
Molecular Taphonomy of Heme: Chemical Degradation of Hemin under Presumed Fossilization Conditions

 ,
,  , , and
, , and
Abstract
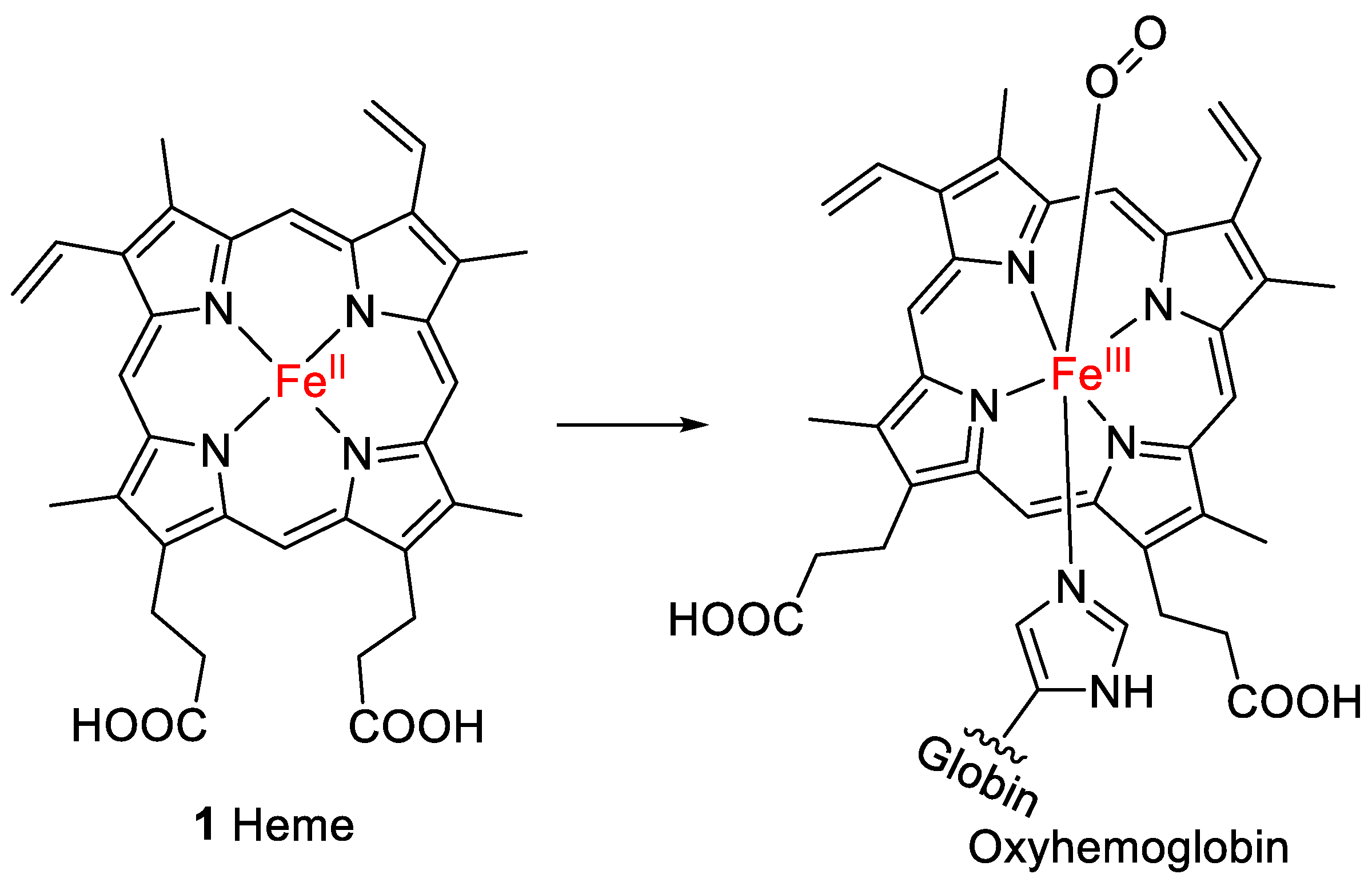
1. Introduction
2. Results and Discussion
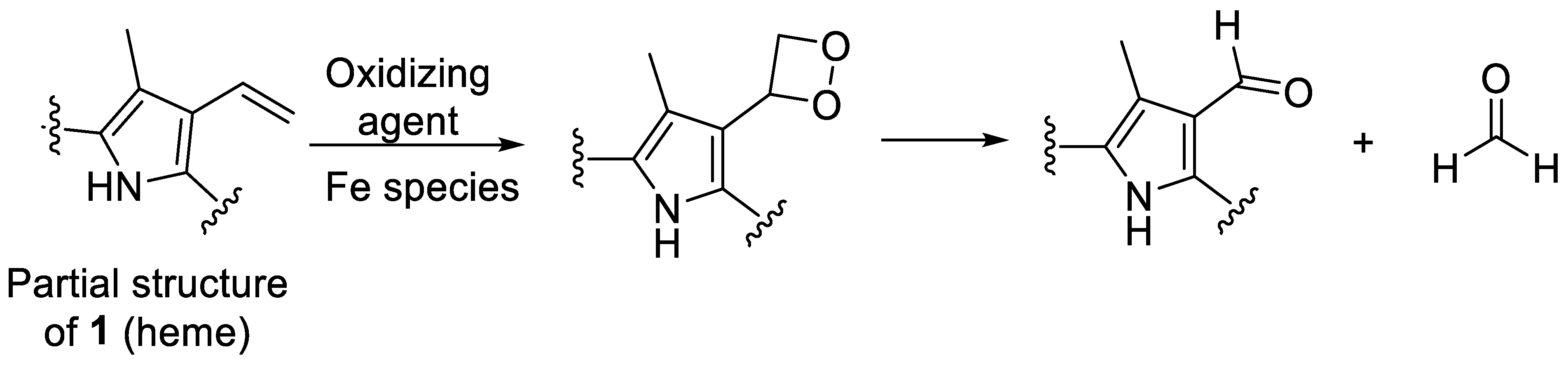
2.1. Oxidative Conditions Lead to Hematinic Acid
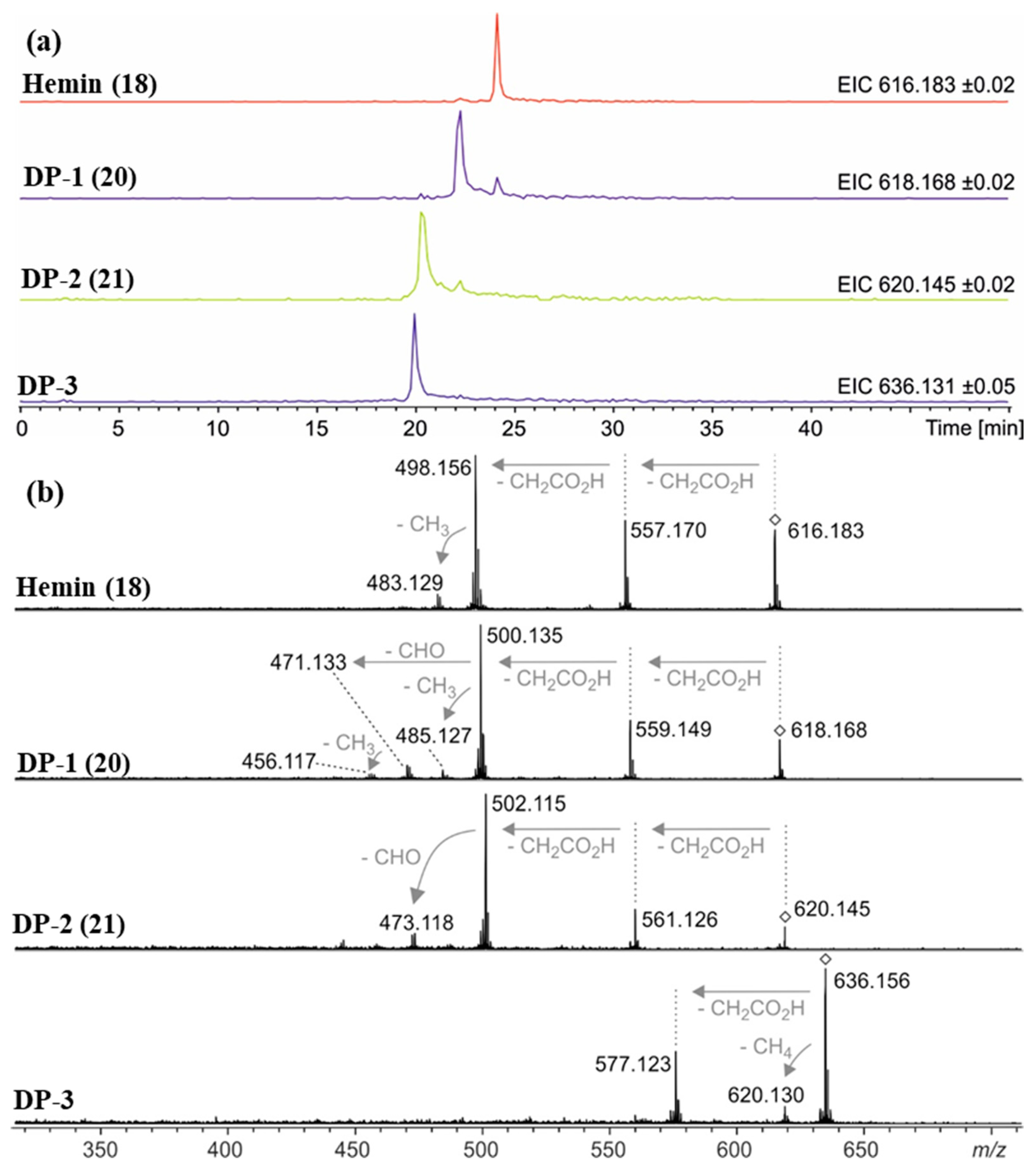
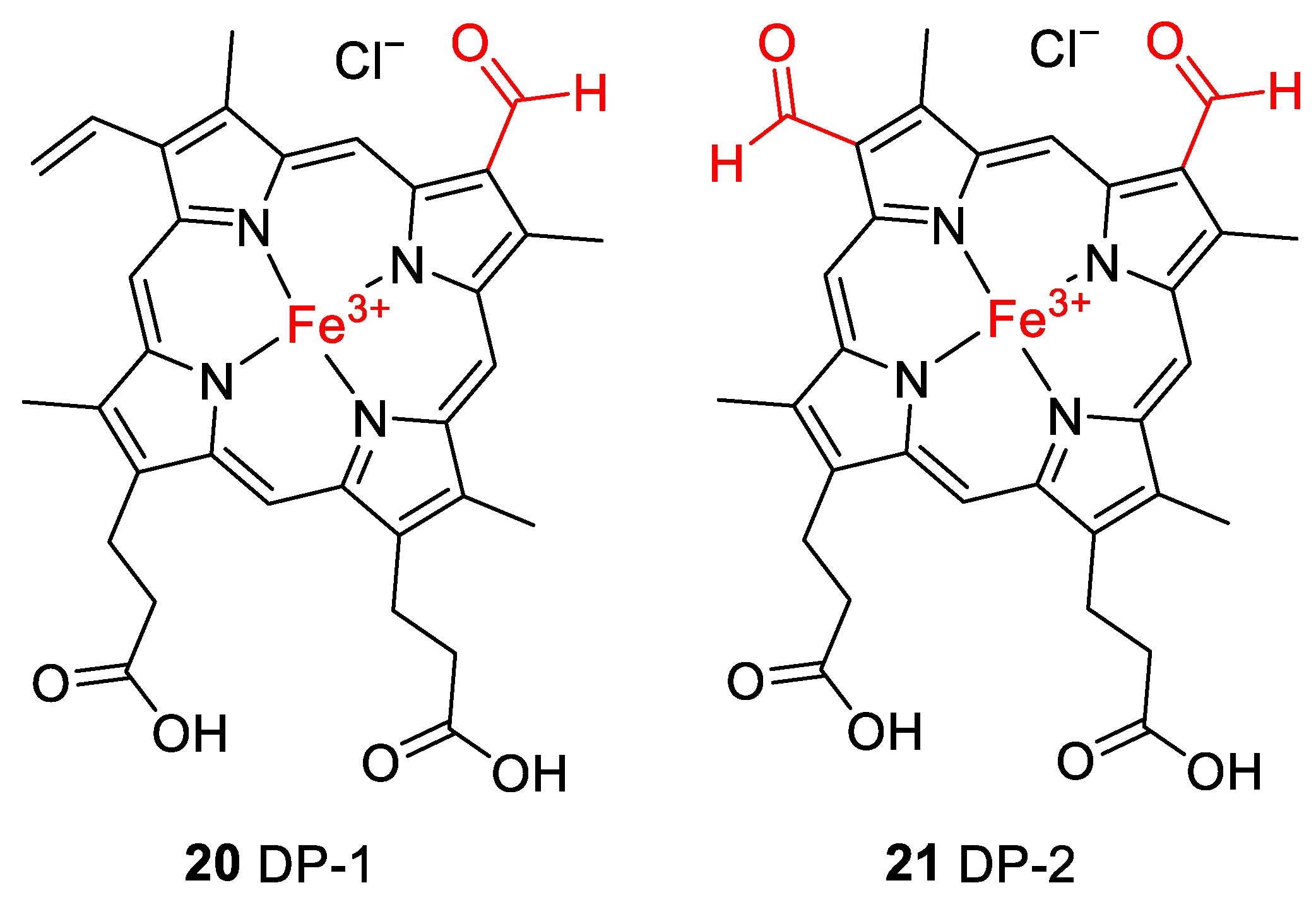
2.2. Thermal Degradation and Structural Elucidation of Intermediate Degradation Products
2.3. Control Experiments Reveal Site of Degradation and Role of Iron
2.4. Reductive Conditions
3. Materials and Methods
3.1. Materials
3.2. Oxidative Degradation (Conditions A–C, Table 1)
3.2.1. Oxidation under Alkaline Methanolic Conditions (Condition A, Table 1)
3.2.2. Oxidation under Alkaline Aqueous Conditions (Condition B, Table 1)
3.2.3. Oxidation at Physiological pH Value (Condition C, Table 1)
3.3. Thermal Degradation
3.3.1. Thermal Degradation at Physiological pH Value (Conditions D–F, Table 1)
3.3.2. Thermal Degradation at Alkaline pH under an Argon Atmosphere (Condition G, Table 1)
3.4. Reductive Conditions (Condition H, Table 1)
3.5. Preparative Reversed-Phase HPLC for Isolation of Hematinic Acid
3.6. Analysis by HPLC-(DAD-UV)-MS
3.7. Analysis by HPLC-Coupled High-Resolution Mass Spectrometry
3.8. Analysis of Hematinic Acid by Nuclear Magnetic Resonance Spectroscopy (NMR)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Giardina, B.; Messana, I.; Scatena, R.; Castagnola, M. The multiple functions of hemoglobin. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 165–196. [Google Scholar] [CrossRef]
- Tahoun, M.; Gee, C.T.; McCoy, V.E.; Sander, P.M.; Müller, C.E. Chemistry of porphyrins in fossil plants and animals. RSC Adv. 2021, 11, 7552–7563. [Google Scholar] [CrossRef]
- Kundu, S.; Trent, J.T.; Hargrove, M.S. Plants, humans and hemoglobins. Trends Plant Sci. 2003, 8, 387–393. [Google Scholar] [CrossRef]
- Yoshida, T.; Migita, C.T. Mechanism of heme degradation by heme oxygenase. J. Inorg. Biochem. 2000, 82, 33–41. [Google Scholar] [CrossRef]
- Kikuchi, G.; Yoshida, T.; Noguchi, M. Heme oxygenase and heme degradation. Biochem. Biophys. Res. Commun. 2005, 338, 558–567. [Google Scholar] [CrossRef]
- Terry, M.J.; Linley, P.J.; Kohchi, T. Making light of it: The role of plant haem oxygenases in phytochrome chromophore synthesis. Biochem. Soc. Trans. 2002, 30, 604–609. [Google Scholar] [CrossRef]
- Cornejo, J.; Beale, S.I. Phycobilin biosynthetic reactions in extracts of cyanobacteria. Photosynth. Res. 1997, 51, 223–230. [Google Scholar] [CrossRef]
- Beale, S.I. Biosynthesis of phycobilins. Chem. Rev. 1993, 93, 785–802. [Google Scholar] [CrossRef]
- Pendrak, M.L.; Chao, M.P.; Yan, S.S.; Roberts, D.D. Heme oxygenase in Candida albicans is regulated by hemoglobin and is necessary for metabolism of exogenous heme and hemoglobin to α-biliverdin. J. Biol. Chem. 2004, 279, 3426–3433. [Google Scholar] [CrossRef]
- Schmitt, M.P. Utilization of host iron sources by Corynebacterium diphtheriae: Identification of a gene whose product is homologous to eukaryotic heme oxygenases and is required for acquisition of iron from heme and hemoglobin. J. Bacteriol. 1997, 179, 838–845. [Google Scholar] [CrossRef]
- Wilks, A.; Schmitt, M.P. Expression and characterization of a heme oxygenase (Hmu O) from Corynebacterium diphtheriae. J. Biol. Chem. 1998, 273, 837–841. [Google Scholar] [CrossRef] [PubMed]
- Melanie, R.; Wenming, Z.; Rahul, D.; Angela, W.; Igor, S. Homologues of neisserial heme oxygenase in Gram-negative bacteria: Degradation of heme by the product of the pigA gene of Pseudomonas aeruginosa. J. Bacteriol. 2001, 183, 6394–6403. [Google Scholar] [CrossRef]
- Zhu, W.; Wilks, A.; Stojiljkovic, I. Degradation of heme in Gram-negative bacteria: The product of the hemO gene of neisseriae is a heme oxygenase. J. Bacteriol. 2000, 182, 6783–6790. [Google Scholar] [CrossRef]
- Zhu, W.; Hunt, D.J.; Richardson, A.R.; Stojiljkovic, I. Use of heme compounds as iron sources by pathogenic neisseriae requires the product of the hemO gene. J. Bacteriol. 2000, 182, 439–447. [Google Scholar] [CrossRef]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef]
- Bonnett, R.; McDonagh, A.F. The meso-reactivity of porphyrins and related compounds. Part VI. Oxidative cleavage of the haem system. The four isomeric biliverdins of the IX series. J. Chem. Soc. Perkin Trans. 1 1973, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Giulivi, C.; Cadenas, E. The reaction of ascorbic acid with different heme iron redox states of myoglobin. FEBS Lett. 1993, 332, 287–290. [Google Scholar] [CrossRef]
- Schaefer, W.H.; Harris, T.M.; Guengerich, F.P. Characterization of the enzymic and nonenzymic peroxidative degradation of iron porphyrins and cytochrome P-450 heme. Biochemistry 1985, 24, 3254–3263. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Destruction of heme and hemoproteins mediated by liver microsomal reduced nicotinamide adenine dinucleotide phosphate-cytochrome P-450 reductase. Biochemistry 1978, 17, 3633–3639. [Google Scholar] [CrossRef]
- Ouellet, Y.H.; Ndiaye, C.T.; Gagné, S.M.; Sebilo, A.; Suits, M.D.L.; Jubinville, É.; Jia, Z.; Ivancich, A.; Couture, M. An alternative reaction for heme degradation catalyzed by the Escherichia coli O157:H7 ChuS protein: Release of hematinic acid, tripyrrole and Fe(III). J. Inorg. Biochem. 2016, 154, 103–113. [Google Scholar] [CrossRef]
- Groves, J.T.; Haushalter, R.C.; Nakamura, M.; Nemo, T.E.; Evans, B.J. High-valent iron-porphyrin complexes related to peroxidase and cytochrome P-450. J. Am. Chem. Soc. 1981, 103, 2884–2886. [Google Scholar] [CrossRef]
- Takahashi, A.; Kurahashi, T.; Fujii, H. Redox potentials of oxoiron(IV) porphyrin π-cation radical complexes: Participation of electron transfer process in oxygenation reactions. Inorg. Chem. 2011, 50, 6922–6928. [Google Scholar] [CrossRef]
- Ritter, M.; Oetama, V.S.P.; Schulze, D.; Muetzlaff, K.; Meents, A.K.; Seidel, R.A.; Görls, H.; Westerhausen, M.; Boland, W.; Pohnert, G. Pyrrolic and dipyrrolic chlorophyll degradation products in plants and herbivores. Chem.-Eur. J. 2020, 26, 6205–6213. [Google Scholar] [CrossRef] [PubMed]
- Lightner, D.A.; Quistad, G.B. Hematinic acid and propentdyopents from bilirubin photo-oxidation in vitro. FEBS Lett. 1972, 25, 94–96. [Google Scholar] [CrossRef] [PubMed]
- Lightner, D.A.; Linnane, W.P.; Ahlfors, C.E. Bilirubin photooxidation products in the urine of jaundiced neonates receiving phototherapy. Pediatr. Res. 1984, 18, 696–700. [Google Scholar] [CrossRef]
- Rüdiger, W. Recent chemistry and biochemistry of bile pigments. Angew. Chem. Int. Ed. Engl. 1970, 9, 473–480. [Google Scholar] [CrossRef]
- Muir, H.M.; Neuberger, A. The biogenesis of porphyrins. 2. The origin of the methyne carbon atoms. Biochem. J. 1950, 47, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Sasaki, K. Production of hematinic acid by the reaction of hemoglobin with phenylhydrazine: Evidence for the oxidative cleavage of heme. Biol. Pharm. Bull. 1994, 17, 856–858. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Shioi, Y. Detection of chlorophyll breakdown products in the senescent leaves of higher plants. Plant Cell Physiol. 1999, 40, 909–915. [Google Scholar] [CrossRef]
- Schweitzer, M.H.; Marshall, M.; Carron, K.; Bohle, D.S.; Busse, S.C.; Arnold, E.V.; Barnard, D.; Horner, J.R.; Starkey, J.R. Heme compounds in dinosaur trabecular bone. Proc. Natl. Acad. Sci. USA 1997, 94, 6291–6296. [Google Scholar] [CrossRef]
- Greenwalt, D.E.; Goreva, Y.S.; Siljeström, S.M.; Rose, T.; Harbach, R.E. Hemoglobin-derived porphyrins preserved in a Middle Eocene blood-engorged mosquito. Proc. Natl. Acad. Sci. USA 2013, 110, 18496–18500. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, J.; Kuriyama, T.; Madsen, H.; Sjövall, P.; Zheng, W.; Uvdal, P.; Engdahl, A.; Moyer, A.E.; Gren, J.A.; Kamezaki, N.; et al. Biochemistry and adaptive colouration of an exceptionally preserved juvenile fossil sea turtle. Sci. Rep. 2017, 7, 13324. [Google Scholar] [CrossRef]
- Wiemann, J.; Yang, T.R.; Sander, P.N.; Schneider, M.; Engeser, M.; Kath-Schorr, S.; Müller, C.E.; Sander, P.M. Dinosaur origin of egg color: Oviraptors laid blue-green eggs. PeerJ 2017, 5, e3706. [Google Scholar] [CrossRef] [PubMed]
- Wiersma, K.; Läbe, S.; Sander, P.M. Organic phase preservation in fossil dinosaur and other tetrapod bone from deep time. In Fossilization: Understanding the Material Nature of Ancient Plants and Animals; Gee, C.T., McCoy, V.E., Sander, P.M., Eds.; Johns Hopkins University Press: Baltimore, MD, USA, 2021; pp. 16–45. ISBN 9781421440217. [Google Scholar]
- Schweitzer, M.H.; Wittmeyer, J.L.; Horner, J.R. Soft tissue and cellular preservation in vertebrate skeletal elements from the Cretaceous to the present. Proc. R. Soc. B Biol. Sci. 2007, 274, 183–197. [Google Scholar] [CrossRef]
- Schweitzer, M.H. Soft tissue preservation in terrestrial Mesozoic vertebrates. Annu. Rev. Earth Planet. Sci. 2011, 39, 187–216. [Google Scholar] [CrossRef]
- Tahoun, M.; Engeser, M.; Namasivayam, V.; Sander, P.M.; Müller, C.E. Chemistry and analysis of organic compounds in dinosaurs. Biology 2022, 11, 670. [Google Scholar] [CrossRef]
- Behrensmeyer, A.K.; Kidwell, S.M. Taphonomy’s contributions to paleobiology. Paleobiology 1985, 11, 105–119. [Google Scholar] [CrossRef]
- Behrensmeyer, A.K.; Kidwell, S.M.; Gastaldo, R.A. Taphonomy and paleobiology. Paleobiology 2000, 26, 103–147. [Google Scholar] [CrossRef]
- Gifford, D.P. Taphonomy and paleoecology: A critical review of archaeology’s sister disciplines. In Advances in Archaeological Method and Theory; Academic Press: New York, NY, USA, 1981; pp. 365–438. [Google Scholar]
- Schweitzer, M.H.; Zheng, W.; Cleland, T.P.; Goodwin, M.B.; Boatman, E.; Theil, E.; Marcus, M.A.; Fakra, S.C. A role for iron and oxygen chemistry in preserving soft tissues, cells and molecules from deep time. Proc. R. Soc. B Biol. Sci. 2014, 281, 20132741. [Google Scholar] [CrossRef] [PubMed]
- Eglinton, G.; Logan, G.A. Molecular preservation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1991, 333, 315–328. [Google Scholar] [CrossRef]
- Latham, K.E.; Miller, J.J. DNA recovery and analysis from skeletal material in modern forensic contexts. Forensic Sci. Res. 2019, 4, 51–59. [Google Scholar] [CrossRef]
- Retallack, G.J. Organisms. In Soils of the Past: An Introduction to Paleopedology, 2nd ed.; Blackwell Science Ltd.: Oxford, UK, 2001; pp. 128–159. [Google Scholar]
- Pfretzschner, H.-U. Fossilization of Haversian bone in aquatic environments. Comptes Rendus Palevol 2004, 3, 605–616. [Google Scholar] [CrossRef]
- Von Endt, D.W.; Ortner, D.J. Experimental effects of bone size and temperature on bone diagenesis. J. Archaeol. Sci. 1984, 11, 247–253. [Google Scholar] [CrossRef]
- Briggs, D.E.G.; McMahon, S. The role of experiments in investigating the taphonomy of exceptional preservation. Palaeontology 2016, 59, 1–11. [Google Scholar] [CrossRef]
- Brynjelsen, S.E.; Doty, M.; Poss, M.J. Facile synthesis of hematinic acid. Synth. Commun. 1998, 28, 1885–1889. [Google Scholar] [CrossRef]
- Tomat, E. Propentdyopents: Brief history of a family of dipyrrolic pigments. J. Porphyr. Phthalocyanines 2019, 23, 1265–1272. [Google Scholar] [CrossRef]
- Charkin, O.P.; Klimenko, N.M.; Nguyen, P.T.; Charkin, D.O.; Mebel, A.M.; Lin, S.H.; Wang, Y.-S.; Wei, S.-C.; Chang, H.-C. Fragmentation of heme and hemin+ with sequential loss of carboxymethyl groups: A DFT and mass-spectrometry study. Chem. Phys. Lett. 2005, 415, 362–369. [Google Scholar] [CrossRef]
- Siegert, S.W.K.; Holt, R.J. Physicochemical properties, pharmacokinetics, and pharmacodynamics of intravenous hematin: A literature review. Adv. Ther. 2008, 25, 842–857. [Google Scholar] [CrossRef]
- Rothschild, M.-L.; Cosi, L.; Myers, L.S. Effect of gamma-radiation on ferriprotoporphyrin. Nature 1958, 182, 316. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.H.; Kenner, G.W.; Wass, J. Pyrroles and related compounds. Part XXV. Pemptoporphyrin, isopemptoporphyrin, and chlorocruoroporphyrin (Spirographis porphyrin). J. Chem. Soc. Perkin Trans. 1 1974, 480–490. [Google Scholar] [CrossRef]
- Inhoffen, H.H.; Bliesener, C.; Brockmann, H. Zur weiteren Kenntnis des Chlorophylls und des Hämins, VIII.: Umwandlung von Protoporphyrin IX über Photoprotoporphyrin in Spirographis- und Isospirographisporphyrin. Tetrahedron Lett. 1966, 7, 3779–3783. [Google Scholar] [CrossRef]
- Fischer, H.; Wecker, G. Synthese des Spirographisporphyrins. Hoppe. Seylers. Z. Physiol. Chem. 1942, 272, 1–22. [Google Scholar] [CrossRef]
- Fischer, H.; Deilmann, K.-O. Überführung von Hämin IX in Spirographisporphyrin und über einige Derivate des Deuteroporphyrins. Hoppe. Seylers. Z. Physiol. Chem. 1944, 280, 186–216. [Google Scholar] [CrossRef]
- Tsubaki, M.; Nagai, K.; Kitagawa, T. Resonance Raman spectra of myoglobins reconstituted with spirographis and isospirographis hemes and iron 2, 4-diformylprotoporphyrin. Biochemistry 1980, 19, 379–385. [Google Scholar] [CrossRef]
- Sono, M.; Asakura, T. Separation and properties of spirographis and isospirographis porphyrin dimethyl esters. Biochemistry 1974, 13, 4386–4394. [Google Scholar] [CrossRef]
- Inhoffen, H.H.; Brockmann, H., Jr.; Bliesener, K.-M. Zur weiteren Kenntnis des Chlorophylls und des Hämins, XXX Photoprotoporphyrine und ihre Umwandlung in Spirographis-sowie Isospirographis-porphyrin). Justus Liebigs Ann. Chem. 1969, 730, 173–185. [Google Scholar] [CrossRef]
- Horsey, B.E.; Whitten, D.G. Photochemical reactions in organized monolayer assemblies. 8. Environmental effects on photochemical reactions: Contrasts in the photooxidation behavior of protoporphyrin IX in solution, monolayer films, organized monolayer assemblies, and micelles. J. Am. Chem. Soc. 1978, 100, 1293–1295. [Google Scholar] [CrossRef]
- Drabkin, D.L. Spectrophotometric studies: X. Structural interpretation of the spectra of cyanide, pyridine, and carbon monoxide derivatives of cytochrome c and hemoglobin. J. Biol. Chem. 1942, 146, 605–617. [Google Scholar] [CrossRef]
- Rothschild, M.-L.; Myers, L.S. The spontaneous change of ferriprotoporphyrin in alkaline solution. Nature 1958, 182, 1671–1672. [Google Scholar] [CrossRef] [PubMed]
- Rothschild, M.-L. The reaction of ferriprotoporphyrin with hydrogen peroxide in alkaline solutions. Arch. Biochem. Biophys. 1960, 90, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.; Deilmann, K.-O. Überführung von Hämin in Deuteroporphyrin-2,4-dicarbonsäure-tetramethylester und von Hämatoporphyrin in Diacetyl-deuteroporphyrin. Justus Liebigs Ann. Chem. 1940, 545, 22–27. [Google Scholar] [CrossRef]
- Cox, G.S.; Whitten, D.G. Mechanisms for the photooxidation of protoporphyrin IX in solution. J. Am. Chem. Soc. 1982, 104, 516–521. [Google Scholar] [CrossRef]
- Gonzalez-de-Castro, A.; Xiao, J. Green and efficient: Iron-catalyzed selective oxidation of olefins to carbonyls with O2. J. Am. Chem. Soc. 2015, 137, 8206–8218. [Google Scholar] [CrossRef]
- Li, X.; Liu, T.; Li, F.; Zhang, W.; Zhou, S.; Li, Y. Reduction of structural Fe(III) in oxyhydroxides by Shewanella decolorationis S12 and characterization of the surface properties of iron minerals. J. Soils Sediments 2012, 12, 217–227. [Google Scholar] [CrossRef]
- Yan, B.; Wrenn, B.A.; Basak, S.; Biswas, P.; Giammar, D.E. Microbial reduction of Fe(III) in hematite nanoparticles by Geobacter sulfurreducens. Environ. Sci. Technol. 2008, 42, 6526–6531. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Medium | Concentration a | Temperature | Observed Half-Life of Hemin | Identified Degradation Product(s) |
|---|---|---|---|---|---|
| Oxidation under alkaline conditions | |||||
| A | 1% NH3 in methanol + aq. H2O2 (final conc. 5%) | 5 mg/mL hemin | Room temperature | ≤1 min | Hematinic acid (15) |
| B | 1% NH3 in water (pH 10.5) + aq. H2O2 (final conc. 5%) | 5.5 mg/mL hemin | 60 °C for the initial 30 min, then room temperature | ≤1 min | Hematinic acid (15) |
| Oxidation at physiological pH value | |||||
| C | Phosphate-buffered saline (PBS pH 7.4) + aq. H2O2 (final conc. 3%) | 1.3 mg/mL hemin | Room temperature | ≤1 min | Hematinic acid (15) |
| Heating at physiological pH value | |||||
| D | PBS (pH 7.4) in the presence of air | 1.3 mg/mL hemin | 75 °C | 2.6 days | Hematinic acid (15) DP-1 (20) |
| E | PBS (pH 7.4) in the presence of air | 1.3 mg/mL hemin | 95 °C | 0.73 days | Hematinic acid (15) DP-1 (20) |
| F | PBS (pH 7.4) under an argon atmosphere | 1.3 mg/mL hemin | 75 °C | n.d. b | Hematinic acid (15) DP-1 (20) |
| Heating under alkaline conditions | |||||
| G | 0.1N aq. NaOH (pH 8) under an argon atmosphere (control experiments using compounds 18 and 20) | 12 mg/mL hemin, 1.8 mg/mL ferric mesoporphyrin IX (19), or 12 mg/mL protoporphyrin IX (17) | 70 °C | 5.5 days (17: minor degradation; 19: no degradation) | DP-1 (20) DP-2 (21) DP-3 |
| H | 0.1 N aq. NaOH (pH 8) + 0.9% Na2S2O4 under an argon atmosphere | 12 mg/mL hemin | 9.5 days | Unknown degradation product (mass of 650 Da) | |
| Compound a | Molecular Ion (Calculated Mass) | Molecular Formula | Accurate Mass Determination (HRMS) |
|---|---|---|---|
| Hemin (18) | 616.1768 | C34H32N4O4Fe | 616.1785 |
| DP-1 (20) | 618.1561 | C33H30N4O5Fe | 618.1576 |
| DP-2 (21) | 620.1353 | C32H28N4O6Fe | 620.1332 |
| DP-3 | 636.1302 636.1666 | C32H28N4O7Fe or C33H32N4O6Fe | 636.137–636.159 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tahoun, M.; Engeser, M.; Svolacchia, L.; Sander, P.M.; Müller, C.E. Molecular Taphonomy of Heme: Chemical Degradation of Hemin under Presumed Fossilization Conditions. Molecules 2023, 28, 4887. https://doi.org/10.3390/molecules28134887
Tahoun M, Engeser M, Svolacchia L, Sander PM, Müller CE. Molecular Taphonomy of Heme: Chemical Degradation of Hemin under Presumed Fossilization Conditions. Molecules. 2023; 28(13):4887. https://doi.org/10.3390/molecules28134887
Chicago/Turabian StyleTahoun, Mariam, Marianne Engeser, Luca Svolacchia, Paul Martin Sander, and Christa E. Müller. 2023. "Molecular Taphonomy of Heme: Chemical Degradation of Hemin under Presumed Fossilization Conditions" Molecules 28, no. 13: 4887. https://doi.org/10.3390/molecules28134887
APA StyleTahoun, M., Engeser, M., Svolacchia, L., Sander, P. M., & Müller, C. E. (2023). Molecular Taphonomy of Heme: Chemical Degradation of Hemin under Presumed Fossilization Conditions. Molecules, 28(13), 4887. https://doi.org/10.3390/molecules28134887