Abstract
Pynegabine, an antiepileptic drug candidate in phase I clinical trials, is a structural analog of the marketed drug retigabine with improved chemical stability, strong efficacy, and a better safety margin. The reported shortest synthetic route for pynegabine contains six steps and involves the manipulation of highly toxic methyl chloroformate and dangerous hydrogen gas. To improve the feasibility of drug production, we developed a concise, three-step process using unconventional methoxycarbonylation and highly efficient Buchwald–Hartwig cross coupling. The new synthetic route generated pynegabine at the decagram scale without column chromatographic purification and avoided the dangerous manipulation of hazardous reagents.
1. Introduction
Epilepsy, a chronic disorder of the brain, is one of the most common neurological diseases, affecting around 50 million people globally [1]. Clinical antiepileptic drugs (AEDs) are only effective in about 70% of patients with epilepsy, making the discovery of new AEDs with novel mechanisms and/or low side effects critically important [2,3]. Retigabine (Figure 1), an AED approved by the U.S. Food and Drug Administration (FDA) in 2011, is the first and only marketed drug functioning as an agonist of KCNQ2 voltage-gated potassium ion channels [4]. However, in 2013, the FDA added warnings to the drug label emphasizing the serious risks associated with the use of retigabine, which included skin discoloration and retinal pigment abnormalities [5]. Subsequently, the production of retigabine was permanently discontinued in 2017 due to the declining market for the medication [6]. Several pigmented dimers of retigabine have been identified in an in vivo study, suggesting the correlation between the discoloration and the triaminobenzene segment of retigabine [7]. Therefore, the above adverse events of retigabine seem to originate from its chemical structure but not from the KCNQ2 target [8]. Accordingly, great efforts are made to search for new KCNQ2 agonists for AED candidates based on the optimization of retigabine structure [9,10,11,12,13,14,15,16].
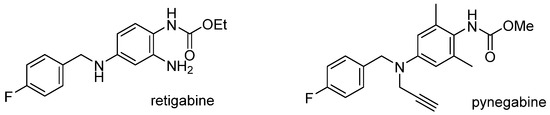
Figure 1.
Chemical structures of retigabine and pynegabine.
Pynegabine, also known as HN37, is a structural analog of retigabine (Figure 1) [17]. Its ratios of the brain to blood exposure are 10 times more than those of retigabine in mice and rats. The EC50 of pynegabine for KCNQ2 channels and KCNQ2/KCNQ3 channels are also 55- and 125-fold more potent than those of retigabine. In a maximal electroshock seizure model test, the TD50/ED50 ratio of pynegabine was at least eight-fold bigger than those of retigabine and two other AEDs (levetiracetam and topiramate) which show a better safety margin. More importantly, by change of the triaminobenzene segment in retigabine, pynegabine exhibited much better stability for temperature, humidity, and light, implying a lower risk of discoloration [18]. Pynegabine has progressed into phase I clinical trials for epilepsy treatment in China (ChinaDrugTrials Identifier: CTR20201676, CTR20222616).
There are two synthetic routes reported for pynegabine, both starting from 2,6-dimethylaniline (1, Scheme 1). The first-generation route prepared pynegabine at the milligram scale with a 30% total yield in 10 steps, out of which four steps applied column chromatography for purification [17,18]. Since this route is not feasible for large-scale production, a second-generation route was developed, achieving the synthesis of 10 g of pynegabine with a 30% total yield over six steps [17]. Although the latter synthetic route eliminated the unfavorable chromatographic purification procedure, the requirements for hazardous reagents and conditions such as highly toxic methyl chloroformate [19] for the preparation of compound 11 and the dangerous manipulation of hydrogen gas [20] still remained. Therefore, the development of a more efficient and safer synthetic route for the manufacture of pynegabine is highly desirable. Herein, we describe a concise, three-step synthetic route for the preparation of pynegabine at the 10 g scale with easily handling reagents and conditions.
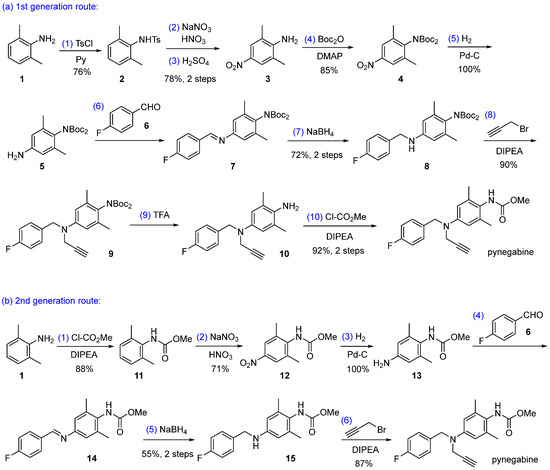
Scheme 1.
Previously reported synthetic routes for pynegabine. Ts = p-toluenesulfonyl; Boc = tert-butoxycarbonyl; DMAP = 4-(dimethylamino)pyridine; DIPEA = diisopropylethylamine.
2. Results and Discussions
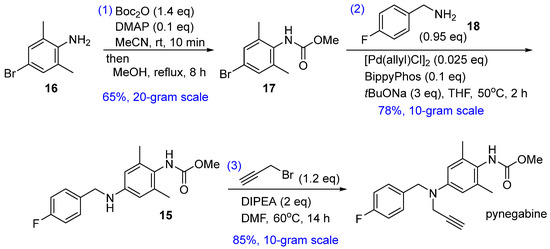
Our proposed new route is shown in Scheme 2. The core reaction of this three-step strategy is the palladium-catalyzed Buchwald–Hartwig cross coupling of bromide 17 and commercially available p-fluorobenzylamine (18). The Buchwald–Hartwig reaction is a highly versatile protocol used to forge aryl C–N bonds [21]. In the present case, this reaction would considerably shorten the synthesis route by replacing the two-step reductive amination (from 13 to 15) and omitting the nitrogen-installation steps (nitration and hydrogenation from 11 to 13). Bromide 17 can be prepared from commercially available 2,6-dimethyl-4-bromoaniline (16), and the known compound 15 can be transformed to pynegabine by a process similar to that applied in the original route (Scheme 1).
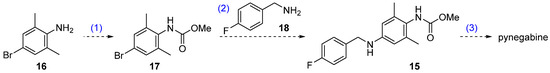
Scheme 2.
Proposed synthetic route for pynegabine.
2.1. Methoxycarbonylation of Aniline 16
To avoid the toxic methyl chloroformate (from 10 to pynegabine and from 1 to 11 in Scheme 1), we applied an unusual method [22] for the methoxycarbonylation of aniline 16 (Table 1). Treatment of 16 with di-tert-butyl dicarbonate (Boc2O) and 4-(dimethylamino)pyridine (DMAP) in MeCN afforded isocyanate 19 within 10 min at room temperature. The addition of MeOH to the reaction mixture, followed by heating to reflux, provided carbamate 17 in full conversion. The purification of 17 was simply achieved without column chromatography through acidic aqueous washing to remove DMAP, followed by recrystallization. Using the 0.1 equivalent of DMAP instead of the stoichiometric amount in the literature [22] did not affect the yield (Table 1, entry 2) and the reaction could be scaled up to 20 g scale (Table 1, entry 3). Notably, in this reaction, MeOH should be added immediately after the complete formation of 19 because of the easy oligomerization property of isocyanate [23]. We observed a bulk of precipitate (presumably polyisocyanate) when extending the isocyanation step to 30 min before MeOH addition in a preliminary experiment. Additionally, the use of MeOH instead of MeCN at the beginning of synthesis (Table 1, entry 4) resulted in low conversion, thereby confirming the importance of MeCN as the initial solvent.

Table 1.
Methoxycarbonylation of aniline 16.
2.2. Buchwald–Hartwig Cross Coupling of the Compounds 17 and 18
For the key Buchwald–Hartwig cross coupling between the compounds 17 and 18, we initially screened the reaction conditions using allylpalladium (II) chloride dimer as the precatalyst and BrettPhos as the ligand [24]. With Cs2CO3 as the base (Table 2, entries 1–4), ether-type solvent gave the optimal conversion at 95 °C, and a higher yield was obtained with a stronger base (tBuONa) in tetrahydrofuran (THF) at a lower temperature (Table 2, entry 5). Further ligand screening (Table 2, entries 6–12) revealed that BippyPhos [25] was most suitable for this coupling reaction (Table 2, entry 12). However, since substrate 18 and product 15 were both amines, the excess 18 which could not be fully consumed need to be removed by column chromatography (Table 2, entry 13). Therefore, using a slight excess of 17 was tested. Under this condition, 18 was completely consumed and pure 15 was successfully obtained through acid–base extraction and recrystallization without column chromatography (Table 2, entry 14). Further optimization of the reaction temperature and time (Table 2, entry 15) showed that a slightly higher temperature (50 °C) gave better yield and reduced the reaction time to a few hours. Scale-up of the reaction to decagram levels maintained the yield level (Table 2, entries 16 and 17).
Table 2.
Screening of the reaction conditions for Buchwald–Hartwig cross coupling of 17 and 18.
Interestingly, 1H-NMR spectra of compound 15 revealed two sets of peaks (integration ratio: ~2:1) for OMe and amide NH groups in CDCl3 (Figure 2a). This might be due to the slow rotation of the hindered carbamate C–N bond [26]. Changing the solvent to d6-DMSO altered the integration ratio to ~4:1, confirming its origination from the cis-trans isomerization of the amide group. The 13C-NMR spectra also showed a similar peak pattern (see Supporting Information).
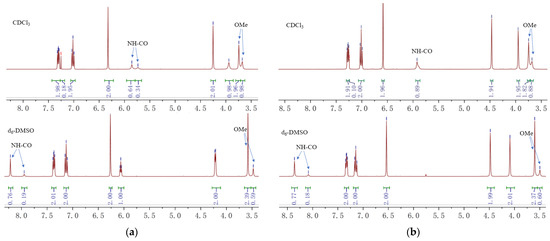
Figure 2.
Localized 1H-NMR spectra (400 MHz) of the compounds 15 (a) and pynegabine (b).
2.3. Propargylation of Compound 15
The third step in pynegabine synthesis involves the propargylation of compound 15. We reproduced this reaction successfully at a 10 g scale with an 85% yield using a similar procedure as that reported previously (Scheme 1b). Additionally, we found that the target pynegabine also showed amide-isomeric peaks in the NMR spectra using CDCl3 and d6-DMSO as the solvents (Figure 2b).
3. Materials and Methods
3.1. General Information
Reagents were used as received from commercial suppliers unless otherwise indicated. Reaction solvents (MeCN, MeOH, THF, DMF) were purchased as anhydrous solvents (H2O ≤ 50 ppm) and stored with active molecular sieves 3A or 4A under Ar. All solvents for work-up procedures were used as received. Analytical thin-layer chromatography (TLC) was performed using glass TLC plates (silica gel HSGF254 plates, Yantai Huayang New Material Technology Co., Ltd., Yantai, China). Visualization was accomplished with UV light or staining with 5% (w/v) phosphomolybdic acid/EtOH followed by heating.
Melting points were uncorrected and measured with a Yanaco MP-J3 melting point apparatus. 1H- and 13C-NMR spectrum were acquired on a Bruker AVANCE III-400 spectrometer. Chemical shifts are indicated in parts per million (ppm) downfield from tetramethylsilane (TMS, δ = 0.00) with residual undeuterated solvent peaks used as internal references for 1H-NMR and deuterated solvent peak shifts for 13C-NMR. Multiplicities are reported as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br (broad), or combinations of those. Mass spectra were generated using electrospray ionization (ESI) and measured on a Thermo-Fisher Accela liquid chromatography system coupled with an Exactive Plus Orbitrap mass spectrometer.
3.2. Synthesis of Methyl (4-Bromo-2,6-dimethylphenyl)carbamate (17)
DMAP (1.22 g, 10.0 mmol) and 4-bromo-2,6-dimethylaniline (compound 16, 20.0 g, 100 mmol) were successively added to a solution of Boc2O (30.5 g, 140 mmol) and MeCN (200 mL) in a 500-mL flask at room temperature under an Ar atmosphere. The reaction mixture was stirred for 10 min, followed by the addition of MeOH (50 mL) and reflux at 82 °C for 8 h. After cooling to room temperature, the reaction mixture was concentrated under reduced pressure. CH2Cl2 (40 mL) was then added to the solid residue, and the organic phase was washed with 1 mol/L hydrochloric acid (40 mL × 3), dried with Na2SO4, filtered, and concentrated under reduced pressure. The solid residue was then dissolved in CH2Cl2 (60 mL), and petroleum ether (150 mL) was added dropwise under stirring to induce precipitation. The precipitate was filtered to obtain compound 17 (16.90 g, 65% yield) as a white solid. m.p. 108.0–109.5 °C. 1H-NMR (400 MHz, CDCl3) δ 7.22 (s, 2H), 5.97 (s, 1H), 3.76 (s, 3H), 2.23 (s, 6H). 13C-NMR (101 MHz, CDCl3) δ 154.7, 138.0 (2C), 132.7, 131.0 (2C), 120.7, 52.7, 18.2 (2C). HRMS (ESI): m/z 258.0126 [M+H]+ (calcd for C10H13BrNO2+: 258.0124).
3.3. Synthesis of Methyl (4-(4-Fluorobenzylamino)-2,6-dimethylphenyl)carbamate (15)
THF (94 mL) and 4-fluorobenzylamine (compound 18, 5.26 mL, 5.76 g, 46.0 mmol) were successively added to the mixture of compound 17 (12.5 g, 48.4 mmol), [Pd(allyl)Cl]2 (0.443 g, 1.21 mmol), and BippyPhos (2.45 g, 4.83 mmol) in a 350 mL flask at room temperature under an Ar atmosphere. The reaction mixture was stirred for 10 min, followed by the addition of tBuONa (1 mol/L in THF, 143 mL, 143 mmol) and stirring at 50 °C for 2 h. After cooling to room temperature, 36% hydrochloric acid (7 mL) was added to adjust the pH to 6, and the mixture was concentrated under reduced pressure. Methyl tert-butyl ether (100 mL) was added to the solid residue, and the organic phase was extracted with 0.1 mol/L hydrochloric acid (100 mL × 15). The combined aqueous layer was then adjusted to pH 13 with 1 mol/L aqueous NaOH and extracted with CH2Cl2 (750 mL × 2). The combined organic layer was then dried with Na2SO4, filtered, and concentrated under reduced pressure. Petroleum ether was added dropwise to the residue under stirring to induce precipitation. The precipitate was filtered to give compound 15 (10.92 g, 78% yield) as a white solid. m.p. 123.6–124.9 °C. 1H-NMR (400 MHz, CDCl3) δ 7.31 (dd, J = 8.8, 5.6 Hz, 2H), 7.02 (dd, J = 8.8, 8.8 Hz, 2H), 6.33 (s, 2H), 5.86 (s, 0.66H), 5.73 (s, 0.34H), 4.25 (s, 2H), 3.94 (s, 1H), 3.74 (s, 2H), 3.68 (s, 1H), 2.16 (s, 6H). 1H-NMR (400 MHz, d6-DMSO) δ 8.22 (s, 0.8H), 7.95 (s, 0.2H), 7.37 (dd, J = 8.4, 5.6 Hz, 2H), 7.13 (dd, J = 8.8, 8.4 Hz, 2H), 6.26 (s, 2H), 6.06 (t, J = 6.0 Hz, 1H), 4.22 (d, J = 6.0 Hz, 2H), 3.58 (s, 2.4H), 3.48 (s, 0.6H), 1.99 (s, 6H). 13C-NMR (101 MHz, CDCl3) δ 162.0 (d, J = 245.1 Hz), 156.8 (minor), 155.6 (major), 147.0, 137.2 (2C), 135.1, 129.0 (d, J = 7.9 Hz, 2C), 124.3 (minor), 123.9 (major), 115.4 (d, J = 21.3 Hz, 2C), 112.3 (major, 2C), 112.0 (minor, 2C), 52.8 (minor), 52.4 (major), 47.6, 18.5 (2C). 13C-NMR (101 MHz, d6-DMSO) δ 161.0 (d, J = 241.4 Hz), 155.8 (minor), 155.4 (major), 146.8, 136.5 (d, J = 2.9 Hz), 136.0 (major, 2C), 135.8 (minor, 2C), 128.9 (d, J = 7.9 Hz, 2C), 124.1 (minor), 123.8 (major), 115.0 (d, J = 21.2 Hz, 2C), 111.4 (2C), 51.6 (minor), 51.4 (major), 45.6, 18.3 (2C). HRMS (ESI): m/z 303.1508 [M + H]+ (calcd for C17H20FN2O2+: 303.1503).
3.4. Synthesis of Pynegabine
Diisopropylethylamine (12.1 mL, 8.98 g, 69.5 mmol) and propargyl bromide (3.59 mL, 4.95 g, 41.6 mmol) were successively added to a solution of compound 15 (10.5 g, 34.7 mmol) and DMF (84 mL) in a 500 mL flask at room temperature under an Ar atmosphere. The reaction mixture was then stirred at 60 °C for 14 h and after cooling to room temperature, water (350 mL) was added to induce precipitation. Filtration gave a solid residue that was dried and dissolved in CH2Cl2 (30 mL). Petroleum ether (90 mL) was added dropwise under stirring to induce precipitation, and the precipitate was filtered to give pynegabine (10.02 g, 85% yield) as a white solid. m.p. 136.2–137.6 °C. 1H-NMR (400 MHz, CDCl3) δ 7.27 (dd, J = 8.8, 5.2 Hz, 2H), 7.01 (dd, J = 8.8, 8.8 Hz, 2H), 6.59 (s, 2H), 5.92 (br, 1H), 4.47 (s, 2H), 3.95 (s, 2H), 3.75 (s, 2H), 3.68 (s, 1H), 2.20 (brs, 7H). 1H-NMR (400 MHz, d6-DMSO) δ 8.36 (s, 0.8H), 8.08 (s, 0.2H), 7.33 (dd, J = 8.8, 6.0 Hz, 2H), 7.15 (dd, J = 8.8, 8.8 Hz, 2H), 6.54 (s, 2H), 4.48 (s, 2H), 4.09 (s, 2H), 3.60 (s, 2.4H), 3.50 (s, 0.6H), 3.14 (s, 1H), 2.06 (s, 6H). 13C-NMR (101 MHz, CDCl3) δ 162.0 (d, J = 244.9 Hz), 156.7 (minor), 155.5 (major), 147.6, 136.9 (2C), 134.0, 128.8 (d, J = 7.9 Hz, 2C), 125.2 (minor), 124.9 (major), 115.4 (d, J = 21.3 Hz, 2C), 113.9 (major, 2C), 113.6 (minor, 2C), 79.5, 72.3, 54.3, 52.8 (minor), 52.4 (major), 39.7, 18.8 (2C). 13C-NMR (101 MHz, d6-DMSO) δ 161.2 (d, J = 242.1 Hz), 155.7 (minor), 155.2 (major), 146.1, 136.0 (major, 2C), 135.9 (minor, 2C), 134.9 (d, J = 2.6 Hz), 129.0 (d, J = 8.0 Hz, 2C), 125.8 (minor), 125.5 (major), 115.1 (d, J = 21.2 Hz, 2C), 113.2 (2C), 80.5, 74.6, 53.4, 51.7 (minor), 51.5 (major), 40.1, 18.5 (2C). Elemental analysis calcd for C20H21FN2O2: C, 70.57; H, 6.22; N, 8.23. Found: C, 69.61; H, 6.09; N, 8.23. HRMS (ESI): m/z 341.1667 [M + H]+ (calcd for C20H22FN2O2+: 341.1660).
4. Conclusions
We successfully developed a concise, three-step route for the decagram-scale synthesis of the AED candidate pynegabine (Scheme 3). This route provided a 41% total yield without column chromatographic purification and avoided the original dangerous manipulation of methyl chloroformate and hydrogen gas, thereby making this method more suitable for pynegabine manufacture in the subsequent phases of clinical trials and future marketing.
Scheme 3.
Successful and scalable three-step route for synthesizing pynegabine. DMF = N,N-dimethylformamide.
5. Patents
S.X., Y.-J.S., S.-P.Z., Y.-L.G. and S.-C.L. are inventors on patent application 202310142629.9 submitted by the Institute of Materia Medica, Chinese Academy of Medical Sciences, that covers a part of the work of this manuscript.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/molecules28134888/s1, 1H- and 13C-NMR spectra of compounds 17 (CDCl3), 15 (CDCl3 and d6-DMSO), and pynegabine (CDCl3 and d6-DMSO).
Author Contributions
Conceptualization, S.-P.Z. and S.X.; methodology, S.-P.Z. and S.X.; validation, Y.-L.G. and S.-P.Z.; investigation, Y.-J.S. and S.-P.Z.; writing—original draft preparation, Y.-J.S. and S.X.; writing—review and editing, Y.-L.G. and S.-C.L.; supervision and project administration, S.X.; funding acquisition, S.-C.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by Beijing natural science foundation (No. 2232032) and the CAMS Innovation Fund for Medical Sciences (CIFMS, 2021-I2M-1-054).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the Materials and Methods section and the Supplementary Material of this article.
Acknowledgments
We are thankful to Haibo Yu of the Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College for helpful discussion.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds 15, 17, and pynegabine are available from the authors.
References
- World Health Organization. Epilepsy, 9 February 2023. Available online: https://www.who.int/en/news-room/fact-sheets/detail/epilepsy (accessed on 27 May 2023).
- Elkommos, S.; Mula, M. Current and future pharmacotherapy options for drug-resistant epilepsy. Expert Opin. Pharmacother. 2022, 23, 2023–2034. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, K.; Chu, Y.; Li, C.; Zhang, T.; Liu, P.; Sun, T.; Jiang, C. ROS-removing nano-medicine for navigating inflammatory microenvironment to enhance anti-epileptic therapy. Acta Pharm. Sin. B 2023, 13, 1246–1261. [Google Scholar] [CrossRef] [PubMed]
- Stafstrom, C.E.; Grippon, S.; Kirkpatrick, P. Ezogabine (retigabine). Nat. Rev. Drug Discov. 2011, 10, 729–730. [Google Scholar] [CrossRef]
- Clark, S.; Antell, A.; Kaufman, K. New antiepileptic medication linked to blue discoloration of the skin and eyes. Ther. Adv. Drug Saf. 2015, 6, 15–19. [Google Scholar] [CrossRef] [PubMed]
- GlaxoSmithKline: Trobalt®/Potiga® Discontinuation—Important Reminder. Available online: https://www.ilae.org/files/dmfile/GSK_RetigabineTrobalt-Reminder.pdf (accessed on 27 May 2023).
- Groseclose, M.R.; Castellino, S. An investigation into retigabine (ezogabine) associated dyspigmentation in rat eyes by MALDI imaging mass spectrometry. Chem. Res. Toxicol. 2019, 32, 294–303. [Google Scholar] [CrossRef]
- Bock, C.; Link, A. How to replace the lost keys? Strategies toward safer KV7 channel openers. Future Med. Chem. 2019, 11, 337–355. [Google Scholar] [CrossRef]
- Borgini, M.; Mondal, P.; Liu, R.; Wipf, P. Chemical modulation of Kv7 potassium channels. RSC Med. Chem. 2021, 12, 483–537. [Google Scholar] [CrossRef]
- Hernandez, C.C.; Tarfa, R.A.; Limcaoco, J.M.I.; Liu, R.; Mondal, P.; Hill, C.; Duncan, R.K.; Tzounopoulos, T.; Stephenson, C.R.J.; O’Meara, M.J.; et al. Development of an automated screen for Kv7.2 potassium channels and discovery of a new agonist chemotype. Bioorg. Med. Chem. Lett. 2022, 71, 128841. [Google Scholar] [CrossRef]
- Liu, S.; Guo, P.; Wang, K.; Zhang, S.; Li, Y.; Shen, J.; Mei, L.; Ye, Y.; Zhang, Q.; Yang, H. General pharmacological activation mechanism of K+ channels bypassing channel gates. J. Med. Chem. 2022, 65, 10285–10299. [Google Scholar] [CrossRef]
- Musella, S.; Carotenuto, L.; Iraci, N.; Baroli, G.; Ciaglia, T.; Nappi, P.; Basilicata, M.G.; Salviati, E.; Barrese, V.; Vestuto, V.; et al. Beyond retigabine: Design, synthesis, and pharmacological characterization of a potent and chemically stable neuronal Kv7 channel activator with anticonvulsant activity. J. Med. Chem. 2022, 65, 11340–11364. [Google Scholar] [CrossRef]
- Wang, T.; Krauss, G.L. XEN1101: A novel potassium channel modulator for the potential treatment of focal epilepsy in adults. Touchrev. Neurol. 2022, 18, 2–4. [Google Scholar] [CrossRef]
- Wurm, K.W.; Bartz, F.-M.; Schulig, L.; Bodtke, A.; Bednarski, P.J.; Link, A. Modifications of the triaminoaryl metabophore of flupirtine and retigabine aimed at avoiding quinone diimine formation. ACS Omega 2022, 7, 7989–8012. [Google Scholar] [CrossRef]
- Wurm, K.W.; Bartz, F.-M.; Schulig, L.; Bodtke, A.; Bednarski, P.J.; Link, A. Carba analogues of flupirtine and retigabine with improved oxidation resistance and reduced risk of quinoid metabolite formation. ChemMedChem 2022, 17, e202200262. [Google Scholar] [CrossRef]
- Wurm, K.W.; Bartz, F.-M.; Schulig, L.; Bodtke, A.; Bednarski, P.J.; Link, A. Replacing the oxidation-sensitive triaminoaryl chemotype of problematic KV7 channel openers: Exploration of a nicotinamide scaffold. Arch. Pharm. 2023, 356, e2200473. [Google Scholar] [CrossRef]
- Nan, F.-J.; Li, M.; Gao, Z.-B.; Zhang, Y.-M.; Hu, H.-N.; Xu, H.-Y.; Liu, H.-N.; Pi, X.-P. Novel KCNQ Potassium Channel Agonist and Preparation Method and Application Thereof. CN201410175315, 29 June 2018. [Google Scholar]
- Zhang, Y.-M.; Xu, H.-Y.; Hu, H.-N.; Tian, F.-Y.; Chen, F.; Liu, H.-N.; Zhan, L.; Pi, X.-P.; Liu, J.; Gao, Z.-B.; et al. Discovery of HN37 as a potent and chemically stable antiepileptic drug candidate. J. Med. Chem. 2021, 64, 5816–5837. [Google Scholar] [CrossRef]
- Rudén, C.; Hansson, S.O. How accurate are the European Union’s classifications of chemical substances. Toxicol. Lett. 2003, 144, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Hachoose, R.C. How to handle hydrogen in process plants. Chem. Eng. 2006, 113, 54–59. [Google Scholar]
- Heravi, M.M.; Zadsirjan, V.; Malmir, M.; Mohammadi, L. Buchwald–Hartwig reaction: An update. Monatsh. Chem. 2021, 152, 1127–1171. [Google Scholar] [CrossRef]
- Knölker, H.-J.; Braxmeier, T. Isocyanates—Part 3. Synthesis of carbamates by DMAP-catalyzed reaction of amines with di-tert-butyldicarbonate and alcohols. Tetrahedron Lett. 1996, 37, 5861–5864. [Google Scholar] [CrossRef]
- Bak, I.G.; Chae, C.-G.; Lee, J.-S. Synthetic control of helical polyisocyanates by living anionic polymerization toward peptide mimicry. Macromolecules 2022, 55, 1923–1945. [Google Scholar] [CrossRef]
- Maiti, D.; Fors, B.P.; Henderson, J.L.; Nakamura, Y.; Buchwald, S.L. Palladium-catalyzed coupling of functionalized primary and secondary amines with aryl and heteroaryl halides: Two ligands suffice in most cases. Chem. Sci. 2011, 2, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Singer, R.A. BippyPhos: A highly versatile ligand for Pd-catalyzed C-N, C-O and C-C couplings. Isr. J. Chem. 2020, 60, 294–302. [Google Scholar] [CrossRef]
- Stewart, W.E.; Siddall, T.H., III. Nuclear magnetic resonance studies of amides. Chem. Rev. 1970, 70, 517–551. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).