
Characterization of Metabolic Correlations of Ursodeoxycholic Acid with Other Bile Acid Species through In Vitro Sequential Metabolism and Isomer-Focused Identification

 ,
, 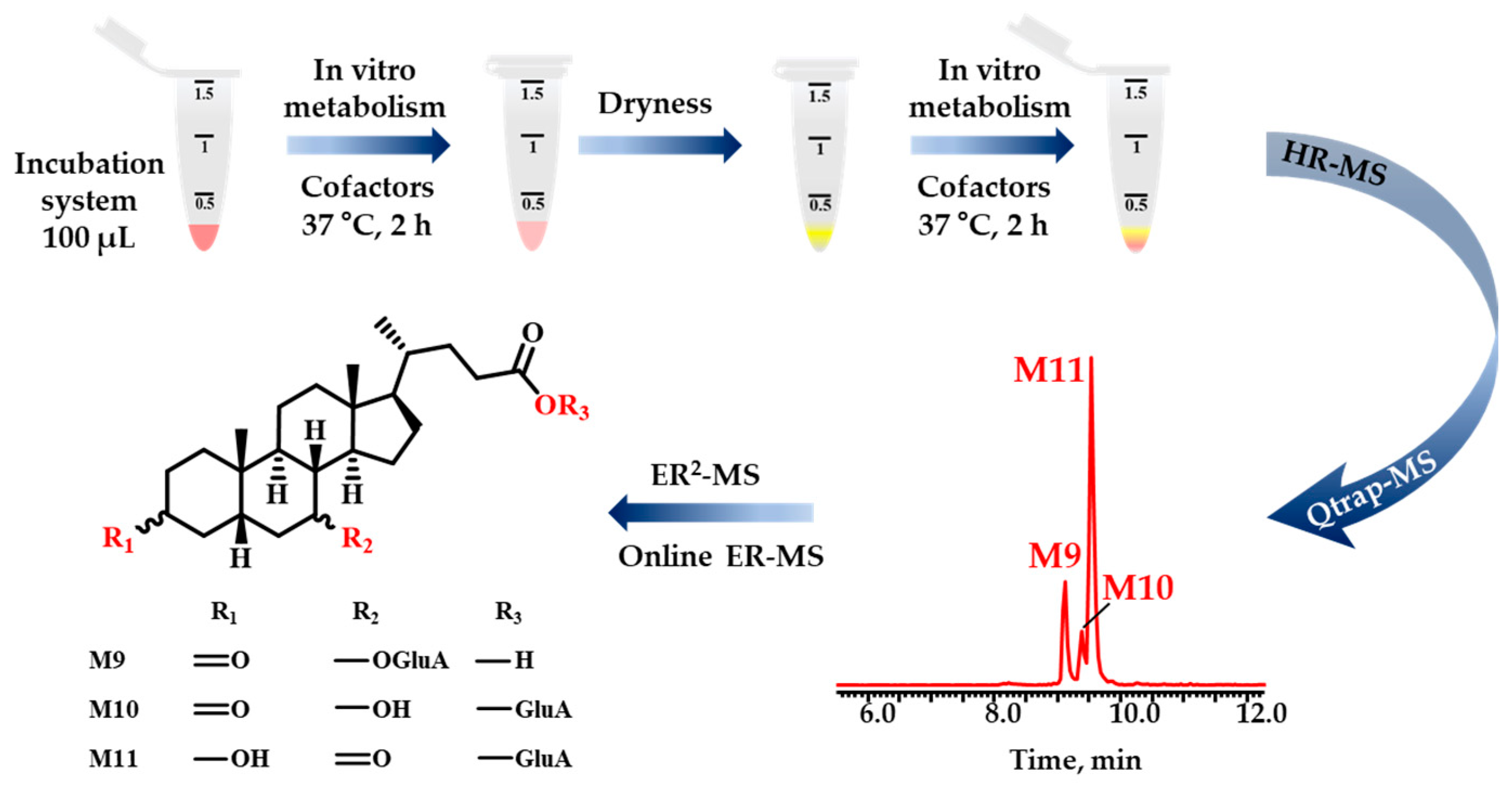{kind=link}
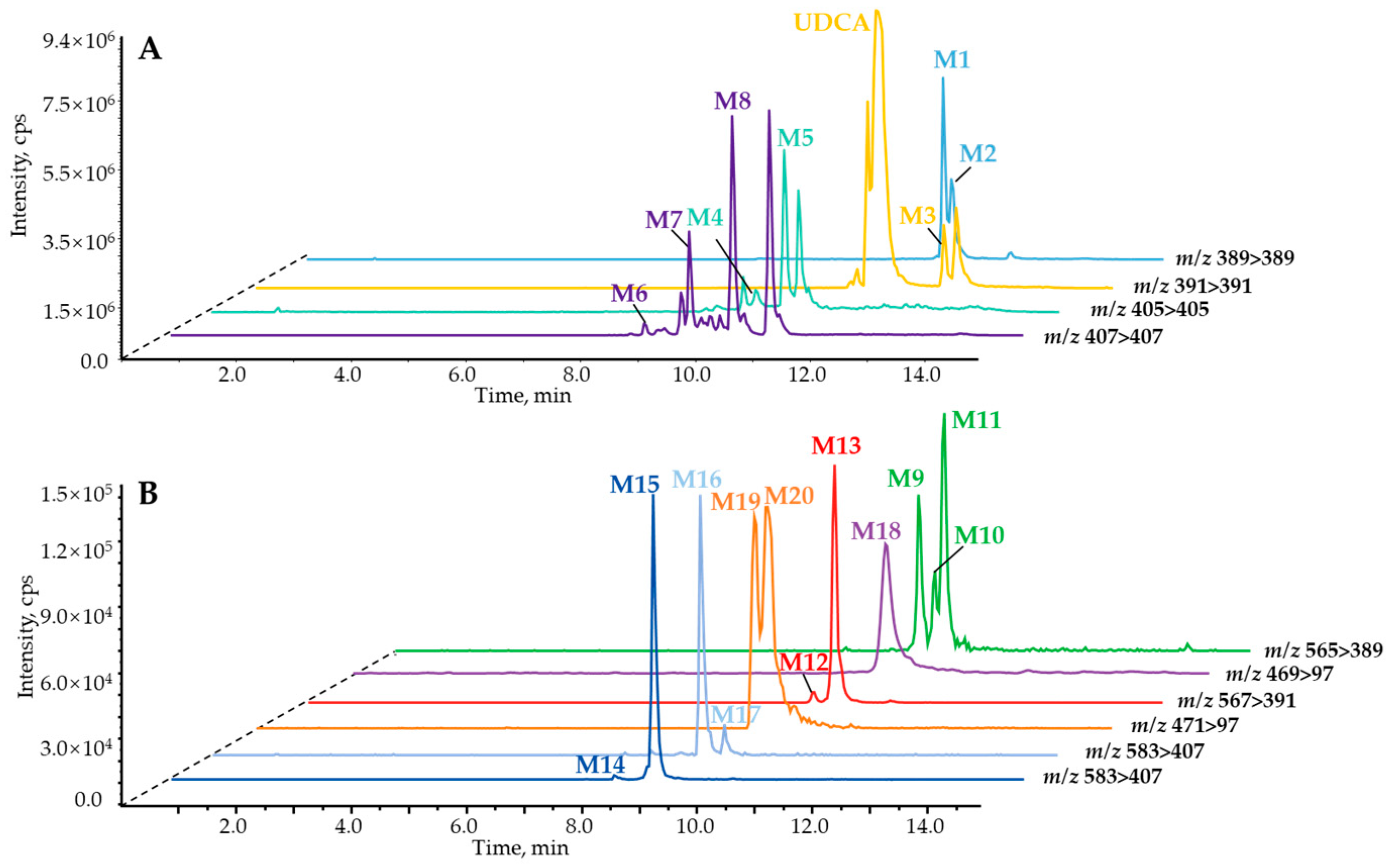{kind=link}
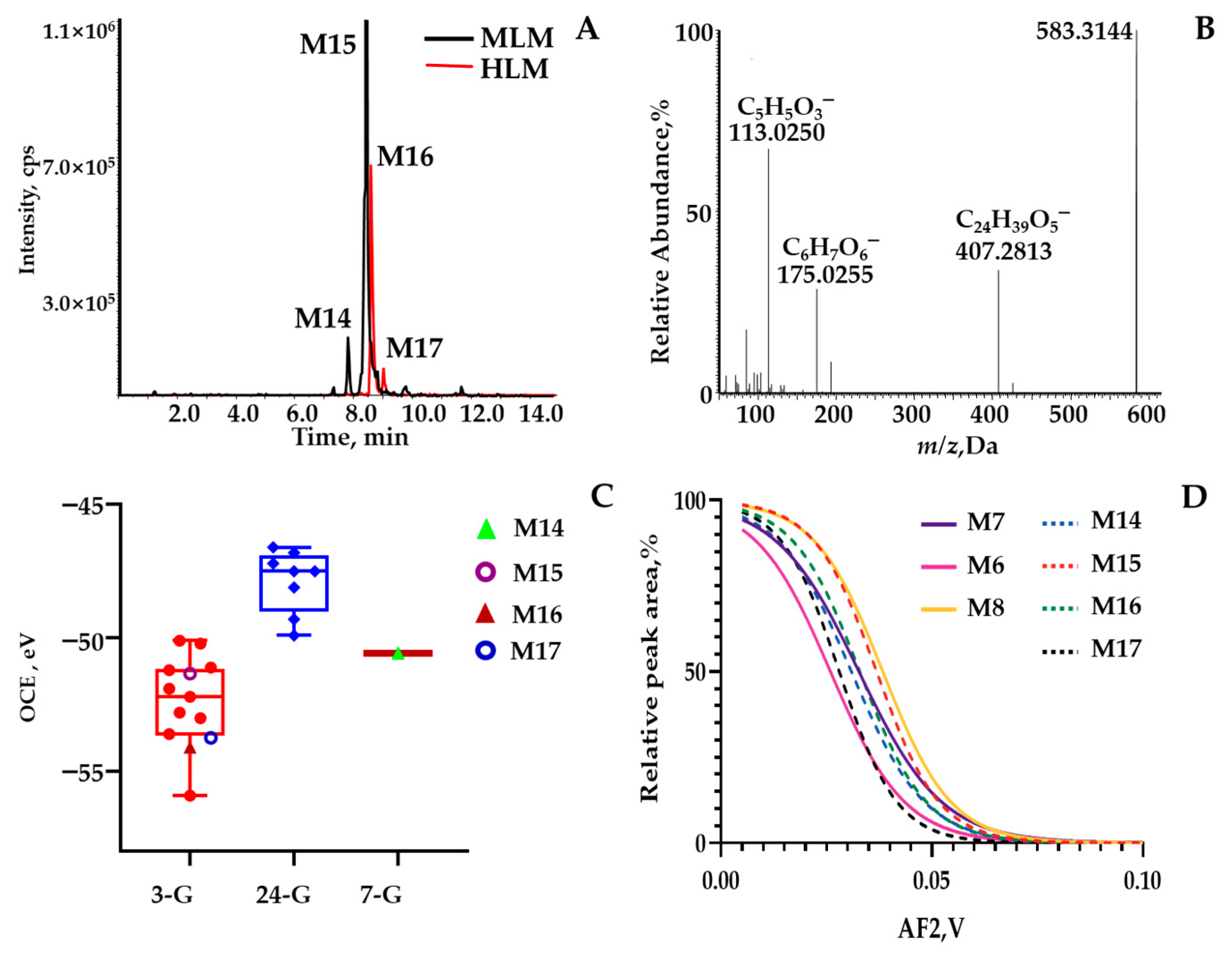{kind=link}
{kind=link}
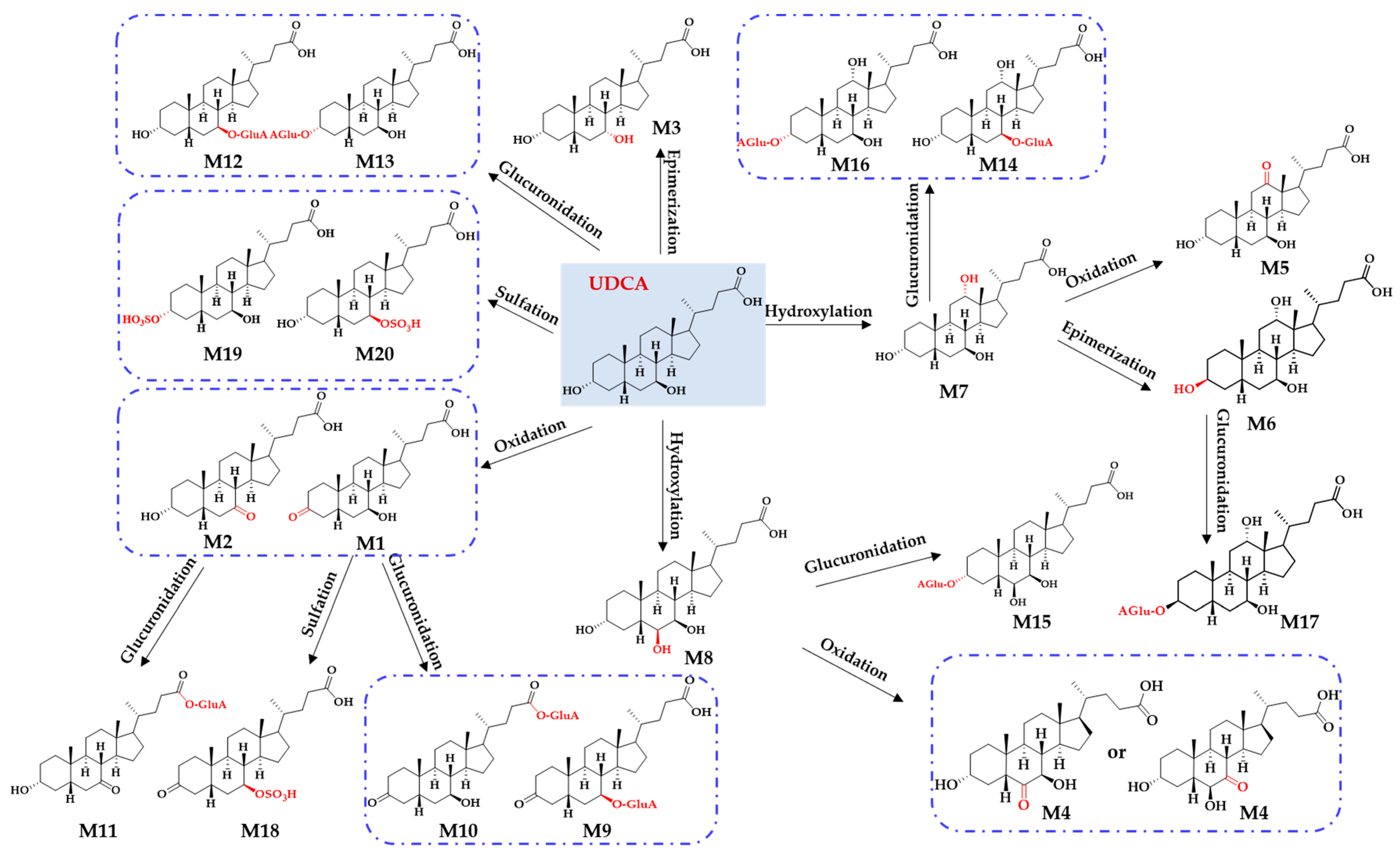{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. Identification of In Vitro Sequential Metabolites of UDCA
2.2. Phase II Metabolites’ Identification by ER2-MS
3. Materials and Methods
3.1. Materials and Chemicals
3.2. Liver Microsomes Incubation Condition
3.3. Qualitative Characterization
3.3.1. Qualitative Characterization for LC–MS/MS
3.3.2. Isomeric Identification through ER2-MS Strategy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Hofmann, A.F.; Hagey, L.R. Bile Acids: Chemistry, Pathochemistry, Biology, Pathobiology, and Therapeutics. Cell Mol. Life Sci. 2008, 65, 2461–2483. [Google Scholar] [CrossRef]
- Hofmann, A.F. Bile acids: Trying to understand their chemistry and biology with the hope of helping patients. Hepatology 2009, 49, 1403–1418. [Google Scholar] [CrossRef] [PubMed]
- de Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F.; Hagey, L.R. Key discoveries in bile acid chemistry and biology and their clinical applications: History of the last eight decades. J. Lipid Res. 2014, 55, 1553–1595. [Google Scholar] [CrossRef]
- Kumari, A.; Pal Pathak, D.; Asthana, S. Bile acids mediated potential functional interaction between FXR and FATP5 in the regulation of lipid metabolism. Int. J. Biol. Sci. 2020, 16, 2308–2322. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Houten, S.M.; Mataki, C.; Christoffolete, M.A.; Kim, B.W.; Sato, H.; Messaddeq, N.; Harney, J.W.; Ezaki, O.; Kodama, T.; et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 2006, 439, 484–489. [Google Scholar] [CrossRef]
- Thomas, C.; Pellicciari, R.; Pruzanski, M.; Auwerx, J.; Schoonjans, K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 2008, 7, 678–693. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef]
- Tore Midtvedt, M.D. Microbial bile acid transformation. Am. J. Clin. Nutr. 1974, 27, 1341–1347. [Google Scholar] [CrossRef]
- Gerard, P. Metabolism of cholesterol and bile acids by the gut microbiota. Pathogens 2013, 3, 14–24. [Google Scholar] [CrossRef]
- Begley, M.; Gahan, C.G.; Hill, C. The interaction between bacteria and bile. FEMS Microbiol. Rev. 2005, 29, 625–651. [Google Scholar] [CrossRef]
- Zhu, Q.F.; Wang, Y.Z.; An, N.; Hao, J.D.; Mei, P.C.; Bai, Y.L.; Hu, Y.N.; Bai, P.R.; Feng, Y.Q. Alternating dual-collision energy scanning mass spectrometry approach: Discovery of novel microbial bile-acid conjugates. Anal. Chem. 2022, 94, 2655–2664. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Z.; Mei, P.C.; Bai, P.R.; An, N.; He, J.G.; Wang, J.; Zhu, Q.F.; Feng, Y.Q. A strategy for screening and identification of new amino acid-conjugated bile acids with high coverage by liquid chromatography-mass spectrometry. Anal. Chim. Acta 2023, 1239, 340691. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, Y.; Fan, J.; Zhang, M.; Hasan, A.; Yi, Y.; Yu, R.; Zhou, X.; Ye, M.; Qiao, X. Expanding the scope of targeted metabolomics by one-pot microscale synthesis and tailored metabolite profiling: Investigation of bile acid–amino acid conjugates. Anal. Chem. 2022, 94, 16596–16603. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Q.; Carey, M.C. Therapeutic uses of animal biles in traditional Chinese medicine: An ethnopharmacological, biophysical chemical and medicinal review. World J. Gastroenterol. 2014, 20, 9952–9975. [Google Scholar] [CrossRef]
- Hagey, L.R.; Crombie, D.L.; Espinosa, E.; Carey, M.C.; Igimi, H.; Hofmann, A.F. Ursodeoxycholic acid in the Ursidae: Biliary bile acids of bears, pandas, and related carnivores. J. Lipid Res. 1993, 34, 1911–1917. [Google Scholar] [CrossRef]
- Xuan, G.Y.; Ding, D.W.; Liu, N.; Hu, Y.N.; Yang, F.F.; Tian, S.Y.; Sun, H.; Yang, J.Q.; Xu, A.; Guo, G.Y.; et al. Efficacy and safety of fenofibrate add-on therapy in patients with primary biliary cholangitis refractory to ursodeoxycholic acid: A retrospective study and updated meta-analysis. Front. Pharmacol. 2022, 13, 948362. [Google Scholar]
- Ovadia, C.; Perdones-Montero, A.; Fan, H.M.; Mullish, B.H.; McDonald, J.A.K.; Papacleovoulou, G.; Wahlström, A.; Ståhlman, M.; Tsakmaki, A.; Clarke, L.C.D.; et al. Ursodeoxycholic acid enriches intestinal bile salt hydrolase-expressing Bacteroidetes in cholestatic pregnancy. Sci. Rep. 2020, 10, 3895. [Google Scholar] [CrossRef]
- Brevini, T.; Maes, M.; Webb, G.J.; John, B.V.; Fuchs, C.D.; Buescher, G.; Wang, L.; Griffiths, C.; Brown, M.L., 3rd; Scott, W.E.; et al. FXR inhibition may protect from SARS-CoV-2 infection by reducing ACE2. Nature 2023, 615, 134–142. [Google Scholar] [CrossRef]
- Song, Q.Q.; Li, J.; Huo, H.X.; Cao, Y.; Wang, Y.T.; Song, Y.L.; Tu, P.F. Retention time and optimal collision energy advance structural annotation relied on LC-MS/MS: An application in metabolite identification of an antidementia agent namely echinacoside. Anal. Chem. 2019, 91, 15040–15048. [Google Scholar] [CrossRef]
- Song, Y.L.; Song, Q.Q.; Li, J.; Zheng, J.; Li, C.; Zhang, Y.; Zhang, L.L.; Jiang, Y.; Tu, P.F. An integrated platform for directly widely-targeted quantitative analysis of feces part II: An application for steroids, eicosanoids, and porphyrins profiling. J. Chromatogr. A 2016, 1460, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Chen, X.; Wang, Z.; Wang, D.; Zhang, J.L. Global profiling and identification of bile acids by multi-dimensional data mining to reveal a way of eliminating abnormal bile acids. Anal. Chim. Acta 2020, 1132, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Su, M.; Xie, G.; Ferslew, B.C.; Brouwer, K.L.; Rajani, C.; Liu, C.; Jia, W. Key role for the 12-hydroxy group in the negative ion fragmentation of unconjugated C24 bile acids. Anal. Chem. 2016, 88, 7041–7048. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Cao, Y.; Song, X.; Zhang, Y.; Li, J.; Wang, Y.; Wu, X.; Qi, X. BAFinder: A Software for unknown bile acid identification using accurate mass LC-MS/MS in positive and negative modes. Anal. Chem. 2022, 94, 6242–6250. [Google Scholar] [CrossRef]
- Cao, Y.; Li, W.; Chen, W.; Niu, X.Y.; Wu, N.; Wang, Y.T.; Li, J.; Tu, P.F.; Zheng, J.; Song, Y.L. Squared energy-resolved mass spectrometry advances quantitative bile acid submetabolome characterization. Anal. Chem. 2022, 94, 15395–15404. [Google Scholar] [CrossRef]
- Yu, Z.Q.; Yang, J.Y.; Xiang, D.; Li, G.D.; Liu, D.; Zhang, C.L. Circadian rhythms and bile acid homeostasis: A comprehensive review. Chronobiol. Int. 2020, 37, 618–628. [Google Scholar] [CrossRef]
- Song, Y.L.; Song, Q.Q.; Liu, W.J.; Li, J.; Tu, P.F. High-confidence structural identification of metabolites relying on tandem mass spectrometry through isomeric identification: A tutorial. Trends Analyt. Chem. 2023, 160, 116982. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, J.Y.; Han, D.Q.; Yao, Z.P. Recent advances in differentiation of isomers by ion mobility mass spectrometry—ScienceDirect. Trends Analyt. Chem. 2020, 124, 115801. [Google Scholar] [CrossRef]
- Thakare, R.; Alamoudi, J.A.; Gautam, N.; Rodrigues, A.D.; Alnouti, Y. Species differences in bile acids II. Bile acid metabolism. J. Appl. Toxicol. 2018, 38, 1336–1352. [Google Scholar] [CrossRef]
- Deo, A.K.; Bandiera, S.M. Identification of human hepatic cytochrome p450 enzymes involved in the biotransformation of cholic and chenodeoxycholic acid. Drug Metab. Dispos. 2008, 36, 1983–1991. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Chen, W.; Niu, X.; Zhao, C.; Tu, P.; Li, J.; Liu, W.; Song, Y. Characterization of Metabolic Correlations of Ursodeoxycholic Acid with Other Bile Acid Species through In Vitro Sequential Metabolism and Isomer-Focused Identification. Molecules 2023, 28, 4801. https://doi.org/10.3390/molecules28124801
Li W, Chen W, Niu X, Zhao C, Tu P, Li J, Liu W, Song Y. Characterization of Metabolic Correlations of Ursodeoxycholic Acid with Other Bile Acid Species through In Vitro Sequential Metabolism and Isomer-Focused Identification. Molecules. 2023; 28(12):4801. https://doi.org/10.3390/molecules28124801
Chicago/Turabian StyleLi, Wei, Wei Chen, Xiaoya Niu, Chen Zhao, Pengfei Tu, Jun Li, Wenjing Liu, and Yuelin Song. 2023. "Characterization of Metabolic Correlations of Ursodeoxycholic Acid with Other Bile Acid Species through In Vitro Sequential Metabolism and Isomer-Focused Identification" Molecules 28, no. 12: 4801. https://doi.org/10.3390/molecules28124801
APA StyleLi, W., Chen, W., Niu, X., Zhao, C., Tu, P., Li, J., Liu, W., & Song, Y. (2023). Characterization of Metabolic Correlations of Ursodeoxycholic Acid with Other Bile Acid Species through In Vitro Sequential Metabolism and Isomer-Focused Identification. Molecules, 28(12), 4801. https://doi.org/10.3390/molecules28124801