
Recent Advances in Chemical Synthesis of Amino Sugars


{kind=link}
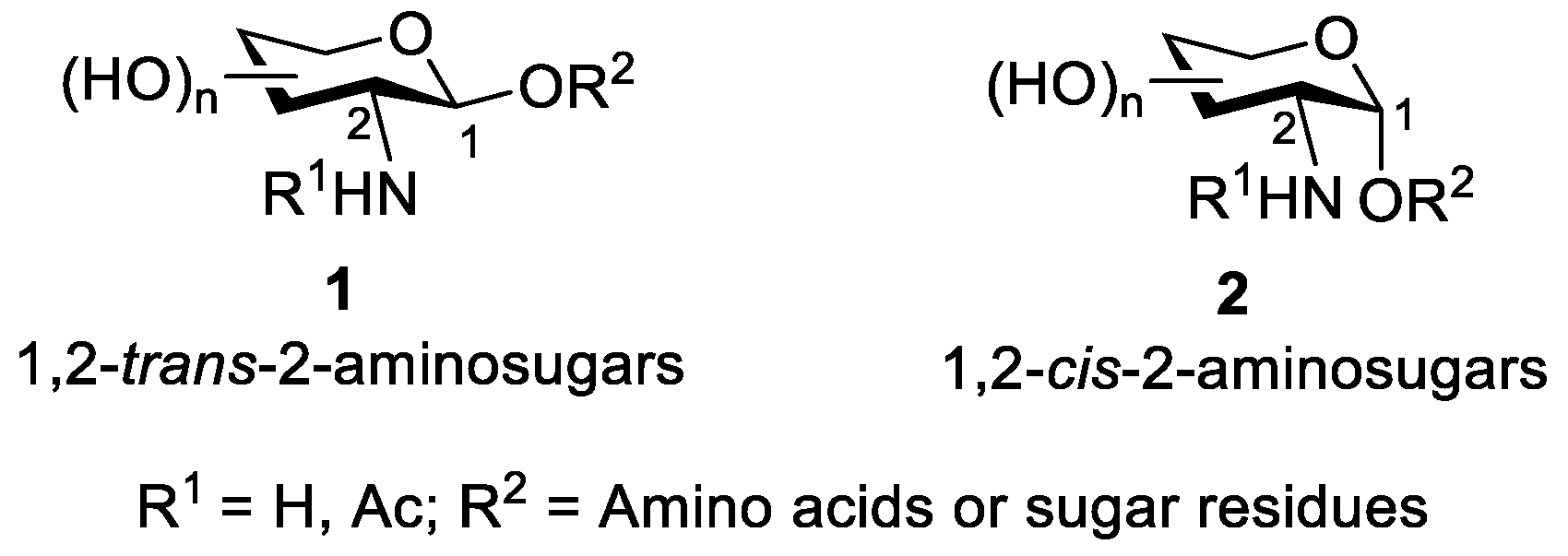{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
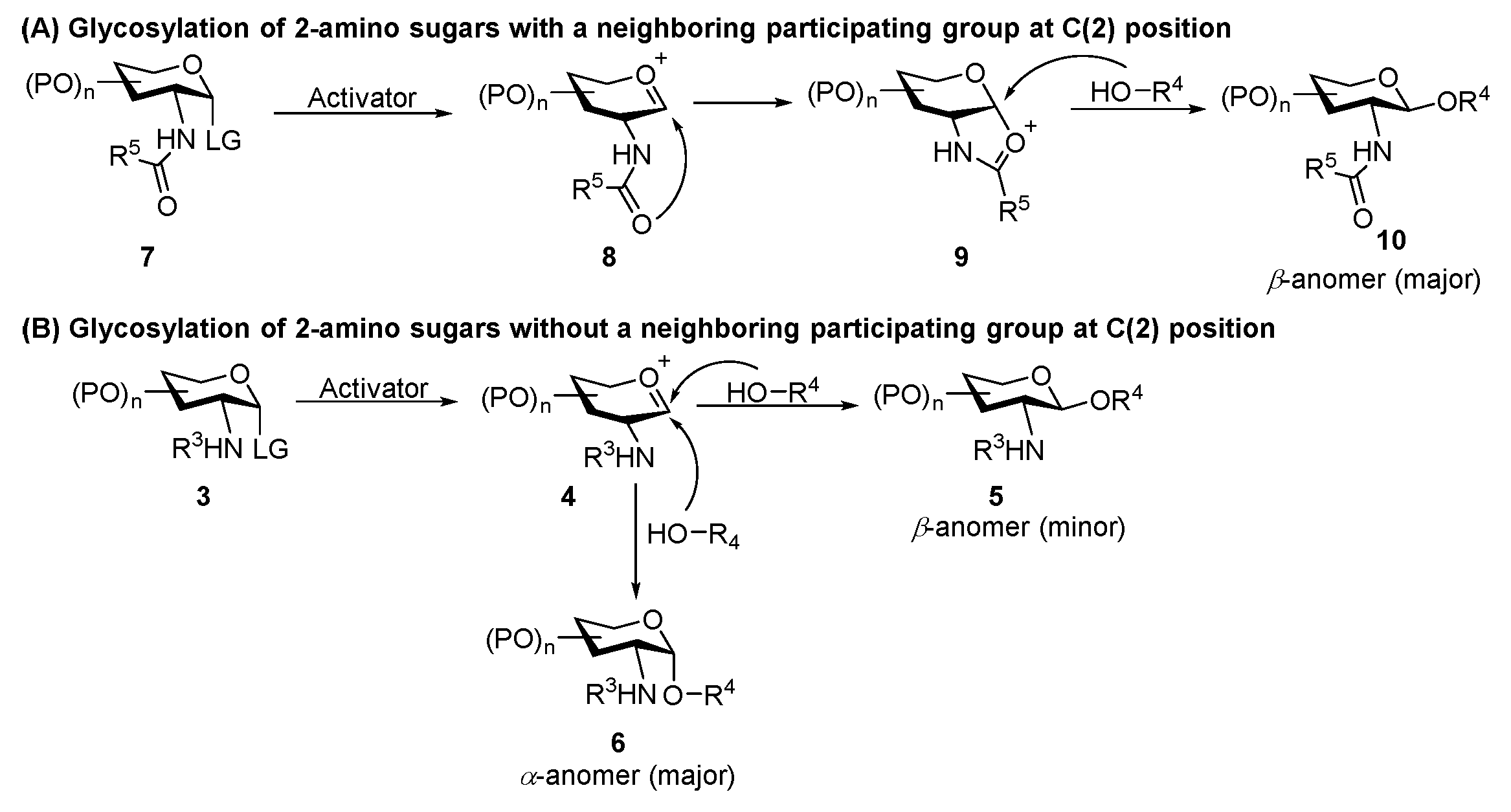{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
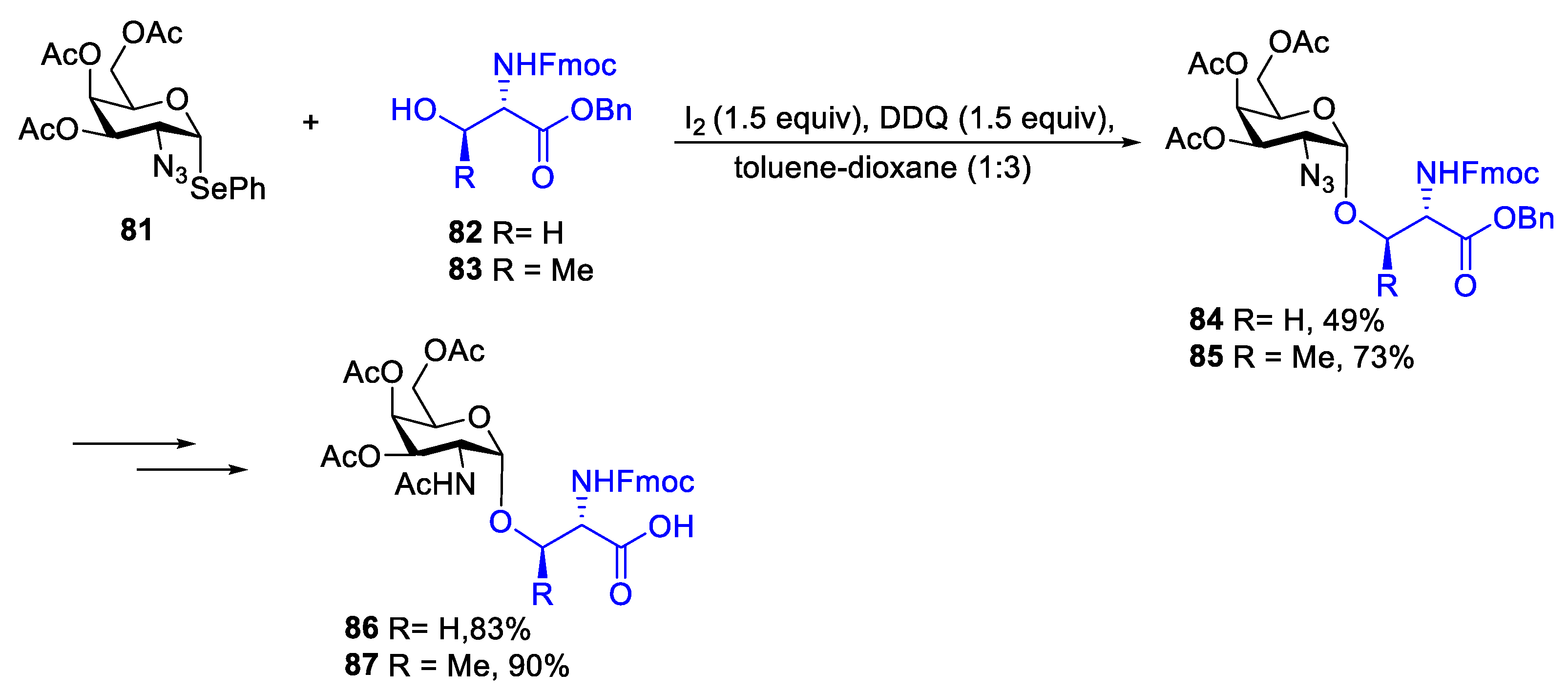{kind=link}
{kind=link}
{kind=link}
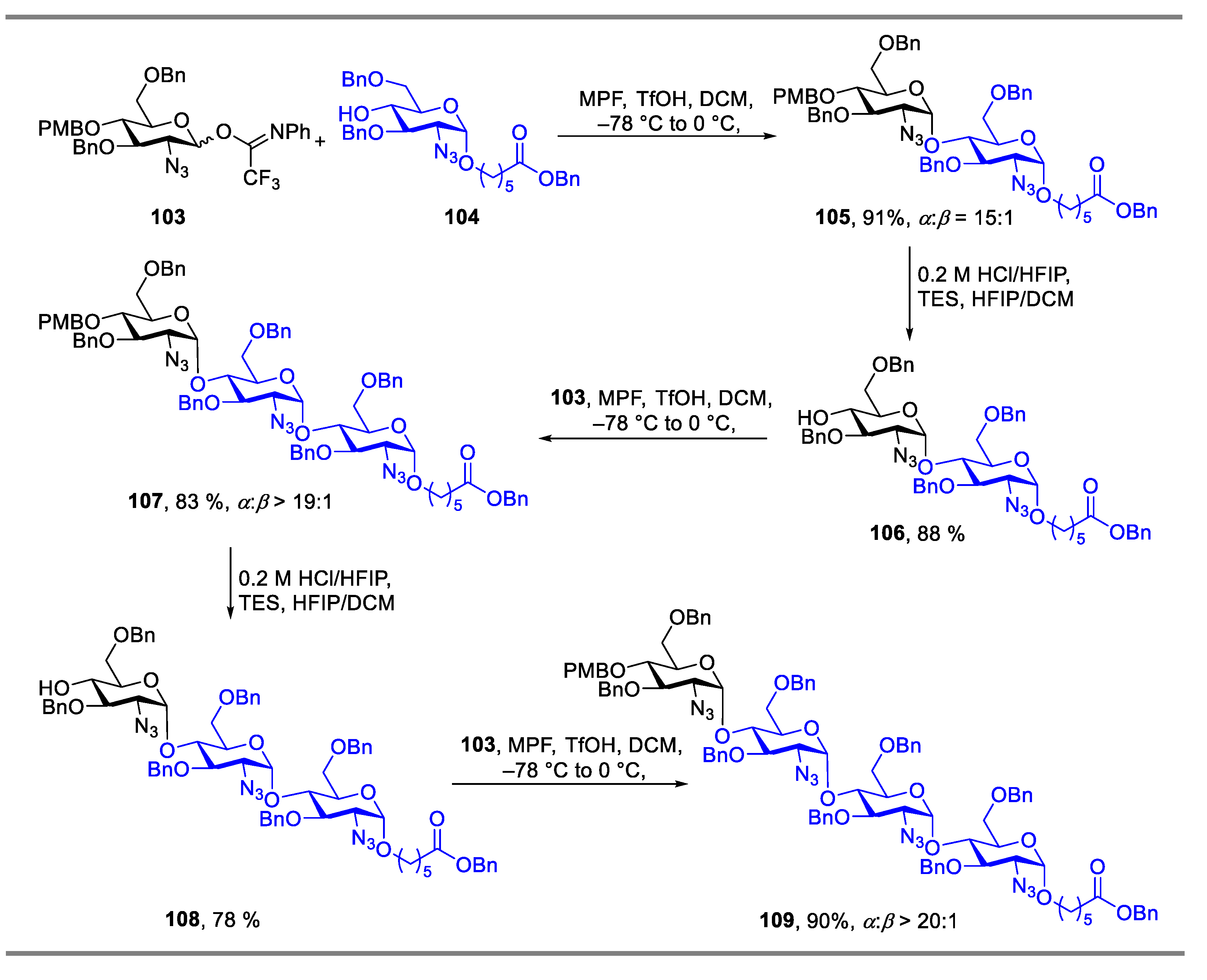{kind=link}
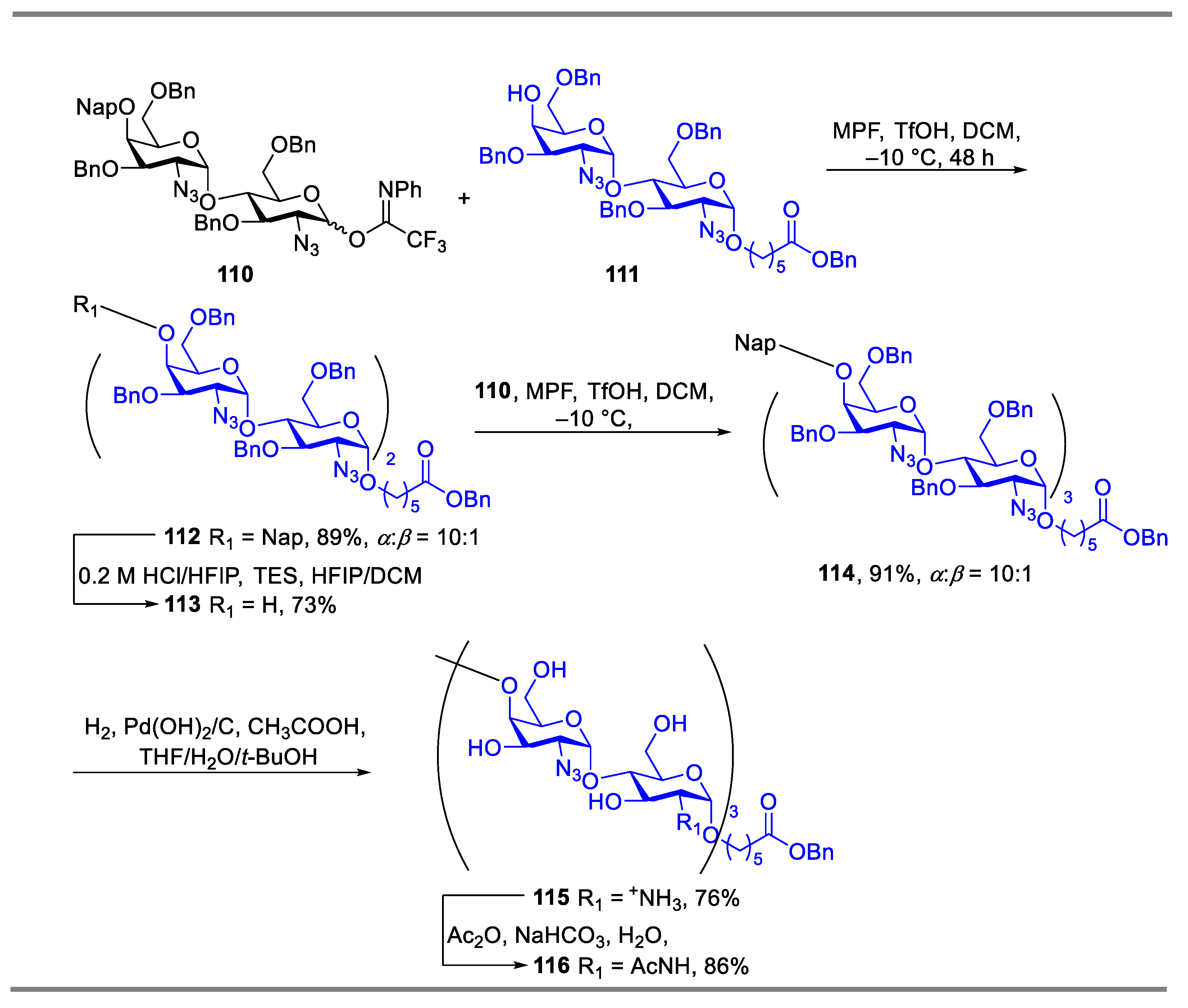{kind=link}
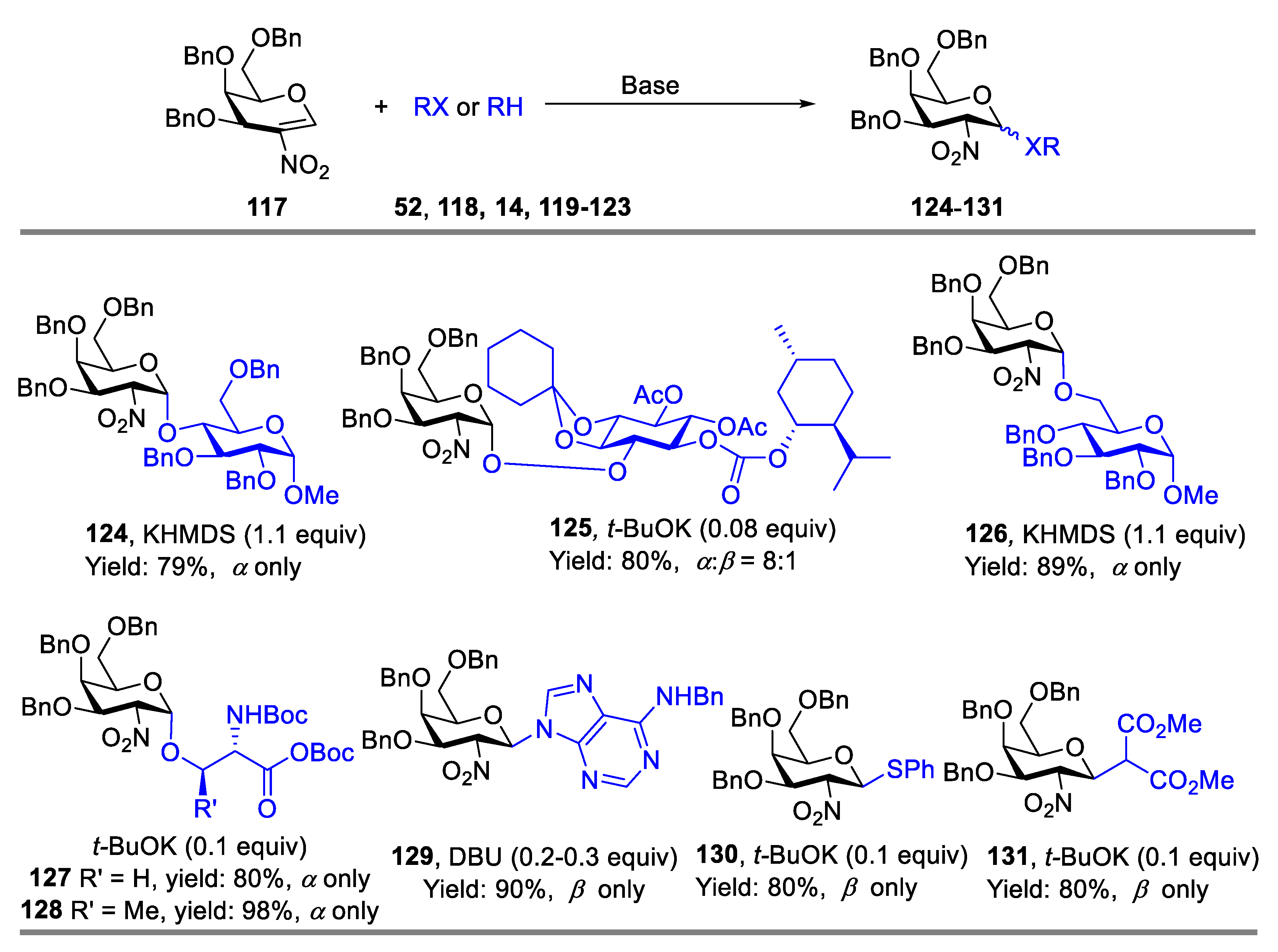{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
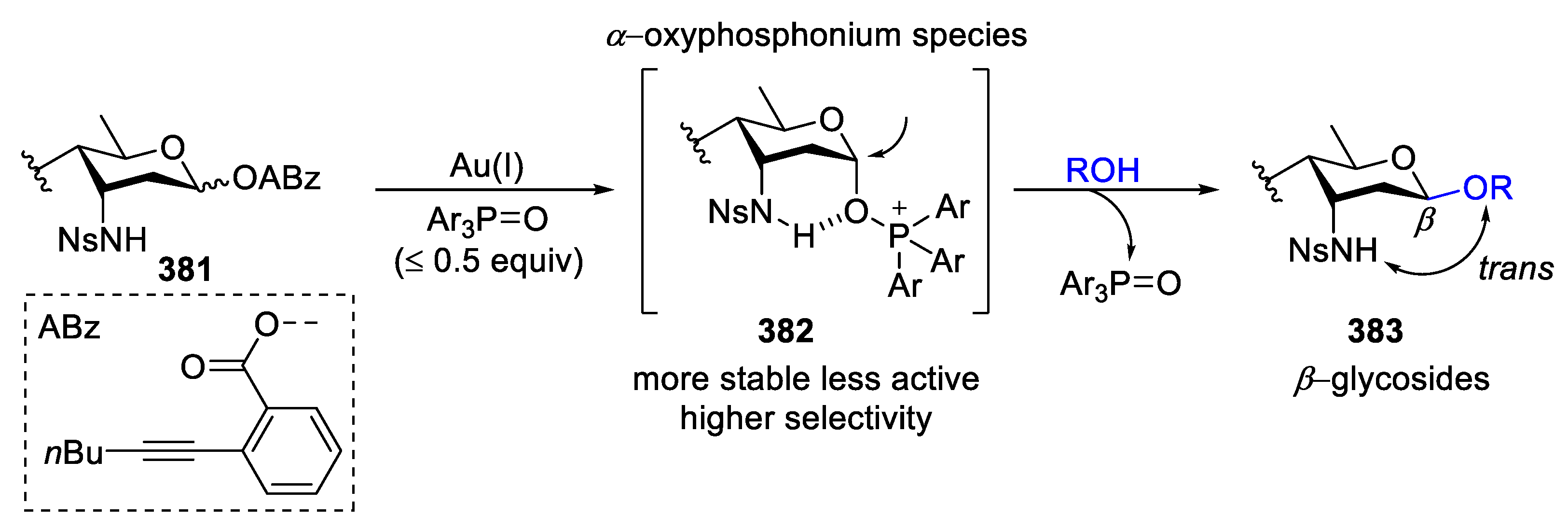{kind=link}
{kind=link}
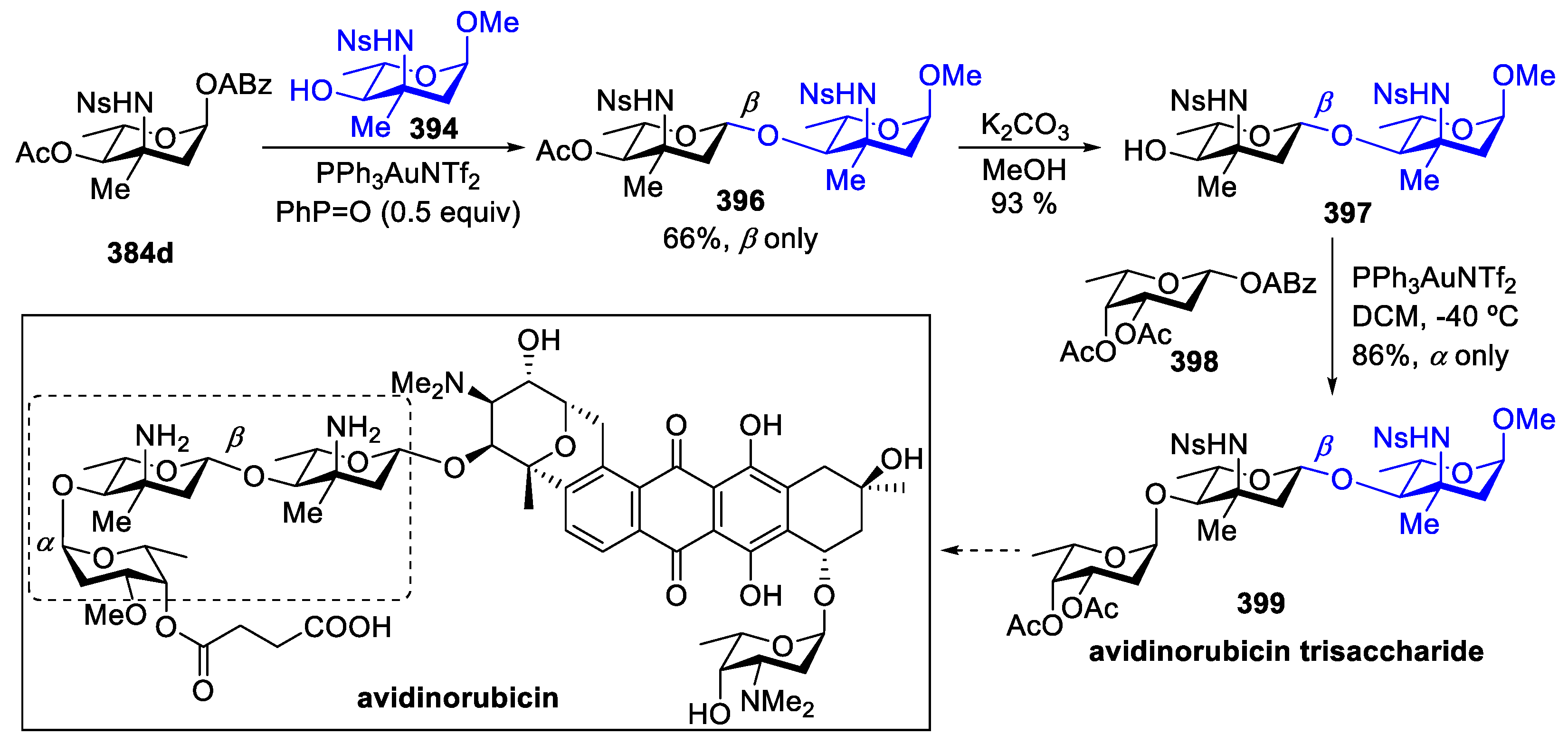{kind=link}
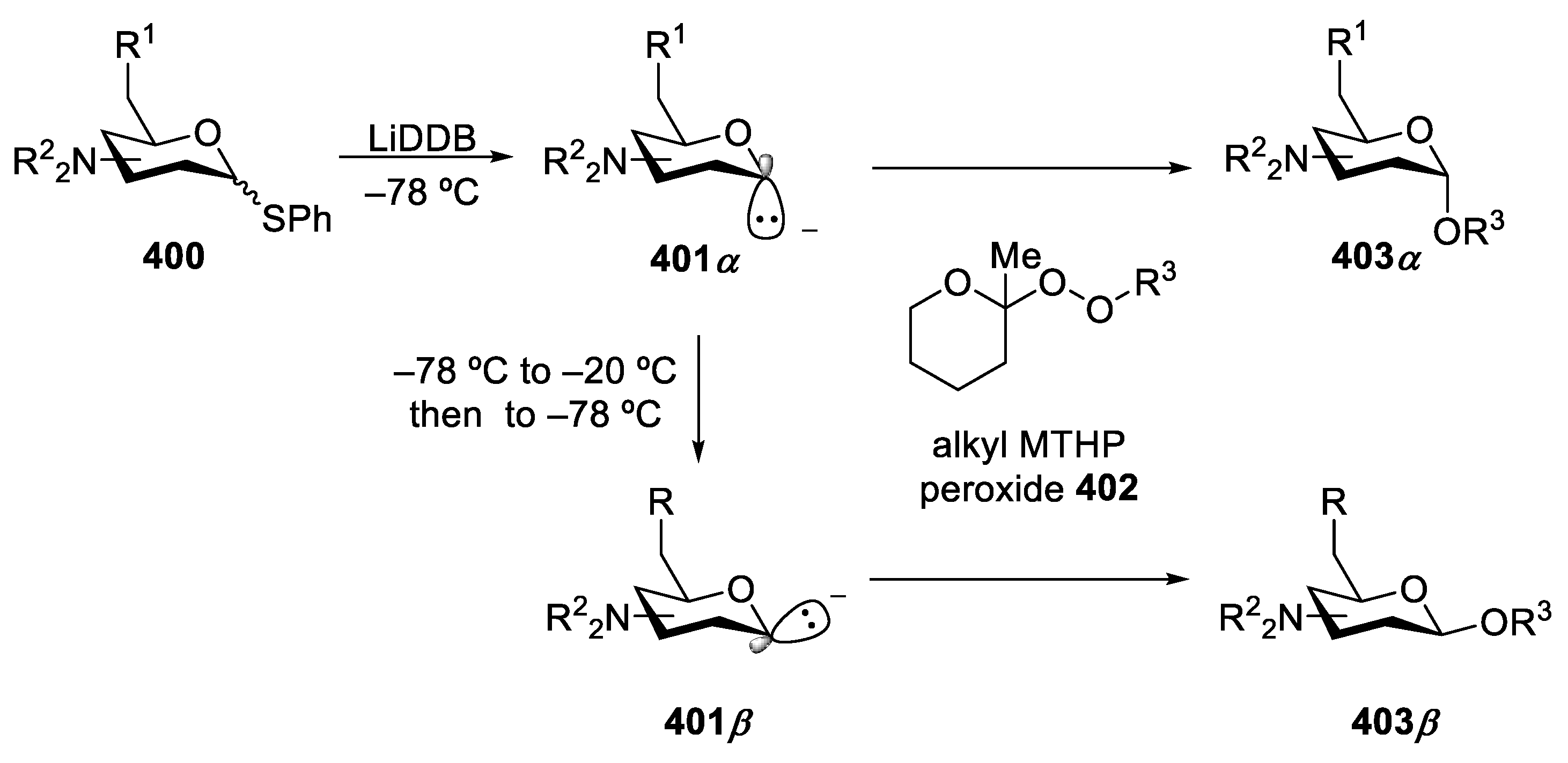{kind=link}
{kind=link}
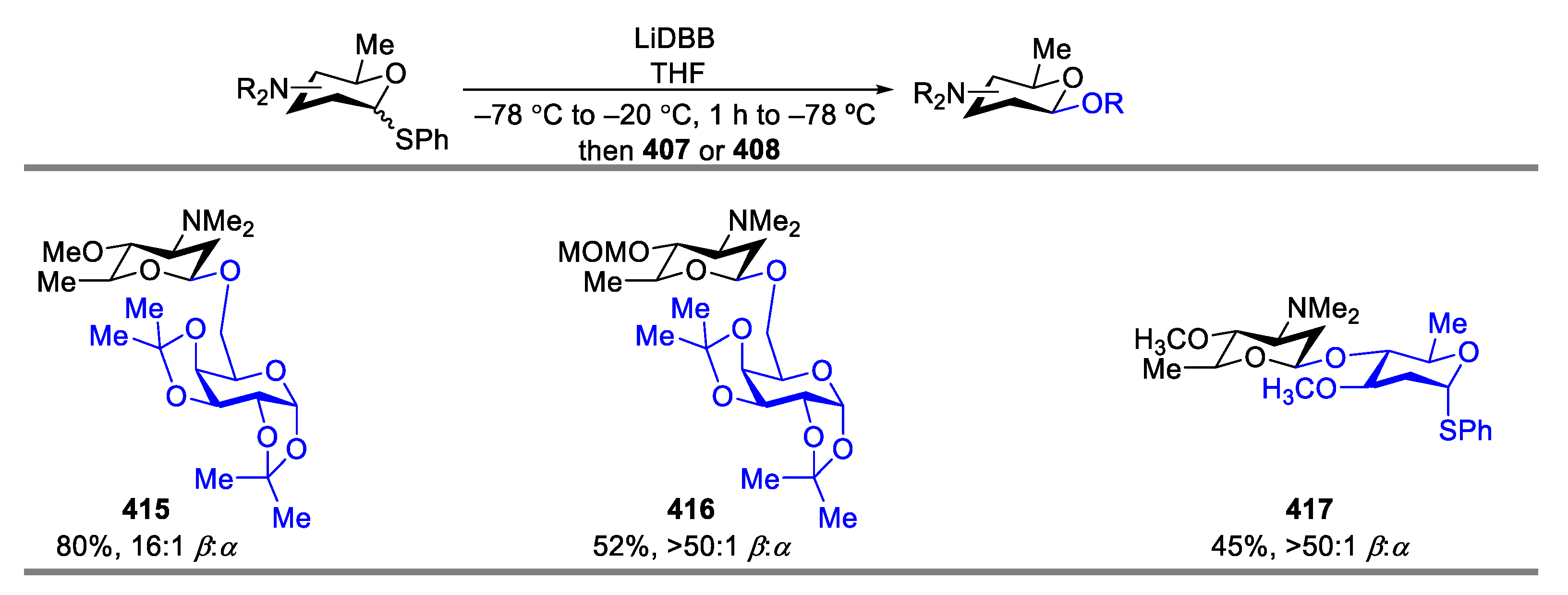{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
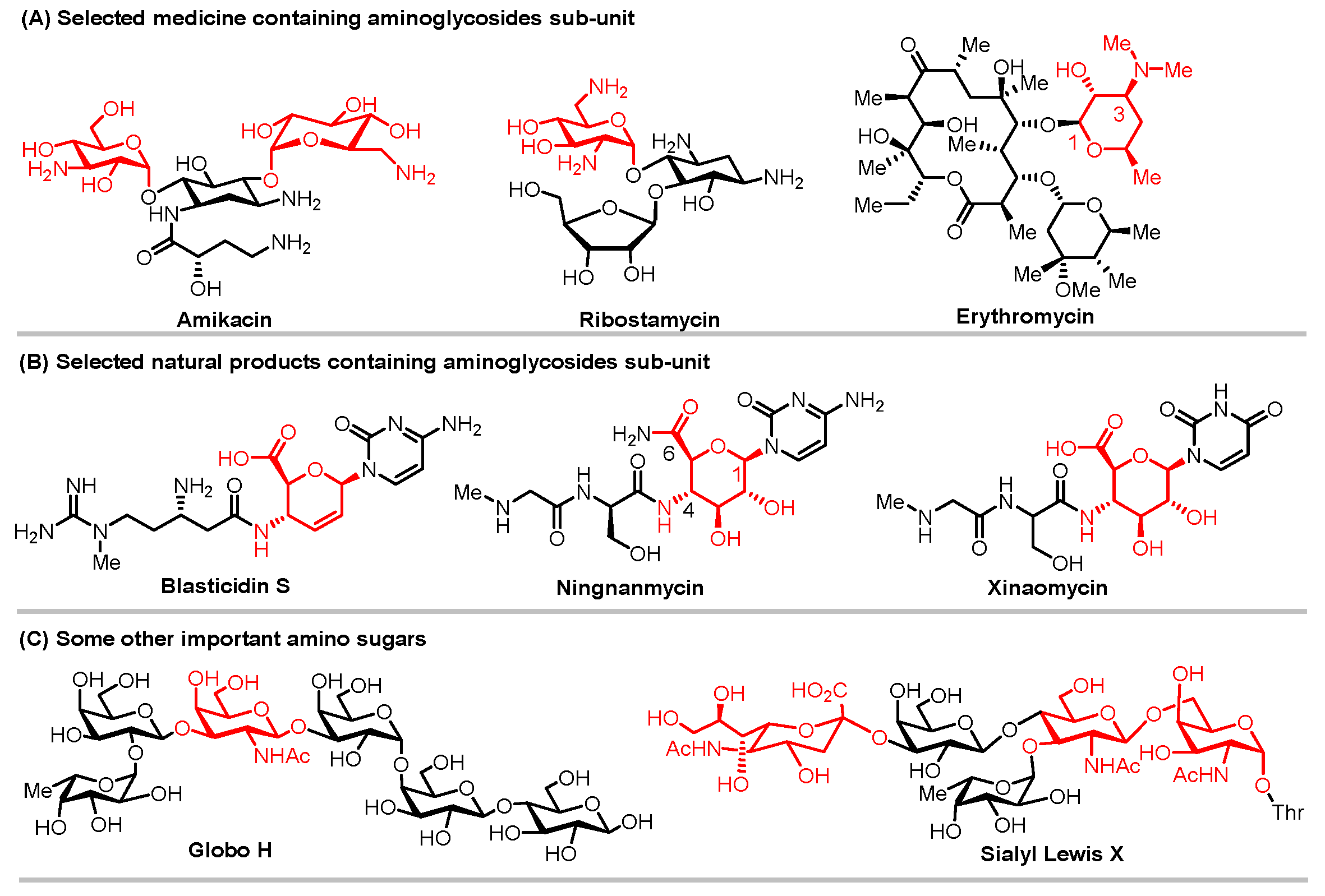
1. Introduction
2. Glycosylation of 2-Amino-2-Deoxysugars
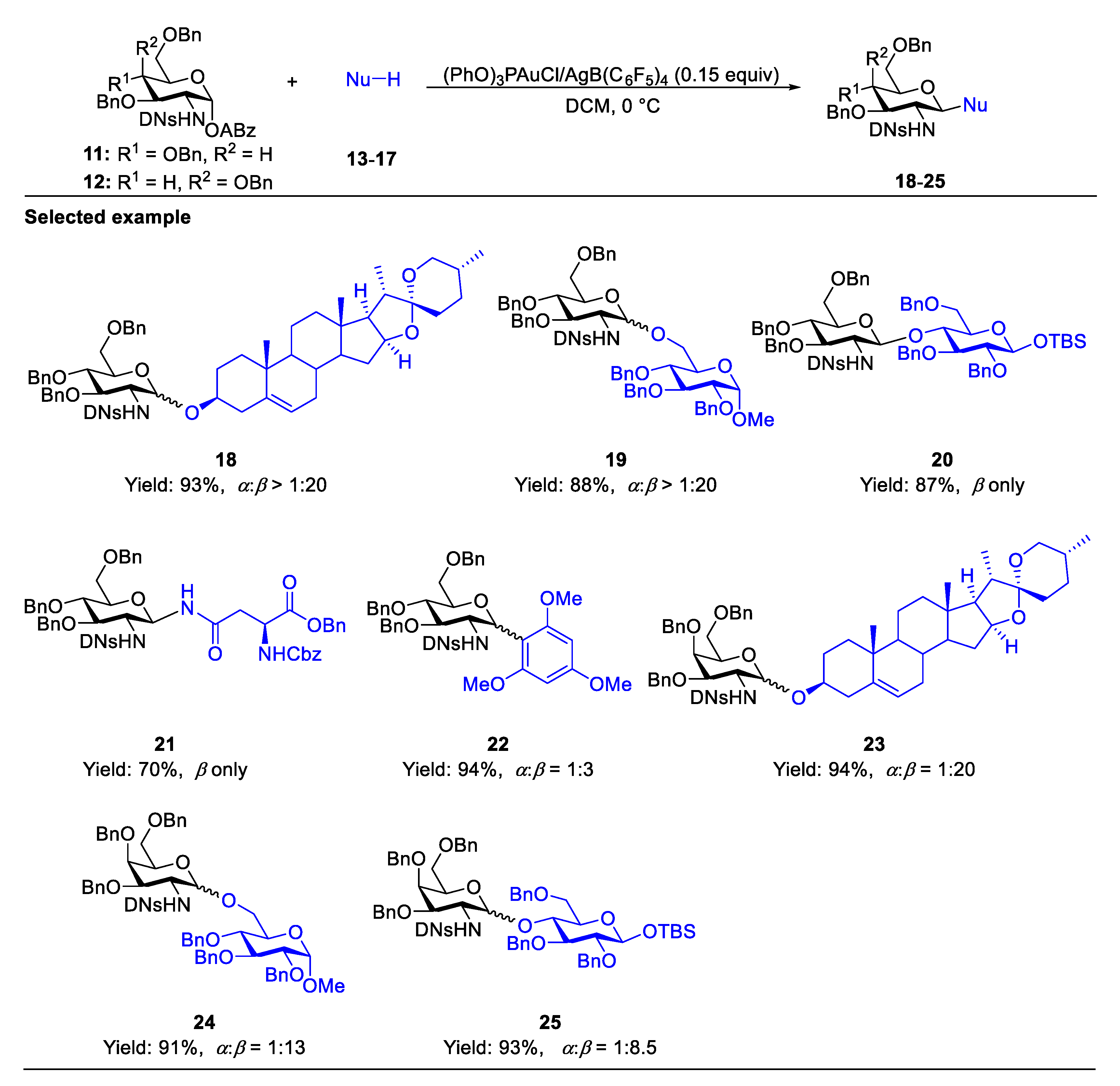
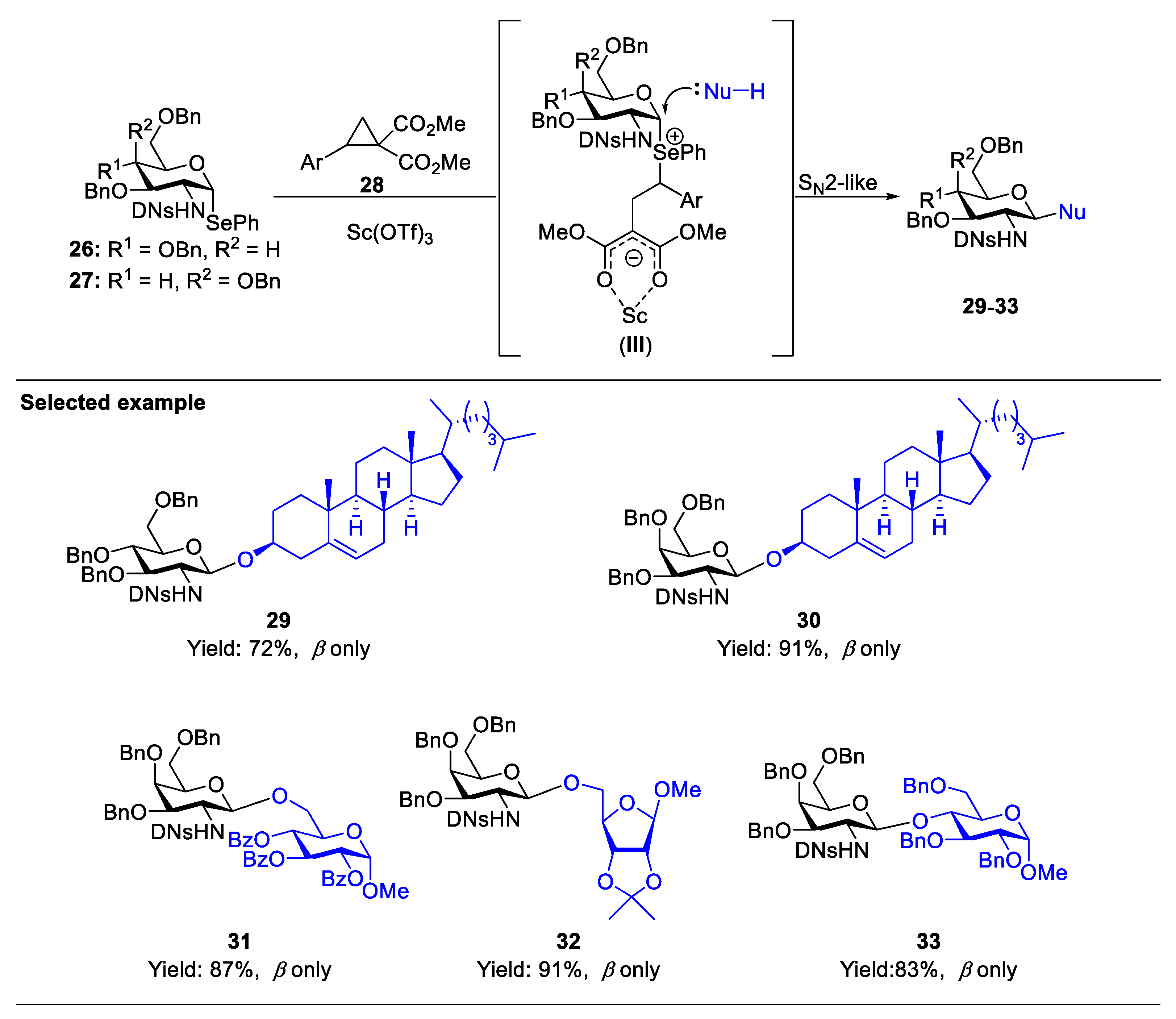
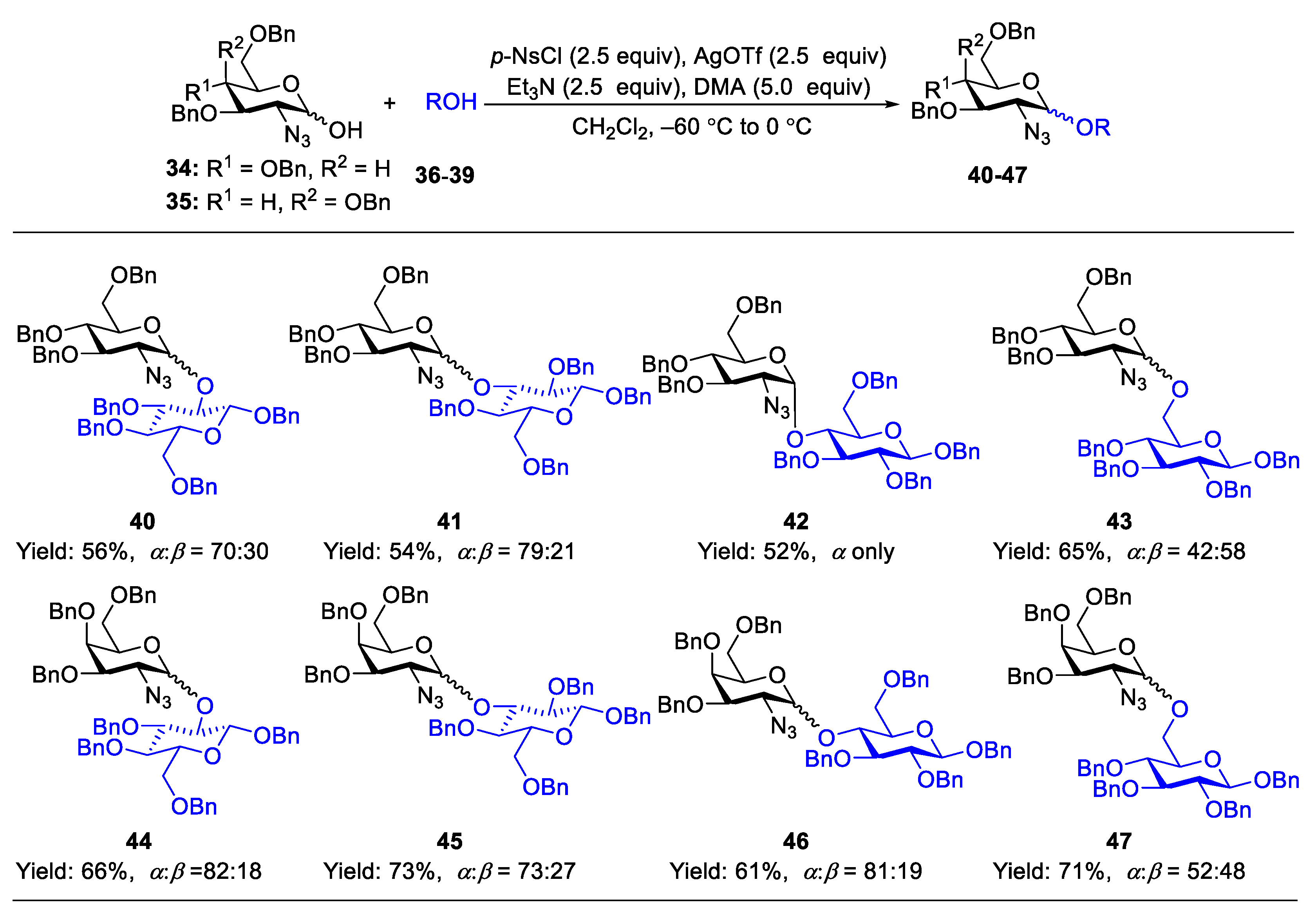
2.1. Glycosylation with C(2)-Azido Donors
2.2. Glycosylation with 2-Nitroglycals
2.3. Glycosylation with Ring-Fused 2,3-Oxazolidinone Thioglycosides
2.4. Glycosylation with C(2)-Benzylidenamino Donors
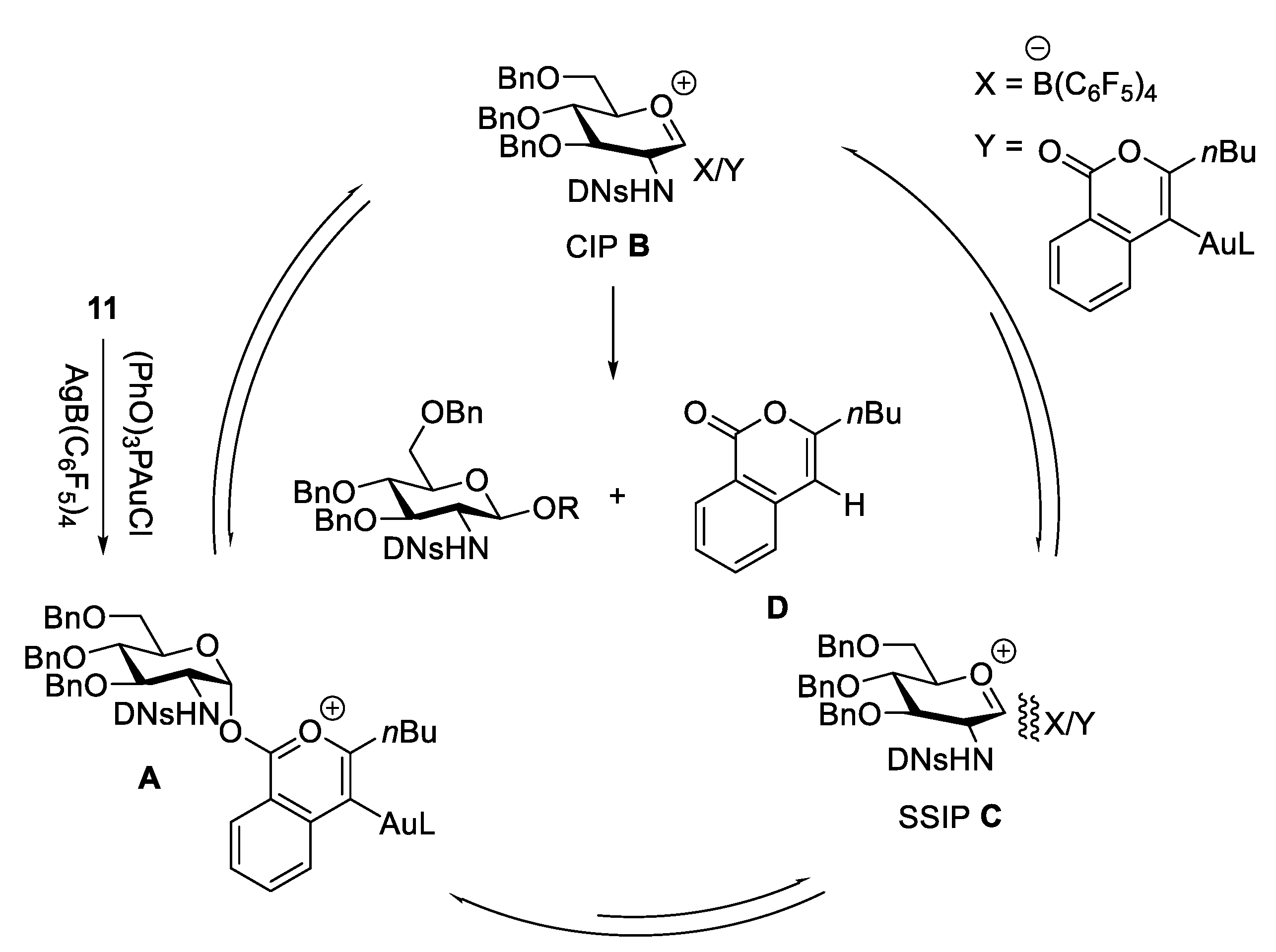
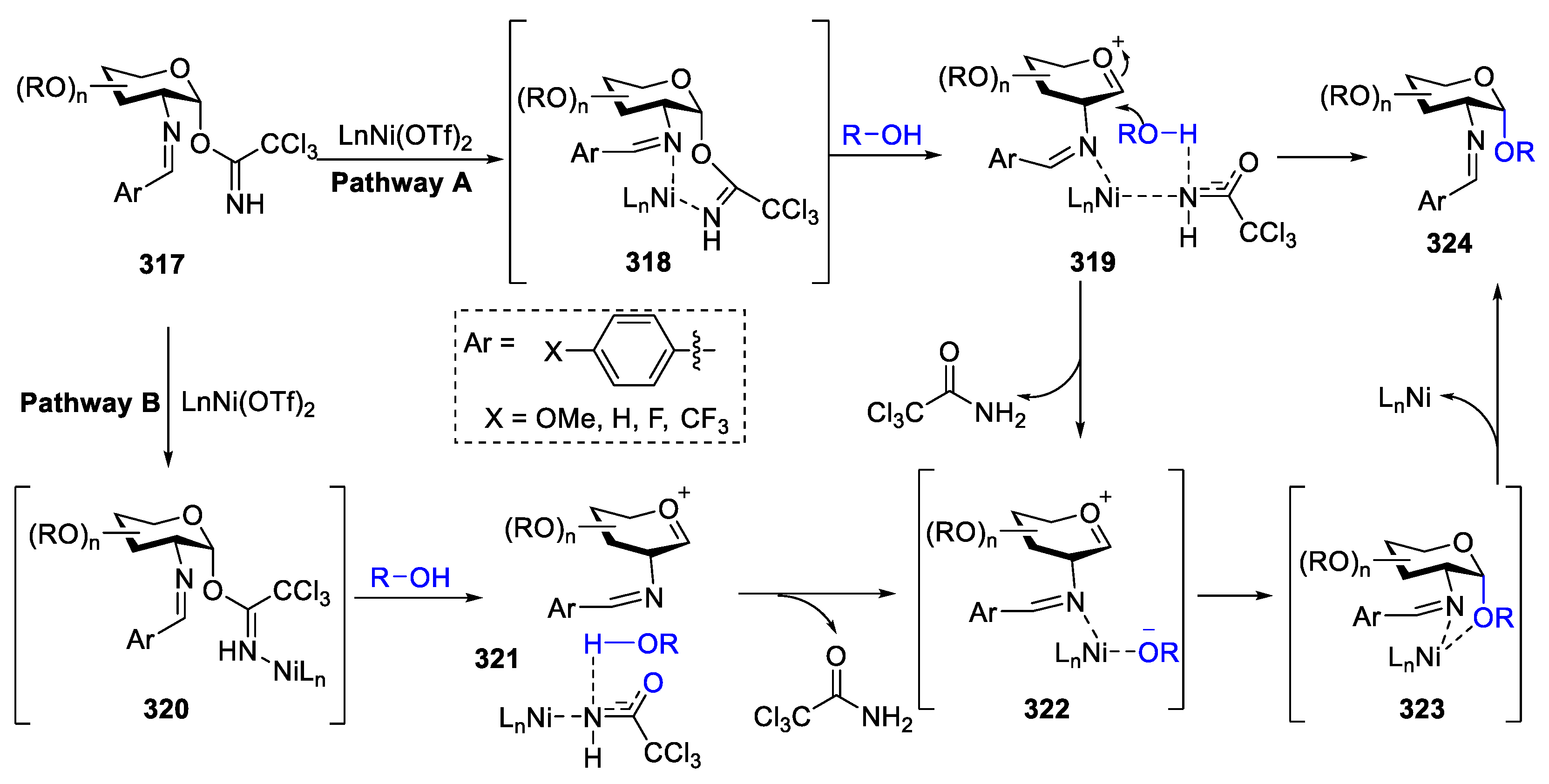
2.4.1. The Development of Nickel-Catalyzed Stereoselective Glycosylation
2.4.2. Synthesis of Biologically Active Glycans via Nickel-Catalyzed Glycosylation
Synthesis of Mycothiol
Synthesis of GPI Anchor
Synthesis of TN Antigen
3. Glycosylation of 3-Amino-3-Deoxysugars and 4-Amino-4-Deoxysugars
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Ac | acetyl |
| All | allyl |
| Ar | ary (substituted aromatic ring) |
| ABz | o-hexynylbenzoic acid |
| Bn | benzyl |
| Boc | t-butoxycarbonyl |
| BSM | benzenesulfinyl morpholine |
| BSP | 1-benzensulfinylpiperidine |
| Bz | benzoyl |
| nBu | n-butyl |
| tBu | t-butyl |
| Cbz | benzyloxycarbonyl |
| DCM | dichloromethane |
| DNs | 2,4-dinitrobenzenesulfonyl |
| Et | ethyl |
| Fmoc | 9-fluorenylmethoxycarbonyl |
| HFIP | 1,1,1,3,3,3-hexafluoro-2-propanol |
| KHMDS | potassium bis(trimethylsilyl)amide |
| Me | methyl |
| NIS | N-iodosuccinimide |
| Ns | p-nitrobenzene sulfonyl |
| Phth | phthaloyl |
| PMB (MP) | p-methoxybenzyl |
| iPr | isopropyl |
| Ser | L-serine |
| TBAF | tetrabutylammonium fluoride |
| TBS | t-butyldimethylsilyl |
| TES | triethylsilane |
| Tf | trifluoromethanesulfony |
| Thr | L-threonine |
| TIPS | triisopropylsilyl |
References
- Gannett, C.; Banks, P.; Chuong, C.; Weger-Lucarelli, J.; Mevers, E.; Lowell, A.N. Semisynthetic Blasticidin S Ester Derivatives Show Enhanced Antibiotic Activity. RSC Med. Chem. 2023, 14, 782–789. [Google Scholar] [CrossRef] [PubMed]
- An, M.; Zhou, T.; Guo, Y.; Zhao, X.; Wu, Y. Molecular Regulation of Host Defense Responses Mediated by Biological Anti-TMV Agent Ningnanmycin. Viruses 2019, 11, 815. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.-H.; Luo, D.; Shu, D.; Zhong, J.; Tan, H. Development of an Intergeneric Conjugal Transfer System for Xinaomycins-Producing Streptomyces noursei Xinao-4. Int. J. Mol. Sci. 2014, 15, 12217–12230. [Google Scholar] [CrossRef] [PubMed]
- Flatt, P.M.; Mahmud, T. Biosynthesis of Aminocyclitol-Aminoglycoside Antibiotics and Related Compounds. Nat. Prod. Rep. 2007, 24, 358–392. [Google Scholar] [CrossRef] [PubMed]
- Behera, A.; Kulkarni, S. Chemical Synthesis of Rare, Deoxy-Amino Sugars Containing Bacterial Glycoconjugates as Potential Vaccine Candidates. Molecules 2018, 23, 1997. [Google Scholar] [CrossRef]
- Bennett, C.S.; Galan, M.C. Methods for 2-Deoxyglycoside Synthesis. Chem. Rev. 2018, 118, 7931–7985. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Seitz, S.P.; Pavia, M.R. Carbohydrates in Organic Synthesis. Synthesis of 16-Membered-Ring Macrolide Antibiotics. 6. Total Synthesis of O-Mycinosyltylonolide: Coupling of Key Intermediates and Macrocyclization. J. Am. Chem. Soc. 1982, 104, 2030–2031. [Google Scholar] [CrossRef]
- Suzuki, K.; Maeta, H.; Matsumoto, T.; Tsuchihashi, L.G.-I. New Glycosidation Reaction 2. Preparation of 1-Fluoro-D-Desosamine Derivative and Its Efficient Glycosidation by the Use of Cp2HfCl2-AgClO4 as the Activator. Tetrahedron Lett. 1988, 29, 3571–3574. [Google Scholar] [CrossRef]
- Bai, Y.; Shen, X.; Li, Y.; Dai, M. Total Synthesis of (-)-Spinosyn A via Carbonylative Macrolactonization. J. Am. Chem. Soc. 2016, 138, 10838–10841. [Google Scholar] [CrossRef]
- Balthaser, B.R.; McDonald, F.E. Brønsted Acid-Promoted Glycosylations of Disaccharide Glycal Substructures of the Saccharomicins. Org. Lett. 2009, 11, 4850–4853. [Google Scholar] [CrossRef]
- Bylsma, M.; Bennett, C.S. Stereospecific Synthesis of the Saccharosamine-Rhamnose-Fucose Fragment Present in Saccharomicin B. Org. Lett. 2018, 20, 4695–4698. [Google Scholar] [CrossRef]
- Skarbek, K.; Milewska, M.J. Biosynthetic and Synthetic Access to Amino Sugars. Carbohydr. Res. 2016, 434, 44–71. [Google Scholar] [CrossRef]
- Wang, Y.; Yao, H.; Hua, M.; Jiao, Y.; He, H.; Liu, M.; Huang, N.; Zou, K. Direct N-Glycosylation of Amides/Amines with Glycal Donors. J. Org. Chem. 2020, 85, 7485–7493. [Google Scholar] [CrossRef]
- Sangwan, R.; Khanam, A.; Mandal, P.K. An Overview on the Chemical N-Functionalization of Sugars and Formation of N-Glycosides. Eur. J. Org. Chem. 2020, 2020, 5949–5977. [Google Scholar] [CrossRef]
- Manabe, S. Chapter Twenty—The Synthesis of 1,2-cis-Amino Containing Oligosaccharides toward Biological Investigation. In Methods in Enzymology; Fukuda, M., Ed.; Academic Press: Cambridge, MA, USA, 2010; pp. 413–435. [Google Scholar]
- Bongat, A.F.G.; Demchenko, A.V. Recent Trends in the Synthesis of O-glycoside of 2-Amino-2-Deoxysugars. Carbohydr. Res. 2007, 342, 374–406. [Google Scholar] [CrossRef]
- Petitou, M.; van Boeckel, C.A.A. A Synthetic Antithrombin III Binding Pentasaccharide Is Now a Drug! What Comes Next? Angew. Chem. Int. Ed. 2004, 43, 3118–3133. [Google Scholar] [CrossRef]
- Hakomori, S.-I. Tumor-Associated Carbohydrate Antigens. Amnu. Rev. Immunol. 1984, 2, 103–126. [Google Scholar] [CrossRef]
- Danishefsky, S.J.; Allen, J.R. From the Laboratory to the Clinic: A Retrospective on Fully Synthetic Carbohydrate-Based Anticancer Vaccines. Angew. Chem. Int. Ed. 2000, 39, 836–863. [Google Scholar] [CrossRef]
- Dwek, R.A. Glycobiology: Toward Understanding the Function of Sugars. Chem. Rev. 1996, 96, 683–720. [Google Scholar] [CrossRef]
- Werz, D.B.; Ranzinger, R.; Herget, S.; Adibekian, A.; von der Lieth, C.-W.; Seeberger, P.H. Exploring the Structural Diversity of Mammalian Carbohydrates (“Glycospace”) by Statistical Databank Analysis. ACS Chem. Biol. 2007, 2, 685–691. [Google Scholar] [CrossRef]
- Hart, G.W.; Housley, M.P.; Slawson, C. Cycling of O-linked β-N-Acetylglucosamine on Nucleocytoplasmic Proteins. Nature 2007, 446, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Banoub, J.; Boullanger, P.; Lafont, D. Synthesis of Oligosaccharides of 2-Amino-2-Deoxy Sugars. Chem. Rev. 1992, 92, 1167–1195. [Google Scholar] [CrossRef]
- Kerns, R.J.; Wei, P. Synthetic Methods to Incorporate α-Linked 2-Amino-2-Deoxy-D-Glucopyranoside and 2-Amino-2-Deoxy-D-Galactopyranoside Residues into Glycoconjugate Structures. In Glycobiology and Drug Design; American Chemical Society: Washington, DC, USA, 2012; pp. 235–263. [Google Scholar]
- Beau, J.M.; Boyer, F.D.; Norsikian, S.; Urban, D.; Vauzeilles, B.; Xolin, A. Glycosylation: The Direct Synthesis of 2-Acetamido-2-Deoxy-Sugar glycoside. Eur. J. Org. Chem. 2018, 2018, 5795–5814. [Google Scholar] [CrossRef]
- Wang, X.; Wang, P.; Li, D.; Li, M. 2,4-Dinitrobenzenesulfonamide-Directed SN2-Type Displacement Reaction Enables Synthesis of β-D-Glycosaminosides. Org. Lett. 2019, 21, 2402–2407. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Ding, H.; Peng, L.-C.; Fang, X.-Y.; Qin, Y.-Y.; Mu, Q.-Q.; Liu, X.-W. Sweet Strain Release: Donor–Acceptor Cyclopropane Mediated Glycosylation. CCS Chem. 2023; in press. [Google Scholar] [CrossRef]
- Orgueira, H.A.; Bartolozzi, A.; Schell, P.; Litjens, R.; Palmacci, E.R.; Seeberger, P.H. Modular Synthesis of Heparin Oligosaccharides. Chem. Eur. J. 2003, 9, 140–169. [Google Scholar] [CrossRef]
- Tingoli, M.; Tiecco, M.; Chianelli, D.; Balducci, R.; Temperini, A. Novel Azido-Phenylselenenylation of Double Bonds. Evidence for a free-radical process. J. Org. Chem. 1991, 56, 809–6813. [Google Scholar] [CrossRef]
- Koto, S.; Asami, K.; Hirooka, M.; Nagura, K.; Takizawa, M.; Yamamoto, S.; Okamoto, N.; Sato, M.; Tajima, H.; Yoshida, T.; et al. Glycosylation using 2-Azido-3,4,6-Tri-O-Benzyl-2-Deoxy-D-Glucose, -Galactose, and -Mannose with the Aid of p-Nitrobenzenesulfonyl Chloride Silver Trifluoromethanesulfonate Triethylamine System. Bull. Chem. Soc. Jpn. 1999, 72, 765–777. [Google Scholar] [CrossRef]
- Singh, Y.; Wang, T.H.; Demchenko, A.V. Direct Glycosidation of 2-Azido-2-Deoxyglycosyl Nitrates. Eur. J. Org. Chem. 2019, 2019, 6413–6416. [Google Scholar] [CrossRef]
- Jeanneret, R.A.; Johnson, S.E.; Galan, M.C. Conformationally Constrained Glycosyl Donors as Tools to Control Glycosylation Outcomes. J. Org. Chem. 2020, 85, 15801–15826. [Google Scholar] [CrossRef]
- Bousquet, E.; Khitri, M.; Lay, L.; Nicotra, F.; Panza, L.; Russo, G. Capsular Polysaccharide of Streptococcus Pneumoniae Type 19F: Synthesis of the Repeating Unit. Carbohydr. Res. 1998, 311, 171–181. [Google Scholar] [CrossRef]
- Crich, D.; Cai, W. Chemistry of 4,6-O-Benzylidene-D-glycopyranosyl Triflates: Contrasting Behavior between the Gluco and Manno Series. J. Org. Chem. 1999, 64, 4926–4930. [Google Scholar] [CrossRef]
- van der Vorm, S.; Overkleeft, H.S.; van der Marel, G.A.; Codée, J.D.C. Stereoselectivity of Conformationally Restricted Glucosazide Donors. J. Org. Chem. 2017, 82, 4793–4811. [Google Scholar] [CrossRef]
- Xue, J.; Guo, Z. Convergent Synthesis of An Inner Core GPI of Sperm CD52. Bioorg. Med. Chem. Lett. 2002, 12, 2015–2018. [Google Scholar] [CrossRef]
- Plattner, C.; Höfener, M.; Sewald, N. One-Pot Azidochlorination of Glycals. Org. Lett. 2011, 13, 545–547. [Google Scholar] [CrossRef]
- Lemieux, R.U.; Ratcliffe, R.M. The Azidonitration of Tri-O-Acetyl-D-Galactal. Can. J. Chem. 1979, 57, 1244–1251. [Google Scholar] [CrossRef]
- Broddefalk, J.; Nilsson, U.; Kihlberg, J. An Improved Synthesis of 3,4,6-Tri-O-Acetyl-2-Azido-2-Deoxy-α-D-Galactopyranosyl Bromide: A Key Component for Synthesis of Glycopeptides and Glycolipids. J. Carbohydr. Chem. 1994, 13, 129–132. [Google Scholar] [CrossRef]
- Capila, I.; Linhardt, R.J. Heparin–Protein Interactions. Angew. Chem. Int. Ed. 2002, 41, 390–412. [Google Scholar] [CrossRef]
- Varki, A. Biological Roles of Oligosaccharides: All of the Theories are Correct. Glycobiology 1993, 3, 97–130. [Google Scholar] [CrossRef]
- Orgueira, H.A.; Bartolozzi, A.; Schell, P.; Seeberger, P.H. Conformational Locking of the Glycosyl Acceptor for Stereocontrol in the Key Step in the Synthesis of Heparin. Angew. Chem. Int. Ed. 2002, 41, 2128–2131. [Google Scholar] [CrossRef]
- Park, J.; Kawatkar, S.; Kim, J.-H.; Boons, G.-J. Stereoselective Glycosylations of 2-Azido-2-Deoxy-Glucosides using Intermediate Sulfonium Ions. Org. Lett. 2007, 9, 1959–1962. [Google Scholar] [CrossRef] [PubMed]
- Kärkkäinen, T.S.; Ravindranathan Kartha, K.P.; MacMillan, D.; Field, R.A. Iodine-Mediated Glycosylation En Route to Mucin-Related Glyco-Aminoacids and Glycopeptides. Carbohydr. Res. 2008, 343, 1830–1834. [Google Scholar] [CrossRef] [PubMed]
- Kurfiřt, M.; Lucie, Č.Š.A.; Cuřínová, P.; Hamala, V.; Karban, J. Development of α-Selective Glycosylation for the Synthesis of Deoxyfluorinated TN Antigen Analogues. J. Org. Chem. 2021, 86, 5073–5090. [Google Scholar] [CrossRef] [PubMed]
- Li, Z. Computational Study of the Influence of Cyclic Protecting Groups in Stereoselectivity of Glycosylation Reactions. Carbohydr. Res. 2010, 345, 1952–1957. [Google Scholar] [CrossRef] [PubMed]
- Kalikanda, J.; Li, Z. Study of the Stereoselectivity of 2-Azido-2-Deoxygalactosyl Donors: Remote Protecting Group Effects and Temperature Dependency. J. Org. Chem. 2011, 76, 5207–5218. [Google Scholar] [CrossRef]
- Ngoje, G.; Li, Z. Study of the Stereoselectivity of 2-Azido-2-Deoxyglucosyl Donors: Protecting Group Effects. Org. Biomol. Chem. 2013, 11, 1879–1886. [Google Scholar] [CrossRef]
- Ngoje, G.; Addae, J.; Kaur, H.; Li, Z. Development of Highly Stereoselective GalN3 Donors and Their Application in the Chemical Synthesis of Precursors of Tn Antigen. Org. Biomol. Chem. 2011, 9, 6825–6831. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Y.; Overkleeft, H.S.; van der Marel, G.A.; Codée, J.D.C. Reagent Controlled Glycosylations for the Assembly of Well-Defined Pel Oligosaccharides. J. Org. Chem. 2020, 85, 15872–15884. [Google Scholar] [CrossRef]
- Lemieux, R.U.; Nagabhushan, T.L.; O’Neill, I.K. The Reactions of Nitrosyl Chloride and Dinitrogen Tetroxide with Acetylated Glycals. Acetylated 2-Deoxy-2-Nitroso-α-D-Hexopyranosyl Chlorides and Titrates and Acetylated 2-Nitroglycals. Can. J. Chem. 1968, 46, 413–418. [Google Scholar] [CrossRef]
- Khodair, A.I.; Winterfeld, G.A.; Schmidt, R.R. Conjugate Addition of Phenols to 2-Nitrogalactal—Synthesis of O-(2-Acetamido-2-Deoxygalactosyl)Tyrosine. Eur. J. Org. Chem. 2003, 2003, 1847–1852. [Google Scholar] [CrossRef]
- Das, J.; Schmidt, R.R. Convenient Glycoside Synthesis of Amino Sugars: Michael-Type Addition to 2-Nnitro-D-Galactal. Eur. J. Org. Chem. 1998, 1998, 1609–1613. [Google Scholar] [CrossRef]
- Winterfeld, G.A.; Ito, Y.; Ogawa, T.; Schmidt, R.R. A Novel and Efficient Route towards α-GalNAc-Ser and α-GalNAc-Thr Building Blocks for Glycopeptide Synthesis. Eur. J. Org. Chem. 1999, 1999, 1167–1171. [Google Scholar] [CrossRef]
- Winterfeld, G.A.; Schmidt, R.R. Nitroglycal Concatenation: A Broadly Applicable and Efficient Approach to the Synthesis of Complex O-Glycans. Angew. Chem. Int. Ed. 2001, 40, 2654–2657. [Google Scholar] [CrossRef]
- Winterfeld, G.A.; Khodair, A.I.; Schmidt, R.R. O-glycosyl Amino Acids by 2-Nitrogalactal Concatenation—Synthesis of a Mucin-Type O-Glycan. Eur. J. Org. Chem. 2003, 2003, 1009–1021. [Google Scholar] [CrossRef]
- Winterfeld, G.A.; Das, J.; Schmidt, R.R. Convenient Synthesis of Nucleosides of 2-Deoxy-2-Nitro-D-Galactose and N-Acetyl-D-Galactosamine. Eur. J. Org. Chem. 2000, 2000, 3047–3050. [Google Scholar] [CrossRef]
- Shigeno, M.; Hayashi, K.; Nozawa-Kumada, K.; Kondo, Y. Phosphazene Base t-Bu-P4 Catalyzed Methoxy–Alkoxy Exchange Reaction on (Hetero)Arenes. Chem. Eur. J. 2019, 25, 6077–6081. [Google Scholar] [CrossRef]
- Shigeno, M.; Nakamura, R.; Hayashi, K.; Nozawa-Kumada, K.; Kondo, Y. Catalytic Amination of β-(Hetero)Arylethyl Ethers by Phosphazene Base t-Bu-P4. Org. Lett. 2019, 21, 6695–6699. [Google Scholar] [CrossRef]
- Pal, K.B.; Guo, A.; Das, M.; Báti, G.; Liu, X.-W. Superbase-Catalyzed Stereo- and Regioselective Glycosylation with 2-Nitroglycals: Facile Access to 2-Amino-2-deoxy-O-glycoside. ACS Catal. 2020, 10, 6707–6715. [Google Scholar] [CrossRef]
- Medina, S.; Harper, M.J.; Balmond, E.I.; Miranda, S.; Crisenza, G.E.M.; Coe, D.M.; McGarrigle, E.M.; Galan, M.C. Stereoselective Glycosylation of 2-Nitrogalactals Catalyzed by a Bifunctional Organocatalyst. Org. Lett. 2016, 18, 4222–4225. [Google Scholar] [CrossRef]
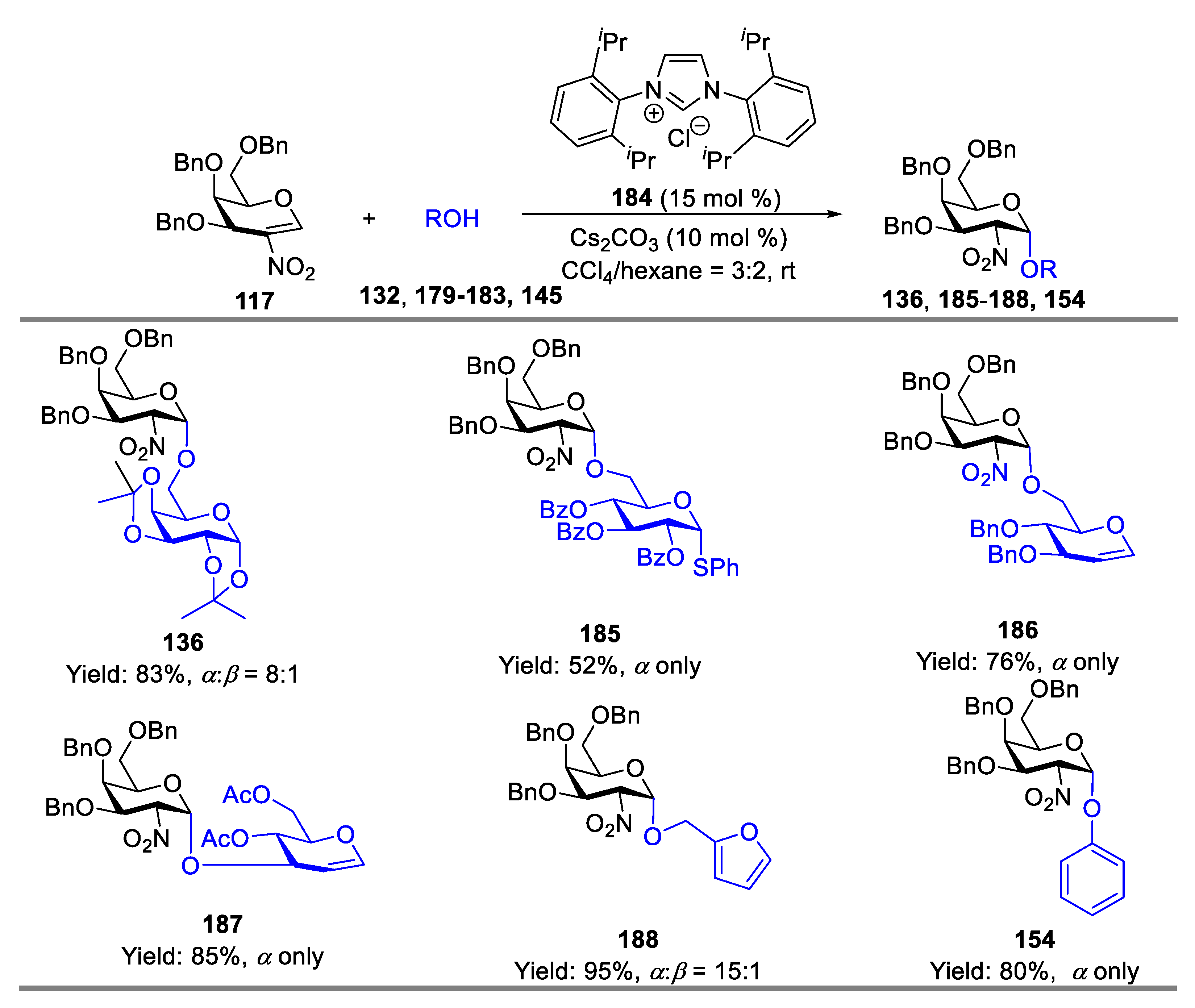
- Liu, J.-L.; Zhang, Y.-T.; Liu, H.-F.; Zhou, L.; Chen, J. N-Heterocyclic Carbene Catalyzed Stereoselective Glycosylation of 2-Nitrogalactals. Org. Lett. 2017, 19, 5272–5275. [Google Scholar] [CrossRef]
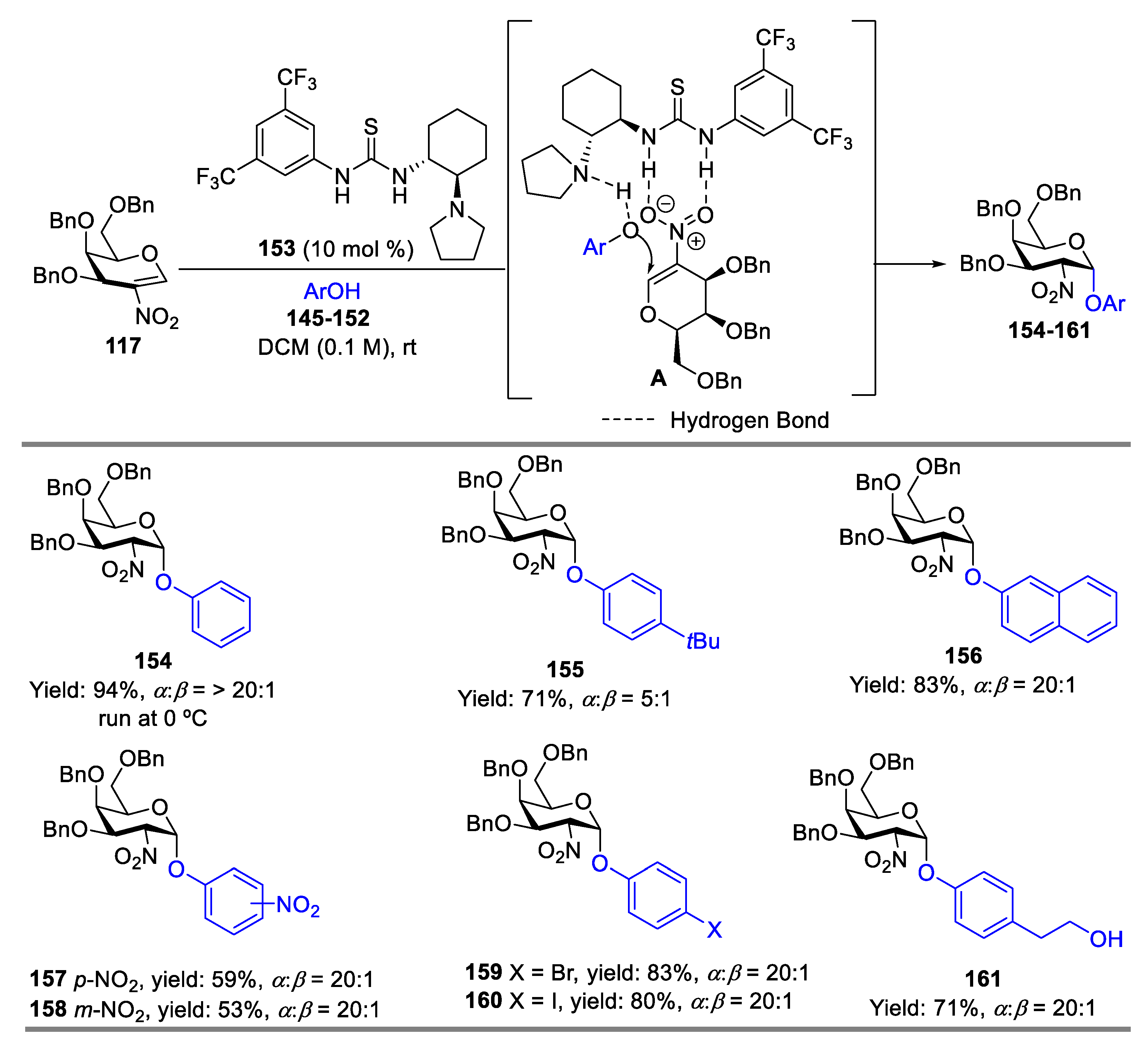
- Yoshida, K.; Kanoko, Y.; Takao, K. Kinetically Controlled α-Selective O-Glycosylation of Phenol Derivatives Using 2-Nitroglycals by a Bifunctional Chiral Thiourea Catalyst. Asian. J. Org. Chem. 2016, 5, 1230–1236. [Google Scholar] [CrossRef]
- Wan, Y.; Wu, X.; Xue, Y.; Lin, X.-E.; Wang, L.; Sun, J.-S.; Zhang, Q. Stereoselective Glycosylation with Conformation-Constrained 2-Nitroglycals as Donors and Bifunctional Thiourea as Catalyst. J. Carbohydr. Chem. 2021, 40, 535–557. [Google Scholar] [CrossRef]
- Enders, D.; Balensiefer, T. Nucleophilic Carbenes in Asymmetric Organocatalysis. Acc. Chem. Res. 2004, 37, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.; Bindu, S.; Sreekumar, V. N-Heterocyclic Carbenes: Reagents, Not Just Ligands! Angew. Chem. Int. Ed. 2004, 43, 5130–5135. [Google Scholar] [CrossRef]
- Enders, D.; Niemeier, O.; Henseler, A. Organocatalysis by N-Heterocyclic Carbenes. Chem. Rev. 2007, 107, 5606–5655. [Google Scholar] [CrossRef]
- Biju, A.T.; Kuhl, N.; Glorius, F. Extending NHC-Catalysis: Coupling Aldehydes with Unconventional Reaction Partners. Acc. Chem. Res. 2011, 44, 1182–1195. [Google Scholar] [CrossRef]
- Flanigan, D.M.; Romanov-Michailidis, F.; White, N.A.; Rovis, T. Organocatalytic Reactions Enabled by N-heterocyclic Carbenes. Chem. Rev. 2015, 115, 9307–9387. [Google Scholar] [CrossRef]
- Paul, M.; Peckelsen, K.; Thomulka, T.; Martens, J.; Berden, G.; Oomens, J.; Neudörfl, J.-M.; Breugst, M.; Meijer, A.J.H.M.; Schäfer, M.; et al. Breslow Intermediates (Amino Enols) and Their Keto Tautomers: First Gas-Phase Characterization by IR Ion Spectroscopy. Chem.Eur. J. 2021, 27, 2662–2669. [Google Scholar] [CrossRef]
- Movassaghi, M.; Schmidt, M.A. N-Heterocyclic Carbene-Catalyzed Amidation of Unactivated Esters with Amino Alcohols. Org. Lett. 2005, 7, 2453–2456. [Google Scholar] [CrossRef]
- Phillips, E.M.; Riedrich, M.; Scheidt, K.A. N-Heterocyclic Carbene-Catalyzed Conjugate Additions of Alcohols. J. Am. Chem. Soc. 2010, 132, 13179–13181. [Google Scholar] [CrossRef]
- He, L.; Guo, H.; Li, Y.-Z.; Du, G.-F.; Dai, B. N-Heterocyclic Carbene-Catalyzed Formal Cross-Coupling Reaction of α-Haloenals with Thiols: Organocatalytic Construction of sp2 Carbon–Sulfur Bonds. Chem. Commun. 2014, 50, 3719–3721. [Google Scholar] [CrossRef]
- Chen, J.; Meng, S.; Wang, L.; Tang, H.; Huang, Y. Highly Enantioselective Sulfa-Michael Addition Reactions using N-Heterocyclic Carbene as A Non-Covalent Organocatalyst. Chem. Sci. 2015, 6, 4184–4189. [Google Scholar] [CrossRef]
- Nie, Q.; Deng, L.; Tu, Y.; Liu, H.; Sun, J.-S.; Wang, L.; Zhang, Q. Stereoselective Synthesis of 1,1′-2-Amino Thiodisaccharides by Organocatalysis. Eur. J. Org. Chem. 2022, 2022, e202201019. [Google Scholar] [CrossRef]
- Wan, Y.; Zhou, M.; Wang, L.; Hu, K.; Liu, D.; Liu, H.; Sun, J.-S.; Codée, J.D.C.; Zhang, Q. Regio- and Stereoselective Organocatalyzed Relay Glycosylations to Synthesize 2-Amino-2-deoxy-1,3-dithioglycosides. Org. Lett. 2023, 25, 3611–3617. [Google Scholar] [CrossRef]
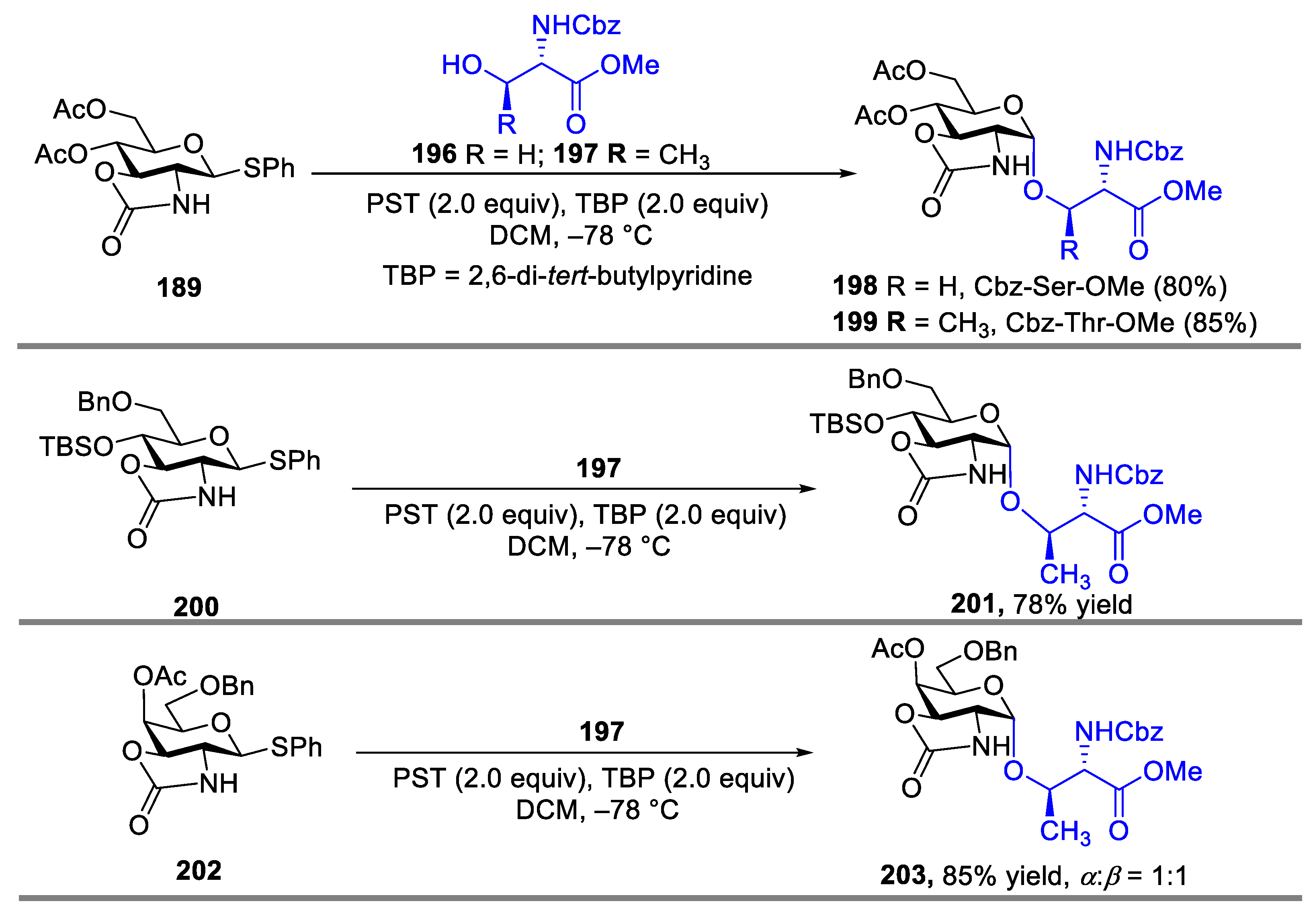
- Benakli, K.; Zha, C.; Kerns, R.J. Oxazolidinone Protected 2-Amino-2-Deoxy-D-Glucose Derivatives as Versatile Intermediates in Stereoselective Oligosaccharide Synthesis and the Formation of α-Linked glycoside. J. Am. Chem. Soc. 2001, 123, 9461–9462. [Google Scholar] [CrossRef]
- Kerns, R.J.; Zha, C.; Benakli, K.; Liang, Y.-Z. Extended Applications and Potential Limitations of Ring-Fused 2,3-Oxazolidinone Thioglycoside in Glycoconjugate Synthesis. Tetrahedron Lett. 2003, 44, 8069–8072. [Google Scholar] [CrossRef]
- Andreotti, A.H.; Kahne, D. The effects of Glycosylation on Peptide Backbone Conformation. J. Am. Chem. Soc. 1993, 115, 3352–3353. [Google Scholar] [CrossRef]
- Jiaang, W.-T.; Chang, M.-Y.; Tseng, P.-H.; Chen, S.-T. A Concise Synthesis of the O-Glycosylated Amino acid Building Block; Using Phenyl Selenoglycoside as A Glycosyl Donor. Tetrahedron Lett. 2000, 41, 3127–3130. [Google Scholar] [CrossRef]
- Miyajima, K.; Nekado, T.; Ikeda, K.; Achiwa, K. Synthesis of Tn, Sialyl Tn and HIV-1-Derived Peptide Antigen Conjugates Having a Lipid A Analog as an Immunoadjuvant for Synthetic VAaccines. Chem. Pharm. Bull. 1998, 46, 1676–1682. [Google Scholar] [CrossRef]
- Wei, P.; Kerns, R.J. Factors Affecting Stereocontrol during Glycosidation of 2,3-Oxazolidinone-Protected 1-Tolylthio-N-Acetyl-D-Glucosamine. J. Org. Chem. 2005, 70, 4195–4198. [Google Scholar] [CrossRef]
- Boysen, M.; Gemma, E.; Lahmann, M.; Oscarson, S. Ethyl 2-Acetamido-4,6-Di-O-Benzyl-2,3-N,O-Carbonyl-2-Deoxy-1-Thio-β-D-Glycopyranoside as a Versatile GlcNAc Donor. Chem. Commun. 2005, 92, 3044–3046. [Google Scholar] [CrossRef] [PubMed]
- Olsson, J.D.M.; Eriksson, L.; Lahmann, M.; Oscarson, S. Investigations of Glycosylation Reactions with 2-N-Acetyl-2N,3O-Oxazolidinone-Protected Glucosamine donors. J. Org. Chem. 2008, 73, 7181–7188. [Google Scholar] [CrossRef] [PubMed]
- Manabe, S.; Ishii, K.; Ito, Y. N-benzyl-2,3-Oxazolidinone as A Glycosyl Donor for Selective α-Glycosylation and One-Pot Oligosaccharide Synthesis Involving 1,2-cis-Glycosylation. J. Am. Chem. Soc. 2006, 128, 10666–10667. [Google Scholar] [CrossRef] [PubMed]
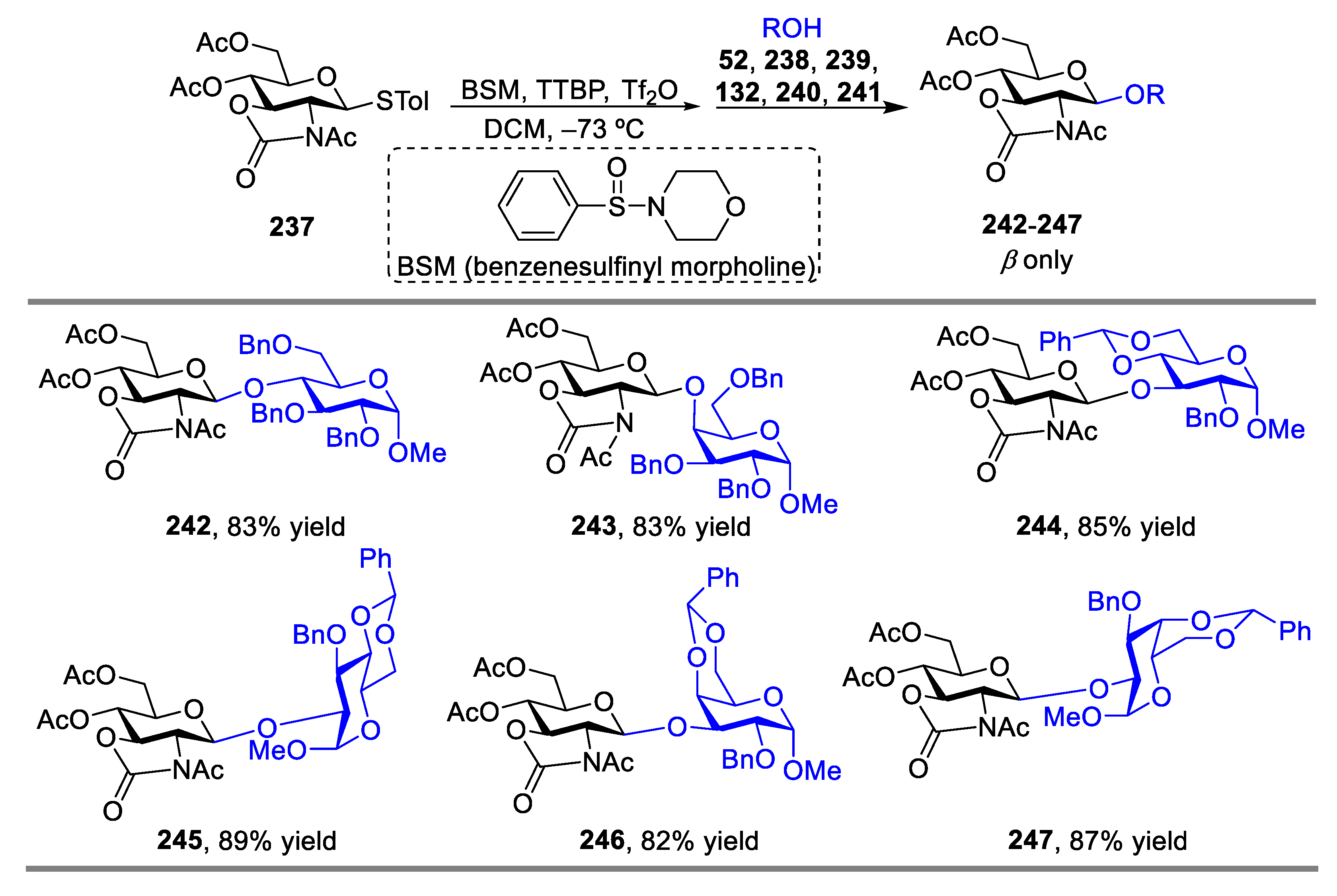
- Geng, Y.; Zhang, L.-H.; Ye, X.-S. Pre-activation Protocol Leading to Highly Stereoselectivity-Controllable Glycosylations of Oxazolidinone Protected Glucosamines. Chem. Commun. 2008, 5, 597–599. [Google Scholar] [CrossRef]
- Crich, D.; Smith, M.; Yao, Q.; Picione, J. 2,4,6-Tri-tert-butylpyrimidine (TTBP): A Cost Effective, Readily Available Alternative to the Hindered Base 2,6-Di-Tert-Butylpyridine and its 4-Substituted Derivatives in Glycosylation and Other Reactions. Synthesis 2001, 2001, 0323–0326. [Google Scholar] [CrossRef]
- Geng, Y.; Zhang, L.-H.; Ye, X.-S. Stereoselectivity Investigation on Glycosylation of Oxazolidinone Protected 2-Amino-2-Deoxy-D-Glucose Donors Based on Pre-Activation Protocol. Tetrahedron 2008, 64, 4949–4958. [Google Scholar] [CrossRef]
- Mensah, E.A.; Nguyen, H.M. Nickel-Catalyzed Stereoselective Formation of α-2-Deoxy-2-Amino glycoside. J. Am. Chem. Soc. 2009, 131, 8778–8780. [Google Scholar] [CrossRef]
- Mensah, E.A.; Yu, F.; Nguyen, H.M. Nickel-Catalyzed Stereoselective Glycosylation with C(2)-N-Substituted Benzylidene D-Glucosamine and Galactosamine Trichloroacetimidates for the Formation of 1,2-cis-2-Amino glycoside. Applications to the Synthesis of Heparin Disaccharides, GPI Anchor Pseudodisaccharides, and α-GalNAc. J. Am. Chem. Soc. 2010, 132, 14288–14302. [Google Scholar]
- Mensah, E.A.; Azzarelli, J.M.; Nguyen, H.M. Palladium-Controlled β-Selective Glycosylation in the Absence of the C(2)-Ester Participatory Group. J. Org. Chem. 2009, 74, 1650–1657. [Google Scholar]
- Marra, A.; Sinaÿ, P. N-p-methoxybenzylidene Derivatives of 2-Amino-2-Deoxy-D-Glucose as Glycosyl Donors: A Reinvestigation. Carbohydr. Res. 1990, 200, 319–337. [Google Scholar] [CrossRef]
- van Boeckel, C.A.A.; Petitou, M. The Unique Antithrombin III Binding Domain of Heparin: A Lead to New Synthetic Antithrombotics. Angew. Chem. Int. Ed. 1993, 32, 1671–1690. [Google Scholar] [CrossRef]
- Arungundram, S.; Al-Mafraji, K.; Asong, J.; Leach, F.E.; Amster, I.J.; Venot, A.; Turnbull, J.E.; Boons, G.-J. Modular Synthesis of Heparan Sulfate Oligosaccharides for Structure−Activity Relationship Studies. J. Am. Chem. Soc. 2009, 131, 17394–17405. [Google Scholar] [CrossRef]
- Paulick, M.G.; Bertozzi, C.R. The Glycosylphosphatidylinositol Anchor: A Complex Membrane-Anchoring Structure for Proteins. Biochemistry 2008, 47, 6991–7000. [Google Scholar] [CrossRef]
- Ferguson, M.A. The Structure, Biosynthesis and Functions of Glycosylphosphatidylinositol Anchors, and the Contributions of Trypanosome Research. J. Cell Sci. 1999, 112, 2799–2809. [Google Scholar] [CrossRef]
- Tiede, A.; Bastisch, I.; Schubert, J.; Orlean, P.; Schmidt, R.E. Biosynthesis of Glycosylphosphatidylinositols in Mammals and Unicellular Microbes. Biol. Chem. 1999, 380, 503–524. [Google Scholar] [CrossRef]
- Nosjean, O.; Briolay, A.; Roux, B. Mammalian GPI Proteins: Sorting, Membrane Residence and Functions. Biochim. Biophys. Acta 1997, 1331, 153–186. [Google Scholar] [CrossRef]
- Chesebro, B.; Trifilo, M.; Race, R.; Meade-White, K.; Tang, C.; LaCasse, R.; Raymond, L.; Favara, C.; Baron, G.; Priola, S.; et al. Anchorless Prion Protein Pesults in Infectious Amyloid Disease without Clinical Scrapie. Science 2005, 308, 1435–1439. [Google Scholar] [CrossRef]
- Guo, Z.; Bishop, L. Chemical synthesis of GPIs and GPI-Anchored Glycopeptides. Eur. J. Org. Chem. 2004, 2004, 3585–3596. [Google Scholar] [CrossRef]
- Udodong, U.E.; Madsen, R.; Roberts, C.; Fraser-Reid, B. A Ready, Convergent Synthesis of the Heptasaccharide GPI Membrane Anchor of Rat Brain Thy-1 Glycoprotein. J. Am. Chem. Soc. 1993, 115, 7886–7887. [Google Scholar] [CrossRef]
- Baeschlin, D.K.; Chaperon, A.R.; Green, L.G.; Hahn, M.G.; Ince, S.J.; Ley, S.V. 1,2-Diacetals in Synthesis: Total Synthesis of a Glycosylphosphatidylinositol Anchor of Trypanosoma Brucei. Chem. Eur. J. 2000, 6, 172–186. [Google Scholar] [CrossRef]
- Liu, X.; Kwon, Y.-U.; Seeberger, P.H. Convergent Synthesis of a Fully Lipidated Glycosylphosphatidylinositol Anchor of Plasmodium Falciparum. J. Am. Chem. Soc. 2005, 127, 5004–5005. [Google Scholar] [CrossRef] [PubMed]
- Swarts, B.M.; Guo, Z. Synthesis of a Glycosylphosphatidylinositol Anchor Bearing Unsaturated Lipid Chains. J. Am. Chem. Soc. 2010, 132, 6648–6650. [Google Scholar] [CrossRef] [PubMed]
- Kihlberg, J.; Eichler, E.; Bundle, D.R. The Design and Synthesis of Antibody Binding Site Probes: Three Pentasaccharide Analogues of the Brucella a Antigen Prepared by Activation in Situ of Thioglycoside with Bromine. Carbohydr. Res. 1991, 211, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Knapp, S.; Nandan, S.R. Synthesis of Capuramycin. J. Org. Chem. 1994, 59, 281–283. [Google Scholar] [CrossRef]
- Leigh, D.A.; Smart, J.P.; Truscello, A.M. Intermolecular Aglycon Transfer of Ethyl 1-Thiorhamnopyranosides under Koenigs—Knorr and Helferich Glycosylation Conditions. Carbohydr. Res. 1995, 276, 417–424. [Google Scholar] [CrossRef]
- Belot, F.; Jacquinet, J.-C. Intermolecular Aglycon Transfer of a Phenyl 1-Thiogalactosaminide Derivative under Trichloroacetimidate Glycosylation Conditions. Carbohydr. Res. 1996, 290, 79–86. [Google Scholar] [CrossRef]
- Du, Y.; Lin, J.; Linhardt, R.J. Regioselective Synthesis of L-Idopyranuronic Acid Derivatives: Intermolecular Aglycon Transfer of Dithioacetal Under Standard Glycosylation Conditions. J. Carbohydr. Chem. 1997, 16, 1327–1344. [Google Scholar] [CrossRef]
- Yu, H.; Yu, B.; Wu, X.; Hui, Y.; Han, X. Synthesis of a Group of Diosgenyl Saponins with Combined Use of Glycosyl Trichloroacetimidate and Thioglycoside Donors. J. Chem. Soc. Perkin Trans. 1 2000, 1445–1453. [Google Scholar] [CrossRef]
- Zhu, T.; Boons, G.-J. Intermolecular Aglycon Transfer of Rthyl Thioglycoside Can be Prevented by Judicious Choice of Protecting Groups. Carbohydr. Res. 2000, 329, 709–715. [Google Scholar] [CrossRef]
- Sherman, A.A.; Yudina, O.N.; Mironov, Y.V.; Sukhova, E.V.; Shashkov, A.S.; Menshov, V.M.; Nifantiev, N.E. Study of Glycosylation with N-trichloroacetyl-D-Glucosamine Derivatives in the Syntheses of the Spacer-Armed Pentasaccharides Sialyl Lacto-N-Neotetraose and Sialyl Lacto-N-Tetraose, Their Fragments, and Aanalogues. Carbohydr. Res. 2001, 336, 13–46. [Google Scholar] [CrossRef]
- Cheshev, P.E.; Kononov, L.O.; Tsvetkov, Y.E.; Shashkov, A.S.; Nifantiev, N.E. Syntheses of α- and β-Glycosyl Donors with a Disaccharide β-D-Gal-(1→3)-D-GalNAc Backbone. Russ. J. Bioorg. Chem. 2002, 28, 419–429. [Google Scholar]
- Geurtsen, R.; Boons, G.-J. Chemoselective Glycosylations of Sterically Hindered Glycosyl Acceptors. Tetrahedron Lett. 2002, 43, 9429–9431. [Google Scholar] [CrossRef]
- Tanaka, H.; Adachi, M.; Takahashi, T. Efficient Synthesis of Core 2 Class Glycosyl Amino Acids by One-Pot Glycosylation Approach. Tetrahedron Lett. 2004, 45, 1433–1436. [Google Scholar] [CrossRef]
- Xue, J.; Khaja, S.D.; Locke, R.D.; Matta, K.L. A Concise Synthesis of N-Trichloroethoxycarbonyl Lactosamine Trichloroacetimidate Donor and its Application in the Synthesis of Gal(β1-4)GlcNAc(β1-3)L-Fuc(α-OAll). Synlett 2004, 2004, 861–865. [Google Scholar] [CrossRef]
- Codée, J.D.C.; Stubba, B.; Schiattarella, M.; Overkleeft, H.S.; van Boeckel, C.A.A.; van Boom, J.H.; van der Marel, G.A. A Modular Strategy toward the Synthesis of Heparin-like Oligosaccharides Using Monomeric Building Blocks in a Sequential Glycosylation Strategy. J. Am. Chem. Soc. 2005, 127, 3767–3773. [Google Scholar] [CrossRef]
- Sun, J.; Han, X.; Yu, B. Synthesis of Anemoclemoside B, the First Natural Product with an Open-Chain Cyclic Acetal Glycosidic Linkage. Org. Lett. 2005, 7, 1935–1938. [Google Scholar] [CrossRef]
- Li, Z.; Gildersleeve, J.C. An Armed-Disarmed Approach for Blocking Aglycon Transfer of Thioglycoside. Tetrahedron Lett. 2007, 48, 559–562. [Google Scholar] [CrossRef]
- Kato, M.; Hirai, G.; Sodeoka, M. Studies on the Selectivity between Glycosylation and Intermolecular Aglycone Transfer of Thioglucoside in Synthesis of Lactose Derivatives. Chem. Lett. 2011, 40, 877–879. [Google Scholar] [CrossRef]
- Yu, B.; Tao, H. Glycosyl Trifluoroacetimidates. Part 1: Preparation and Application as New Glycosyl Donors. Tetrahedron Lett. 2001, 42, 2405–2407. [Google Scholar] [CrossRef]
- Cai, S.; Yu, B. Efficient Sialylation with Phenyltrifluoroacetimidates as Leaving Groups. Org. Lett. 2003, 5, 3827–3830. [Google Scholar] [CrossRef]
- Tanaka, H.; Iwata, Y.; Takahashi, D.; Adachi, M.; Takahashi, T. Efficient Stereoselective Synthesis of γ-N-Glycosyl Asparagines by N-Glycosylation of Primary Amide Groups. J. Am. Chem. Soc. 2005, 127, 1630–1631. [Google Scholar] [PubMed]
- Yu, F.; Nguyen, H.M. Studies on the Selectivity Between Nickel-Catalyzed 1,2-cis-2-Amin Glycosylation of Hydroxyl Groups of Thioglycoside Acceptors with C(2)-Substituted Benzylidene N-Phenyl Trifluoroacetimidates and Intermolecular Aglycon Transfer of the Sulfide Group. J. Org. Chem. 2012, 77, 7330–7343. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-T.; Gildersleeve, J.C. Mechanistic Studies and Methods to Prevent Aglycon Transfer of Thioglycoside. J. Am. Chem. Soc. 2006, 128, 11612–11619. [Google Scholar] [PubMed]
- Sakuda, S.; Zhou, Z.-Y.; Yamada, Y. Structure of a Novel Disulfide of 2-(N-Acetylcysteinyl)amido-2-Deoxy-α-D-Glucopyran-Osyl-Myo-Inositol Produced by Streptomyces sp. Biosci. Biotechnol. Biochem. 1994, 58, 1347–1348. [Google Scholar] [CrossRef] [PubMed]
- Newton, G.L.; Fahey, R.C. Mycothiol Biochemistry. Arch. Microbiol. 2002, 178, 388–394. [Google Scholar] [CrossRef]
- Newton, G.L.; Av-Gay, Y.; Fahey, R.C. A Novel Mycothiol-Dependent Detoxification Pathway in Mycobacteria Involving Mycothiol S-conjugate Amidase. Biochemistry 2000, 39, 10739. [Google Scholar]
- Ajayi, K.; Thakur, V.V.; Lapo, R.C.; Knapp, S. Intramolecular α-glucosaminidation: Synthesis of Mycothiol. Org. Lett. 2010, 12, 2630–2633. [Google Scholar]
- Chung, C.C.; Zulueta, M.M.; Padiyar, L.T.; Hung, S.C. Desymmetrization of 2,4,5,6-Tetra-O-Benzyl-D-Myo-Inositol for the Synthesis of Mycothiol. Org. Lett. 2011, 13, 5496–5499. [Google Scholar]
- Lee, S.; Rosazza, J.P. First Total Synthesis of Mycothiol and Mycothiol Disulfide. Org. Lett. 2004, 6, 365–368. [Google Scholar]
- Nicholas, G.M.; Kovác, P.; Bewley, C.A. Total Synthesis and Proof of Structure of Mycothiol Bimane. J. Am. Chem. Soc. 2002, 124, 3492–3493. [Google Scholar]
- Jardine, M.A.; Spies, H.S.; Nkambule, C.M.; Gammon, D.W.; Steenkamp, D.J. Synthesis of Mycothiol, 1D-1-O-(2-[N-acetyl-L-Cysteinyl]amino-2-Deoxy-α-D-Glucopyranosyl)-MyoInositol, Principal Low Molecular Mass Thiol in the Actinomycetes. Bioorg. Med. Chem. 2002, 10, 875–881. [Google Scholar] [CrossRef]
- McConnell, M.S.; Yu, F.; Nguyen, H.M. Nickel-Catalyzed α-glycosylation of C(1)-Hydroxyl D-Myo-Inositol: A Formal Synthesis of Mycothiol. Chem. Commun. 2013, 49, 4313–4315. [Google Scholar] [CrossRef]
- Hu, Y.-P.; Lin, S.-Y.; Huang, C.-Y.; Zulueta, M.M.L.; Liu, J.-Y.; Chang, W.; Hung, S.-C. Synthesis of 3-O-sulfonated Heparan Sulfate Octasaccharides that Inhibit the Herpes Simplex Virus Type 1 Host-Cell Interaction. Nat. Chem. 2011, 3, 557–563. [Google Scholar] [CrossRef]
- Goel, M.; Azev, V.N.; d’Alarcao, M. The Biological Activity of Structurally Defined Inositol Glycans. Future Med. Chem. 2009, 1, 95–118. [Google Scholar] [CrossRef]
- Kasahara, K.; Sanai, Y. Functional Roles of Glycosphingolipids in Signal Transduction via Lipid Rafts. Glycoconj. J. 2000, 17, 153–162. [Google Scholar] [CrossRef]
- Paulick, M.G.; Forstner, M.B.; Groves, J.T.; Bertozzi, C.R. A Chemical Approach to Unraveling the Biological Function of the Glycosylphosphatidylinositol Anchor. Proc. Natl. Acad. Sci. USA 2007, 104, 20332–20337. [Google Scholar] [CrossRef]
- McConnell, M.S.; Mensah, E.A.; Nguyen, H.M. Stereoselective α-glycosylation of C(6)-Hydroxyl Myo-Inositols via Nickel Catalysis-Application to the Synthesis of GPI Anchor Pseudo-Oligosaccharides. Carbohydr. Res. 2013, 381, 146–152. [Google Scholar] [CrossRef]
- Tsai, Y.-H.; Götze, S.; Vilotijevic, I.; Grube, M.; Silva, D.V.; Seeberger, P.H. A General and Convergent Synthesis of Diverse Glycosylphosphatidylinositol Glycolipids. Chem. Sci. 2013, 4, 468–481. [Google Scholar] [CrossRef]
- Swarts, B.M.; Guo, Z. Chapter 4—Chemical Synthesis of Glycosylphosphatidylinositol Anchors. In Advances in Carbohydrate Chemistry and Biochemistry; Horton, D., Ed.; Academic Press: Cambridge, MA, USA, 2012; pp. 137–219. [Google Scholar]
- Homans, S.W.; Ferguson, M.A.J.; Dwek, R.A.; Rademacher, T.W.; Anand, R.; Williams, A.F. Complete Structure of the Glycosyl Phosphatidylinositol Membrane Anchor of Rat Brain Thy-1 Glycoprotein. Nature 1988, 333, 269–272. [Google Scholar] [CrossRef]
- Pratt, M.R.; Bertozzi, C.R. Synthetic Glycopeptides and Glycoproteins as Tools for Biology. Chem. Soc. Rev. 2005, 34, 58–68. [Google Scholar] [CrossRef]
- Slovin, S.F.; Ragupathi, G.; Musselli, C.; Olkiewicz, K.; Verbel, D.; Kuduk, S.D.; Schwarz, J.B.; Sames, D.; Danishefsky, S.; Livingston, P.O.; et al. Fully Synthetic Carbohydrate-Based Vaccines in Biochemically Relapsed Prostate Cancer: Clinical Trial Results with α-N-Acetylgalactosamine-O-Serine/Threonine Conjugate Vaccine. J. Clin. Oncol. 2003, 21, 4292–4298. [Google Scholar] [CrossRef]
- Springer, G.F. Immunoreactive T and Tn Epitopes in Cancer Diagnosis, Prognosis, and Immunotherapy. J. Mol. Med. 1997, 75, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Dziadek, S.; Hobel, A.; Schmitt, E.; Kunz, H. A Fully Synthetic Vaccine Consisting of a Tumor-Associated Glycopeptide Antigen and a T-Cell Epitope for the Induction of a Highly Specific Humoral Immune Response. Angew. Chem. Int. Ed. 2005, 44, 7630–7635. [Google Scholar] [CrossRef] [PubMed]
- Kuduk, S.D.; Schwarz, J.B.; Chen, X.-T.; Glunz, P.W.; Sames, D.; Ragupathi, G.; Livingston, P.O.; Danishefsky, S.J. Synthetic and Immunological Studies on Clustered Modes of Mucin-Related TN and TF O-Linked Antigens: The Preparation of a Glycopeptide-Based Vaccine for Clinical Trials against Prostate Cancer. J. Am. Chem. Soc. 1998, 120, 12474–12485. [Google Scholar] [CrossRef]
- Yu, F.; McConnell, M.S.; Nguyen, H.M. Scalable Synthesis of Fmoc-Protected GalNAc-Threonine Amino Acid and TN Antigen via Nickel Catalysis. Org. Lett. 2015, 17, 2018–2021. [Google Scholar] [CrossRef] [PubMed]
- Sletten, E.T.; Ramadugu, S.K.; Nguyen, H.M. Utilization of Bench-Stable and Readily Available Nickel(II) Triflate for Access to 1,2-cis-2-Aminoglycoside. Carbohydr. Res. 2016, 435, 195–207. [Google Scholar] [CrossRef]
- Zhu, S.; Samala, G.; Sletten, E.T.; Stockdill, J.L.; Nguyen, H.M. Facile Triflic Acid-Catalyzed α-1,2-cis-Thio Glycosylations: Scope and Application to the Synthesis of S-linked Oligosaccharides, Glycolipids, Sublancin Glycopeptides, and TN/TF Antigens. Chem. Sci. 2019, 10, 10475–10480. [Google Scholar] [CrossRef]
- Zeng, J.; Sun, G.; Yao, W.; Zhu, Y.; Wang, R.; Cai, L.; Liu, K.; Zhang, Q.; Liu, X.-W.; Wan, Q. 3-Aminodeoxypyranoses in Glycosylation: Diversity-Oriented Synthesis and Assembly in Oligosaccharides. Angew. Chem. Int. Ed. 2017, 56, 5227–5231. [Google Scholar] [CrossRef]
- Zeng, J.; Sun, G.; Wang, R.; Zhang, S.; Teng, S.; Liao, Z.; Meng, L.; Wan, Q. Gold-Catalyzed Diversified Synthesis of 3-Aminosugar Analogues of Digitoxin and Digoxin. Org. Chem. Front. 2017, 4, 2450–2454. [Google Scholar] [CrossRef]
- Zeng, J.; Wang, R.; Yao, W.; Zhang, S.; Sun, G.; Liao, Z.; Meng, L.; Wan, Q. Diversified Synthesis and α-Selective Glycosylation of 3-Amino-2,3,6-Trideoxy Sugars. Org. Chem. Front. 2018, 5, 3391–3395. [Google Scholar] [CrossRef]
- Zeng, J.; Wang, R.; Zhang, S.; Fang, J.; Liu, S.; Sun, G.; Xu, B.; Xiao, Y.; Fu, D.; Zhang, W.; et al. Hydrogen-Bonding-Assisted Exogenous Nucleophilic Reagent Effect for β-Selective Glycosylation of Rare 3-Amino Sugars. J. Am. Chem. Soc. 2019, 141, 8509–8515. [Google Scholar] [CrossRef]
- Aoki, M.; Shirai, H.; Nakayama, N.; Itezono, Y.; Mori, M.; Satoh, T.; Ohshima, S.; Watanabe, J.; Yokose, K.; Seto, H. Structural Studies on Avidinorubicin, a Novel Anthracycline with Platelet Aggregation Inhibitory Activity. J. Antibiot. 1991, 44, 635–645. [Google Scholar] [CrossRef]
- Hoang, K.M.; Lees, N.R.; Herzon, S.B. General Method for the Synthesis of α- or β-Deoxyaminoglycoside Bearing Basic Nitrogen. J. Am. Chem. Soc. 2021, 143, 2777–2783. [Google Scholar] [CrossRef] [PubMed]
- Hoang, K.M.; Lees, N.R.; Herzon, S.B. Programmable Synthesis of 2-Deoxyglycoside. J. Am. Chem. Soc. 2019, 141, 8098–8103. [Google Scholar] [CrossRef]
- Hill, R.R.; Rychnovsky, S.D. Generation, Stability, and Utility of Lithium 4,4′-Di-Tert-Butylbiphenylide (LiDB). J. Org. Chem. 2016, 81, 10707–10714. [Google Scholar] [CrossRef]
- Cohen, T.; Lin, M.T. Two-Flask Preparation of Alpha.-Lithio Cyclic Ethers from Gamma.- and .Delta.-Lactones. Reductive Lithiation as a Route, via Radical Intermediates, to Axial 2-Lithiotetrahydropyrans and Their Equilibration to the Equatorial Isomers. J. Am. Chem. Soc. 1984, 106, 1130–1131. [Google Scholar] [CrossRef]
- Cohen, T.; Bhupathy, M. Organoalkali Compounds by Radical Anion Induced Reductive Metalation of Phenyl Thioethers. Acc. Chem. Res. 1989, 22, 152–161. [Google Scholar] [CrossRef]
- Rychnovsky, S.D.; Mickus, D.E. Preparation of 2-Lithiotetrahydropyrans: Kinetic and Thermodynamic Generation of Alkyllithium Reagents. Tetrahedron Lett. 1989, 30, 3011–3014. [Google Scholar] [CrossRef]
- Baryal, K.N.; Zhu, D.; Li, X.; Zhu, J. Umpolung Reactivity in the Stereoselective Synthesis of S-linked 2-Deoxyglycoside. Angew. Chem. Int. Ed. 2013, 52, 8012–8016. [Google Scholar] [CrossRef]
- Kyasa, S.; Meier, R.N.; Pardini, R.A.; Truttmann, T.K.; Kuwata, K.T.; Dussault, P.H. Synthesis of Ethers via Reaction of Carbanions and Monoperoxyacetals. J. Org. Chem. 2015, 80, 12100–12114. [Google Scholar] [CrossRef]
- Stich, H.F.; Stich, W.; Acton, A.B. Mutagenicity of Fecal Extracts from Carnivorous and Herbivorous Animals. Mutat. Res. 1980, 78, 105–112. [Google Scholar] [CrossRef]
- Lehn, J.M.; Wipff, G. Stereoelectronic Properties, Stereospecificity, and Stabilization of α-Oxa and α-Thia Carbanions. J. Am. Chem. Soc. 1976, 98, 7498–7505. [Google Scholar] [CrossRef]
- Liu, D.M.; Wang, H.L.; Lei, J.C.; Zhou, X.Y.; Yang, J.S. A Highly α-Stereoselective Sialylation Method Using 4-O-4-Nitropicoloyl Thiosialoside Donor. Eur. J. Org. Chem. 2020, 2020, 575–585. [Google Scholar] [CrossRef]
- Yu, C.S.; Niikura, K.; Lin, C.C.; Wong, C.H. The Thioglycoside and Glycosyl Phosphite of 5-Azido Sialic Acid: Excellent Donors for the α-Glycosylation of Primary Hydroxy Groups. Angew. Chem. Int. Ed. 2001, 40, 2900–2903. [Google Scholar] [CrossRef]
- Mandhapati, A.R.; Rajender, S.; Shaw, J.; Crich, D. The Isothiocyanato Moiety: An Ideal Protecting Group for the Stereoselective Synthesis of Sialic Acid glycoside and Subsequent Diversification. Angew. Chem. Int. Ed. 2015, 54, 1275–1278. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Ishiwata, A.; Ito, Y. Stereodivergent Mannosylation Using 2- O-(ortho-Tosylamido)benzyl Group. Org. Lett. 2018, 20, 4833–4837. [Google Scholar] [CrossRef]
- Escopy, S.; Geringer, S.A.; de Meo, C. Combined Effect of the Picoloyl Protecting Group and Triflic Acid in Sialylation. Org. Lett. 2017, 19, 2638–2641. [Google Scholar] [CrossRef]
- Jones, B.; Behm, A.; Shadrick, M.; Geringer, S.A.; Escopy, S.; Lohman, M.; de Meo, C. Comparative Study on the Effects of Picoloyl Groups in Sialylations Based on Their Substitution Pattern. J. Org. Chem. 2019, 84, 15052–15062. [Google Scholar] [CrossRef]
- Asressu, K.H.; Chang, C.W.; Lam, S.; Wang, C.C. Donor-Reactivity-Controlled Sialylation Reactions. Eur. J. Org. Chem. 2021, 2021, 4525–4530. [Google Scholar] [CrossRef]
- Hayashi, T.; Axer, A.; Kehr, G.; Bergander, K.; Gilmour, R. Halogen-Directed Chemical Sialylation: Pseudo-Stereodivergent Access to Marine Ganglioside Epitopes. Chem. Sci. 2020, 11, 6527–6531. [Google Scholar] [CrossRef]
- Kurimoto, K.; Yamamura, H.; Miyagawa, A. Sialylation with Systematically Protected Allyl Galactoside Acceptors Using Sialyl Phosphate Donors. Tetrahedron Lett. 2016, 57, 1442–1445. [Google Scholar] [CrossRef]
- Wang, J.; Lou, Q.; Rong, J.; Yang, Y. Gold(I)-Promoted α-selective Sialylation of Glycosyl ortho-Hexynylbenzoates for the Latent-Active Ssynthesis of Oligosialic Acids. Org. Biomol. Chem. 2019, 17, 6580–6584. [Google Scholar] [CrossRef]
- Boons, G.J.; Demchenko, A.V. Recent Advances in O-sialylation. Chem. Rev. 2000, 100, 4539–4565. [Google Scholar] [CrossRef]
- Sun, B. Recent Developments of Highly Selective Sialylation. Curr. Org. Chem. 2016, 20, 1465–1476. [Google Scholar] [CrossRef]
- Vibhute, A.M.; Komura, N.; Tanaka, H.N.; Imamura, A.; Ando, H. Advanced Chemical Methods for Stereoselective Sialylation and Their Applications in Sialoglycan Syntheses. Chem. Rec. 2021, 21, 3194–3223. [Google Scholar] [CrossRef]






























































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Xie, D.; Ma, X. Recent Advances in Chemical Synthesis of Amino Sugars. Molecules 2023, 28, 4724. https://doi.org/10.3390/molecules28124724
Yang J, Xie D, Ma X. Recent Advances in Chemical Synthesis of Amino Sugars. Molecules. 2023; 28(12):4724. https://doi.org/10.3390/molecules28124724
Chicago/Turabian StyleYang, Jian, Demeng Xie, and Xiaofeng Ma. 2023. "Recent Advances in Chemical Synthesis of Amino Sugars" Molecules 28, no. 12: 4724. https://doi.org/10.3390/molecules28124724
APA StyleYang, J., Xie, D., & Ma, X. (2023). Recent Advances in Chemical Synthesis of Amino Sugars. Molecules, 28(12), 4724. https://doi.org/10.3390/molecules28124724