Abstract
A novel anionic heptamethine cyanine (HMC) dye with two trifluoromethyl groups that selectively absorb near-infrared light is synthesized. When contrasted with previously studied anionic HMC dyes with substituents such as methyl, phenyl, and pentafluorophenyl groups, the trifluoromethylated dye displays a red-shifted maximum absorption wavelength (for instance, 948 nm in CH2Cl2) along with enhanced photostability. Furthermore, HMC dyes with broad absorption in the near-infrared region are synthesized by combining a trifluoromethylated anionic HMC dye with a cationic HMC dye as a counterion.
1. Introduction
Near-infrared (NIR)-absorbing organic dyes have garnered considerable attention for their potential applications in biology [1,2,3], transparent solar cells [4,5,6,7,8,9,10,11,12,13,14,15], optical sensors [16,17,18], and optical communications [19,20,21]. These dyes are particularly important as they allow us to take advantage of NIR light, a resource that has yet to be fully exploited. Among them, heptamethine cyanine (HMC) dye [22,23,24,25,26,27,28] is an organic dye that absorbs NIR light with excellent optical properties, such as selective absorption in the NIR region and a high molar absorption coefficient. Figure 1 illustrates the two categories of HMC dyes—cationic and anionic. Notably, extensive literature is available detailing the synthesis and various applications of cationic HMC dyes [29,30,31,32]. However, reports on anionic HMC dyes are limited [33,34,35]. Generally, anionic HMC dyes exhibit superior optical properties, including a red-shifted maximum absorption wavelength (λmax) relative to their cationic counterparts. However, comparatively lower photostability hinders their application [36,37].
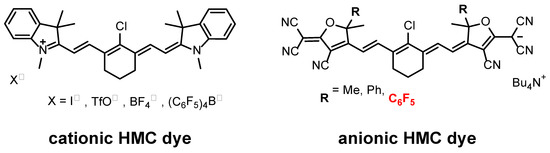
Figure 1.
Structures of HMC dyes.
We previously reported that introducing perfluorophenyl groups into an anionic HMC dye resulted in more red-shifted λmax and higher photostability of the dye than those with methyl or phenyl groups [37]. Since the mechanism of photolysis of HMC dyes is based on the addition of an electrophilic singlet oxygen to a double bond, the introduction of electron-withdrawing properties into the HMC dye backbone should be the most powerful means of improving its photostability. Among them, the introduction of the CF3 group, one of the most electron-withdrawing substituents, can be expected to be an effective means of improving the photostability of the dye. Herein, we report the synthesis and optical properties of novel anionic HMC-dye-bearing trifluoromethyl groups. Specifically, the trifluoromethylated dye achieved a more red-shifted λmax, lower highest occupied molecular orbital (HOMO) level, and higher photostability than the previously reported anionic HMC dyes.
On the other hand, the synthesis of cyanine–cyanine mixed dyes consisting of a cationic HMC skeleton and an anionic HMC skeleton to broaden the narrow absorption range of HMC dyes is an excellent method, but the optical properties and applications of cyanine–cyanine mixed dyes composed of cationic and anionic HMC skeletons to photoelectric conversion devices have been scarcely reported recently [38,39,40]. Additionally, their photostability has not been investigated so far. Therefore, a novel mixture of trifluoromethylated anionic HMC dye and cationic HMC dye was also synthesized and its absorption properties and photostability were investigated.
2. Results
2.1. Synthesis of the Anionic Dye with Trifluoromethyl Groups 5a and 8a
Anionic HMC dyes with trifluoromethyl groups were prepared as follows. First, tricyanofurans bearing trifluoromethyl groups were synthesized (Scheme 1). 2-Hydroxy-2-(trifluoromethyl)propionitrile and methylmagnesium bromide were treated in dehydrated diethyl ether (Et2O) at −20 °C. The temperature was gradually increased to 0 °C and stirred for 30 min. The reaction was quenched with 10% HCl and stirred at 25 °C for 1 h, resulting in the production of 4,4,4-trifluoro-3-hydroxy-3-methylbutan-2-one (1a), which returned a 38% yield as determined by 19F nuclear magnetic resonance (NMR) analysis [41]. A couple of reasons for the low yield of 1a may be due to the reaction not progressing sufficiently and the high volatility of 1a when the extraction solvent was removed. The ketone 1a was used in the following reaction without further purification. A mixture of 1a and 2 equiv. of malononitrile and a catalytic amount of lithium ethoxide in dehydrated THF was stirred under reflux for 8 h to obtain 2-(2-cyano-3,4-dimethyl-4-(trifluoromethyl)cyclopent-2-en-1-ylidene)malononitrile 2a at 25% yield [42]. In the synthesis of various tricyanofurans, it has been reported that only low yields can be obtained when hydroxyketones with electron-withdrawing substituents are used [43]. In the present study, the extremely strong electron-withdrawing CF3 group seems to have a significant influence on the low yield of tricyanofuran 2a.
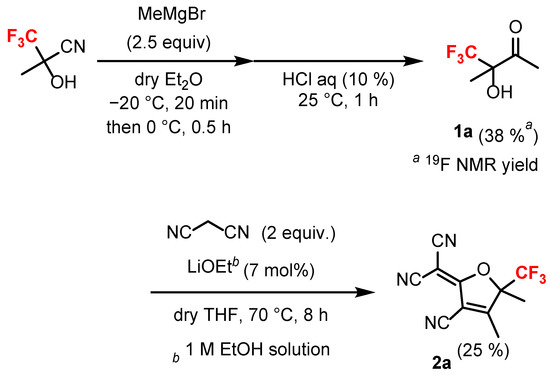
Scheme 1.
Synthesis of trifluoromethylated tricyanofuran 2a.
The sodium salt of anionic HMC dye 4a was obtained via the reaction of dialdehyde 3 [44,45] with two equiv. of trifluoromethylated tricyanofuran 2a in sodium acetate in anhydrous acetic acid at 25 °C overnight (Scheme 2). The crude sodium salt of the HMC dye 4a was used in subsequent reactions without further purification. A mixture of crude 4a and tetrabutylammonium iodide (Bu4N+I−) was stirred at 25 °C to afford the trifluoromethylated anionic HMC dye 5a with tetrabutylammonium cation. The overall yield of tricyanofuran 2a to trifluoromethylated anionic HMC dye 5a was 13% from 3. The low yield may be due to the reaction to synthesize the sodium salt 4a not progressing sufficiently.
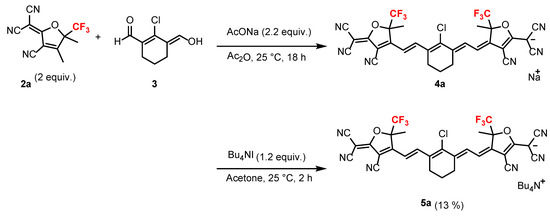
Scheme 2.
Synthesis of the trifluoromethylated anionic HMC dye 5a.
The tetrabutylammonium salt of the anionic HMC dye 5a was stirred with 1.2 equiv. of the cationic HMC dye 7 [46], which was prepared via the reaction of indolenium salt 6 [47] with dialdehyde 3 in acetone overnight at 25 °C to afford the cyanine–cyanine mixed dye 8a with a yield of 92% (Scheme 3).
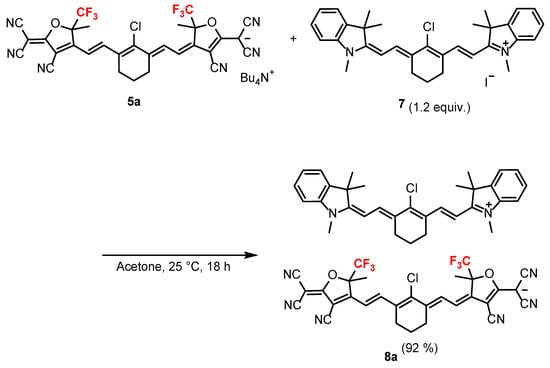
Scheme 3.
Synthesis of the cyanine–cyanine mixed dye 8a.
2.2. UV–vis–NIR Spectra and CV Measurements of Anionic HMC Dye 5a and Cyanine–Cyanine Mixed Dye 8a
The ultraviolet–visible–NIR (UV–vis–NIR) absorption spectra of the prepared trifluoromethylated anionic HMC dye 5a with Bu4N+ cations and cyanine–cyanine mixed dye 8a in a dichloromethane (CH2Cl2) solution are shown in Figure 2a for the prepared trifluoromethylated anionic HMC dye 5a and cationic HMC dye. Figure 2b shows the cyanine–cyanine mixed dye 8a. Table 1 summarizes λmax, molar absorption coefficient (ε), oxidation potential (Eox), HOMO, and lowest unoccupied molecular orbital (LUMO) levels of the previously synthesized anionic HMC dyes.
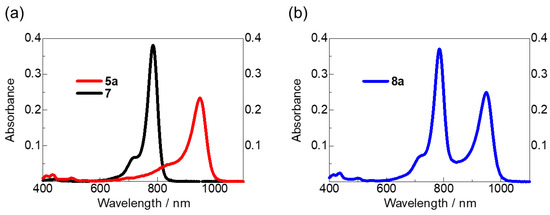
Figure 2.
UV–vis–NIR absorption spectra of the prepared anionic HMC dye 5a and cationic HMC dye 7 (a), and cyanine–cyanine dye 8a (b) in CH2Cl2 (1 × 10−6 M).

Table 1.
UV–vis–NIR absorption spectra of anionic HMC dyes 5a–d, cationic HMC dye 7, cyanine–cyanine mixed dye 8a in CH2Cl2, and electrochemical properties of each anionic HMC dye 5a–d in acetonitrile.
As a result, the λmax of the dye 5a was observed at 948 nm, with negligible absorption in the visible region. However, cyanine–cyanine mixed dye 8a showed absorption from the cationic and anionic HMC skeletons at 785 and 949 nm, respectively, giving a broader absorption range than that of anionic HMC dye 5a. Compared with the various anionic HMC dyes synthesized, 5a showed a significant red shift in λmax, stabilization of each energy level, and decreased HOMO−LUMO energy gap. These results can be attributed to the trifluoromethyl groups being stronger electron-withdrawing substituents than the substituents of other anionic HMC dyes.
2.3. The Photostabilities of Anionic HMC Dye 5a and Cyanine−Cyanine Mixed Dye 8a
The photostabilities of the anionic HMC dye 5a and cyanine–cyanine mixed dye 8a were evaluated by irradiating them with a white LED light (8.5 W, emitting blue LED + yellow phosphor, peak wavelength: 440 nm) in a CH2Cl2 solution (1.0 × 10−6 M) at 25 °C in a constant temperature chamber. The residual rates of dyes 5a and 8a, calculated from the change in absorbance at λmax in the UV–vis–NIR spectra, are illustrated in Figure 3a–d, respectively, and are compared with those of previous HMC dyes (Table 2).
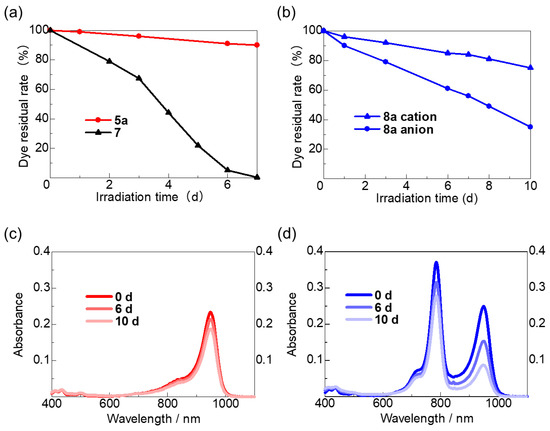
Figure 3.
Photostabilities of the prepared anionic HMC dye 5a and cyanine–cyanine mixed dye 8a in CH2Cl2 (1 × 10−6 M) under white LED irradiation (8.5 W) in an incubator at 25 °C. Changes in the residual rates of (a) 5a and cationic HMC dye 7, and (b) 8a, respectively. Changes in the absorption spectra of (c) 5a and (d) 8a over time.
Table 2.
Residual rates of anionic HMC dyes 5a–d, cationic HMC dye 7, and cyanine–cyanine mixed dye 8a in CH2Cl2 after 10 d.
The residual rate of anionic HMC dye 5a after 10 days of light irradiation was 80%. This dye showed the best photostability compared to the anionic and cationic HMC dyes we synthesized previously. The observed results can be ascribed to the enhanced electron-withdrawing properties of the trifluoromethyl groups. These groups significantly suppress the electrophilic addition of singlet oxygen to the methine chain [37].
Furthermore, when photostability tests were carried out on the cyanine–cyanine mixed dye 8a under identical conditions, it was observed that the absorption peaks originating from the anionic HMC structure diminished more rapidly compared to those from the cationic HMC structure. This is because the methine chain of the anionic HMC skeleton, which is more electron-rich than the cationic skeleton, is more readily affected by the electrophilic addition to singlet oxygen, which matches the reported photodegradation mechanism. Interestingly, the photostability of the anionic dye in the cyanine–cyanine mixed dye 8a was much lower than that of anionic HMC 5a alone. In contrast, the photostability of the cationic dye in cyanine–cyanine mixed dye 8a was significantly improved compared to that of cationic HMC 7 alone.
3. Experimental Section
3.1. Measurements
The 1H NMR spectra of the compounds were obtained at 392 or 400 MHz in CDCl3, hexadeuteroacetone ((CD3)2CO), or hexadeuterodimethyl sulfoxide ((CD3)2SO) solutions using the residual solvent as the internal standard and a JEOL ECS-400 or ECX-400P Fourier transform NMR (FT-NMR) spectrometer. The 13C NMR spectra of the compounds were obtained at 99 or 101 MHz in CDCl3, (CD3)2CO, or (CD3)2SO solutions, using the residual solvent as the internal standard, and a JEOL ECS-400 or ECX-400P FT-NMR spectrometer. The 19F NMR spectra of the compounds were obtained at 369 or 376 MHz in CDCl3 or (CD3)2CO solutions, respectively, using CFCl3 as the external standard and a JEOL ECS-400 or ECX-400P FT-NMR spectrometer. The data were reported as follows: (s = singlet, t = triplet, q = quartet, m = multiplet, br s = broad singlet, coupling constant(s), and integration). The melting points of the compounds were obtained using a Yanagimoto MP-S3 micro melting point apparatus and are uncorrected. The compounds’ infrared (IR) spectra were recorded on a Shimadzu IR Affinity-1 instrument. Electrospray ionization-mass spectroscopy (ESI–MS) and HRMS measurements were performed using a Waters Xevo quadrupole time-of-flight (QTOF) mass spectrometer. The UV–vis–NIR absorption spectra of the dyes in solution were recorded using a Hitachi U-4100 instrument. The CV profiles were obtained using an HSV-110 automatic polarization system. TG–DTA experiments were performed using an SII EXSTAR 6000 thermogravimetry differential thermal analysis (TG/DTA) 6300 apparatus under a nitrogen atmosphere after heating to 80 °C under vacuum for 18 h; the measured values were uncorrected.
3.2. Materials
Diacetyl, 2-hydroxy-2-(trifluoromethyl)propionitrile, iodomethane, malononitrile, tetrabutylammonium iodide, cyclohexanone, and 2,3,3-trimethyl-3H-indole were purchased from TCI Fine Chemicals. Bromopentafluorobenzene, N,N-dimethylformamide (DMF), phosphoryl chloride, super-dehydrated diethyl ether, and acetic anhydride were purchased from Wako Pure Chemicals. Hydrogen chloride (ca. 12 mol/L in water) and sodium acetate (AcONa) were purchased from NACALAI TESQUE, Inc., Shiga, Japan. Acetone was purchased from KANTO CHEMICAL CO., Japan. Isopropylmagnesium chloride lithium chloride complex solution, methylmagnesium bromide solution (ca. 3.0 mol/L in diethyl ether (Et2O)), and lithium ethoxide were purchased from Sigma-Aldrich Co. LLC, Tokyo, Japan.
Pure products were isolated via column chromatography using silica gel 60 (spherical, 270–325 mesh, Kanto Chemical Co., Inc.,Tokyo, Japan) or Wakogel® C-200 (100–200 mesh, FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan). Analytical thin-layer chromatography (TLC) was performed using Merck pre-coated (0.25 mm) silica gel 60 F254 plates. All chemicals were reagent-grade and purified before use if needed.
3.3. Synthesis of 4,4,4-Trifluoro-3-hydroxy-3-methylbutan-2-one (1a) [41]
2-Hydroxy-2-(trifluoromethyl)propionitrile (1.467 g, 10.125 mmol) was dissolved in Et2O super-dehydrated (14 mL) under an argon atmosphere. After that, 8.42 mL of methylmagnesium bromide (3 M in Et2O), which was cooled to −20 °C, was added dropwise to the solution. After the reaction mixture was warmed to 0 °C, it was added to 10% hydrochloric acid (60 mL) and stirred at 25 °C for 1 h. Subsequently, the reaction mixture was neutralized by adding saturated Na2CO3 aq (65 mL), and the blend was extracted with Et2O (30 mL × 3). Next, the combined organic layers were dried over anhydrous sodium sulfate (Na2SO4). The organic solvent was concentrated to obtain 4,4,4-trifluoro-3-hydroxy-3-methylbutan-2-one (1a) (1.368 g), used in subsequent reactions without further purification.
3.4. Synthesis of 2-(2-Cyano-3,4-dimethyl-4-(trifluoromethyl)cyclopent-2-en-1-ylidene)malononitrile (2a) [42]
- First, a mixture of 1a (0.601 g, 3.847 mmol) and malononitrile (0.510 g, 7.724 mmol) was dissolved in an anhydrous THF (4 mL) under an argon atmosphere. Subsequently, 0.269 mL of lithium ethoxide (1.0 M in EtOH, 0.269 mmol) was added dropwise to the solution. The reaction mixture was stirred under reflux for 8 h and then concentrated using a rotary evaporator. The residue was extracted with CH2Cl2 (30 mL × 3), washed with brine, and the combined organic layers were dried over anhydrous Na2SO4. After the organic solvent was concentrated, the crude product was purified via column chromatography on silica gel using CH2Cl2 as the solvent, followed by washing with methanol to produce 2-(2-cyano-3,4-dimethyl-4-(trifluoromethyl)cyclopent-2-en-1-ylidene)malononitrile (2a) with a yield of 25% (0.239 g).
- White solid; Yield 25%; m.p. = 160.1–161.0 °C; Rf 0.49 (CH2Cl2); IR (KBr) 2233 (C≡N) cm−1; HRMS (ESI) found: m/z 254.0555. Calc. for C16H12N3O: 254.0541; 1H NMR (CDCl3) δ 1.84 (s, 3H, –CH3), 2.46 (s, 3H, –CH3); 13C NMR (CDCl3) δ 14.7 (s), 17.6 (s), 62.6 (s), 96.2 (q, J = 32.89 Hz), 107.9 (s), 109.1 (s), 109.5 (s), 109.7 (s), 121.6 (q, J = 285.94 Hz), 172.0 (s), 173.8 (s); 19F NMR (CDCl3) δ −77.23 (s, 3F) (see S2–S4 in the Supplementary Materials).
3.5. Synthesis of (E)-2-Chloro-3-(hydroxymethylene)cyclohex-1-ene-1-carbaldehyde (3) [44,48]
- Phosphoryl chloride (1.90 mL, 20.119 mmol) was added dropwise to DMF (4 mL) at 0 °C under an argon atmosphere, and the reaction mixture was stirred for 30 min. A DMF (1 mL) solution of cyclohexanone (0.514 g, 5.080 mmol) was slowly added to the mixture at 0 °C, followed by stirring for 30 min. After the reaction mixture was heated to 55 °C, it was stirred for 4 h. Next, 150 mL of water and crushed ice was added to the reaction mixture, and the blend was stored in a refrigerator overnight. The precipitate obtained was filtered and washed with water. The yield of the prepared solid, name (E)-2-chloro-3-(hydroxymethylene)cyclohex-1-ene-1-carbaldehyde (3), was 67% (0.595 g).
- Yellow solid; Yield 67%; m.p. = 143.0–145.0 °C; IR (KBr) 1620 (C=O) cm−1; HRMS (ESI) found: m/z 173.0370. Calcd for C8H9ClO2: 173.0369; 1H NMR (DMSO-d6) δ 1.57 (quin, J = 6.17 Hz, 2H, –CH2CH2CH2–), 2.35 (s, 4H, –CH2CH2CH2–), 7.56 (br s, 1H, vinly H), 10.1 (br s, 1H, –CHO), 10.8 (br s, 1H, –OH); 13C NMR (DMSO-d6) δ 20.0 (s), 23.7 (s), 146.1 (s) (see S5 and S6 in the Supplementary Materials).
3.6. Synthesis of Sodium ((Z)-4-((E)-2-(2-Chloro-3-((E)-2-(4-cyano-5-(dicyanomethylene)-2-methyl-2-(trifluoromethyl)-2,5-dihydrofuran-3-yl)vinyl)cyclohex-2-en-1-ylidene)ethylidene)-3-cyano-5-methyl-5-(trifluoromethyl)-4,5-dihydrofuran-2-yl)dicyanomethanide (4a)
- A mixture of 2a (0.205 g, 0.810 mmol), 3 (0.069 g, 0.401 mmol), and sodium acetate (0.075 g, 0.890 mmol) in acetic anhydride (8 mL) was stirred at 25 ℃ overnight under an argon atmosphere. The reaction mixture was added to hexane (200 mL) and Et2O (15 mL), and the precipitate was filtered. The dark green solid crude product, namely sodium ((Z)-4-((E)-2-(2-chloro-3-((E)-2-(4-cyano-5-(dicyanomethylene)-2-methyl-2-(trifluoromethyl)-2,5-dihydrofuran-3-yl)vinyl)cyclohex-2-en-1-ylidene)ethylidene)-3-cyano-5-methyl-5-(trifluoromethyl)-4,5-dihydrofuran-2-yl)dicyanomethanide (4a) (0.273 g), was used for subsequent reactions without further purification.
3.7. Synthesis of Tetrabutylammonium ((Z)-4-((E)-2-(2-Chloro-3-((E)-2-(4-cyano-5-(dicyanomethylene)-2-methyl-2-(trifluoromethyl)-2,5-dihydrofuran-3-yl)vinyl)cyclohex-2-en-1-ylidene)ethylidene)-3-cyano-5-methyl-5-(trifluoromethyl)-4,5-dihydrofuran-2-yl)dicyanomethanide (5a)
- A mixture of 4a (0.273 g, 0.411 mmol) and tetrabutylammonium iodide (0.039 g, 0.452 mmol) in acetone (7 mL) was stirred at 25 °C for 2 h under an argon atmosphere. After the solvent was removed under low pressure, the residue was purified via column chromatography on silica gel using a CH2Cl2–methanol (100:1 (v/v)) solvent to obtain tetrabutylammonium ((Z)-4-((E)-2-(2-chloro-3-((E)-2-(4-cyano-5-(dicyanomethylene)-2-methyl-2-(trifluoromethyl)-2,5-dihydrofuran-3-yl)vinyl)cyclohex-2-en-1-ylidene)ethylidene)-3-cyano-5-methyl-5-(trifluoromethyl)-4,5-dihydrofuran-2-yl)dicyanomethanide (5a) with a yield of 13% (0.047 g).
- Dark green solid; Yield 13%; Tdt 238.3 °C; Rf 0.46 (CH2Cl2/methanol = 20/1); IR (KBr) 2214 (C≡N) cm−1; HRMS (ESI) found: m/z 641.0933. Calc. for C30H16ClF6N6O2: Bu4N, 641.0927; 1H NMR (Acetone-d6) δ 0.98 (td, J = 7.31, 1.35 Hz, 12H, –CH2CH2CH2CH3 × 4), 1.44 (sex, J = 7.31 Hz, 8H, –CH2CH2CH2CH3 × 4), 1.77–1.80 (m, 2H, –CH2CH2CH2–), 1.80–1.86 (m, 8H, –CH2CH2CH2CH3 × 4), 1.93 (s, 6H, –CH3 × 2), 2.59–2.73 (m, 4H, –CH2CH2CH2–), 3.41–3.45 (m, 8H, –CH2CH2CH2CH3 × 4), 6.24 (d, J = 13.91 Hz, 2H, vinyl H), 8.58 (d, J = 13.91 Hz, 2H, vinyl H); 13C NMR (Acetone-d6) δ 13.9 (s), 19.7 (s), 20.4 (s), 21.6 (s), 24.4 (s), 26.9 (s), 49.6 (s), 59.4 (s), 85.7 (s), 92.9 (q, J = 31.63 Hz), 109.4 (s), 114.0 (s), 114.6 (s), 124.0 (q, J = 282.50 Hz), 131.4(s), 137.9 (s), 141.7 (s), 149.4 (s), 156.6 (s), 177.2 (s); 19F NMR (Acetone-d6) δ −80.29 (s, 6F) (see S7–S9 and S18 in the Supplementary Materials).
3.8. Synthesis of 1,2,3,3-Tetramethyl-3H-indol-1-ium Iodide (6) [47]
- To an acetonitrile solution (5 mL) of 2,3,3-trimethyl-3H-indole (0.7883 g, 4.950 mmol), iodomethane was added (1.417 g, 9.980 mmol), and the mixture was stirred at 40 °C for 1 day. After the reaction mixture was poured into diethyl ether (75 mL), the precipitate was filtered to 1,2,3,3-tetramethyl-3H-indol-1-ium iodide (3) (1.180 g, 82%).
- Pale pink solid; Yield 82%; m.p. = 248.0–252.0 °C; IR (KBr) 1609 (C=N) cm−1; HRMS (ESI) found: m/z 174.1283 Calc. for C12H16IN: [M-I]+, 174.1254; 1H NMR (DMSO-d6) δ 1.54 (s, 6H, –CH(CH3)(CH3)), 2.79–2.80 (m, 3H, CH3), 3.99 (s, 3H, -NCH3), 7.58–7.65 (m, 2H, aryl H × 2), 7.81–7.88 (m, 1H, aryl H), 7.89–7.95 (m, 1H, aryl H); 13C NMR (DMSO-d6) δ 14.5 (s), 21.7 (s), 34.9 (s), 53.9 (s), 115.1 (s), 123.3 (s), 128.8 (s), 129.2 (s), 141.6 (s), 142.1 (s), 195.9 (s) (see S10 and S11 in the Supplementary Materials).
3.9. Synthesis of 2-((E)-2-((E)-2-Chloro-3-(2-((E)-1,3,3-trimethylindolin-2-ylidene)ethylidene)cyclohex-1-en-1-yl)vinyl)-1,3,3-trimethyl-3H-indol-1-ium Iodide (7) [46]
- (E)-2-chloro-3-(hydroxymethylene)cyclohex-1-ene-1-carbaldehyde (3) (0.085 g, 0.492 mmol) was added to a N,N-dimethylformamide solution (3 mL) of 1,2,3,3-tetramethyl-3H-indol-1-ium iodide (6) (0.3036 g, 1.001 mmol), and the mixture was stirred at 120 °C for 5 h. The reaction mixture was poured into ice water (100 mL), stirred for 30 min, and thereafter refrigerated for 30 min. The resulting precipitate was collected using suction filtration. The residue was purified using silica gel chromatography (dichloromethane/methanol = 25/1) to yield 2-((E)-2-((E)-2-chloro-3-(2-((E)-1,3,3-trimethylindolin-2-ylidene)ethylidene)cyclohex-1-en-1-yl)vinyl)-1,3,3-trimethyl-3H-indol-1-ium iodide (7) (0.229 g, 76%).
- Yellow-green solid; Yield 76%; Tdt 244.0 °C; Rf 0.25 (CH2Cl2/methanol = 25/1); IR (KBr) 1550 (C=N) cm−1; HRMS (ESI) found: m/z 483.2561 Calc. for C32H36N2Cl: [M − I−]+, 483.2567; 1H NMR (CDCl3) δ 1.77 (s, 12H, –C(CH3)2 × 2), 1.96–1.99 (m, 2H, –CH2CH2CH2–), 2.75–2.81 (m, 4H, –CH2CH2CH2–), 3.76 (s, 6H, NCH3 × 2), 6.25 (d, J = 13.91 Hz, 2H, vinyl H×2), 7.17–7.24 (m, 4H, Aryl H), 7.34–7.39 (m, 4H, Aryl H), 8.34 (d, J = 13.91 Hz, 2H, vinyl H×2); 13C NMR (CDCl3) δ 20.8 (s), 27.0 (s), 28.2 (s), 32.8 (s), 49.3 (s), 102.0 (s), 110.9 (s), 122.2 (s), 125.4 (s), 128.1 (s), 129.0 (s), 141.1 (s), 143.0 (s), 144.4 (s), 150.6 (s), 173.0 (s) (see S12, S13 and S18 in the Supplementary Materials).
3.10. Synthesis of 2-((E)-2-((E)-2-Chloro-3-(2-((E)-1,3,3-trimethylindolin-2-ylidene)ethylidene)cyclohex-1-en-1-yl)vinyl)-1,3,3-trimethyl-3H-indol-1-ium ((Z)-4-((E)-2-(2-chloro-3-((E)-2-(4-cyano-5-(dicyanomethylene)-2-methyl-2-(trifluoromethyl)-2,5-dihydrofuran-3-yl)vinyl)cyclohex-2-en-1-ylidene)ethylidene)-3-cyano-5-methyl-5-(trifluoromethyl)-4,5-dihydrofuran-2-yl)dicyanomethanide (8a)
- A mixture of 7 (0.008 g, 0.013 mmol) and 5a (0.009 g, 0.0010 mmol) in acetone (2 mL) was stirred at 25 ℃ overnight under an argon atmosphere. After the solvent was removed under low pressure, the residue was purified via column chromatography on silica gel using a CH2Cl2–methanol (200:1 (v/v)) solvent to obtain 2-((E)-2-((E)-2-chloro-3-(2-((E)-1,3,3-trimethylindolin-2-ylidene)ethylidene)cyclohex-1-en-1-yl)vinyl)-1,3,3-trimethyl-3H-indol-1-ium ((Z)-4-((E)-2-(2-chloro-3-((E)-2-(4-cyano-5-(dicyanomethylene)-2-methyl-2-(trifluoromethyl)-2,5-dihydrofuran-3-yl)vinyl)cyclohex-2-en-1-ylidene)ethylidene)-3-cyano-5-methyl-5-(trifluoromethyl)-4,5-dihydrofuran-2-yl)dicyanomethanide (8a) with a yield of 92% (0.011 g).
- Red glossy solid; Yield 92%; Tdt 201.3 °C; Rf 0.55 (CH2Cl2/methanol = 10/1); IR (KBr) 1550 (C=N), 2214 (C≡N) cm−1; HRMS (ESI) found: m/z 483.2566. Calc. for C32H36ClN2: [M]+, 483.2567, m/z 641.0931. Calc. for C30H16ClN6O2F6: [M]−, 641.0927; 1H NMR (Acetone-d6) δ 1.78 (s, 12H, –CH3 × 4), 1.83–1.88 (m, 2H, –CH2CH2CH2–), 1.93 (m, 6H, –CH3 × 2), 1.94–1.99 (m, 2H, –CH2CH2CH2–), 2.55–2.71 (m, 4H, –CH2CH2CH2–), 2.77 (t, J = 5.17 Hz, 4H, –CH2CH2CH2–), 3.80 (s, 3H, -NCH3 × 2), 6.24 (d, J = 13.91 Hz, 2H, vinyl H), 6.41 (d, J = 14.39 Hz, 2H, vinyl H), 7.32 (t, J = 6.73 Hz, 2H, aryl H), 7.41–7.49 (m, 4H, aryl H), 7.62 (d, J = 7.18 Hz, 2H, aryl H), 8.44 (d, J = 13.91 Hz, 2H, vinyl H), 8.55 (d, J = 14.39 Hz, 2H, vinyl H); 13C NMR (Acetone-d6) δ 19.6 (s), 21.6 (s), 26.9 (s), 27.0 (s), 28.1 (s), 32.0 (s), 49.6 (s), 50.1 (s), 85.8 (s), 92.8 (q, J = 31.32 Hz), 102.5 (s), 109.3 (s), 112.0 (s), 113.9 (s), 114.5 (s), 114.6 (s), 123.2 (s), 123.9 (q, J = 285.67 Hz), 126.2 (s), 127.4 (s), 130.0 (s), 131.3 (s), 141.6 (s), 142.1 (s), 144.1 (s), 144.7 (s), 149.4 (s), 150.1 (s), 156.7 (s), 174.3 (s), 177.1 (s); 19F NMR (Acetone-d6) δ −80.25 (s, 6F) (see S14–S16 and S18 in the Supplementary Materials).
3.11. Electrochemical Measurements of the Dyes
Electrochemical measurements of the dyes were performed in MeCN solutions (1.0 × 10−3 M) containing Bu4NClO4 (0.1 M). The Eox values were measured using three small electrodes. A silver quasi-reference electrode, a platinum wire, and a carbon electrode were used as the reference, counter, and working electrodes, respectively. All the electrode potentials were calibrated concerning the Fc/ferrocenium redox couple. Electrochemical measurements were performed at a scan rate of 200 mV s−1. The Eox value of Fc vs. SCE was 0.380 V [48]. The Eox values vs. SCE were determined using the observed Eox (V vs. Ag) values of the dyes in MeCN solutions as follows:
Eox (V vs. SCE) = E (V vs. Ag, observed value) + 0.380 − (measured Eox value of Fc for Ag in the MeCN solution).
The energy of the HOMO (eV) was obtained using the Eox (V vs. SCE) values as follows [48]:
HOMO (eV) = −(Eox (V vs. SCE) + 4.4)
The band gap (E0-0) and energy of the LUMO (eV) were calculated using the λonsetabs value as follows:
E0-0 (eV) = 1240/λonsetabs (nm)
LUMO (eV) = HOMO (eV) − E0-0 (eV)
3.12. Methods for Evaluating Photostability
CH2Cl2 solutions of the dyes were maintained in an incubator at 25 °C and irradiated with white LED light (8.5 W).
4. Conclusions
We synthesized a novel anionic HMC dye 5a with trifluoromethyl groups in the dye skeleton and compared its properties with those of our previously synthesized anionic HMC dyes. The new anionic HMC dye 5a showed a more red-shifted absorption wavelength and improved photostability than our previously synthesized anionic HMC dyes. These properties are attributed to the electron-withdrawing characteristics of the trifluoromethyl groups of 5a, which are more potent than the substituents of the previously synthesized dyes.
We proceeded to synthesize a cyanine–cyanine mixed dye, 8a, composed of an anionic HMC skeleton bearing trifluoromethyl groups and a cationic HMC skeleton. Subsequent investigations into its absorption properties and photostability were conducted. Our findings revealed that the unique properties of both the anionic and cationic HMC skeletons were distinctly represented in this compound. In the cyanine–cyanine mixed dye 8a, the photostability of the cationic HMC skeleton was enhanced by the photodegradation of the anionic HMC skeleton. The enhanced properties of compounds 5a and 8a indicated their potential suitability for photovoltaic devices, thus presenting a significant advantage.
We are currently investigating organic solar cells that utilize only near-infrared light using HMC dyes with CF3 groups.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28124650/s1, NMR, IR, HRMS, Cyclic voltammograms, and TG–DTA.
Author Contributions
Conceptualization, K.F.; Methodology, H.M.; Validation, Y.K., T.I. and K.F.; Formal analysis, H.M.; Investigation, H.M.; Writing—original draft, H.M.; Writing—review & editing, K.F.; Supervision, K.F.; Project administration, K.F. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partially supported by JSPS KAKENHI Grant Numbers JP20K05647 and JST Adaptable and Seamless Technology transfer Program through Target-driven R&D (A-STEP) Grant Number JPMJTM22CP to K.F.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
There are no conflict of interest to declare.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Tian, Y.; Yin, D.; Yan, L. J-aggregation strategy of organic dyes for near-infrared bioimaging and fluorescent image-guided phototherapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2023, 15, e1831. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Han, X.; Wang, M.; Li, C.; Jia, T.; Zhao, X. Recent advances in the development of near-infrared organic photothermal agents. Chem. Eng. J. 2021, 417, 128844. [Google Scholar] [CrossRef]
- Li, B.; Zhao, M.; Zhang, F. Rational design of near-infrared-ii organic molecular dyes for bioimaging and biosensing. ACS Materials Lett. 2020, 2, 905–917. [Google Scholar] [CrossRef]
- Meng, D.; Zheng, R.; Zhao, Y.; Zhang, E.; Dou, L.; Yang, Y. Near-infrared materials: The turning point of organic photovoltaics. Adv. Mater. 2022, 34, 2107330. [Google Scholar] [CrossRef]
- Bar, N.; Chowdhury, P. A brief review on advances in rhodamine b based chromic materials and their prospects. ACS Appl. Electron. Mater. 2022, 4, 3749–3771. [Google Scholar] [CrossRef]
- Uno, K.; Kim, D.; Bucevicius, J.; Bossi, M.L.; Belov, V.N.; Hell, S.W. Synthesis, structure–property relationships and absorbance modulation of highly asymmetric photochromes with variable oxidation and substitution patterns. Org. Chem. Front. 2022, 9, 6295–6304. [Google Scholar] [CrossRef]
- Ochi, J.; Tanaka, K.; Chujo, Y. Recent progress in the development of solid-state luminescent o-carboranes with stimuli responsivity. Angew. Chem. Int. Ed. 2020, 59, 9841–9855. [Google Scholar] [CrossRef]
- Abdollahi, A.; Mamaqani, H.R.; Razavi, B.; Kalajahi, M.S. Photoluminescent and chromic nanomaterials for anticounterfeiting technologies: Recent advances and future challenges. ACS Nano 2020, 14, 14417–14492. [Google Scholar] [CrossRef]
- Peng, Z.; Lin, Q.; Tai, Y.-A.A.; Wang, Y. Applications of Cellulose Nanomaterials in Stimuli-Responsive Optics. J. Agric. Food Chem. 2020, 68, 12940–12955. [Google Scholar] [CrossRef]
- Naren, G.; Li, S.; Andreasson, J. A simplicity-guided cocktail approach toward multicolor fluorescent systems. Chem. Commun. 2020, 56, 3377–3380. [Google Scholar] [CrossRef]
- Ikeya, M.; Katada, G.; Ito, S. Tunable mechanochromic luminescence of 2-alkyl-4-(pyren-1-yl)thiophenes: Controlling the self-recovering properties and the range of chromism. Chem. Commun. 2019, 55, 12296–12299. [Google Scholar] [CrossRef] [PubMed]
- Traverse, C.J.; Young, M.; Suddard-Bangsund, J.; Partrick, T.; Bates, M.; Chen, P.; Wingate, B.; Lunt, S.Y.; Anctil, A.; Lunt, R.R. Anions for near-infrared selective organic salt photovoltaics. Sci. Rep. 2017, 7, 16399. [Google Scholar] [CrossRef] [PubMed]
- Young, M.; Suddard-Bangsund, J.; Patrick, T.J.; Pajares, N.; Traverse, C.J.; Barr, M.C.; Lunt, S.Y.; Lunt, R.R. Organic heptamethine salts for photovoltaics and detectors with near-infrared photoresponse up to 1600 nm. Adv. Optical Mater. 2016, 4, 1028–1033. [Google Scholar] [CrossRef]
- Funabiki, K.; Mase, H.; Hibino, A.; Tanaka, N.; Mizuhata, N.; Sakuragi, Y.; Nakashima, A.; Yoshida, T.; Kubota, Y.; Matsui, M. Synthesis of a novel heptamethine–cyanine dye for use in near-infrared active dye-sensitized solar cells with porous zinc oxide prepared at low temperature. Energy Environ. Sci. 2011, 4, 2186–2192. [Google Scholar] [CrossRef]
- Peters, A.; Branda, N.R. Enantioselective michael reaction of malonates to nitroolefins catalyzed by bifunctional organocatalysts. J. Am. Chem. Soc. 2003, 125, 3404–3405. [Google Scholar] [CrossRef]
- Wang, Y.; Kublitski, J.; Xing, S.; Dollinger, F.; Spoltore, D.; Benduhn, J.; Leo, K. Narrowband organic photodetectors-towards miniaturized, spectroscopic sensing. Mater. Horiz. 2022, 9, 220–251. [Google Scholar] [CrossRef]
- Zhang, H.; Jenatsch, S.; Jonghe, J.D.; Nuesch, F.; Steim, R.; Veron, A.C.; Hany, R. Transparent organic photodetector using a near-infrared absorbing cyanine dye. Sci. Rep. 2015, 5, 9439. [Google Scholar] [CrossRef]
- Wang, S.; Yan, X.; Cheng, Z.; Zhang, H.; Liu, Y.; Wang, Y. Highly efficient near-infrared delayed fluorescence organic light emitting diodes using a phenanthrene-based charge-transfer compound. Angew. Chem. Int. Ed. 2015, 54, 13068–13072. [Google Scholar] [CrossRef]
- Hao, Q.; Li, Z.J.; Bai, B.; Zhang, X.; Zhong, Y.W.; Wan, L.J.; Wang, D. A covalent organic framework film for three-state near-infrared electrochromism and a molecular logic gate. Angew. Chem. Int. Ed. 2021, 60, 12498–12503. [Google Scholar] [CrossRef]
- Cheng, X.; Wang, K.; Huang, S.; Zhang, H.; Wang, Y.Y. Organic crystals with near-infrared amplified spontaneous emissions based on 2’-hydroxychalcone derivatives: Subtle structure modification but great property change. Angew. Chem. Int. Ed. 2015, 54, 8369–8373. [Google Scholar] [CrossRef]
- Cai, K.; Xie, J.; Zhao, D. Equivalent circuits of a self-assembled monolayer-based tunnel junction determined by impedance spectroscopy. J. Am. Chem. Soc. 2014, 136, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, N.G.; Braga, C.A.; Camera, V.S.; Duarte, R.C.; Rodembusch, F.S. Near-infrared fluorophores based on heptamethine cyanine dyes: From their synthesis and photophysical properties to recent optical sensing and bioimaging applications. Asian J. Org. Chem. 2022, 11, e202200095. [Google Scholar] [CrossRef]
- Feng, L.; Chen, W.; Ma, X.; Liu, S.H.; Yin, J. Near-infrared heptamethine cyanines (Cy7): From structure, property to application. Org. Biomol. Chem. 2020, 18, 9385–9397. [Google Scholar] [CrossRef]
- Broadwater, D.; Medeiros, H.C.D.; Bates, M.; Roshanzadeh, A.; Teoh, S.T.; Ogrodzinski, M.P.; Borhan, B.; Luont, R.R.; Lunt, S.Y. Counterion tuning of near-infrared organic salts dictates phototoxicity to inhibit tumor growth. ACS Appl. Mater. Interfaces 2022, 14, 53511–53522. [Google Scholar] [CrossRef]
- She, Z.; Chen, J.; Sun, L.; Zeng, F.; Wu, S. An NO-responsive probe for detecting acute inflammation using NIR-II fluorescence/optoacoustic imaging. Chem. Commun. 2022, 58, 13123–13126. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, H.; Matikonda, S.S.; Usama, S.M.; Furusawa, A.; Kato, T.; Stackova, L.; Klan, P.; Kobayashi, H.; Schnermann, M.J. Cyanine phototruncation enables spatiotemporal cell labeling. J. Am. Chem. Soc. 2022, 144, 11075–11080. [Google Scholar] [CrossRef]
- Zhao, X.; Zhao, H.; Wang, S.; Fan, Z.; Ma, Y.; Yin, Y.; Wang, W.; Xi, R.; Meng, M. A tumor-targeting near-infrared heptamethine cyanine photosensitizer with twisted molecular structure for enhanced imaging-guided cancer phototherapy. J. Am. Chem. Soc. 2021, 143, 20828–20836. [Google Scholar] [CrossRef]
- Naim, W.; Novelli, V.; Nikolinakos, I.; Barbero, N.; Dzeba, I.; Grifoni, F.; Ren, Y.; Alnasser, T.; Velardo, A.; Borrelli, R.; et al. Transparent and colorless dye-sensitized solar cells exceeding 75% average visible transmittance. JACS Au. 2021, 1, 409–426. [Google Scholar] [CrossRef]
- Li, D.H.; Smith, B.D. Supramolecular mitigation of the cyanine limit problem. J. Org. Chem. 2022, 87, 5893–5903. [Google Scholar] [CrossRef]
- Sato, R.; Machida, S.; Sohmiya, M.; Sugahara, Y.; Guegan, R. Intercalation of a cationic cyanine dye assisted by anionic surfactants within Mg−Al layered double hydroxide. ACS Omega 2021, 6, 23837–23845. [Google Scholar] [CrossRef]
- Qi, Y.; Ndaleh, D.; Meador, W.E.; Delcamp, J.H.; Hill, G.; Pradhan, N.R.; Dai, Q. Interface passivation of inverted perovskite solar cells by dye molecules. ACS Appl. Energy Mater. 2021, 4, 9525–9533. [Google Scholar] [CrossRef]
- Guralchuk, G.Y.; Katrunov, I.K.; Grynyov, R.S.; Sorokin, A.V.; Yefimova, S.L.; Borovoy, I.A.; Malyukin, Y.V. Anomalous surfactant-induced enhancement of luminescence quantum yield of cyanine dye J-aggregates. J. Phys. Chem. C 2008, 112, 14762–14768. [Google Scholar] [CrossRef]
- Li, Z.; Syed, A.A.; Zhao, P.; Yang, J.C.; Sharma, R.; Ensley, T.R.; Matichak, J.D.; Davydenko, I.; Jang, S.H.; Hagan, D.J.; et al. Cationic polyelectrolyte for anionic cyanines: An efficient way to translate molecular properties into material properties. J. Am. Chem. Soc. 2019, 141, 17331–17336. [Google Scholar] [CrossRef] [PubMed]
- Villegas, C.; Krokos, E.; Bouit, P.A.; Delgaro, J.L.; Guldi, D.M.; Martin, N. Efficient light harvesting anionic heptamethine cyanine–[60] and [70]fullerene hybrids. Energy Environ. Sci. 2011, 4, 679–684. [Google Scholar] [CrossRef]
- Bouit, P.A.; Piazza, E.D.; Rigaut, S.; Guennic, B.L.; Aronica, C.; Toupet, L.; Andraud, C.; Maury, O. Stable near-infrared anionic polymethine dyes: Structure, photophysical, and redox properties. Org. Lett. 2008, 10, 4159–4162. [Google Scholar] [CrossRef]
- Li, Y.; Ma, T.; Jiang, H.; Li, W.; Tian, D.; Zhu, J.; Li, Z. Anionic cyanine J-type aggregate nanoparticles with enhanced photosensitization for mitochondria-targeting tumor phototherapy. Angew. Chem. Int. Ed. 2022, 61, e202203093. [Google Scholar]
- Arisawa, Y.; Kubota, Y.; Inuzuka, T.; Funabiki, K. Photostability and halochromic properties of near-infrared absorbing anionic heptamethine cyanine dyes. ChemistrySelect 2022, 7, e202104213. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, P.; Tofighi, S.; Sharma, R.; Ensley, T.R.; Jang, S.H.; Hagan, D.J.; Stryland, E.W.V.; Jen, A.K.-Y. Zwitterionic cyanine–cyanine salt: Structure and optical properties. J. Phys. Chem. C 2016, 120, 15378–15384. [Google Scholar] [CrossRef]
- Li, Z.; Mukhopadhyay, S.; Jang, S.H.; Bredas, J.L.; Jen, A.K.-Y. Supramolecular assembly of complementary cyanine salt J-aggregates. J. Am. Chem. Soc. 2015, 137, 11920–11923. [Google Scholar] [CrossRef]
- Bouit, P.-A.; Rauh, D.; Neugebauer, S.; Delgado, J.L.; Pizza, E.D.; Rigaut, S.; Maury, O.; Andraud, C.; Dyakonov, V.; Martin, N.A. A “cyanine−cyanine” salt exhibiting photovoltaic properties. Org. Lett. 2009, 11, 4806–4809. [Google Scholar] [CrossRef]
- Tarrant, P.; Taylor, R.E. Fluoroolefins. VII. The synthesis of 2-trifluoromethyl-1,3-butadiene1. J. Org. Chem. 1959, 24, 1888–1890. [Google Scholar] [CrossRef]
- Liu, S.; Haller, M.A.; Ma, H.; Dalton, L.R.; Jang, S.-H.; Jen, A.K.-Y. Focused microwave-assisted synthesis of 2,5-dihydrofuran derivatives as electron acceptors for highly efficient nonlinear optical chromophores. Adv. Mater. 2003, 15, 603–607. [Google Scholar] [CrossRef]
- He, M.; Leslie, T.M.; Sinicrop, J.A. α-Hydroxy Ketone Precursors Leading to a Novel Class of Electro-optic Acceptors. Chem. Mater. 2002, 14, 2393–2400. [Google Scholar] [CrossRef]
- Carrera, M.; De Coen, L.; Coppens, M.; Dermaut, W.; Stevens, C.V. A Vilsmeier Chloroformylation by Continuous Flow Chemistry. Org. Process Res. Dev. 2020, 24, 2260–2265. [Google Scholar] [CrossRef]
- Mukherjee, A.; Saha, P.C.; Das, R.S.; Bera, T.; Guha, S. Acidic pH-Activatable Visible to Near-Infrared Switchable Ratiometric Fluorescent Probe for Live-Cell Lysosome Targeted Imaging. ACS Sens. 2021, 6, 2141–2146. [Google Scholar] [CrossRef] [PubMed]
- Shibayama, M.; Uehashi, Y.; Ajioka, S.; Kubota, Y.; Inuzuka, T.; Funabiki, K. Vapochromism of indolenine-based heptamethine cyanine dye adsorbed on silica gel. New J. Chem. 2023, 47, 5262–5269. [Google Scholar] [CrossRef]
- Burdette, M.K.; Jenkins, R.; Bandera, Y.P.; Jones, H.; Foulger, I.K.; Dickey, A.; Nieminen, A.-L.; Foulger, S.H. Click-Engineered, Bioresponsive, and Versatile Particle–Protein–Dye System. ACS Appl. Bio Mater. 2019, 2, 3183–3193. [Google Scholar] [CrossRef]
- Connelly, N.G.; Geiger, W.E. Chemical redox agents for organometallic chemistry. Chem. Rev. 1996, 96, 877–910. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).