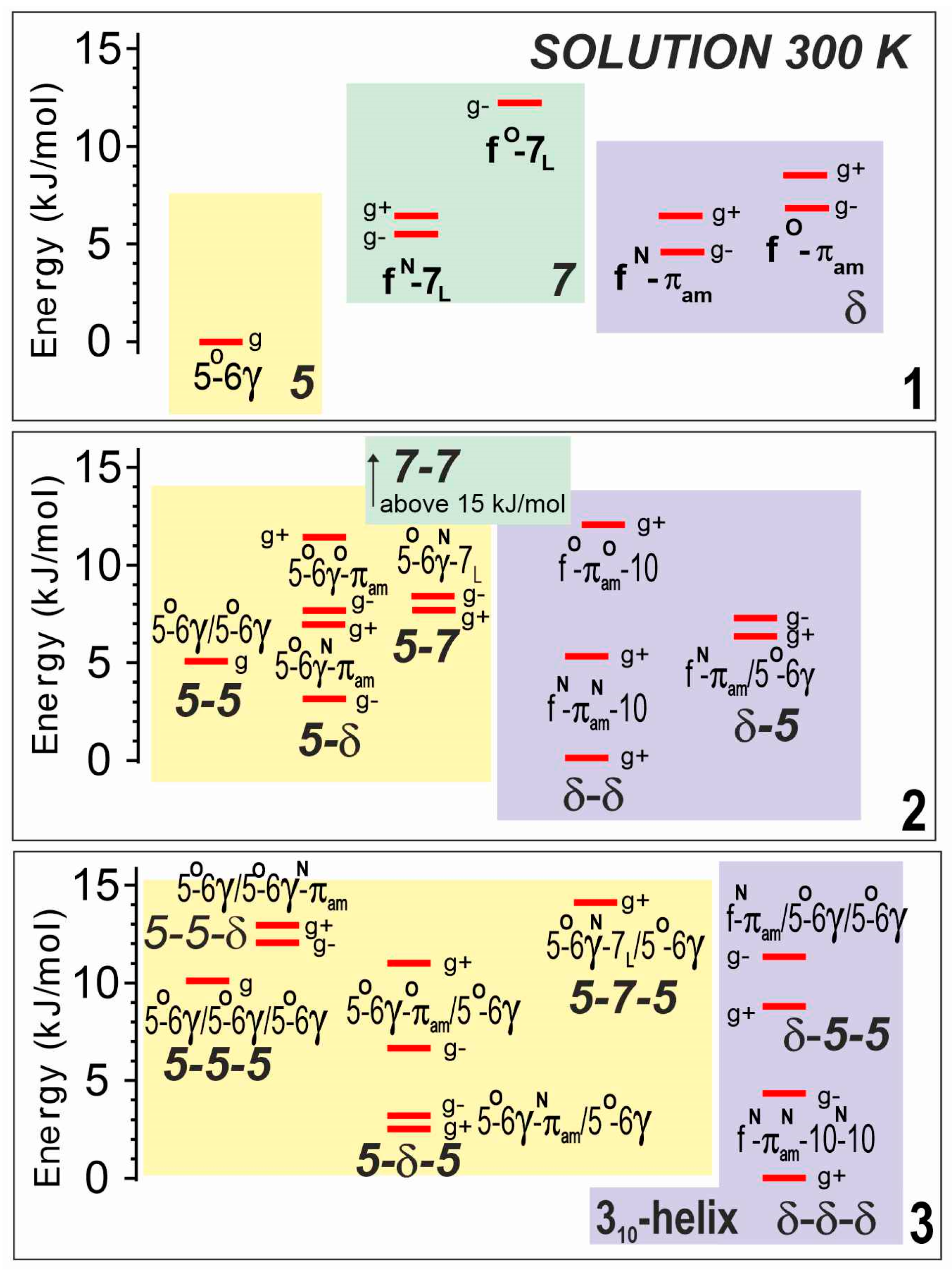
2.2.1. Theoretical Landscapes
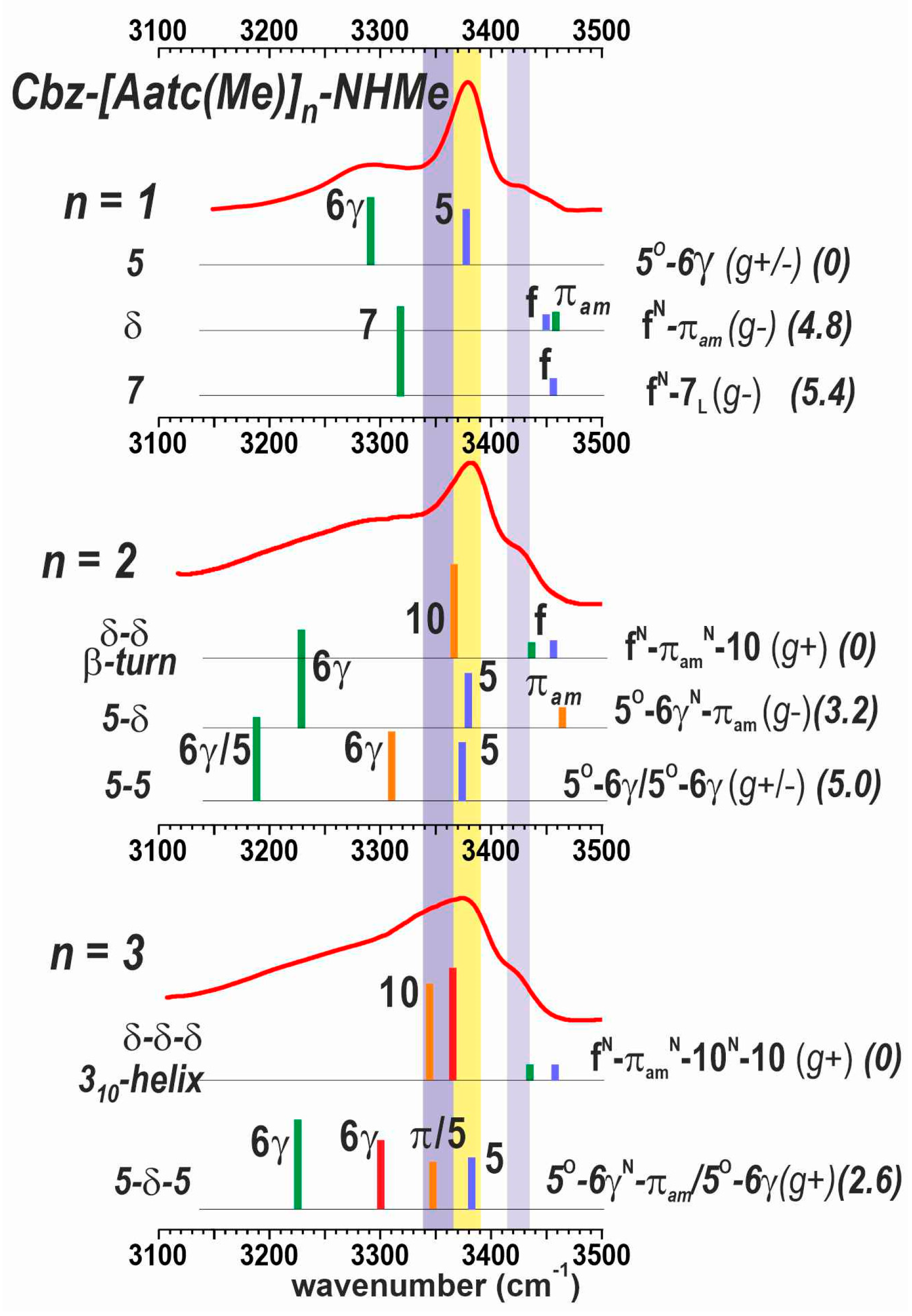
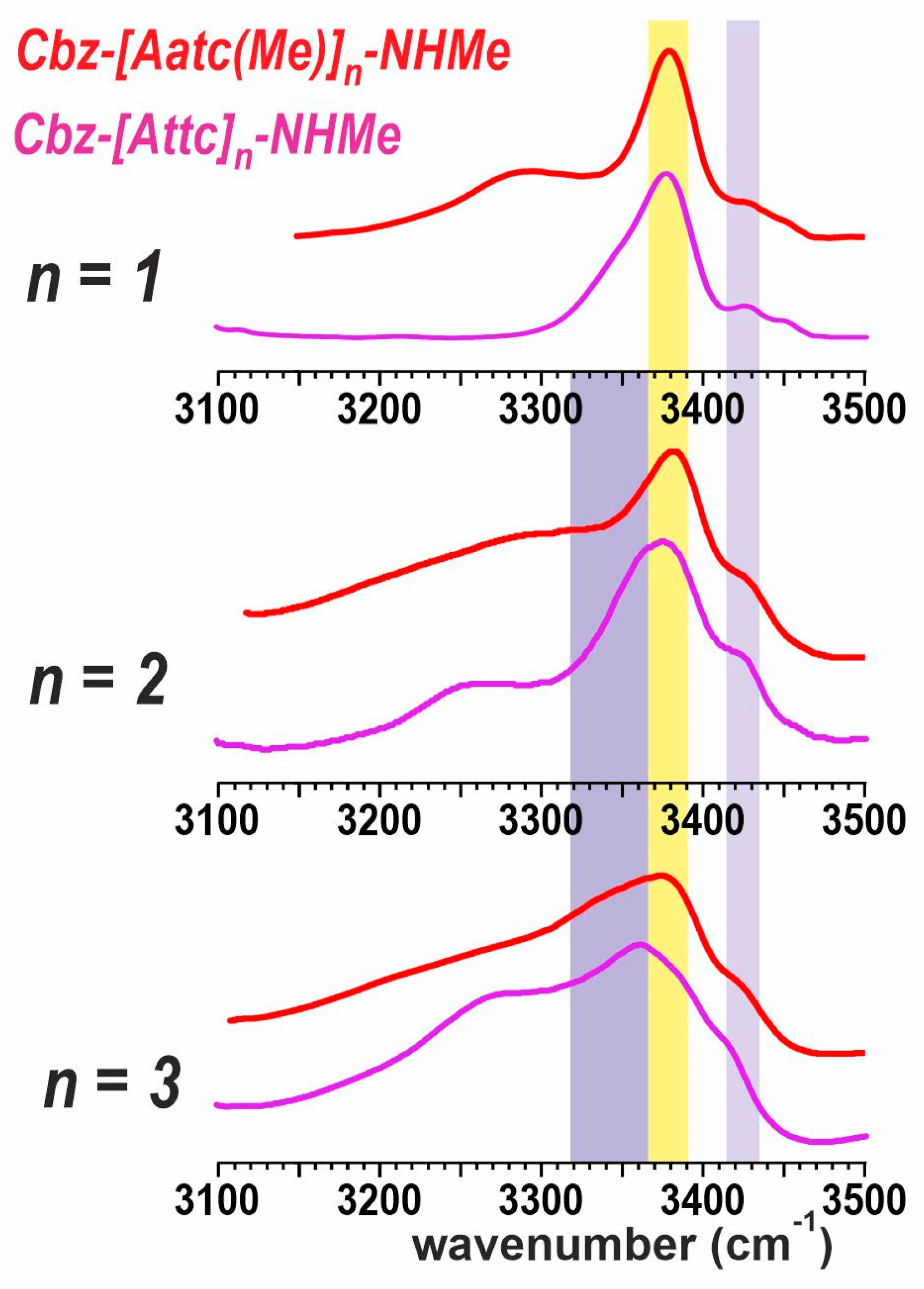
2.2.2. Solution-State IR Spectroscopy and Quantum Chemistry Calculations
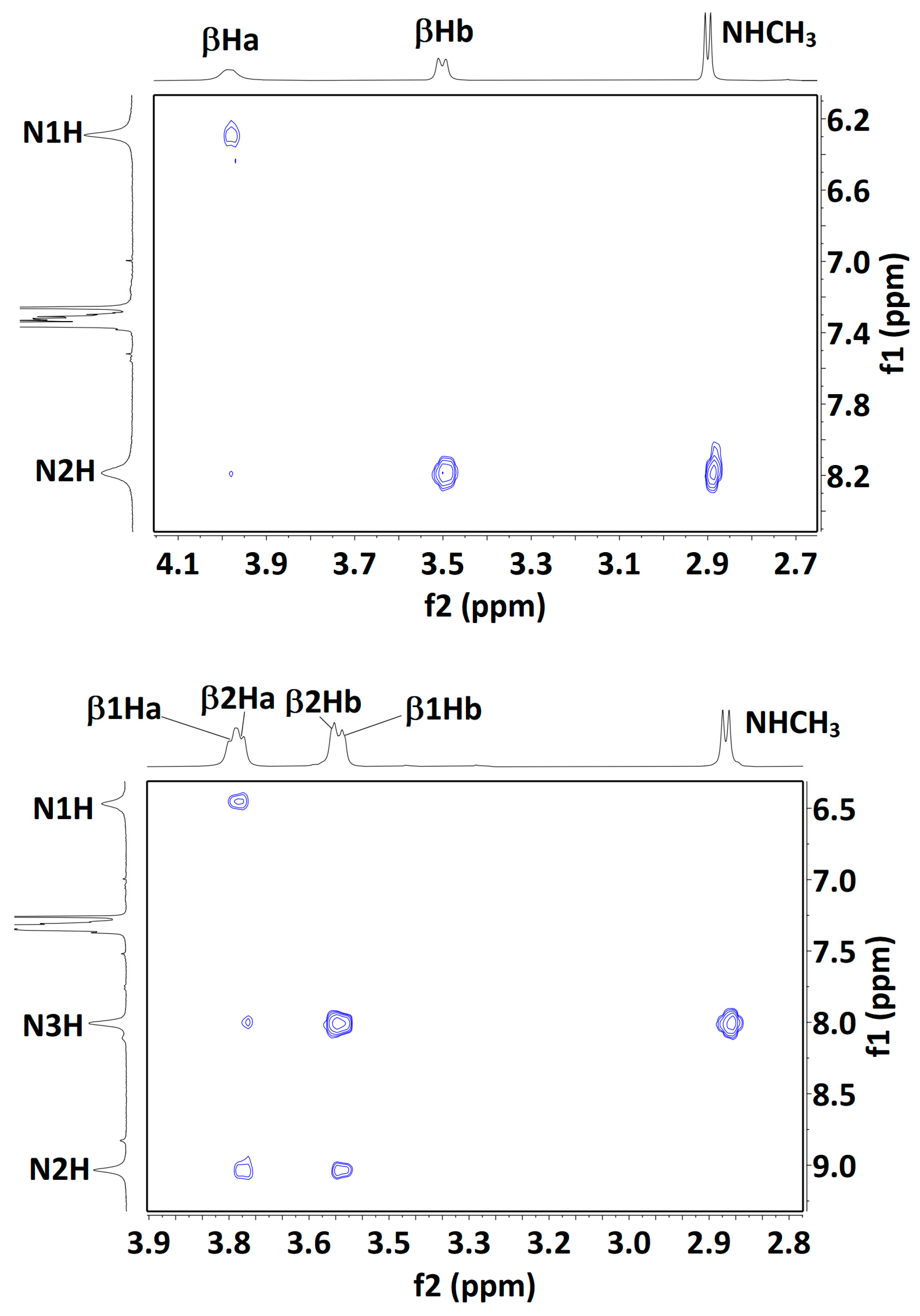
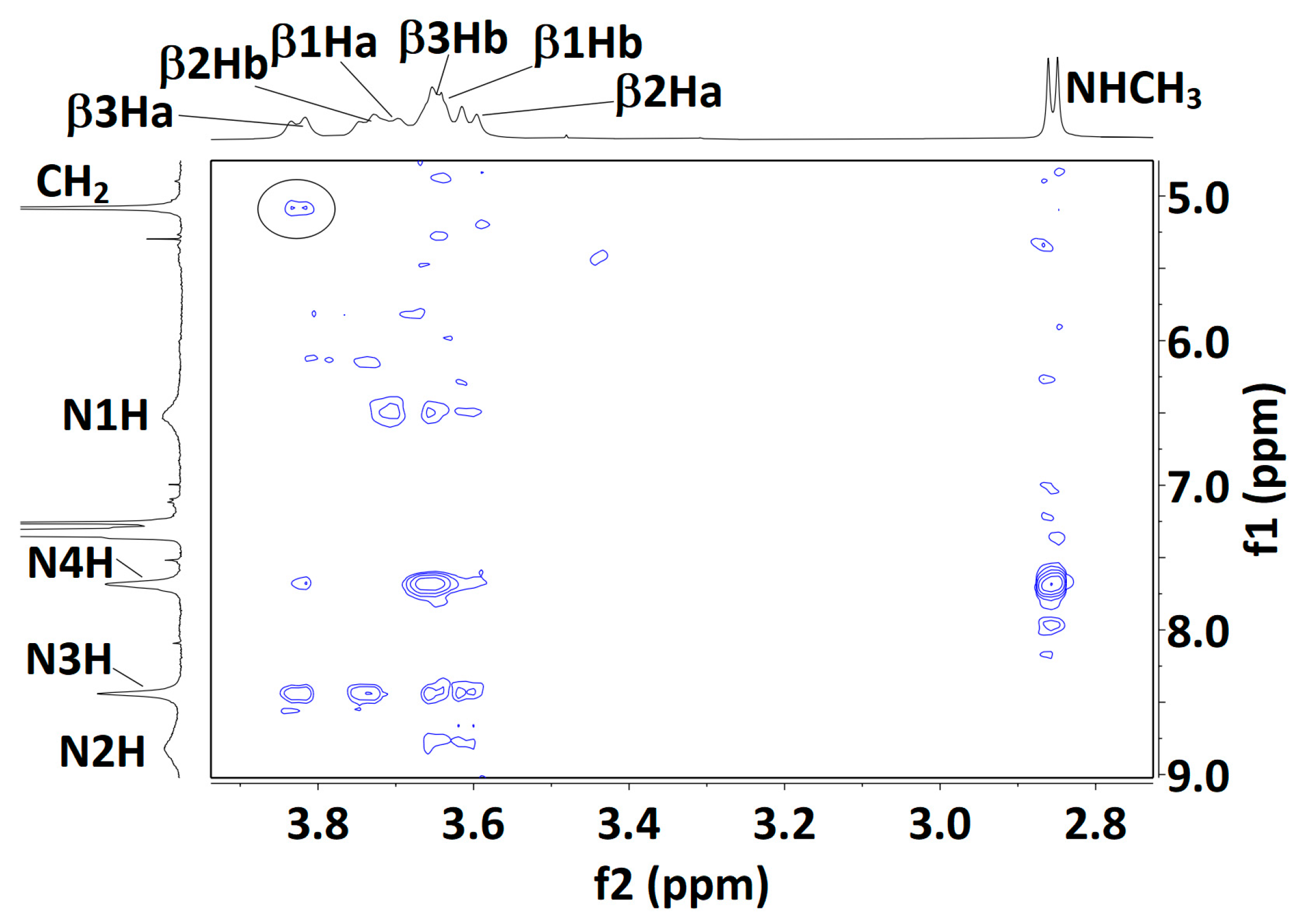
2.2.3. Solution-State 1H NMR Spectroscopy
3. Discussion
4. Materials and Methods
4.1. Theoretical Chemistry
4.2. Gas Phase Experimental Spectroscopy
4.3. Solution-State Spectroscopy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jeffrey, G.A.; Sänger, W. Hydrogen Bonding in Biological Structures; Springer: Berlin, Germany, 1991. [Google Scholar]
- Perrin, C.L.; Nielson, J.B. “Strong” hydrogen bonds in chemistry and biology. Ann. Rev. Phys. Chem. 1997, 48, 511–544. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford University Press: New York, NY, USA, 1999. [Google Scholar]
- Baker, E.N.; Hubbard, R.E. Hydrogen Bonding in Globular Proteins. Prog. Biophys. Mol. Biol. 1984, 44, 97–179. [Google Scholar] [CrossRef] [PubMed]
- Karshikoff, A. Non-Covalent Interactions in Proteins; Imperial College Press: London, UK, 2006. [Google Scholar]
- Rose, G.D.; Gierasch, L.M.; Smith, J.A. Turns in Peptides and Proteins. Adv. Prot. Chem. 1985, 37, 1–109. [Google Scholar] [CrossRef]
- Toniolo, C. Intramolecularly Hydrogen-Bonded Peptide Conformations. Crit. Rev. Biochem. 1980, 9, 1–44. [Google Scholar] [CrossRef] [PubMed]
- Avignon, M.; Huong, P.V.; Lascombe, J.; Marraud, M.; Néel, J. Etude, par Spectroscopie Infra-rouge, de la Conformation de Quelques Composés Peptidiques Modèles. Biopolymers 1969, 8, 69–89. [Google Scholar] [CrossRef]
- Cung, M.T.; Marraud, M.; Néel, J. Etude expérimentale de la conformation de molécules dipeptidiques-Comparaison avec les résultats théoriques. Anal. Chim. 1972, 7, 183–209. [Google Scholar]
- Burgess, A.W.; Scheraga, H.A. Stable Conformations of Dipeptides. Biopolymers 1973, 12, 2177–2183. [Google Scholar] [CrossRef]
- Boussard, G.; Marraud, M.; Néel, J. Étude Expérimentale du Mode de Repliement β dans Quelques Molécules Tripeptidiques Modèles. Interprétation des Spectres d’Absorption Infrarouge. J. Chim. Phys. Phys.-Chim. Biol. 1974, 71, 1081–1091. [Google Scholar] [CrossRef]
- Aubry, A.; Protas, J.; Boussard, G.; Marraud, M.; Néel, J. Experimental Conformational Study of Two Peptides Containing α-Aminoisobutyric Acid-Crystal-Structure of N-Acetyl-α-Aminoisobutyric Acid Methylamide. Biopolymers 1978, 17, 1693–1711. [Google Scholar] [CrossRef]
- Rao, C.P.; Nagaraj, R.; Rao, C.N.R.; Balaram, P. Infrared Studies on the Conformation of Synthetic Alamethicin Fragments and Model Peptides Containing α-Aminoisobutyric Acid. Biochemistry 1980, 19, 425–431. [Google Scholar] [CrossRef]
- Paterson, Y.; Stimson, E.R.; Evans, D.J.; Leach, S.J.; Scheraga, H.A. Solution Conformations of Oligomers of α-Aminoisobutyric Acid. Int. J. Pept. Protein Res. 1982, 20, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.R.; Gellman, S.H. Evaluation of the Conformation-Directing Effects of Secondary Hydrogen-Bonding Interactions in Flexible Tetrapeptide Analogs. J. Am. Chem. Soc. 1995, 117, 10411–10412. [Google Scholar] [CrossRef]
- Broda, M.A.; Rzeszotarska, B.; Smelka, L.; Rospenk, M. Conformational Investigation of α, β-Dehydropeptides. 8. N-Acetyl-α,β-Dehydroamino Acid N’-methylamides: Conformation and Electron Density Perturbation from Infrared and Theoretical Studies. J. Pept. Res. 1997, 50, 342–351. [Google Scholar] [CrossRef]
- Yang, J.H.; Gellman, S.H. Energetic Superiority of Two-Center Hydrogen Bonding Relative to Three-Center Hydrogen Bonding in a Model System. J. Am. Chem. Soc. 1998, 120, 9090–9091. [Google Scholar] [CrossRef]
- Pauling, L.; Corey, R.B. Configurations of Polypeptide Chains with Favored Orientations around Single Bonds-Two New Pleated Sheets. Proc. Natl. Acad. Sci. USA 1951, 37, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Newberry, R.W.; Raines, R.T. A Prevalent Intraresidue Hydrogen Bond Stabilizes Proteins. Nat. Chem. Biol. 2016, 12, 1084. [Google Scholar] [CrossRef]
- Crisma, M.; Formaggio, F.; Alemán, C.; Torras, J.; Ramakrishnan, C.; Kalmankar, N.; Balaram, P.; Toniolo, C. The Fully-Extended Conformation in Peptides and Proteins. Pept. Sci. 2018, 110, e23100. [Google Scholar] [CrossRef]
- Toniolo, C.; Crisma, M.; Formaggio, F.; Peggion, C.; Broxterman, Q.B.; Kaptein, B. Molecular spacers for physicochemical investigations based on novel helical and extended peptide structures. Biopolymers (Pept. Sci.) 2004, 76, 162–176. [Google Scholar] [CrossRef]
- Peggion, C.; Moretto, A.; Formaggio, F.; Crisma, M.; Toniolo, C. Multiple, Consecutive, Fully-Extended 2.05-Helix Peptide Conformation. Biopolymers (Pept. Sci.) 2013, 100, 621–636. [Google Scholar] [CrossRef]
- Toniolo, C.; Benedetti, E. Structures of Polypeptides from α-Amino-Acids Disubstituted at the α-Carbon. Macromolecules 1991, 24, 4004–4009. [Google Scholar] [CrossRef]
- Toniolo, C.; Crisma, M.; Formaggio, F.; Peggion, C. Control of peptide conformation by the Thorpe-Ingold effect (Cα-tetrasubstitution). Biopolymers (Pept. Sci.) 2001, 60, 396–419. [Google Scholar] [CrossRef]
- Prasad, S.; Rao, R.B.; Balaram, P. Contrasting Solution Conformations of Peptides Containing α,α-Dialkylated Residues with Linear and Cyclic Side-Chains. Biopolymers 1995, 35, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Toniolo, C.; Crisma, M.; Formaggio, F.; Benedetti, E.; Santini, A.; Iacovino, R.; Saviano, M.; DiBlasio, B.; Pedone, C.; Kamphuis, J. Preferred conformation of peptides rich in alicyclic Cα,Cα-disubstituted glycines. Biopolymers 1996, 40, 519–522. [Google Scholar] [CrossRef]
- Gatos, M.; Formaggio, F.; Crisma, M.; Toniolo, C.; Bonora, G.M.; Benedetti, Z.; Di Blasio, B.; Iacovino, R.; Santini, A.; Saviano, M.; et al. Conformational characterization of the 1-aminocyclobutane-1-carboxylic acid residue in model peptides. J. Pept. Sci. 1997, 3, 110–122. [Google Scholar] [CrossRef]
- Gord, J.R.; Hewett, D.M.; Hernandez-Castillo, A.O.; Blodgett, K.N.; Rotondaro, M.C.; Varuolo, A.; Kubasik, M.A.; Zwier, T.S. Conformation-specific spectroscopy of capped, gas-phase Aib oligomers: Tests of the Aib residue as a 310-helix former. Phys. Chem. Chem. Phys. 2016, 18, 25512–25527. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.K.C.; Sukumar, M.; Bardi, R.; Piazzesi, A.M.; Valle, G.; Toniolo, C.; Balaram, P. Stereochemically Constrained Peptides-Theoretical and Experimental Studies on the Conformations of Peptides Containing 1-Aminocyclohexanecarboxylic Acid. J. Am. Chem. Soc. 1986, 108, 6363–6370. [Google Scholar] [CrossRef]
- Wolf, W.M.; Stasiak, M.; Leplawy, M.T.; Bianco, A.; Formaggio, F.; Crisma, M.; Toniolo, C. Destabilization of the 310-helix in peptides based on Cα-tetrasubstituted α-amino acids by main-chain to side-chain hydrogen bonds. J. Am. Chem. Soc. 1998, 120, 11558–11566. [Google Scholar] [CrossRef]
- Scaffidi, A.; Skelton, B.W.; Stick, R.V.; White, A.H. Structural Characterization of a Pentapeptide Derived from a Novel Carbohydrate α-amino Acid. Aus. J. Chem. 2007, 60, 93–94. [Google Scholar] [CrossRef]
- Koba, Y.; Ueda, A.; Oba, M.; Doi, M.; Kato, T.; Demizu, Y.; Tanaka, M. Left-Handed Helix of Three-Membered Ring Amino Acid Homopeptide Interrupted by an N-H—Ethereal O-Type Hydrogen Bond. Org. Lett. 2018, 20, 7830–7834. [Google Scholar] [CrossRef]
- Tanda, K.; Eto, R.; Kato, K.; Oba, M.; Ueda, A.; Suemune, H.; Doi, M.; Demizu, Y.; Kurihara, M.; Tanaka, M. Peptide foldamers composed of six-membered ring α,α-disubstituted α-amino acids with two changeable chiral acetal moieties. Tetrahedron 2015, 71, 3909–3914. [Google Scholar] [CrossRef]
- De Zotti, M.; Clayden, J. Extended Diethylglycine Homopeptides Formed by Desulfurization of Their Tetrahydrothiopyran Analogues. Org. Lett. 2019, 21, 2209–2212. [Google Scholar] [CrossRef]
- Imani, Z.; Mundlapati, V.R.; Brenner, V.; Gloaguen, E.; Le Barbu-Debus, K.; Zehnacker-Rentien, A.; Robin, S.; Aitken, D.J.; Mons, M. Non-covalent interactions reveal the protein chain δ conformation in a flexible single-residue model. Chem. Commun. 2023, 59, 1161–1164. [Google Scholar] [CrossRef] [PubMed]
- Imani, Z.; Mundlapati, V.R.; Goldsztejn, G.; Brenner, V.; Gloaguen, E.; Guillot, R.; Baltaze, J.P.; Le Barbu-Debus, K.; Robin, S.; Zehnacker, A.; et al. Conformation Control Through Concurrent N-H···S and N-H···C Hydrogen Bonding and Hyperconjugation Effects. Chem. Sci. 2020, 11, 9191–9197. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Ishida, Y.; Aida, T. Helical Oligopeptides of a Quaternized Amino Acid with Tunable Chiral-Induction Ability and an Anomalous pH Response. Chem. Eur. J. 2017, 23, 4818–4826. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.I.; Tanaka, M.; Sato, S.; Kinbara, K.; Aida, T. Oligo(4-aminopiperidine-4-carboxylic acid): An Unusual Basic Oligopeptide with an Acid-Induced Helical Conformation. J. Am. Chem. Soc. 2010, 132, 13176–13178. [Google Scholar] [CrossRef]
- Yamaberi, Y.; Eto, R.; Umeno, T.; Kato, T.; Doi, M.; Yokoo, H.; Oba, M.; Tanaka, M. Synthesis of (S)-(-)-Cucurbitine and Conformation of Its Homopeptides. Org. Lett. 2021, 23, 4358–4362. [Google Scholar] [CrossRef]
- Mundlapati, V.R.; Imani, Z.; D’mello, V.C.; Brenner, V.; Gloaguen, E.; Baltaze, J.-P.; Robin, S.; Mons, M.; Aitken, D.J. N-H···X interactions stabilize intra-residue C5 hydrogen bonded conformations in heterocyclic α-amino acid derivatives. Chem. Sci. 2021, 12, 14826–14832. [Google Scholar] [CrossRef]
- Mnich, S.J.; Hiebsch, R.R.; Huff, R.M.; Muthian, S. Anti-Inflammatory Properties of CB1-Receptor Antagonist Involves β2 Adrenoceptors. J. Pharmacol. Exp. Ther. 2010, 333, 445–453. [Google Scholar] [CrossRef]
- Žukauskaitė, A.; Mangelinckx, S.; Buinauskaite, V.; Šačkus, A.; De Kimpe, N. Synthesis of new functionalized aziridine-2-and azetidine-3-carboxylic acid derivatives of potential interest for biological and foldameric applications. Amino Acids 2011, 41, 541–558. [Google Scholar] [CrossRef]
- Ivachtchenko, A.V.; Ivanenkov, Y.A.; Mitkin, O.D.; Vorobiev, A.A.; Kuznetsova, I.V.; Shevkun, N.A.; Koryakova, A.G.; Karapetian, R.N.; Trifelenkov, A.S.; Kravchenko, D.V.; et al. Design, synthesis and biological evaluation of novel 5-oxo-2-thioxoimidazolidine derivatives as potent androgen receptor antagonists. Eur. J. Med. Chem. 2015, 99, 51–66. [Google Scholar] [CrossRef]
- Boiteau, J.G.; Ouvry, G.; Arlabosse, J.M.; Astri, S.; Beillard, A.; Bhurruth-Alcor, Y.; Bonnary, L.; Bouix-Peter, C.; Bouquet, K.; Bourotte, M.; et al. Discovery and process development of a novel TACE inhibitor for the topical treatment of psoriasis. Bioorg. Med. Chem. 2018, 26, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Žukauskaitė, A.; Moretto, A.; Peggion, C.; De Zotti, M.; Šačkus, A.; Formaggio, F.; De Kimpe, N.; Mangelinckx, S. Synthesis and Conformational Study of Model Peptides Containing N-Substituted 3-Aminoazetidine-3-Carboxylic Acids. Eur. J. Org. Chem. 2014, 2014, 2312–2321. [Google Scholar] [CrossRef]
- Gloaguen, E.; Mons, M.; Schwing, K.; Gerhards, M. Neutral Peptides in the Gas Phase: Conformation and Aggregation Issues. Chem. Rev. 2020, 120, 12490–12562. [Google Scholar] [CrossRef] [PubMed]
- Mundlapati, V.R.; Imani, Z.; Goldsztejn, G.; Gloaguen, E.; Brenner, V.; Le Barbu-Debus, K.; Zehnacker-Rentien, A.; Baltaze, J.P.; Robin, S.; Mons, M.; et al. A theoretical and experimental case study of the hydrogen bonding predilection of S-methylcysteine. Amino Acids 2021, 53, 621–633. [Google Scholar] [CrossRef]
- Courty, A.; Mons, M.; Le Calvé, J.; Piuzzi, F.; Dimicoli, I. Chemistry of aromatic cations on water clusters: Magic numbers as a mass spectroscopic diagnosis of reactivity. J. Phys. Chem. A 1997, 101, 1445–1450. [Google Scholar] [CrossRef]
- Sobolewski, A.L.; Domcke, W.; Dedonder-Lardeux, C.; Jouvet, C. Excited-state hydrogen detachment and hydrogen transfer driven by repulsive 1πσ* states: A new paradigm for nonradiative decay in aromatic biomolecules. Phys. Chem. Chem. Phys. 2002, 4, 1093–1100. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Rappoport, D.; Furche, F. Property-optimized Gaussian basis sets for molecular response calculations. J. Chem. Phys. 2010, 133, 134105. [Google Scholar] [CrossRef]
- Schafer, A.; Huber, C.; Ahlrichs, R. Fully Optimized Contracted Gaussian-Basis Sets of Triple Zeta Valence Quality For Atoms Li To Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Sierka, M.; Hogekamp, A.; Ahlrichs, R. Fast evaluation of the Coulomb potential for electron densities using multipole accelerated resolution of identity approximation. J. Chem. Phys. 2003, 118, 9136–9148. [Google Scholar] [CrossRef]
- Eichkorn, K.; Treutler, O.; Ohm, H.; Haser, M.; Ahlrichs, R. Auxiliary Basis-Sets To Approximate Coulomb Potentials. Chem. Phys. Lett. 1995, 240, 283–289, Erratum in Chem. Phys. Lett. 1995, 242, 652–660. [Google Scholar] [CrossRef]
- Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theo. Chem. Acc. 1997, 97, 119–124. [Google Scholar] [CrossRef]
- Gloaguen, E.; de Courcy, B.; Piquemal, J.P.; Pilme, J.; Parisel, O.; Pollet, R.; Biswal, H.S.; Piuzzi, F.; Tardivel, B.; Broquier, M.; et al. Gas-Phase Folding of a Two-Residue Model Peptide Chain: On the Importance of an Interplay between Experiment and Theory. J. Am. Chem. Soc. 2010, 132, 11860–11863. [Google Scholar] [CrossRef] [PubMed]
- Loquais, Y.; Gloaguen, E.; Habka, S.; Vaquero-Vara, V.; Brenner, V.; Tardivel, B.; Mons, M. Secondary Structures in Phe-Containing Isolated Dipeptide Chains: Laser Spectroscopy vs Quantum Chemistry. J. Phys. Chem. A 2015, 119, 5932–5941. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A.; Schuurmann, G. COSMO—A New Approach To Dielectric Screening In Solvents With Explicit Expressions For The Screening Energy And Its Gradient. J. Chem. Soc. Perkin Trans. 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Liu, D.; Imani, Z.; Gourson, C.; Guillot, R.; Robin, S.; Aitken, D.J. A Post-Synthetic Modification Strategy for the Preparation of Homooligomers of 3-Amino-1-methylazetidine-3-carboxylic acid. Synlett 2023, 34. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, D.; Bardaud, J.-X.; Imani, Z.; Robin, S.; Gloaguen, E.; Brenner, V.; Aitken, D.J.; Mons, M. Length-Dependent Transition from Extended to Folded Shapes in Short Oligomers of an Azetidine-Based α-Amino Acid: The Critical Role of NH···N H-Bonds. Molecules 2023, 28, 5048. https://doi.org/10.3390/molecules28135048
Liu D, Bardaud J-X, Imani Z, Robin S, Gloaguen E, Brenner V, Aitken DJ, Mons M. Length-Dependent Transition from Extended to Folded Shapes in Short Oligomers of an Azetidine-Based α-Amino Acid: The Critical Role of NH···N H-Bonds. Molecules. 2023; 28(13):5048. https://doi.org/10.3390/molecules28135048
Chicago/Turabian StyleLiu, Dayi, Jean-Xavier Bardaud, Zeynab Imani, Sylvie Robin, Eric Gloaguen, Valérie Brenner, David J. Aitken, and Michel Mons. 2023. "Length-Dependent Transition from Extended to Folded Shapes in Short Oligomers of an Azetidine-Based α-Amino Acid: The Critical Role of NH···N H-Bonds" Molecules 28, no. 13: 5048. https://doi.org/10.3390/molecules28135048
APA StyleLiu, D., Bardaud, J.-X., Imani, Z., Robin, S., Gloaguen, E., Brenner, V., Aitken, D. J., & Mons, M. (2023). Length-Dependent Transition from Extended to Folded Shapes in Short Oligomers of an Azetidine-Based α-Amino Acid: The Critical Role of NH···N H-Bonds. Molecules, 28(13), 5048. https://doi.org/10.3390/molecules28135048