

Phenylketonuria (PKU) Urinary Metabolomic Phenotype Is Defined by Genotype and Metabolite Imbalance: Results in 51 Early Treated Patients Using Ex Vivo 1H-NMR Analysis
 , ,
, , 
Abstract
1. Introduction
2. Results
2.1. Patient Characteristics
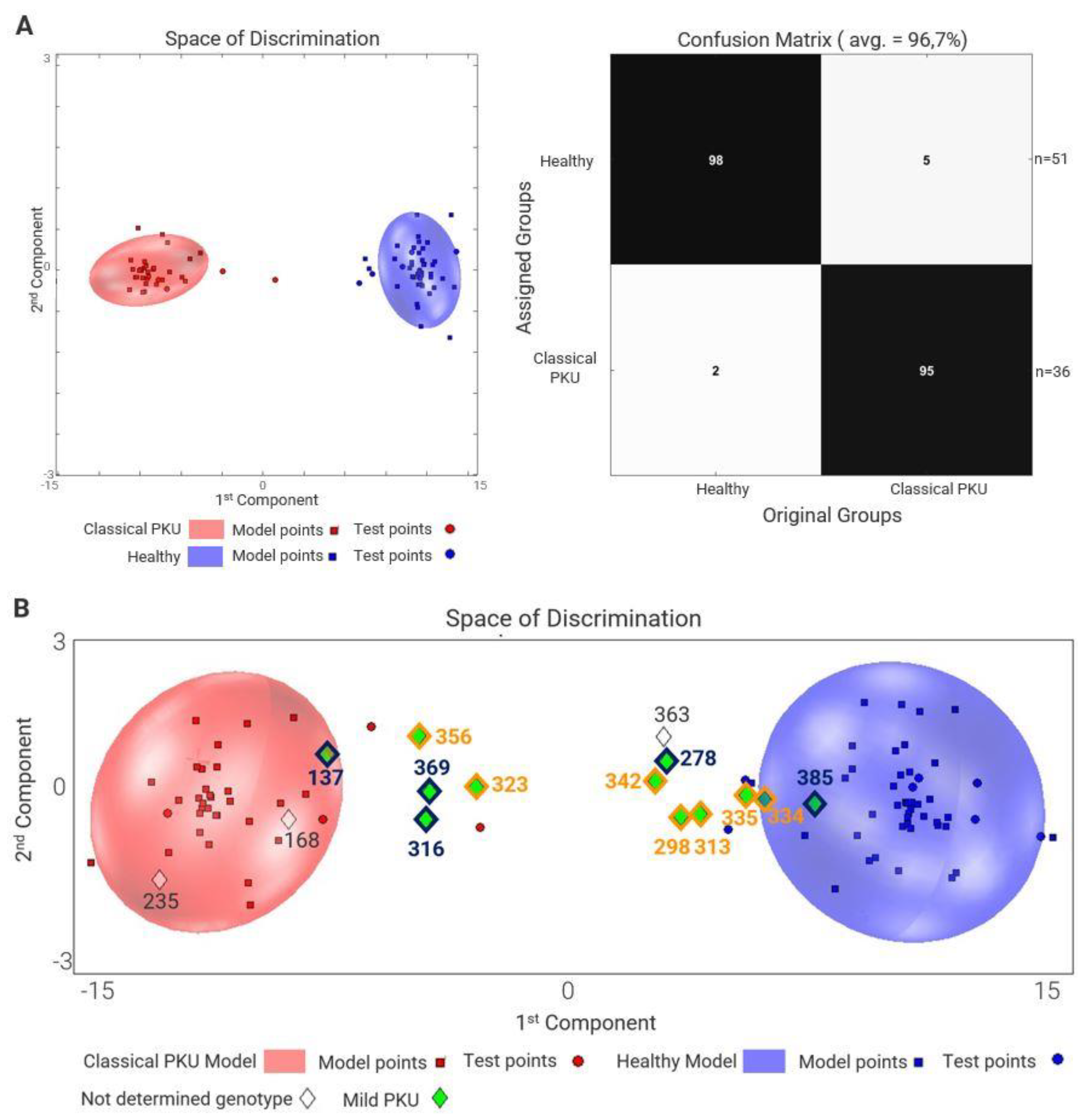
2.2. Metabolome and Genotype
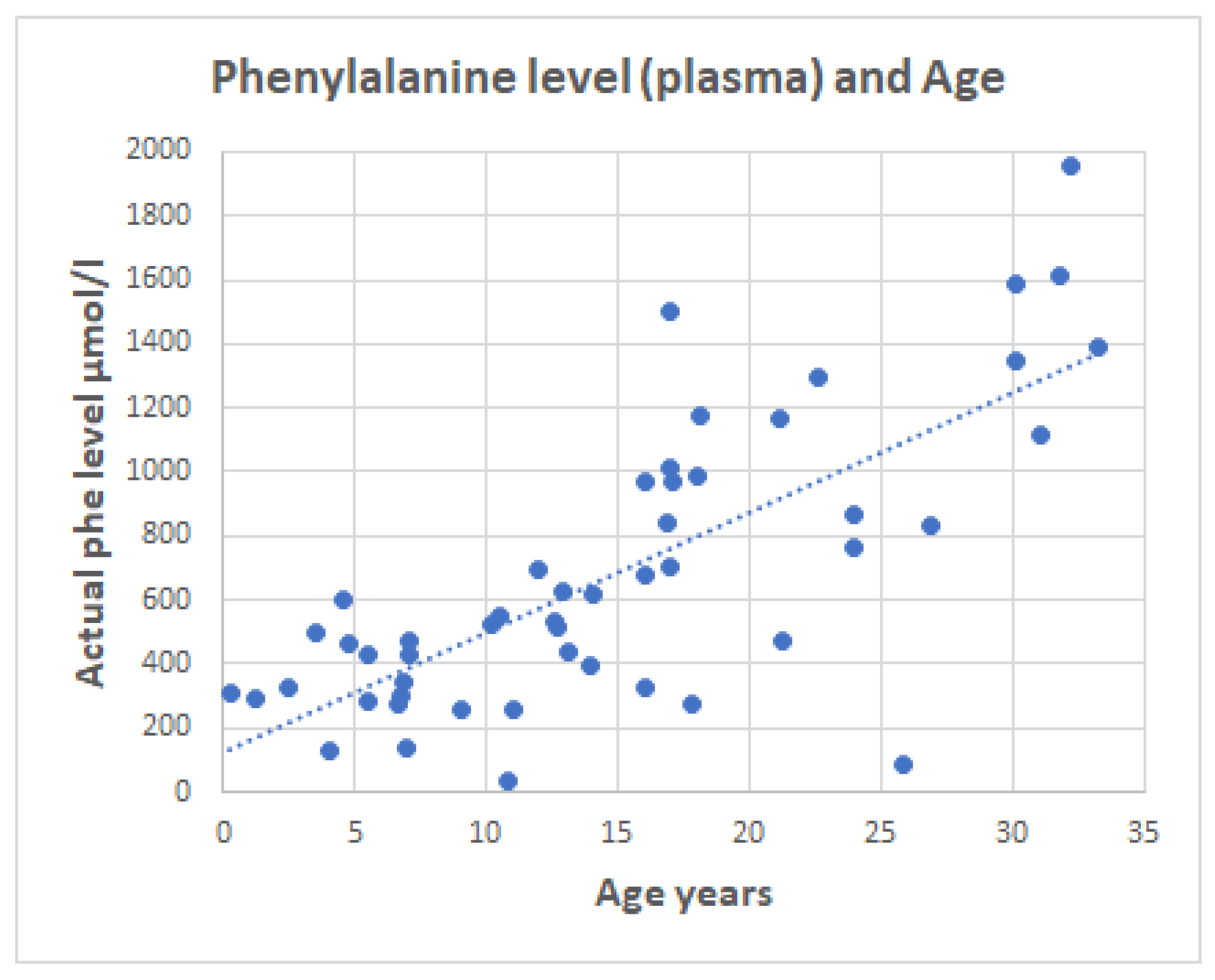
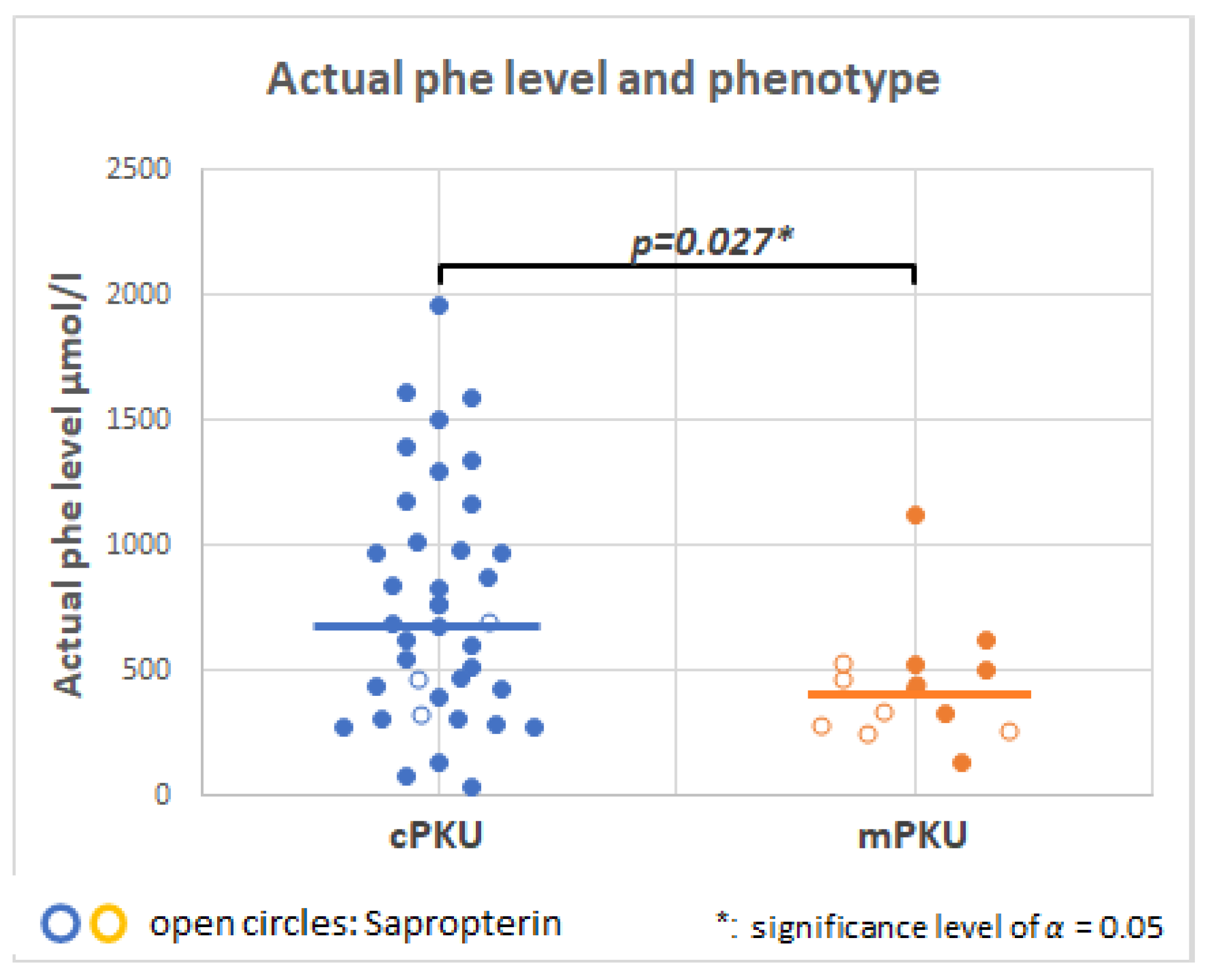
2.3. Serum Phenylalanine in PKU Patients
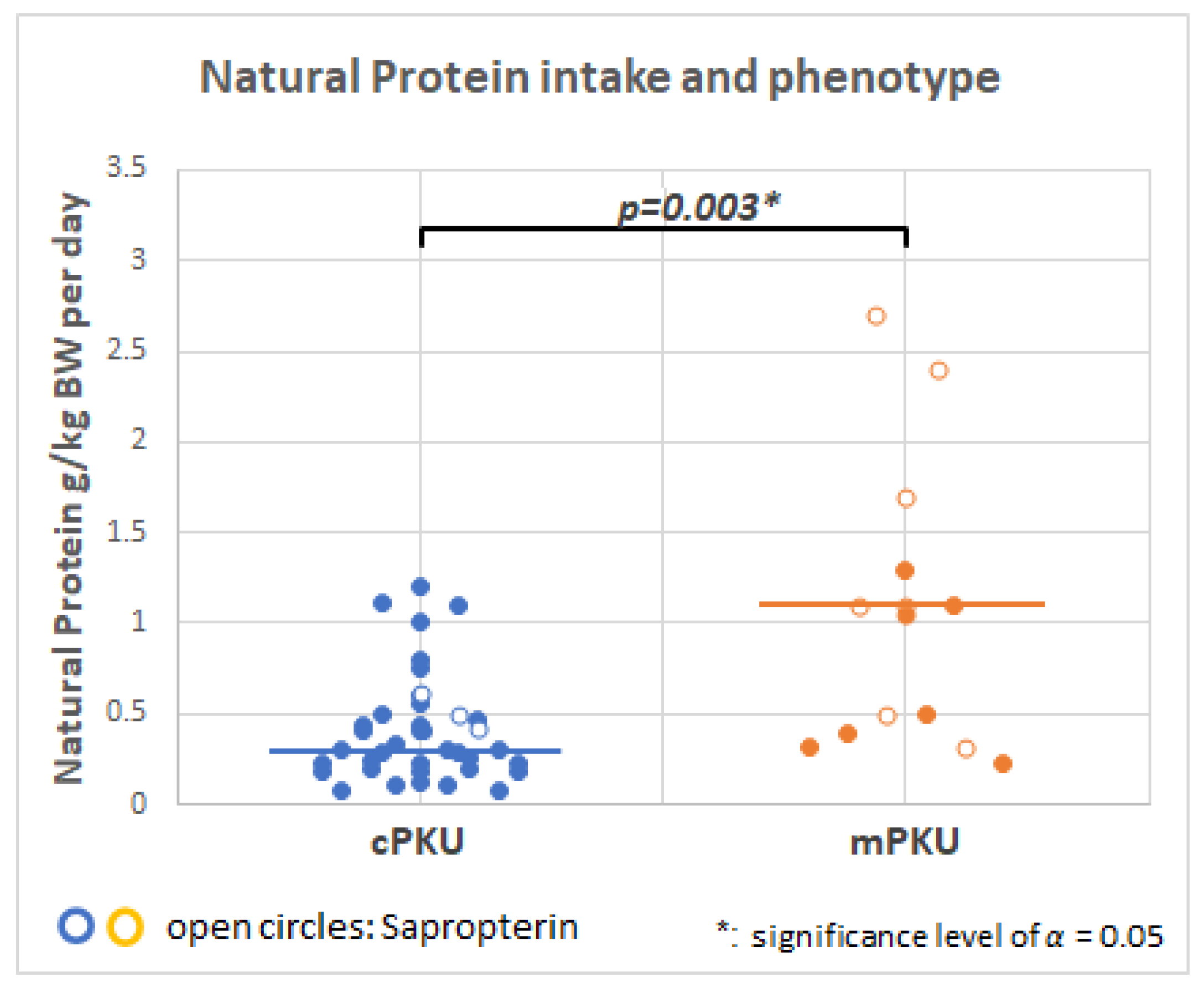
2.4. Dietary Intake
2.5. NMR Targeted Analyses
2.6. Metabolites Linked to Energy Metabolism
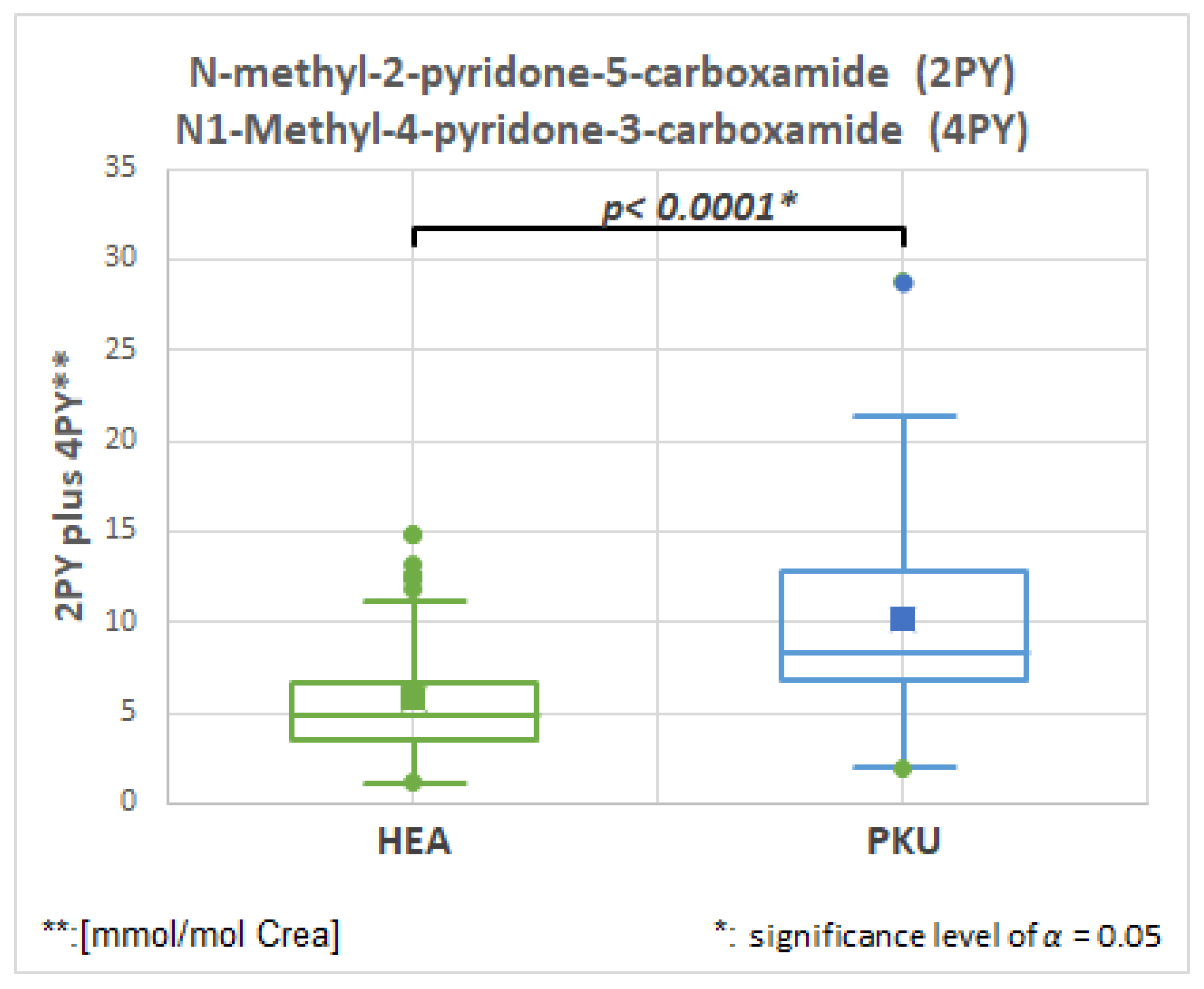
2.6.1. N-Methyl-2-pyridone-5-carboxamide (2PY), HMDB0004193, Fold Change 1.728
2.6.2. 1-N-Methylnicotinamide, HMDB0000699, Fold Change 1.459
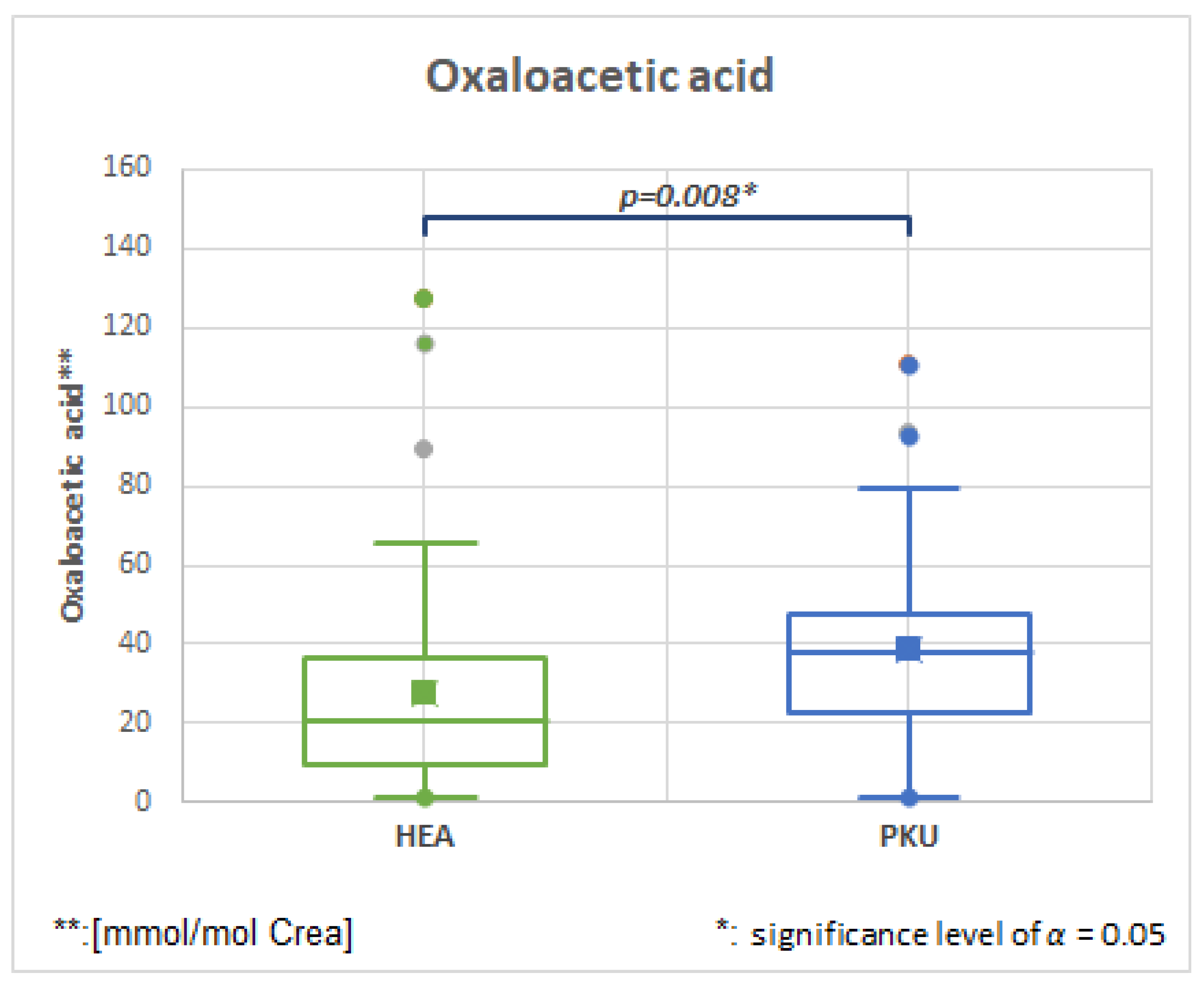
2.6.3. Oxaloacetic, HMDB0000223, Fold Change 1.731
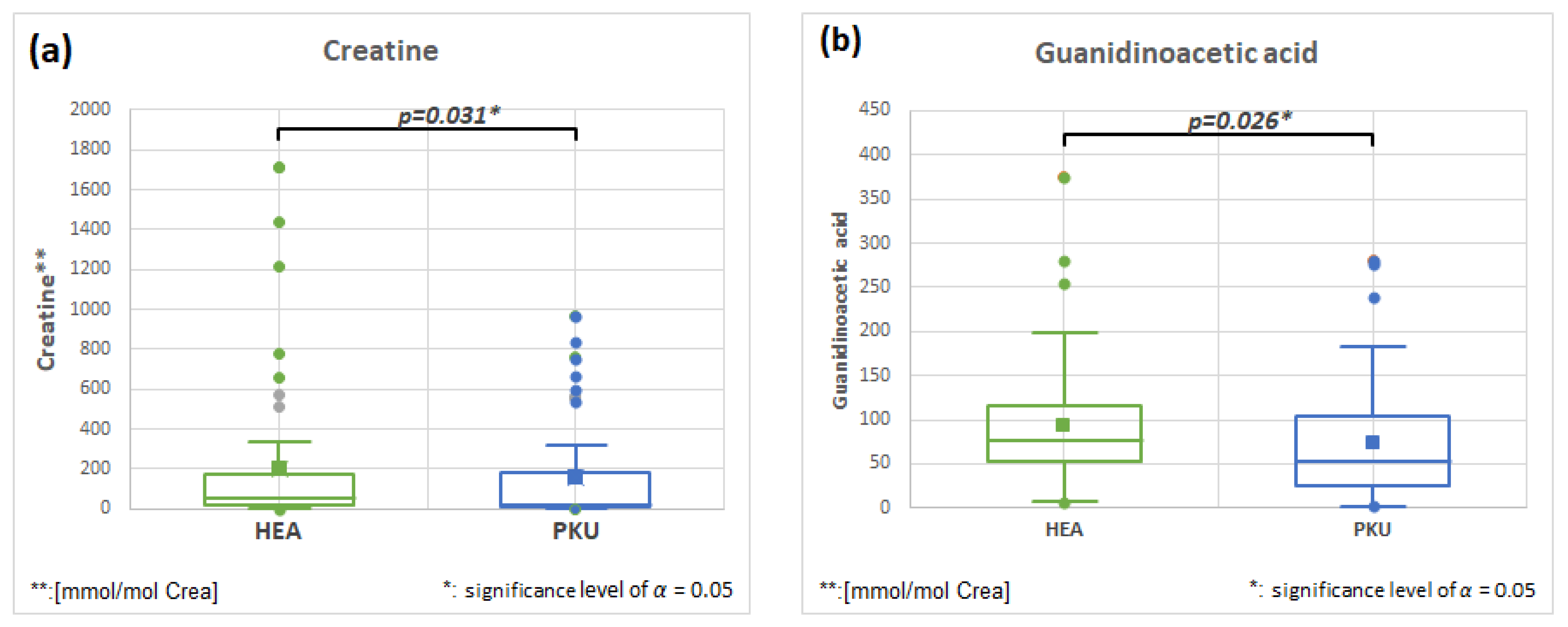
2.6.4. Creatine HMDB0000064 and Guanidinoacetic Acid HMDB0000128, Fold Change 0.796 and 0.796
2.7. Metabolites Related to Gut and Dietary Treatment
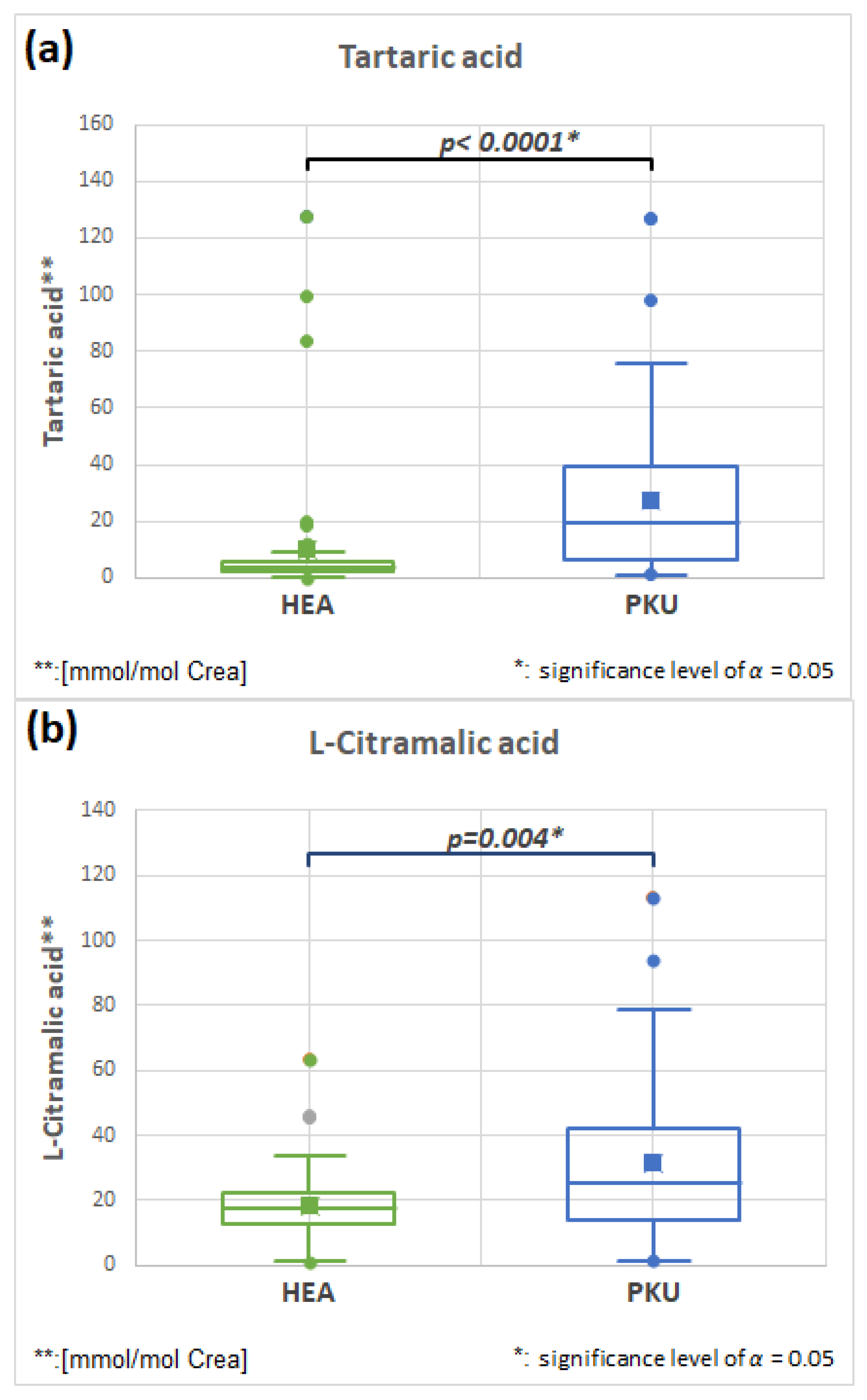
2.7.1. Tartaric Acid HMDB0000956 and L-Citramalic HMDB0000426 Acid (Fold Change 2.665 and 1.721)
2.7.2. Acetic Acid, HMDB0000042, Fold Change 1.931
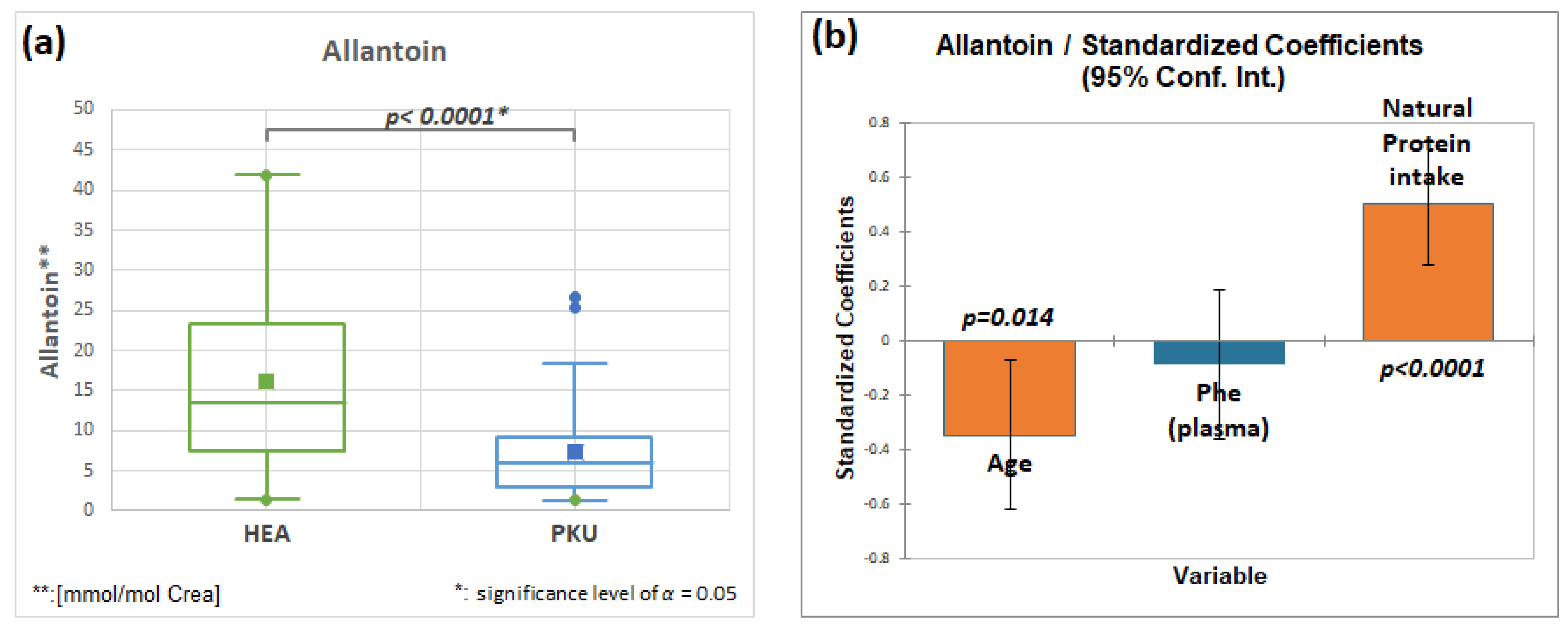
2.7.3. Allantoin, HMDB00462, Fold Change 0.440
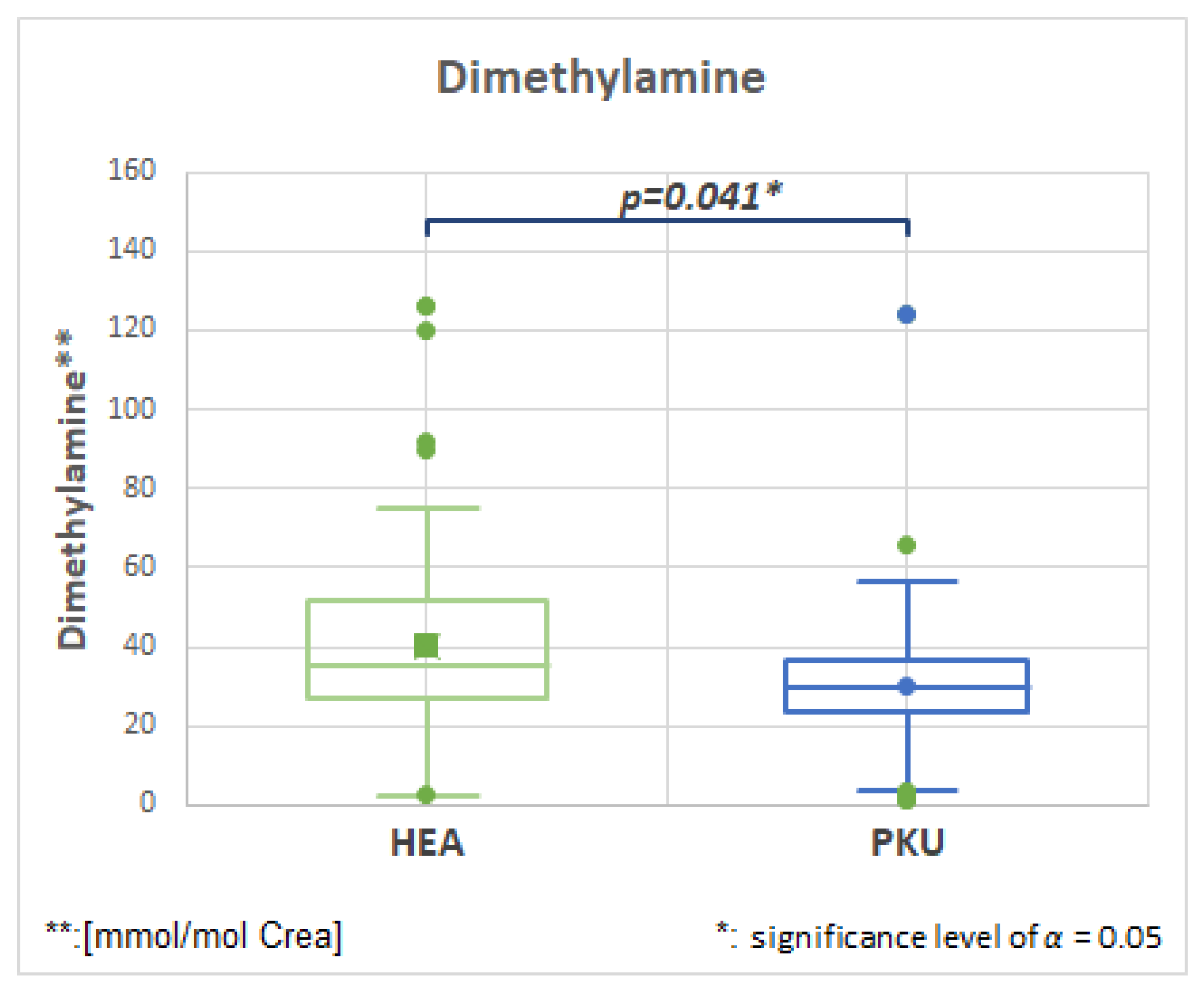
2.7.4. Dimethylamine, HMDB00087, Fold Change 0.753
2.7.5. 2-Furoylglycine, HMDB0000439, Fold Change 0.661
2.8. Other Metabolites Showing Significant Differences (Supplementary Figure S2)
2.8.1. Amino Acids
2.8.2. Organic Acids
2.8.3. Phenylalanine and Phenylalanine Derived Oxidation Products
3. Discussion
3.1. Phenylalanine and Phenylalanine Metabolites
3.2. Role of Natural Low Protein Intake on the Metabolome
3.3. Limitations
4. Materials and Methods
4.1. Study Participants
4.2. Sample Collection
4.3. Blood Phenylalanine Analysis
4.4. NMR Analysis
4.5. Targeted NMR Analysis
4.6. Untargeted NMR Analysis
4.7. Identification of Unknown Metabolites
4.8. Statistical Analysis of Targeted Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Wegberg, A.M.J.; MacDonald, A.; Ahring, K.; Bélanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Giżewska, M.; et al. The complete European guidelines on phenylketonuria: Diagnosis and treatment. Orphanet J. Rare Dis. 2017, 12, 162. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J.; Andersson, H.C.; Antshel, K.M.; Braverman, N.E.; Burton, B.K.; Frazier, D.M.; Mitchell, J.; Smith, W.E.; Thompson, B.H.; Berry, S.A. Phenylalanine hydroxylase deficiency: Diagnosis and management guideline. Genet. Med. 2014, 16, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Pilotto, A.; Blau, N.; Leks, E.; Schulte, C.; Deuschl, C.; Zipser, C.; Piel, D.; Freisinger, P.; Gramer, G.; Kölker, S.; et al. Cerebrospinal fluid biogenic amines depletion and brain atrophy in adult patients with phenylketonuria. J. Inherit. Metab. Dis. 2019, 42, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Pilotto, A.; Zipser, C.M.; Leks, E.; Haas, D.; Gramer, G.; Freisinger, P.; Schaeffer, E.; Liepelt-Scarfone, I.; Brockmann, K.; Maetzler, W.; et al. Phenylalanine effects on brain function in adult phenylketonuria. Neurology 2021, 96, e399–e411. [Google Scholar] [CrossRef] [PubMed]
- Blau, N.; van Spronsen, F.J.; Levy, H.L. Phenylketonuria. Lancet 2010, 376, 1417–1427. [Google Scholar] [CrossRef]
- Gramer, G.; Förl, B.; Springer, C.; Weimer, P.; Haege, G.; Mackensen, F.; Müller, E.; Völcker, H.E.; Hoffmann, G.F.; Lindner, M.; et al. Visual functions in phenylketonuria-evaluating the dopamine and long-chain polyunsaturated fatty acids depletion hypotheses. Mol. Genet. Metab. 2013, 108, 1–7. [Google Scholar] [CrossRef]
- Nowak, J.K.; Kałużny, Ł.; Suda, W.; Bukowska-Posadzy, A.; Adams, A.; Grulkowski, I.; Kocięcki, J.; Walkowiak, J. Retinal nerve fibre layer thickness associates with phenylketonuria control and cognition. J. Med. Genet. 2020, 57, 863–864. [Google Scholar] [CrossRef]
- Hermida-Ameijeiras, A.; Crujeiras, V.; Roca, I.; Calvo, C.; Leis, R.; Couce, M.L. Arterial stiffness assessment in patients with phenylketonuria. Medicine 2017, 96, e9322. [Google Scholar] [CrossRef]
- Azabdaftari, A.; van der Giet, M.; Schuchardt, M.; Hennermann, J.B.; Plöckinger, U.; Querfeld, U. The cardiovascular phenotype of adult patients with phenylketonuria. Orphanet J. Rare Dis. 2019, 14, 213. [Google Scholar] [CrossRef]
- Tanacli, R.; Hassel, J.H.; Gebker, R.; Berger, A.; Gräfe, M.; Schneeweis, C.; Doeblin, P.; Fleck, E.; Stehning, C.; Tacke, F.; et al. Cardiac magnetic resonance reveals incipient cardiomyopathy traits in adult patients with phenylketonuria. J. Am. Heart Assoc. 2021, 10, e020351. [Google Scholar] [CrossRef]
- Dobrowolski, S.F.; Phua, Y.L.; Vockley, J.; Goetzman, E.; Blair, H.C. Phenylketonuria oxidative stress and energy dysregulation: Emerging pathophysiological elements provide interventional opportunity. Mol. Genet. Metab. 2022, 136, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Kirby, T.O.; Ochoa-Reparaz, J.; Roullet, J.B.; Gibson, K.M. Dysbiosis of the intestinal microbiome as a component of pathophysiology in the inborn errors of metabolism. Mol. Genet. Metab. 2021, 132, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Veyrat-Durebex, C.; Bertrand, M.; Patin, F.; Labarthe, F.; Henique, H.; Emond, P.; Andres, C.R.; Antar, C.; Landon, C.; et al. A multiplatform metabolomics approach to characterize plasma levels of phenylalanine and tyrosine in phenylketonuria. JIMD Rep. 2017, 32, 69–79. [Google Scholar] [PubMed]
- Cannet, C.; Pilotto, A.; Rocha, J.C.; Schäfer, H.; Spraul, M.; Berg, D.; Nawroth, P.; Kasperk, C.; Gramer, G.; Haas, D.; et al. Lower plasma cholesterol, LDL-cholesterol and LDL-lipoprotein subclasses in adult phenylketonuria (PKU) patients compared to healthy controls: Results of NMR metabolomics investigation. Orphanet J. Rare Dis. 2020, 15, 61. [Google Scholar] [CrossRef]
- Andrade, F.; López-Suárez, O.; Llarena, M.; Couce, M.L.; Aldámiz-Echevarría, L. Influence of phenylketonuria’s diet on dimethylated arginines and methylation cycle. Medicine 2017, 96, e7392. [Google Scholar] [CrossRef] [PubMed]
- Trefz, K.F.; Muntau, A.C.; Kohlscheen, K.M.; Altevers, J.; Jacob, C.; Braun, S.; Greiner, W.; Jha, A.; Jain, M.; Alvarez, I.; et al. Clinical burden of illness in patients with phenylketonuria (PKU) and associated comorbidities—A retrospective study of German health insurance claims data. Orphanet J. Rare Dis. 2019, 14, 181. [Google Scholar] [CrossRef]
- Mussap, M.; Zaffanello, M.; Fanos, V. Metabolomics: A challenge for detecting and monitoring inborn errors of metabolism. Ann. Transl. Med. 2018, 6, 338. [Google Scholar] [CrossRef]
- Mordaunt, D.; Cox, D.; Fuller, M. Metabolomics to improve the diagnostic efficiency of inborn errors of metabolism. Int. J. Mol. Sci. 2020, 21, 1195. [Google Scholar] [CrossRef]
- Xiong, X.; Sheng, X.; Liu, D.; Zeng, T.; Peng, Y.; Wang, Y. A GC/MS-based metabolomic approach for reliable diagnosis of phenylketonuria. Anal. Bioanal. Chem. 2015, 407, 8825–8833. [Google Scholar] [CrossRef]
- Pulido, N.; Guevara-Morales, J.M.; Rodriguez-López, A.; Pulido, Á.; Díaz, J.; Edrada-Ebel, R.A.; Echeverri-Peña, O.Y. (1)H-Nuclear magnetic resonance analysis of urine as diagnostic tool for organic acidemias and aminoacidopathies. Metabolites 2021, 11, 891. [Google Scholar] [CrossRef]
- Wishart, D.S. NMR metabolomics: A look ahead. J. Magn. Reson. 2019, 306, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Metabolomics for investigating physiological and pathophysiological processes. Physiol. Rev. 2019, 99, 1819–1875. [Google Scholar] [CrossRef] [PubMed]
- Emwas, A.H.; Roy, R.; McKay, R.T.; Tenori, L.; Saccenti, E.; Gowda, G.A.N.; Raftery, D.; Alahmari, F.; Jaremko, L.; Jaremko, M.; et al. NMR spectroscopy for metabolomics research. Metabolites 2019, 9, 123. [Google Scholar] [CrossRef] [PubMed]
- Emwas, A.H.; Szczepski, K.; Poulson, B.G.; Chandra, K.; McKay, R.T.; Dhahri, M.; Alahmari, F.; Jaremko, L.; Lachowicz, J.I.; Jaremko, M. NMR as a “gold standard” method in drug design and discovery. Molecules 2020, 25, 4597. [Google Scholar] [CrossRef] [PubMed]
- Emwas, A.H.; Saccenti, E.; Gao, X.; McKay, R.T.; Dos Santos, V.A.P.M.; Roy, R.; Wishart, D.S. Recommended strategies for spectral processing and post-processing of 1D (1)H-NMR data of biofluids with a particular focus on urine. Metabolomics 2018, 14, 31. [Google Scholar] [CrossRef]
- Bayat, A.; Yasmeen, S.; Lund, A.; Nielsen, J.B.; Møller, L.B. Mutational and phenotypical spectrum of phenylalanine hydroxylase deficiency in Denmark. Clin. Genet. 2016, 90, 247–251. [Google Scholar] [CrossRef]
- Walter, J.H.; White, F.J. Blood phenylalanine control in adolescents with phenylketonuria. Int. J. Adolesc. Med. Health 2004, 16, 41–45. [Google Scholar] [CrossRef]
- Garbade, S.F.; Shen, N.; Himmelreich, N.; Haas, D.; Trefz, F.K.; Hoffmann, G.F.; Burgard, P.; Blau, N. Allelic phenotype values: A model for genotype-based phenotype prediction in phenylketonuria. Genet. Med. 2019, 21, 580–590. [Google Scholar] [CrossRef]
- Embade, N.; Cannet, C.; Diercks, T.; Gil-Redondo, R.; Bruzzone, C.; Ansó, S.; Echevarría, L.R.; Ayucar, M.M.M.; Collazos, L.; Lodoso, B.; et al. NMR-based newborn urine screening for optimized detection of inherited errors of metabolism. Sci. Rep. 2019, 9, 13067. [Google Scholar] [CrossRef]
- Lenglet, A.; Liabeuf, S.; Bodeau, S.; Louvet, L.; Mary, A.; Boullier, A.; Lemaire-Hurtel, A.S.; Jonet, A.; Sonnet, P.; Kamel, S.; et al. N-methyl-2-pyridone-5-carboxamide (2PY)-major metabolite of nicotinamide: An update on an old uremic toxin. Toxins 2016, 8, 339. [Google Scholar] [CrossRef]
- Kumei, S.; Ishioh, M.; Nozu, T.; Okumura, T. Prostaglandin I(2) suppresses the development of gut-brain axis disorder in irritable bowel syndrome in rats. Biochim. Biophys. Acta Gen. Subj. 2023, 1867, 130344. [Google Scholar] [CrossRef] [PubMed]
- Hara, N.; Osago, H.; Hiyoshi, M.; Kobayashi-Miura, M.; Tsuchiya, M. Quantitative analysis of the effects of nicotinamide phosphoribosyltransferase induction on the rates of NAD+ synthesis and breakdown in mammalian cells using stable isotope-labeling combined with mass spectrometry. PLoS ONE 2019, 14, e0214000. [Google Scholar] [CrossRef] [PubMed]
- Gronwald, W.; Klein, M.S.; Zeltner, R.; Schulze, B.D.; Reinhold, S.W.; Deutschmann, M.; Immervoll, A.K.; Böger, C.A.; Banas, B.; Eckardt, K.U.; et al. Detection of autosomal dominant polycystic kidney disease by NMR spectroscopic fingerprinting of urine. Kidney Int. 2011, 79, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolski, S.F.; Phua, Y.L.; Sudano, C.; Spridik, K.; Zinn, P.O.; Wang, Y.; Bharathi, S.; Vockley, J.; Goetzman, E. Comparative metabolomics in the Pah(enu2) classical PKU mouse identifies cerebral energy pathway disruption and oxidative stress. Mol. Genet. Metab. 2022, 136, 38–45. [Google Scholar] [CrossRef]
- Haas, D.; Gan-Schreier, H.; Langhans, C.D.; Anninos, A.; Haege, G.; Burgard, P.; Schulze, A.; Hoffmann, G.F.; Okun, J.G. Diagnosis and therapeutic monitoring of inborn errors of creatine metabolism and transport using liquid chromatography-tandem mass spectrometry in urine, plasma and CSF. Gene 2014, 538, 188–194. [Google Scholar] [CrossRef]
- Verduci, E.; Carbone, M.T.; Fiori, L.; Gualdi, C.; Banderali, G.; Carducci, C.; Leuzzi, V.; Biasucci, G.; Zuccotti, G.V. Creatine levels in patients with phenylketonuria and mild hyperphenylalaninemia: A pilot study. Life 2021, 11, 425. [Google Scholar] [CrossRef]
- Paprotny, L.; Celejewska, A.; Frajberg, M.; Wianowska, D. Development and validation of GC-MS/MS method useful in diagnosing intestinal dysbiosis. J. Chromatogr. B Analyt Technol. Biomed. Life Sci. 2019, 1130–1131, 121822. [Google Scholar] [CrossRef]
- Ma, J.; Liu, Z.; Gao, X.; Bao, Y.; Hong, Y.; He, X.; Zhu, W.; Li, Y.; Huang, W.; Zheng, N.; et al. Gut microbiota remodeling improves natural aging-related disorders through Akkermansia muciniphila and its derived acetic acid. Pharmacol. Res. 2023, 189, 106687. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, H.; Sun, C.Y.; He, Q.Y.; Zhang, R.R.; Luo, B.F.; Zhou, Z.H.; Chen, X.F. Evaluation of two laboratory model methods for diarrheal irritable bowel syndrome. Mol. Med. 2023, 29, 5. [Google Scholar] [CrossRef]
- Lam, C.W.; Law, C.Y.; To, K.K.W.; Cheung, S.K.K.; Lee, K.; Sze, K.H.; Leung, K.F.; Yuen, K.Y. NMR-based metabolomic urinalysis: A rapid screening test for urinary tract infection. Clin. Chim. Acta 2014, 436, 217–223. [Google Scholar] [CrossRef]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.A.; Crowe, F.L.; Appleby, P.N.; Key, T.J.; Travis, R.C. Serum uric acid concentrations in meat eaters, fish eaters, vegetarians and vegans: A cross-sectional analysis in the EPIC-Oxford cohort. PLoS ONE 2013, 8, e56339. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.C.; Zhang, A.Q.; Smith, R.L. Dimethylamine and diet. Food Chem. Toxicol. 2008, 46, 1734–1738. [Google Scholar] [CrossRef]
- Pettersen, J.E.; Jellum, E. The identification and metabolic origin of 2-furoylglycine and 2,5-furandicarboxylic acid in human urine. Clin. Chim. Acta 1972, 41, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.S.; Owen, O.E. Effect of hyperphenylalaninaemia on lipid synthesis from ketone bodies by rat brain. Biochem. J. 1976, 154, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Blau, N.; Hennermann, J.B.; Langenbeck, U.; Lichter-Konecki, U. Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (BH4) deficiencies. Mol. Genet. Metab. 2011, 104, S2–S9. [Google Scholar] [CrossRef]
- Moritz, L.; Klotz, K.; Grünert, S.C.; Hannibal, L.; Spiekerkoetter, U. Metabolic phenotyping in phenylketonuria reveals disease clustering independently of metabolic control. Mol. Genet. Metab. 2023, 138, 107509. [Google Scholar] [CrossRef]
- Montanari, C.; Parolisi, S.; Borghi, E.; Putignani, L.; Bassanini, G.; Zuvadelli, J.; Bonfanti, C.; Tummolo, A.; Dionisi-Vici, C.; Biasucci, G.; et al. Dysbiosis, host metabolism, and non-communicable diseases: Trialogue in the inborn errors of metabolism. Front. Physiol. 2021, 12, 716520. [Google Scholar] [CrossRef]
- Murray, E.R.; Kemp, M.; Nguyen, T.T. The microbiota-gut-brain axis in Alzheimer’s disease: A review of taxonomic alterations and potential avenues for interventions. Arch. Clin. Neuropsychol. 2022, 37, 595–607. [Google Scholar] [CrossRef]
- Dong, S.; Sun, M.; He, C.; Cheng, H. Brain-gut-microbiota axis in Parkinson’s disease: A historical review and future perspective. Brain Res. Bull. 2022, 183, 84–93. [Google Scholar] [CrossRef]
- Cai, Y.; Song, W.; Li, J.; Jing, Y.; Liang, C.; Zhang, L.; Zhang, X.; Zhang, W.; Liu, B.; An, Y.; et al. The landscape of aging. Sci. China Life Sci. 2022, 65, 2354–2454. [Google Scholar] [CrossRef]
- van der Goot, E.; van Spronsen, F.J.; Falcão Salles, J.; van der Zee, E.A. A microbial community ecology perspective on the gut-microbiome-brain axis. Front. Endocrinol. 2020, 11, 611. [Google Scholar] [CrossRef]
- De Oliveira, F.P.; Mendes, R.H.; Dobbler, P.T.; Mai, V.; Pylro, V.S.; Waugh, S.G.; Vairo, F.; Refosco, L.F.; Roesch, L.F.; Schwartz, I.V. Phenylketonuria and gut microbiota: A controlled study based on next-generation sequencing. PLoS ONE 2016, 11, 0157513. [Google Scholar] [CrossRef] [PubMed]
- Verduci, E.; Moretti, F.; Bassanini, G.; Banderali, G.; Rovelli, V.; Casiraghi, M.C.; Morace, G.; Borgo, F.; Borghi, E. Phenylketonuric diet negatively impacts on butyrate production. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Czibik, G.; Mezdari, Z.; Murat Altintas, D.; Bréhat, J.; Pini, M.; d’Humières, T.; Delmont, T.; Radu, C.; Breau, M.; Liang, H.; et al. Dysregulated phenylalanine catabolism plays a key role in the trajectory of cardiac aging. Circulation 2021, 144, 559–574. [Google Scholar] [CrossRef]
- Gramer, G.; Haege, G.; Langhans, C.D.; Schuhmann, V.; Burgard, P.; Hoffmann, G.F. Long-chain polyunsaturated fatty acid status in children, adolescents and adults with phenylketonuria. Prostaglandins Leukot. Essent. Fat. Acids 2016, 109, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Serfozo, C.; Barta, A.G.; Horvath, E.; Sumanszki, C.; Csakany, B.; Resch, M.; Nagy, Z.Z.; Reismann, P. Reduced macular thickness and macular vessel density in early-treated adult patients with PKU. Mol. Genet. Metab. Rep. 2021, 27, 100767. [Google Scholar] [CrossRef] [PubMed]
- Burton, B.K.; Jones, K.B.; Cederbaum, S.; Rohr, F.; Waisbren, S.; Irwin, D.E.; Kim, G.; Lilienstein, J.; Alvarez, I.; Jurecki, E.; et al. Prevalence of comorbid conditions among adult patients diagnosed with phenylketonuria. Mol. Genet. Metab. 2018, 125, 228–234. [Google Scholar] [CrossRef]
- Hennermann, J.B.; Roloff, S.; Gellermann, J.; Vollmer, I.; Windt, E.; Vetter, B.; Plöckinger, U.; Mönch, E.; Querfeld, U. Chronic kidney disease in adolescent and adult patients with phenylketonuria. J. Inherit. Metab. Dis. 2012, 36, 747–756. [Google Scholar] [CrossRef]
- Ney, D.M. Does the PKU diet contribute to impaired renal function? J. Inherit. Metab. Dis. 2013, 36, 903–904. [Google Scholar] [CrossRef]
- Kaufman, S. PHENYLKETONURIA: Biochemical Mechanisms; Agranoff, B.W., Aprison, M.H., Eds.; Advances in Neurochemistry; Plenum Press: New York, NY, USA, 1977; pp. 1–132. [Google Scholar]
- MacDonald, A.; van Wegberg, A.M.J.; Ahring, K.; Beblo, S.; Bélanger-Quintana, A.; Burlina, A.; Campistol, J.; Coşkun, T.; Feillet, F.; Giżewska, M.; et al. PKU dietary handbook to accompany PKU guidelines. Orphanet J. Rare Dis. 2020, 15, 171. [Google Scholar] [CrossRef]
- van Spronsen, F.J.; van Wegberg, A.M.; Ahring, K.; Bélanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Giżewska, M.; et al. Key European guidelines for the diagnosis and management of patients with phenylketonuria. Lancet Diabetes Endocrinol. 2017, 5, 743–756. [Google Scholar] [CrossRef]
- Evers, R.A.F.; van Wegberg, A.M.J.; van Dam, E.; de Vries, M.C.; Janssen, M.C.H.; van Spronsen, F.J. Anthropomorphic measurements and nutritional biomarkers after 5years of BH4 treatment in phenylketonuria patients. Mol. Genet. Metab. 2018, 124, 238–242. [Google Scholar] [CrossRef]
- Trefz, F.; Maillot, F.; Motzfeldt, K.; Schwarz, M. Adult phenylketonuria outcome and management. Mol. Genet. Metab. 2011, 104, S26–S30. [Google Scholar] [CrossRef] [PubMed]
- Wibrand, F. A microplate-based enzymatic assay for the simultaneous determination of phenylalanine and tyrosine in serum. Clin. Chim. Acta 2014, 347, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Dona, A.C.; Jiménez, B.; Schäfer, H.; Humpfer, E.; Spraul, M.; Lewis, M.R.; Pearce, J.T.M.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Precision high-throughput proton NMR spectroscopy of human urine, serum, and plasma for large-scale metabolic phenotyping. Anal. Chem. 2014, 86, 9887–9894. [Google Scholar] [CrossRef] [PubMed]
- Cannet, C.; Frauendienst-Egger, G.; Freisinger, P.; Götz, H.; Götz, M.; Himmelreich, N.; Kock, V.; Spraul, M.; Bus, C.; Biskup, S.; et al. Ex vivo proton spectroscopy((1) H-NMR) analysis of inborn errors of metabolism: Automatic and computer-assisted analyses. NMR Biomed. 2022, 36, e4853. [Google Scholar] [CrossRef] [PubMed]
- Assfalg, M.; Bertini, I.; Colangiuli, D.; Luchinat, C.; Schäfer, H.; Schütz, B.; Spraul, M. Evidence of different metabolic phenotypes in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 1420–1424. [Google Scholar] [CrossRef]
- Bernini, P.; Bertini, I.; Luchinat, C.; Nepi, S.; Saccenti, E.; Schäfer, H.; Schütz, B.; Spraul, M.; Tenori, L. Individual human phenotypes in metabolic space and time. J. Proteome Res. 2009, 8, 4264–4271. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mean (±SD) or Percentage | p-Value | ||
|---|---|---|---|
| Healthy Controls (n = 51) | PKU Patients (n = 51) | ||
| Gender (% female) | 52.9 | 51.0 | |
| Age (years) | 14.9 ± 9.2 | 14.9 ± 8.2 | 0.545 |
| Creatinine [mmol/mol Crea] | 8.2 ± 7.0 | 8.2 ± 7.3 | 0.857 |
| Patient ID | Phe Level (µmol/L Plasma) | Genetic Phenotype Value (GPV) | Phenotype | Allele 1 | Allele 2 | Sapropterin | Dietary Protein Intake (g/kg BW/day) |
|---|---|---|---|---|---|---|---|
| 404 | 305 | 0 | cPKU | IVS12 + 1G>A | p.R408W | no | 0.420 |
| 397 | 289 | 0 | cPKU | p.R408W | p.E280K | no | 0.330 |
| 385 | 325 | mPKU | IVS12 + 1G>A | p.F410I * | no | 0.320 | |
| 369 | 500 | 5.1 | mPKU | IVS12 + 1G>A | p.Y414C | no | 0.230 |
| 363 | 132 | mPKU | IVS10−11G>A | Not detected | no | 0.500 | |
| 362 | 597 | 0 | cPKU | p.R408W | p.R408W | no | 0.240 |
| 359 | 467 | 0 | cPKU | IVS10−11G>A | p.Y386C | no | 0.300 |
| 356 | 287 | 5.1 | mPKU | p.Y356X | p.Y414C | yes | 1.101 |
| 405 | 429 | 0 | cPKU | IVS12 + 1G>A | p.R408W | no | 0.290 |
| 339 | 303 | 0 | cPKU | IVS12 + 1G>A | p.R408W | no | 0.440 |
| 347 | 279 | 0 | cPKU | IVS12 + 1G>A | IVS10−11G>A | no | 0.200 |
| 342 | 344 | 5.0 | mPKU | p.R408W | p.D129Y | yes | 1.700 |
| 336 | 135 | 0 | cPKU | IVS12 + 1G>A | p.R408W | no | 0.401 |
| 334 | 431 | 5.1 | mPKU | p.G46S | p.Y414C | yes | 2.700 |
| 335 | 475 | 5.1 | mPKU | p.G46S | p.Y414C | yes | 2.400 |
| 323 | 261 | 5.1 | mPKU | p.F39L | p.Y414C | yes | 1.102 |
| 316 | 528 | 5.1 | mPKU | IVS12 + 1G>A | p.Y414C | no | 0.390 |
| 314 | 546 | 0 | cPKU | p.R408W | p.T266E | no | 0.180 |
| 333 | 36 | 0 | cPKU | R252W | p.R252W | no | 0.100 |
| 313 | 254 | 5.1 | mPKU | p.Y414C | p.W120X | yes | 0.501 |
| 306 | 692 | 0 | cPKU | p.R158Q | IVS12 + 1G>A | no | 0.400 |
| 298 | 533 | 5.1 | mPKU | p.R408W | p.Y414C | yes | 0.320 |
| 302 | 513 | 0 | cPKU | IVS10nt−11G>A | C.−473−?_168+?du | no | 0.070 |
| 295 | 627 | 0 | cPKU | IVS12 + 1G>A | P281L | no | 0.070 |
| 292 | 434 | 0 | cPKU | IVS12 + 1G>A | IVS12 + 1G>A | no | 0.180 |
| 282 | 392 | 0 | cPKU | IVS12 + 1G>A | IVS10−11G>A | no | 0.200 |
| 278 | 622 | 6.9 | mPKU | p.R408W | p.E390G | no | 1.290 |
| 254 | 327 | 2.0 | cPKU | p.L48S | IVS12 + 1G>A | yes | 0.620 |
| 253 | 968 | 0 | cPKU | IVS12 + 1G>A | E221D222duAG | no | 0.110 |
| 258 | 682 | 0 | cPKU | IVS12 + 1G>A | p.R408W | no | 0.230 |
| 249 | 840 | 0 | cPKU | IVS12 + 1G>A | p.R408W | no | 0.250 |
| 236 | 1504 | 0 | cPKU | IVS10−11G>A | p.Y386C | no | 0.550 |
| 242 | 700 | 1.1 | cPKU | IVS12 + 1G>A | p.I65T | yes | 0.460 |
| 272 | 1010 | 0 | cPKU | IVS10−11G>A | p.R408W | no | 0.301 |
| 243 | 969 | 0 | cPKU | IVS12 + 1G>A | p.R252W | no | 0.502 |
| 237 | 277 | 0 | cPKU | p.R408W | p.R408W | no | 0.290 |
| 238 | 984 | 0 | cPKU | IVS12 + 1G>A | IVS12 + 1G>A | no | 0.120 |
| 235 | 1175 | cPKU | IVS12 + 1G>A | Not detected | no | 0.220 | |
| 211 | 468 | 2.6 | cPKU | p.R408W | p.A104D | yes | 0.490 |
| 212 | 1163 | 0 | cPKU | IVS10−11G>A | p.R408W | no | 0.302 |
| 196 | 1292 | 0 | cPKU | IVS12 + 1G>A | p.E221D | no | 0.750 |
| 190 | 869 | 0 | cPKU | p.P281L | p.R243X | no | 1.103 |
| 191 | 766 | 0 | cPKU | IVS12 + 1G>A | p.R408W | no | 0.440 |
| 176 | 83 | 0 | mPKU | IVS12 + 1G>A | p.R408W | no | 0.180 |
| 168 | 831 | cPKU | Not detected | Not detected | no | 0.230 | |
| 143 | 1590 | cPKU | IVS12 + 1G>A | not detected | no | 1.200 | |
| 145 | 1344 | 2.6 | cPKU | IVS1 + 5G>T | p.A104D | no | 1.000 |
| 137 | 1118 | 5.1 | mPKU | IVS12 + 1G>A | p.Y414C | no | 1.100 |
| 129 | 1613 | cPKU | IVS12 + 1G>A | unclear | no | 0.600 | |
| 126 | 1955 | 0 | cPKU | IVS12 + 1G>A | p.D282N | no | 0.800 |
| 121 | 1390 | 0 | cPKU | IVS12 + 1G>A | p.R158Q | no | 1.104 |
| HEA | PKU | |||||||
|---|---|---|---|---|---|---|---|---|
| n | Mean * | SD | n | Mean * | SD | p-Value | Fold Change | |
| Phenylpyruvic acid | 42 | 10.8 | 10.2 | 46 | 73.9 | 126.6 | 0.003 | 6.853 |
| D-Mandelic acid | 10 | 2.1 | 2.2 | 8 | 9.9 | 8.2 | 0.013 | 4.762 |
| 2-Furoylglycine | 21 | 12.7 | 17.7 | 20 | 37.5 | 39.8 | 0.007 | 2.950 |
| Tartaric acid | 51 | 10.3 | 24.2 | 51 | 27.3 | 26.9 | 0.0001 | 2.665 |
| Phenylacetic acid | 42 | 5.5 | 5.6 | 38 | 13.9 | 14.7 | 0.003 | 2.558 |
| Glycine | 51 | 117.8 | 124.1 | 51 | 282.6 | 342.2 | 0.008 | 2.400 |
| Methionine | 13 | 3.6 | 1.4 | 12 | 8.4 | 8.6 | 0.034 | 2.343 |
| Acetic acid | 51 | 8.9 | 7.2 | 51 | 17.2 | 13.3 | 0.001 | 1.931 |
| Phenylalanine | 42 | 21.0 | 18.3 | 47 | 39.7 | 21.4 | 0.0001 | 1.889 |
| Neopterin | 51 | 3.0 | 5.3 | 51 | 5.2 | 7.6 | 0.022 | 1.750 |
| 2PY ** and 4PY *** | 51 | 5.9 | 3.5 | 51 | 10.1 | 5.3 | 0.0001 | 1.728 |
| L-Citramalic acid | 51 | 18.5 | 12.0 | 51 | 31.8 | 24.9 | 0.004 | 1.721 |
| Maleic acid | 50 | 1.1 | 1.9 | 51 | 1.7 | 2.2 | 0.0001 | 1.639 |
| Adenine | 45 | 2.0 | 2.0 | 50 | 3.2 | 3.7 | 0.03 | 1.570 |
| 1-Methylnicotinamide | 51 | 9.4 | 9.0 | 51 | 13.6 | 16.0 | 0.027 | 1.459 |
| 2-Hydroxyisovaleric acid | 36 | 1.2 | 0.7 | 31 | 1.7 | 1.1 | 0.01 | 1.459 |
| Oxaloacetic acid | 50 | 27.8 | 26.9 | 47 | 38.8 | 25.1 | 0.008 | 1.396 |
| 3-Methylglutaconic acid | 51 | 5.6 | 2.9 | 51 | 7.8 | 4.4 | 0.003 | 1.392 |
| Valine | 51 | 4.6 | 2.9 | 51 | 6.4 | 5.2 | 0.011 | 1.392 |
| Acetoacetic acid | 46 | 11.8 | 9.9 | 48 | 16.1 | 8.5 | 0.006 | 1.371 |
| Guanidinoacetic acid | 51 | 93.1 | 71.5 | 50 | 74.1 | 67.7 | 0.026 | 0.796 |
| Creatine | 49 | 201.4 | 370.1 | 49 | 160.2 | 269.7 | 0.031 | 0.796 |
| Dimethylamine | 51 | 40.2 | 27.2 | 51 | 30.3 | 20.6 | 0.041 | 0.753 |
| Allantoin | 51 | 16.2 | 11.3 | 48 | 7.1 | 5.9 | 0.0001 | 0.440 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cannet, C.; Bayat, A.; Frauendienst-Egger, G.; Freisinger, P.; Spraul, M.; Himmelreich, N.; Kockaya, M.; Ahring, K.; Godejohann, M.; MacDonald, A.; et al. Phenylketonuria (PKU) Urinary Metabolomic Phenotype Is Defined by Genotype and Metabolite Imbalance: Results in 51 Early Treated Patients Using Ex Vivo 1H-NMR Analysis. Molecules 2023, 28, 4916. https://doi.org/10.3390/molecules28134916
Cannet C, Bayat A, Frauendienst-Egger G, Freisinger P, Spraul M, Himmelreich N, Kockaya M, Ahring K, Godejohann M, MacDonald A, et al. Phenylketonuria (PKU) Urinary Metabolomic Phenotype Is Defined by Genotype and Metabolite Imbalance: Results in 51 Early Treated Patients Using Ex Vivo 1H-NMR Analysis. Molecules. 2023; 28(13):4916. https://doi.org/10.3390/molecules28134916
Chicago/Turabian StyleCannet, Claire, Allan Bayat, Georg Frauendienst-Egger, Peter Freisinger, Manfred Spraul, Nastassja Himmelreich, Musa Kockaya, Kirsten Ahring, Markus Godejohann, Anita MacDonald, and et al. 2023. "Phenylketonuria (PKU) Urinary Metabolomic Phenotype Is Defined by Genotype and Metabolite Imbalance: Results in 51 Early Treated Patients Using Ex Vivo 1H-NMR Analysis" Molecules 28, no. 13: 4916. https://doi.org/10.3390/molecules28134916
APA StyleCannet, C., Bayat, A., Frauendienst-Egger, G., Freisinger, P., Spraul, M., Himmelreich, N., Kockaya, M., Ahring, K., Godejohann, M., MacDonald, A., & Trefz, F. (2023). Phenylketonuria (PKU) Urinary Metabolomic Phenotype Is Defined by Genotype and Metabolite Imbalance: Results in 51 Early Treated Patients Using Ex Vivo 1H-NMR Analysis. Molecules, 28(13), 4916. https://doi.org/10.3390/molecules28134916