Abstract
A simple and straightforward addition or defluorination of α-(trifluoromethyl)styrenes with 2-nitroimino-imidazolidine (2a), 2-(nitromethylene)imidazolidine (2b), 2-cyanoimino-thiazolidine (2c), and (E)-1-methyl-2-nitroguanidine (2d), in a controlled manner, was developed. The hydroamination of α-(trifluoromethyl)styrenes with 2a, 2b, 2c, and 2d was completed in the presence of DBN at room temperature within 0.5–6 h, affording structurally diverse β-trifluoromethyl-β-arylethyl analogues of neonicotinoids in moderate to good yields. The γ,γ-difluoro-β-arylallyl analogues of neonicotinoids were also successfully synthesized via defluorination of α-(trifluoromethyl)styrenes, with 2a and 2c using NaH as base at an elevated temperature together with a prolonged reaction time of 12 h. The method features simple reaction setup, mild reaction conditions, broad substrate scope, high functional group compatibility, and easy scalability.
1. Introduction
Neonicotinoid insecticides, which act as insect nicotinic acetylcholine receptor (nAChR) agonists, are one of the most important classes of insecticides, and are used for crop protection and veterinary pest control due to their supreme insecticidal ability, broad insecticidal spectrum, mammalian safety, and unique mode of action [1,2,3,4]. Since the first commercialized neonicotinoid insecticide, Imidacloprid (IMI), was launched in 1991 by Bayer CropScience, considerable efforts have been made on the development of novel neonicotinoid insecticides with high insecticidal activity and low toxicity to mammalians, and thus other first, second, and third generation neonicotinoids have subsequently been taken into the market (Figure 1) [5,6,7,8].
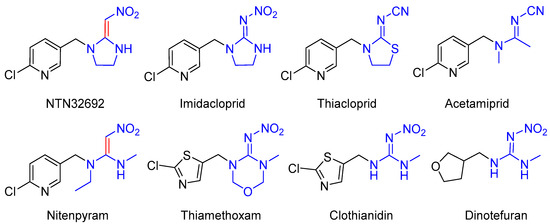
Figure 1.
The lead compound NTN32692 and commercialized neonicotinoid insecticides.
Generally, the structure of neonicotinoid insecticides is composed of four segments: an aromatic heterocycle or heteroalicycle, a nitrogen-containing heteroalicycle or guanidine/amidine, an electron-withdrawing functional group, and flexible linkage such as the methylene group (–CH2–) [9,10,11,12]. Until now, most research has focused overwhelmingly on the modification of the former three moieties, whereas optimization studies on the methylene group (–CH2–) are very scarce and only sporadic examples have been reported (Figure 2) [13,14,15,16,17]. In addition, a survey of the literature reveals that only rare examples of fluorine-containing neonicotinoid insecticides have been described [18].
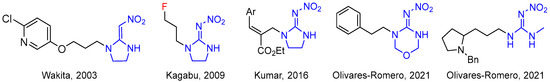
Figure 2.
The modifications of linkages of neonicotinoid insecticides [13,14,15,16,17].
α-(Trifluoromethyl)styrene and its derivatives are useful and versatile trifluoromethyl-containing building blocks for the synthesis of various fluorine-containing organic molecules [19,20,21,22]. With the development of defluorinative functionalization of α-(trifluoromethyl)styrene, a great number of methods for the synthesis of gem-difluoroalkenes have been reported [23,24,25,26]. They became one of the most straightforward approaches to gem-difluoroalkenes, which might be ascribed to the fact that the carbon–fluorine of the α-(trifluoromethyl)styrenes easily undergoes β-fluoride elimination [27,28,29,30]. However, addition reaction of α-(trifluoromethyl)styrenes without fluoride elimination remains largely elusive [31,32,33,34,35,36]. The reaction of α-(trifluoromethyl)styrenes with different nitrogen nucleophiles might proceed via three pathways: the SN2′ type of defluorinative addition/elimination (Scheme 1a) [37], sequential ipso/γ-selective defluorinative amination (Scheme 1b,c) [38,39], or hydroamination (Scheme 1d) [40]. Although conceptually simple, the reaction outcomes of α-(trifluoromethyl)styrenes with nitrogen nucleophiles are still mainly based on empirical analysis and experiences, and no general rule for predicting the products has been established. The reaction pathway, product distribution, and regioselectivity of the reaction are remarkably dependent on the electronic nature and position of the substituents on the phenyl ring of α-(trifluoromethyl)styrenes, the type of nitrogen nucleophiles, and the reaction conditions employed.
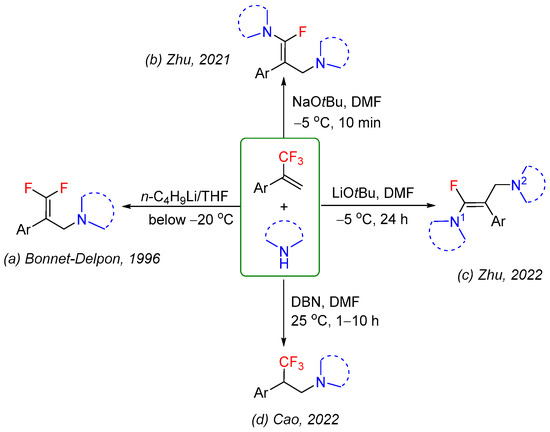
Scheme 1.
The reaction of α-(trifluoromethyl)styrenes with nitrogen nucleophiles under the different reaction conditions [37,38,39,40].
Based on the above-mentioned considerations, we envisaged that the modification of flexible linkage of neonicotinoid insecticide by the incorporation of fluorine-containing groups into neonicotinoid might be realized through the nucleophilic addition or defluorination reaction between α-(trifluoromethyl)styrenes and nitrogen-containing heteroalicycles or guanidine under controlled reaction conditions. In this paper, we developed a facile and practical method for the synthesis of trifluoromethyl or gem-difluorovinyl-containing neonicotinoid analogs via hydroamination or mono-defluorinative amination of α-(trifluoromethyl)styrenes with different nitrogen-containing heteroalicycles or guanidine, respectively, in a controlled manner under basic conditions (Scheme 2).
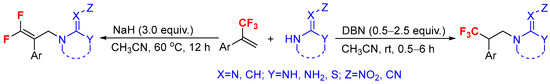
Scheme 2.
Amination of α-(trifluoromethyl)styrenes with nitrogen nucleophiles (this work).
2. Results and Discussion
We began our investigation using the reaction of 4-(3,3,3-trifluoroprop-1-en-2-yl)-1,1′-biphenyl 1a with 2-nitroimino-imidazolidine 2a as the model reaction to optimize the reaction conditions (Table 1). Generally, the product distribution of the amination was highly dependent on the base employed. Thus, our first effort focused on the influence of the base on the outcome of this reaction. A mixture of unidentified byproducts was observed when LiHMDS was used as base (entry 1). Other inorganic bases such as KOH, Cs2CO3, and KOtBu gave a mixture of addition product 3aa and defluorination 4aa (entries 2–4). Among various organic bases examined, only DBN was found to be the most acceptable base for this hydroamination, providing 3aa in 97% yield (entry 13). The competing defluorination reaction was suppressed completely and no defluorinative product 4aa was detected. When the base was changed from DBN to TMG, TBD, and DBU, the product yields of 3aa decreased significantly (entries 10–12). Other organic bases such as Et3N, TMEDA, DIPEA, DMAP, and DABCO, all resulted in no reaction (entries 5–9).

Table 1.
Optimization of reaction conditions a.
Further screening of the solvents indicated that CH3CN, THF, and NMP could afford excellent yields of 3aa (entries 13, 18, and 19), whereas the use of other solvents, such as DMSO, CH2Cl2, and toluene, resulted in lower yields (entries 15–17). When the polar protic solvent CH3OH was used as solvent, only a small amount of the desired product 3aa was formed (entry 14). To our delight, decreasing the amount of DBN from 3.0 to 2.5 equivalents would also provide 3aa in excellent yield (97%, entry 20). Further decreasing the amount of DBN led to significant decrease in the yield (82%, entry 21). In addition, when the reaction was performed at 25 °C using NaH as a base for 12 h, defluorinative amination product 4aa was produced in 30% yield, whereas hydroamination product 3aa was not detected (entry 22). It was pleasing to find that elevating the reaction temperature to 40 °C and 60 °C significantly improved the yield of 4aa, to 43% and 71%, respectively (entries 23 and 24).
With the optimized reaction conditions in hand (Table 1, entry 20), the substrate scope of this novel hydroamination was then investigated. As shown in Scheme 3, when a series of α-(trifluoromethyl)styrenes were treated with 2-nitroimino-imidazolidine 2a, the reactions proceeded smoothly to deliver the corresponding addition products in moderate to good yields. Generally, α-(trifluoromethyl)styrenes bearing electron-withdrawing groups on the phenyl ring are more favorable for this conversion than electron-donating groups (3ca versus 3fa). However, the α-(trifluoromethyl)styrene having a strong electron-donating group such as CH3O was found to be an unsuitable substrate, and only a trace amount of addition product was observed. The reactions exhibited excellent functional group compatibility, and a wide range of functional groups, such as trifluoromethyl, cyano, methylsulfonyl, chloro, methylthio, trifluoromethoxy, nitro, formyl, and ester were well tolerated under the reaction conditions, which may serve as useful reaction handles for further derivatization. The steric effect of an ortho-substituent had an obvious influence on the reaction efficiency. Compared to para- and meta-substituted styrenes, ortho-substituted substrates were unreactive and only small amounts of addition products were observed (3oa and 3pa). In addition, 4-(3,3,3-trifluoroprop-1-en-2-yl)-1,1′-biphenyl 1a and 2-(3,3,3-trifluoroprop-1-en-2-yl)naphthalene 1r were found to be good substrates for the reaction. Importantly, heterocyclic substrates such as 2-(3,3,3-trifluoroprop-1-en-2-yl)thiophene 1s and 2-chloro-5-(3,3,3-trifluoroprop-1-en-2-yl)pyridine 1t were also suitable for this reaction to give the desired products 3sa and 3ta in 71% and 79% yields, respectively. It was worth noting that in most cases, 0.5 or 1.0 equivalents of DBN were enough to make the hydroamination reaction proceed efficiently, and good results were achieved.
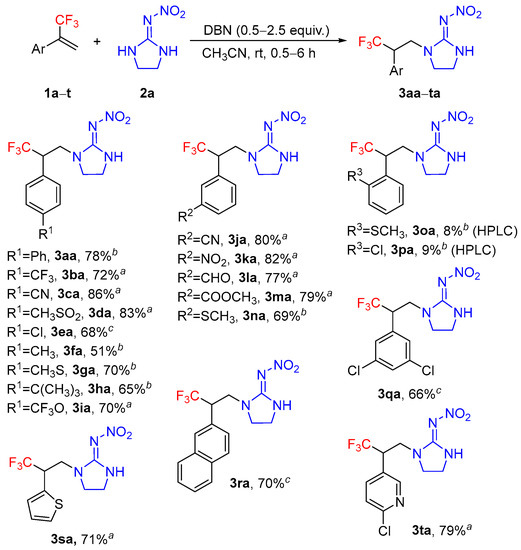
Scheme 3.
Hydroamination of various α-(trifluoromethyl)styrenes with 2-nitroimino-imidazolidine 2a a. a Reaction conditions: 1a–t (1.0 mmol), 2a (1.0 mmol), DBN (0.5 equiv., 0.5 mmol), CH3CN (3 mL), 25 °C, 0.5 h. b 2.5 equiv. of DBN was used, 6 h. c 1.0 equiv. of DBN was used, 3 h. Isolated yields.
To further verify the scope of this novel hydroamination reaction, four nitrogen-containing heteroalicycles (2b, 2c, 2e, and 2f) and two guanidines (2d and 2g) were subjected to the addition reaction with α-(trifluoromethyl)styrenes (1c, 1e, and 1t) under the optimized reaction conditions (Scheme 4). Gratifyingly, without further optimization of the reaction conditions, the reactions of 2-(nitromethylene)imidazolidine 2b, 2-cyanoimino-thiazolidine 2c, and (E)-1-methyl-2-nitroguanidine 2d with α-(trifluoromethyl)styrenes proceeded smoothly and provided corresponding products in moderate to good yields, despite the fact that those substrates are structurally different. However, 2-(nitromethylene)hexahydropyrimidine 2e, 3-methyl-4-nitroimino-tetrahydro-1,3,5-oxadiazine 2f, and 2-cyanoguanidine 2g were poor substrates and failed to furnish the desired products. Therefore, the reaction conditions must be further investigated.
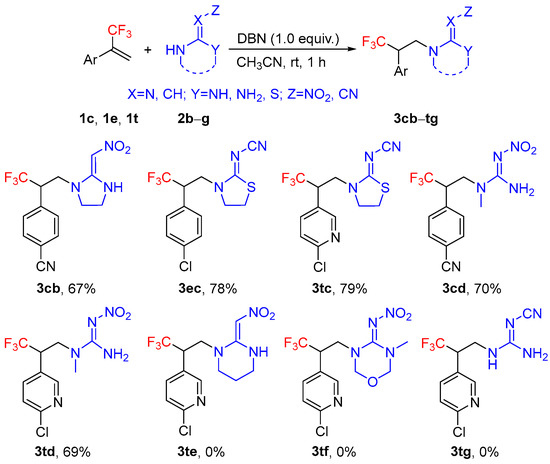
Scheme 4.
Hydroamination of α-(trifluoromethyl)styrenes with different nitrogen-containing heteroalicycles or guanidine. Reaction conditions: 1c, 1e, 1t (1.0 mmol), 2b–g (1.0 mmol), DBN (1.0 equiv., 1.0 mmol), CH3CN (3 mL), 25 °C, 1 h. Isolated yields.
Subsequently, the defluorinative amination of α-(trifluoromethyl)styrenes with different nitrogen-containing heteroalicycles and guanidines under optimal conditions (using NaH in CH3CN at 60 °C for 12 h, Table 1, entry 24) was investigated (Scheme 5). Unfortunately, the scope of the α-(trifluoromethyl)styrenes was rather narrow, where only electron-rich α-(trifluoromethyl)styrenes could furnish gem-difluoroalkenes in acceptable yields. Unexpectedly, 4-(3,3,3-trifluoroprop-1-en-2-yl)dibenzo[b,d]thiophene 1w could be successfully converted to gem-difluoroalkene 4wa in 66% yield. Furthermore, only 2-nitroimino-imidazolidine 2a and 2-cyanoimino-thiazolidine 2c could undergo defluorination reaction efficiently, whereas no reaction occurred when 2-(nitromethylene)imidazolidine 2b, (E)-1-methyl-2-nitroguanidine 2d, 2-(nitromethylene)hexahydropyrimidine 2e, 3-methyl-4-nitroimino-tetrahydro-1,3,5-oxadiazine 2f, and 2-cyanoguanidine 2g were used as substrates. These results further demonstrated that although the reaction conditions seemed the best, this was not the general case for other substrates. The outcomes of the reactions between α-(trifluoromethyl)styrenes and nitrogen nucleophiles are highly dependent on the structures and electronic properties of the substrates and reaction conditions (see Supplementary Materials).
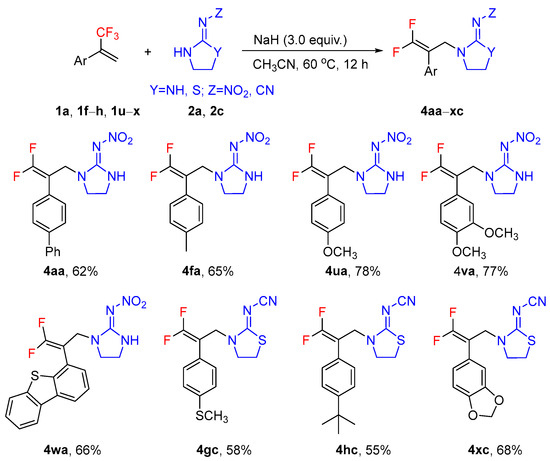
Scheme 5.
Defluorinative reaction of α-(trifluoromethyl)styrenes with 2-nitroimino-imidazolidine 2a and 2-cyanoimino-thiazolidine 2c. Reaction conditions: 1a, 1f–h, 1u–x (1.0 mmol), 2a, 2c (1.0 mmol), NaH (3.0 equiv., 3.0 mmol), CH3CN (3 mL), 60 °C, 12 h. The reaction vial was sealed with a septum. Isolated yields.
To prove the preparative usefulness of the developed methods, three scale-up reactions were performed. All reactions were conducted in 5.0 mmol scale. Without further optimization of reaction conditions, the hydroamination of α-(trifluoromethyl)styrenes 1t with 2a and 2c, and the defluorinative reaction of α-(trifluoromethyl)styrenes 1u with 2a, were easy to scale-up, however, the desired products were obtained in slightly lower yields (3ta, 3tc, and 4ua, Scheme 6).
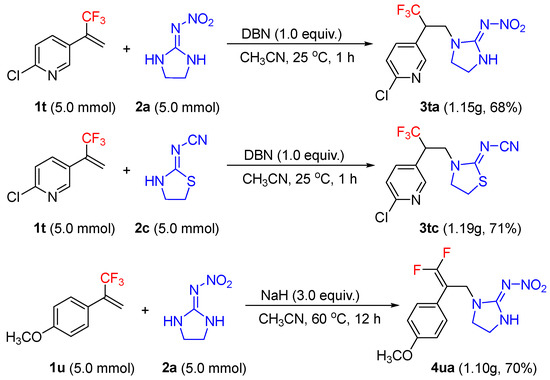
Scheme 6.
Gram-scale synthesis of 3ta, 3tc, and 4ua.
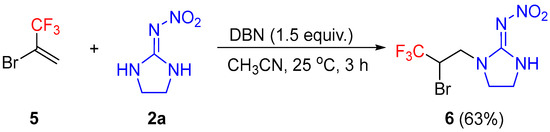
Surprisingly, the reaction of 2-bromo-3,3,3-trifluoroprop-1-ene 5 with 2-nitroimino-imidazolidine 2a also proceeded smoothly, affording the addition product 6 in moderate yield (Scheme 7). Notably, the remaining bromo group in the product offers the opportunity for further downstream diversification.
Scheme 7.
Reaction of 2-bromo-3,3,3-trifluoroprop-1-ene with 2a.
3. Materials and Methods
3.1. General Information
All solvents were of analytical grade, unless otherwise mentioned, and solvents and reagents were received from commercial suppliers and used without further purification. α-(Trifluoromethyl)styrenes 1 [41,42], 2-(nitromethylene)imidazolidine 2b, and 2-(nitromethylene)hexahydropyrimidine 2e were synthesized by previously reported methods [43]. 2-Nitroimino-imidazolidine 2a, 2-cyanoimino-thiazolidine 2c, (E)-1-methyl-2-nitroguanidine 2d, 3-methyl-4-nitroimino-tetrahydro-1,3,5-oxadiazine 2f, and 2-cyanoguanidine 2g were purchased from commercial sources. All the products obtained in this work are new compounds.
Reactions were stirred using Teflon-coated magnetic stir bars. Elevated temperatures were maintained using thermostat-controlled silicone oil baths. 1H NMR and 13C NMR spectra were recorded on a 400 spectrometer (400 MHz for 1H and 100 MHz for 13C, respectively) using TMS as an internal standard. The 19F NMR spectra were obtained on a 600 spectrometer (564 MHz) with CF3COOH as an internal standard. CDCl3, DMSO-d6, or (CD3)2CO were used as the NMR solvents. Data for 1H, 13C, and 19F NMR were recorded as follows: chemical shift (δ, ppm), multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet, q = quartet, dd = double of doublet). Coupling constants are reported in hertz (Hz). High resolution mass spectra (HRMS) were recorded on the EI or ESI mode using a TOF mass analyzer. The melting points were measured on an open capillary using EZ-Melt automated melting point apparatus and were not corrected. HPLC were recorded on a Shimadzu LC-20AT. Silica gel (300–400 mesh size) was used for column chromatography. TLC analysis of reaction mixtures was performed using silica gel plates.
3.2. General Procedure for the Synthesis of the Target Compounds 3aa–td
To a glass tube charged with a stirring bar were added DBN (0.5–2.5 equiv.), α-(trifluoromethyl)styrenes (1a–t, 1.0 mmol), nitrogen nucleophiles 2a–d (1.0 mmol, 1.0 equiv.), and CH3CN (3 mL). The reaction was stirred for 0.5–6 h under room temperature (monitored by TLC). After the completion of reaction, the reaction mixture was quenched with a saturated aqueous solution of NH4Cl (15 mL) and extracted with ethyl acetate (3 × 15 mL). The organic layer was separated and dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by column chromatography on silica gel using n-hexane/ethyl acetate (5/1–1/1) as eluent to afford the target compounds 3aa–td.
(E)-N-(1-(2-([1,1′-Biphenyl]-4-yl)-3,3,3-trifluoropropyl)imidazolidin-2-ylidene)nitramide (3aa). White solid, m.p. 135.3–136.8 °C, yield 78% (295.1 mg); 1H NMR (400 MHz, CDCl3) δ 8.03 (s, 1H), 7.63–7.58 (m, 4H), 7.47–7.42 (m, 4H), 7.39–7.35 (m, 1H), 3.98–3.81 (m, 3H), 3.67–3.56 (m, 2H), 3.55–3.48 (m, 1H), 3.24 (q, J = 9.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 160.2, 140.8, 138.9, 129.9, 129.8, 128.4, 127.9, 126.7, 126.6, 126.0, 124.8 (q, 1JCF = 279.0 Hz), 46.9 (q, 2JCF = 26.2 Hz), 45.6, 43.5 (d, 3JCF = 2.6 Hz), 40.5; 19F NMR (564 MHz, CDCl3) δ −67.9 (d, J = 7.9 Hz, 3F); HRMS (EI): calcd for C18H17F3N4O2 [M]+: 378.1304, found: 378.1301.
(E)-N-(1-(3,3,3-Trifluoro-2-(4-(trifluoromethyl)phenyl)propyl)imidazolidin-2-ylidene)nitramide (3ba). White solid, m.p. 104.4–106.0 °C, yield 72% (266.8 mg); 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.65 (d, J = 8.0 Hz, 2H), 7.51 (d, J = 8.0 Hz, 2H), 4.00–3.94 (m, 1H), 3.92–3.82 (m, 2H), 3.68–3.63 (m, 2H), 3.57–3.51 (m, 1H), 3.27 (q, J = 9.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 161.3, 136.1, 131.3 (q, 2JCF = 32.6 Hz), 129.6, 126.0 (q, 3JCF = 3.6 Hz), 125.4 (q, 1JCF = 278.8 Hz), 123.8 (q, 1JCF = 270.6 Hz), 48.1 (q, 2JCF = 26.4 Hz), 46.7, 44.5 (d, 3JCF = 2.1 Hz), 41.6; 19F NMR (564 MHz, CDCl3) δ −62.8 (s, 3F), −67.9 (d, J = 7.9 Hz, 3F); HRMS (EI): calcd for C13H12F6N4O2 [M]+: 370.0864, found: 370.0868.
(E)-N-(1-(2-(4-Cyanophenyl)-3,3,3-trifluoropropyl)imidazolidin-2-ylidene)nitramide (3ca). White solid, m.p. 126.0–127.4 °C, yield 86% (281.9 mg); 1H NMR (400 MHz, CDCl3) δ 8.05 (s, 1H), 7.67 (d, J = 8.4 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H), 4.00–3.94 (m, 1H), 3.91–3.80 (m, 2H), 3.66 (t, J = 9.0 Hz, 2H), 3.59–3.52 (m, 1H), 3.30 (q, J = 9.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 161.3, 137.3, 132.7, 130.0, 125.4 (q, 1JCF = 279.2 Hz), 118.1, 113.1, 48.2 (q, 2JCF = 26.5 Hz), 46.7, 44.4 (d, 3JCF = 2.4 Hz), 41.7; 19F NMR (564 MHz, CDCl3) δ −67.7 (d, J = 7.9 Hz, 3F); HRMS (EI): calcd for C13H12F3N5O2 [M]+: 327.0943, found: 327.0947.
(E)-N-(1-(3,3,3-Trifluoro-2-(4-(methylsulfonyl)phenyl)propyl)imidazolidin-2-ylidene)nitramide (3da). White solid, m.p. 123.9–145.1 °C, yield 83% (315.6 mg); 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.94 (d, J = 8.0 Hz, 2H), 7.59 (d, J = 8.0 Hz, 2H), 4.03–3.97 (m, 1H), 3.94–3.81 (m, 2H), 3.65 (t, J = 8.8 Hz, 2H), 3.59–3.53 (m, 1H), 3.31 (q, J = 8.8 Hz, 1H), 3.06 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 161.3, 141.3, 138.3, 130.2, 128.1, 125.4 (q, 1JCF = 279.2 Hz), 48.2 (q, 2JCF = 26.6 Hz), 46.7, 44.6 (d, 3JCF = 2.1 Hz), 44.4, 41.7; 19F NMR (564 MHz, CDCl3) δ −67.5 (d, J = 9.0 Hz, 3F); HRMS (EI): calcd for C13H15F3N4O4S [M]+: 380.0766, found: 380.0764.
(E)-N-(1-(2-(4-Chlorophenyl)-3,3,3-trifluoropropyl)imidazolidin-2-ylidene)nitramide (3ea). White solid, m.p. 117.0–118.9 °C, yield 68% (228.7 mg); 1H NMR (400 MHz, CDCl3) δ 8.02 (s, 1H), 7.35 (d, J = 8.4 Hz, 2H), 7.29 (d, J = 8.4 Hz, 2H), 3.89–3.78 (m, 3H), 3.65–3.60 (m, 2H), 3.53–3.46 (m, 1H), 3.24 (q, J = 8.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 161.2, 135.1, 130.5, 130.4, 129.3, 125.6 (q, 1JCF = 279.0 Hz), 47.6 (q, 2JCF = 26.4 Hz), 46.6, 44.4 (d, 3JCF = 2.4 Hz), 41.6; 19F NMR (564 MHz, CDCl3) δ −68.2 (d, J = 7.3 Hz, 3F); HRMS (EI): calcd for C12H12ClF3N4O2 [M]+: 336.0601, found: 336.0600.
(E)-N-(1-(3,3,3-Trifluoro-2-(p-tolyl)propyl)imidazolidin-2-ylidene)nitramide (3fa). White solid, m.p. 121.5–122.9 °C, yield 51% (161.2 mg); 1H NMR (400 MHz, CDCl3) δ 8.01 (s, 1H), 7.23 (d, J = 8.0 Hz, 2H), 7.18 (d, J = 8.0 Hz, 2H), 3.95–3.89 (m, 1H), 3.82–3.73 (m, 2H), 3.63–3.57 (m, 2H), 3.50–3.43 (m, 1H), 3.17 (q, J = 9.2 Hz, 1H), 2.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 161.3, 138.9, 129.7, 128.9, 128.8, 129.5, 129.4, 125.8 (q, 1JCF = 278.8 Hz), 47.9 (q, 2JCF = 26.0 Hz), 46.6, 44.5 (d, 3JCF = 2.4 Hz), 41.5, 21.1; 19F NMR (564 MHz, CDCl3) δ −68.2 (d, J = 7.9 Hz, 3F); HRMS (EI): calcd for C13H15F3N4O2 [M]+: 316.1147, found: 316.1149.
(E)-N-(1-(3,3,3-Trifluoro-2-(4-(methylthio)phenyl)propyl)imidazolidin-2-ylidene)nitramide (3ga). White solid, m.p. 132.4–134.0 °C, yield 70% (243.7 mg); 1H NMR (400 MHz, CDCl3) δ 8.06 (s, 1H), 7.32–7.28 (m, 4H), 3.98–3.91 (m, 1H), 3.87–3.79 (m, 2H), 3.68–3.61 (m, 2H), 3.55–3.48 (m, 1H), 3.26 (q, J = 9.2 Hz, 1H), 2.52 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 161.3, 140.0, 129.4, 128.4, 126.6, 125.7 (q, 1JCF = 278.8 Hz), 47.8 (q, 2JCF = 26.2 Hz), 46.6, 44.4 (d, 3JCF = 2.5 Hz), 41.6, 15.3; 19F NMR (564 MHz, CDCl3) δ −68.2 (d, J = 8.5 Hz, 3F); HRMS (EI): calcd for C13H15F3N4O2S [M]+: 348.0868, found: 348.0864.
(E)-N-(1-(2-(4-(tert-Butyl)phenyl)-3,3,3-trifluoropropyl)imidazolidin-2-ylidene)nitramide (3ha). White solid, m.p. 157.8–159.1 °C, yield 65% (233.0 mg); 1H NMR (400 MHz, CDCl3) δ 8.02 (s, 1H), 7.38 (d, J = 8.4 Hz, 2H), 7.25 (d, J = 8.0 Hz, 2H), 3.95–3.90 (m, 1H), 3.83–3.73 (m, 2H), 3.67–3.55 (m, 2H), 3.52–3.45 (m, 1H), 3.19 (q, J = 9.6 Hz, 1H), 1.30 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 160.2, 151.0, 127.8, 127.6, 124.9, 124.8 (q, 1JCF = 278.8 Hz), 46.7 (q, 2JCF = 26.1 Hz), 45.6, 43.5 (d, 3JCF = 2.7 Hz), 40.5, 33.6, 30.2; 19F NMR (564 MHz, CDCl3) δ −68.1 (d, J = 7.9 Hz, 3F); HRMS (EI): calcd for C16H21F3N4O2 [M]+: 358.1617, found: 358.1615.
(E)-N-(1-(3,3,3-Trifluoro-2-(4-(trifluoromethoxy)phenyl)propyl)imidazolidin-2-ylidene)nitramide (3ia). White solid, m.p. 110.5–112.8 °C, yield 70% (270.1 mg); 1H NMR (400 MHz, CDCl3) δ 8.03 (s, 1H), 7.40 (d, J = 8.8 Hz, 2H), 7.23 (d, J = 8.4 Hz, 2H), 3.94–3.78 (m, 3H), 3.70–3.60 (m, 2H), 3.57–3.50 (m, 1H), 3.26 (q, J = 9.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 161.3, 149.6, 130.6, 125.6 (q, 1JCF = 278.5 Hz), 121.3, 120.4 (q, 1JCF = 256.4 Hz), 47.6 (q, 2JCF = 26.4 Hz), 46.7, 44.6 (d, 3JCF = 2.4 Hz), 41.6; 19F NMR (564 MHz, CDCl3) δ −57.9 (s, 3F), −68.2 (d, J = 9.6 Hz, 3F); HRMS (EI): calcd for C13H12F6N4O3 [M]+: 386.0814, found: 386.0816.
(E)-N-(1-(2-(3-Cyanophenyl)-3,3,3-trifluoropropyl)imidazolidin-2-ylidene)nitramide (3ja). White solid, m.p. 111.5–112.9 °C, yield 80% (262.1 mg); 1H NMR (400 MHz, CDCl3) δ 8.05 (s, 1H), 7.68–7.65 (m, 3H), 7.53 (J = 8.0 Hz, 1H), 3.99–3.80 (m, 3H), 3.71–3.67 (m, 2H), 3.63–3.57 (m, 1H), 3.34 (q, J = 9.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 161.3, 133.7, 133.6, 132.7, 132.6, 130.0, 125.4 (q, 1JCF = 278.9 Hz), 118.1, 113.3, 47.9 (q, 2JCF = 26.5 Hz), 46.7, 44.5 (d, 3JCF = 2.1 Hz), 41.7; 19F NMR (564 MHz, CDCl3) δ −67.9 (d, J = 7.9 Hz, 3F); HRMS (EI): calcd for C13H12F3N5O2 [M]+: 327.0943, found: 327.0940.
(E)-N-(1-(3,3,3-Trifluoro-2-(3-nitrophenyl)propyl)imidazolidin-2-ylidene)nitramide (3ka). White solid, m.p. 121.6–123.2 °C, yield 82% (285.0 mg); 1H NMR (400 MHz, CDCl3) δ 8.23 (t, J = 2.4 Hz, 2H), 8.04 (s, 1H), 7.77 (d, J = 7.6 Hz, 1H), 7.61 (t, J = 8.4 Hz, 1H), 4.08–3.94 (m, 2H), 3.89–3.83 (m, 1H), 3.72–3.60 (m, 3H), 3.43–3.36 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 161.3, 148.4, 135.5, 134.0, 130.2, 125.4 (q, 1JCF = 279.0 Hz), 124.1, 124.0, 47.9 (q, 2JCF = 26.5 Hz), 46.7, 44.3 (d, 3JCF = 2.2 Hz), 41.7; 19F NMR (564 MHz, CDCl3) δ −67.9 (d, J = 9.6 Hz, 3F); HRMS (EI): calcd for C12H12F3N5O4 [M]+: 347.0841, found: 347.0843.
(E)-N-(1-(3,3,3-Trifluoro-2-(3-formylphenyl)propyl)imidazolidin-2-ylidene)nitramide (3la). White solid, m.p. 123.7–124.8 °C, yield 77% (254.4 mg); 1H NMR (400 MHz, CDCl3) δ 10.01 (s, 1H), 8.03 (s, 1H), 7.88 (d, J = 6.4 Hz, 2H), 7.67–7.56 (m, 2H), 4.01–3.95 (m, 1H), 3.92–3.85 (m, 2H), 3.68–3.54 (m, 3H), 3.33 (q, J = 8.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 191.7, 161.3, 136.9, 135.1, 133.3, 130.5, 129.9, 129.8, 125.6 (q, 1JCF = 278.9 Hz), 48.0 (q, 2JCF = 26.4 Hz), 46.6, 44.4 (d, 3JCF = 2.2 Hz), 41.6; 19F NMR (564 MHz, CDCl3) δ −67.9 (d, J = 7.9 Hz, 3F); HRMS (EI): calcd for C13H13F3N4O3 [M]+: 330.0940, found: 330.0945.
Methyl (E)-3-(1,1,1-trifluoro-3-(2-(nitroimino)imidazolidin-1-yl)propan-2-yl)benzoate (3ma). White solid, m.p. 125.5–126.8 °C, yield 79% (284.7 mg). 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 8.02 (s, 2H), 7.57 (d, J = 7.6 Hz, 1H), 7.47 (t, J = 7.6 Hz, 1H), 3.96–3.86 (m, 3H), 3.91 (s, 3H), 3.64–3.57 (m, 2H), 3.55–3.48 (m, 1H), 3.25 (q, J = 8.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 166.4, 161.3, 133.7, 132.5, 131.0, 130.2, 130.1, 129.3, 125.6 (q, 1JCF = 278.8 Hz), 52.4, 48.0 (q, 2JCF = 26.2 Hz), 46.6, 44.3 (d, 3JCF = 2.3 Hz), 41.6; 19F NMR (564 MHz, CDCl3) δ −68.1 (d, J = 7.9 Hz, 3F); HRMS (EI): calcd for C14H15F3N4O4 [M]+: 360.1045, found: 360.1042.
(E)-N-(1-(3,3,3-Trifluoro-2-(3-(methylthio)phenyl)propyl)imidazolidin-2-ylidene)nitramide (3na). White solid, m.p. 142.6–144.0 °C, yield 69% (240.3 mg); 1H NMR (400 MHz, CDCl3) δ 7.97 (s, 1H), 7.23 (d, J = 8.4 Hz, 1H), 7.19–7.15 (m, 2H), 7.05 (d, J = 7.2 Hz, 1H), 3.85–3.73 (m, 3H), 3.60–3.52 (m, 2H), 3.48–3.41 (m, 1H), 3.17 (q, J = 9.2 Hz, 1H), 2.43 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 161.2, 139.9, 132.8, 129.4, 126.8, 126.7, 125.7 (q, 1JCF = 279.0 Hz), 125.5, 48.1 (q, 2JCF = 26.2 Hz), 46.6, 44.4 (d, 3JCF = 2.4 Hz), 41.6, 15.5; 19F NMR (564 MHz, CDCl3) δ −67.9 (d, J = 7.3 Hz, 3F); HRMS (EI): calcd for C13H15F3N4O2S [M]+: 348.0868, found: 348.0864.
(E)-N-(1-(3,3,3-Trifluoro-2-(thiophen-2-yl)propyl)imidazolidin-2-ylidene)nitramide (3qa). White solid, m.p. 114.3–116.2 °C, yield 71% (218.8 mg); 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.33 (d, J = 5.2 Hz, 1H), 7.11 (d, J = 3.2 Hz, 1H), 7.03 (dd, J1 = 5.2 Hz, J2 = 4.0 Hz, 1H), 4.26–4.16 (m, 1H), 4.02–3.97 (m, 1H), 3.71–3.48 (m, 4H), 3.15 (q, J = 9.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 161.2, 133.4, 128.4, 127.4, 126.5, 125.1 (q, 1JCF = 279.0 Hz), 46.9, 45.7 (d, 3JCF = 2.4 Hz), 43.8 (q, 2JCF = 27.6 Hz), 41.7; 19F NMR (564 MHz, CDCl3) δ −74.1 (d, J = 9.6 Hz, 3F); HRMS (EI): calcd for C10H11F3N4O2S [M]+: 308.0555, found: 308.0551.
(E)-N-(1-(3,3,3-Trifluoro-2-(naphthalen-2-yl)propyl)imidazolidin-2-ylidene)nitramide (3ra). White solid, m.p. 128.6–130.1 °C, yield 70% (246.6 mg); 1H NMR (400 MHz, CDCl3) δ 7.97 (s, 1H), 7.88–7.84 (m, 4H), 7.54–7.51 (m, 2H), 7.47 (d, J = 8.4 Hz, 1H), 4.04–4.00 (m, 2H), 3.95–3.88 (m, 1H), 3.56–3.46 (m, 3H), 3.19–3.12 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 161.3, 133.3, 133.2, 129.4, 129.0, 128.9, 128.1, 127.7, 126.9, 126.8, 125.9 (q, 1JCF = 279.2 Hz), 125.8, 48.4 (q, 2JCF = 26.1 Hz), 46.6, 44.5 (d, 3JCF = 2.3 Hz), 41.5; 19F NMR (564 MHz, CDCl3) δ −67.8 (d, J = 7.3 Hz, 3F); HRMS (EI): calcd for C16H15F3N4O2 [M]+: 352.1147, found: 352.1150.
(E)-N-(1-(2-(3,5-Dichlorophenyl)-3,3,3-trifluoropropyl)imidazolidin-2-ylidene)nitramide (3sa). White solid, m.p. 166.4–168.1 °C, yield 66% (244.8 mg); 1H NMR (400 MHz, acetone-d6) δ 8.47 (s, 1H), 7.60 (s, 2H), 7.54 (t, J = 1.6 Hz, 1H), 4.30–4.24 (m, 1H), 4.07–3.93 (m, 2H), 3.74–3.67 (m, 3H), 3.54–3.50 (m, 1H); 13C NMR (100 MHz, acetone-d6) δ 161.4, 136.5, 135.0, 128.8, 128.3, 125.9 (q, 1JCF = 278.3 Hz), 47.1 (q, 2JCF = 26.0 Hz), 46.2, 43.4 (d, 3JCF = 2.6 Hz), 41.8; 19F NMR (564 MHz, acetone-d6) δ −68.9 (d, J = 7.9 Hz, 3F); HRMS (EI): calcd for C12H11Cl2F3N4O2 [M]+: 370.0211, found: 370.0212.
(E)-N-(1-(2-(6-Chloropyridin-3-yl)-3,3,3-trifluoropropyl)imidazolidin-2-ylidene)nitramide (3ta). White solid, m.p. 112.4–113.9 °C, yield 79% (266.5 mg); 1H NMR (400 MHz, CDCl3) δ 8.34 (d, J = 2.0 Hz, 1H), 8.06 (s, 1H), 7.76 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 1H), 7.38 (d, J = 8.4 Hz, 1H), 3.99–3.89 (m, 2H), 3.82–3.77 (m, 1H), 3.73–3.66 (m, 2H), 3.63–3.57 (m, 1H), 3.43–3.36 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 161.3, 152.4, 150.3, 139.1, 126.9, 125.3 (q, 1JCF = 279.0 Hz), 124.7, 46.5, 45.4 (q, 2JCF = 26.8 Hz), 44.1 (d, 3JCF = 1.7 Hz), 41.6; 19F NMR (564 MHz, CDCl3) δ −72.9 (d, J = 7.3 Hz, 3F); HRMS (EI): calcd for C11H11ClF3N5O2 [M]+: 337.0553, found: 337.0554.
(E)-4-(1,1,1-Trifluoro-3-(2-(nitromethylene)imidazolidin-1-yl)propan-2-yl)benzonitrile (3cb). White solid, m.p. 132.4–133.9 °C, yield 67% (218.4 mg); 1H NMR (400 MHz, CDCl3) δ 8.55 (s, 1H), 7.66 (d, J = 8.0 Hz, 2H), 7.46 (d, J = 8.0 Hz, 2H), 6.40 (s, 1H), 3.79–3.71 (m, 2H), 3.63–3.47 (m, 4H), 3.22 (q, J = 8.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 159.1, 137.2, 133.0, 129.8, 125.2 (q, 1JCF = 279.1 Hz), 118.0, 113.4, 96.3, 49.5, 48.6 (q, 2JCF = 26.6 Hz), 45.8 (d, 3JCF = 1.7 Hz), 42.5; 19F NMR (564 MHz, CDCl3) δ −67.7 (d, J = 7.9 Hz, 3F); HRMS (EI): calcd for C14H13F3N4O2 [M]+: 326.0991, found: 326.0993.
(Z)-N-(3-(2-(4-Chlorophenyl)-3,3,3-trifluoropropyl)thiazolidin-2-ylidene)cyanamide (3ec). White solid, m.p. 124.8–126.2 °C, yield 78% (259.9 mg). 1H NMR (400 MHz, CDCl3) δ 7.40 (d, J = 8.4 Hz, 2H), 7.28 (d, J = 8.0 Hz, 2H), 4.04–3.91 (m, 2H), 3.83–3.75 (m, 2H), 3.47–3.41 (m, 1H), 3.32–3.25 (m, 1H), 3.21–3.15 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 174.8, 135.4, 130.4, 130.2, 129.5, 125.5 (q, 1JCF = 278.8 Hz), 116.7, 54.1, 46.9 (q, 2JCF = 26.8 Hz), 46.6 (d, 3JCF = 2.3 Hz), 27.8; 19F NMR (564 MHz, CDCl3) δ −68.1 (d, J = 9.0 Hz, 3F); HRMS (EI): calcd for C13H11ClF3N3S [M]+: 333.0314, found: 333.0311.
(Z)-N-(3-(2-(6-Chloropyridin-3-yl)-3,3,3-trifluoropropyl)thiazolidin-2-ylidene)cyanamide (3tc). White solid, m.p. 122.1–123.7 °C, yield 79% (264.1 mg). 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 7.69 (d, J = 8.4 Hz, 1H), 7.40 (d, J = 8.4 Hz, 1H), 4.02–3.85 (m, 4H), 3.67–3.60 (m, 1H), 3.36–3.25 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 175.3, 152.6, 150.1, 138.9, 126.8, 125.2 (q, 1JCF = 279.0 Hz), 124.9, 116.4, 53.9, 46.1 (d, 3JCF = 2.0 Hz), 44.8 (q, 2JCF = 27.3 Hz), 27.8; 19F NMR (564 MHz, CDCl3) δ −68.0 (d, J = 7.9 Hz, 3F); HRMS (EI): calcd for C12H10ClF3N4S [M]+: 334.0267, found: 334.0269.
(E)-1-(2-(4-Cyanophenyl)-3,3,3-trifluoropropyl)-1-methyl-2-nitroguanidine (3cd). White solid, m.p. 118.9–120.1 °C, yield 70% (220.9 mg); 1H NMR (400 MHz, DMSO-d6) δ 8.36 (s, 2H), 7.88 (d, J = 8.0 Hz, 2H), 7.65 (d, J = 8.0 Hz, 2H), 4.41–4.30 (m, 1H), 4.10–4.00 (m, 2H), 2.79 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 159.1, 138.3, 133.0, 130.9, 126.4 (q, 1JCF = 279.3 Hz), 118.8, 112.1, 48.5, 47.3 (q, 2JCF = 25.9 Hz), 36.7; 19F NMR (564 MHz, DMSO-d6) δ −66.7 (d, J = 9.6 Hz, 3F); HRMS (EI): calcd for C12H12F3N5O2 [M]+: 315.0943, found: 315.0946.
(E)-1-(2-(6-Chloropyridin-3-yl)-3,3,3-trifluoropropyl)-1-methyl-2-nitroguanidine (3td). White solid, m.p. 113.5–114.9 °C, yield 69% (224.3 mg); 1H NMR (400 MHz, DMSO-d6) δ 8.49 (d, J = 2.0 Hz, 1H), 8.37 (s, 2H), 7.97 (dd, J1 = 8.4 Hz, J2 = 2.4 Hz, 1H), 7.59 (d, J = 8.4 Hz, 1H), 4.39–4.33 (m, 1H), 4.12–4.00 (m, 2H), 2.83 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 158.6, 150.8, 150.7, 140.2, 127.8, 125.8 (q, 1JCF = 279.0 Hz), 124.3, 47.6, 43.9 (q, 2JCF = 26.7 Hz), 36.2; 19F NMR (564 MHz, DMSO-d6) δ −62.4 (d, J = 9.6 Hz, 3F); HRMS (EI): calcd for C10H11ClF3N5O2 [M]+: 325.0553, found: 325.0550.
3.3. General Procedure for the Synthesis of the Target Compounds 4aa–xc
To a glass tube charged with a stirring bar were added NaH (3.0 mmol, 3.0 equiv.), α-(trifluoromethyl)styrenes (1a, 1f–h, 1u–x, 1.0 mmol), 2-nitroimino-imidazolidine 2a (1.0 mmol, 1.0 equiv.) or 2-cyanoimino-thiazolidine 2c (1.0 mmol, 1.0 equiv.), and CH3CN (3 mL). The reaction vial was sealed with a rubber septum and then the reaction mixture was stirred at 60 °C in an oil bath for 12 h (monitored by TLC). After the completion of reaction, the reaction mixture was quenched with a saturated aqueous solution of NH4Cl (15 mL) and extracted with ethyl acetate (3 × 15 mL). The organic layer was separated and dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by column chromatography on silica gel using n-hexane/ethyl acetate (5/1–1/1) as eluent to afford the target compounds 4aa–xc.
(E)-N-(1-(2-([1,1′-Biphenyl]-4-yl)-3,3-difluoroallyl)imidazolidin-2-ylidene)nitramide (4aa). White solid, m.p. 168.6–169.0 °C, yield 62% (222.1 mg); 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.60 (t, J = 8.0 Hz, 4H), 7.49–7.42 (m, 4H), 7.36 (t, J = 7.2 Hz, 1H), 4.49 (t, J = 2.0 Hz, 2H), 3.69–3.65 (m, 2H), 3.52–3.48 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 161.3, 155.2 (dd, 1JCF = 294.3, 290.5 Hz), 140.9, 140.1, 129.5, 129.1 (t, 3JCF = 3.2 Hz), 128.9, 128.5 (t, 4JCF = 3.2 Hz), 127.7, 127.4, 127.0, 88.3 (dd, 2JCF = 17.7, 14.0 Hz), 44.8, 41.5 (d, 3JCF = 3.0 Hz), 41.3; 19F NMR (564 MHz, CDCl3) δ −84.6 (d, J = 46.8 Hz, 1F), −86.6 (d, J = 47.4 Hz, 1F); HRMS (ESI): calcd for C18H16F2N4O2Na [M + Na]+: 381.1139, found: 381.1135.
(E)-N-(1-(3,3-Difluoro-2-(p-tolyl)allyl)imidazolidin-2-ylidene)nitramide (4fa). White solid, m.p. 109.9–110.6 °C, yield 65% (193.0 mg); 1H NMR (400 MHz, CDCl3) δ 8.02 (s, 1H), 7.27 (d, J = 7.2 Hz, 2H), 7.17 (d, J = 8.0 Hz, 2H), 4.42 (t, J = 2.0 Hz, 2H), 3.66–3.62 (m, 2H), 3.48–3.44 (m, 2H), 2.33 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 161.3, 155.0 (dd, 1JCF = 293.5, 289.8 Hz), 138.2, 129.5, 127.9 (t, 3JCF = 3.2 Hz), 127.2 (t, 4JCF = 3.4 Hz), 88.4 (dd, 2JCF = 17.2, 14.7 Hz), 44.7, 41.5 (d, 3JCF = 2.3 Hz), 41.3, 21.2; 19F NMR (564 MHz, CDCl3) δ −85.8 (d, J = 33.3 Hz, 1F), −87.8 (d, J = 33.8 Hz, 1F); HRMS (ESI): calcd for C13H14F2N4O2Na [M + Na]+: 319.0983, found: 319.0981.
(E)-N-(1-(3,3-Difluoro-2-(4-methoxyphenyl)allyl)imidazolidin-2-ylidene)nitramide (4ua). White solid, m.p. 128.8–130.2 °C, yield 78% (243.9 mg); 1H NMR (400 MHz, acetone-d6) δ 8.47 (s, 1H), 7.43 (d, J = 8.4 Hz, 2H), 6.68 (d, J = 8.4 Hz, 2H), 4.41 (t, J = 2.0 Hz, 2H), 3.83 (s, 3H), 3.74–3.70 (m, 2H), 3.62–3.57 (m, 2H); 13C NMR (100 MHz, acetone-d6) δ 161.3, 159.5, 154.7 (dd, 1JCF = 288.8, 288.2 Hz), 129.7 (t, 3JCF = 3.0 Hz), 122.9 (t, 4JCF = 3.2 Hz), 113.9, 88.9 (dd, 2JCF = 17.9, 14.7 Hz), 54.7, 44.8, 41.6 (d, 3JCF = 3.0 Hz), 41.4; 19F NMR (564 MHz, acetone-d6) δ −89.9 (d, J = 40.0 Hz, 1F), −91.4 (d, J = 41.2 Hz, 1F); HRMS (ESI): calcd for C13H14F2N4O3Na [M + Na]+: 335.0932, found: 335.0935.
(E)-N-(1-(2-(3,4-Dimethoxyphenyl)-3,3-difluoroallyl)imidazolidin-2-ylidene)nitramide (4va). White solid, m.p. 123.9–124.6 °C, yield 77% (263.5 mg); 1H NMR (400 MHz, CDCl3) δ 7.98 (s, 1H), 6.96–6.94 (m, 2H), 6.83 (d, J = 8.4 Hz, 1H), 4.42 (t, J = 2.2 Hz, 2H), 3.85 (s, 3H), 3.84 (s, 3H), 3.66–3.61 (m, 2H), 3.48–3.43 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 161.1, 154.9 (dd, 1JCF = 293.4, 289.1 Hz), 149.0, 132.0, 128.6, 122.4 (t, 3JCF = 3.3 Hz), 120.8 (dd, 4JCF = 5.0, 2.8 Hz), 111.2, 111.0 (t, 4JCF = 3.1 Hz), 88.3 (dd, 2JCF = 17.6, 13.7 Hz), 56.0, 55.9, 44.7, 41.3 (d, 3JCF = 4.5 Hz); 19F NMR (564 MHz, CDCl3) δ −85.7 (d, J = 35.5 Hz, 1F), −88.1 (d, J = 35.5 Hz, 1F); HRMS (ESI): calcd for C14H16F2N4O4Na [M + Na]+: 365.1038, found: 365.1035.
(E)-N-(1-(2-(Dibenzo[b,d]thiophen-4-yl)-3,3-difluoroallyl)imidazolidin-2-ylidene)nitramide (4wa). White solid, m.p. 181.9–183.0 °C, yield 66% (256.7 mg); 1H NMR (400 MHz, CDCl3) δ 8.14 (s, 1H), 8.13 (d, J = 7.6 Hz, 1H), 7.92–7.84 (m, 2H), 7.52–7.41 (m, 4H), 4.50 (s, 2H), 3.54 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 156.5, 149.6 (t, 1JCF = 293.4 Hz), 135.0, 133.9, 131.4, 131.0, 123.0, 122.5, 120.4, 120.1, 118.0, 117.2, 83.0 (dd, 2JCF = 19.5, 17.9 Hz), 40.7, 37.8 (d, 3JCF = 4.5 Hz), 36.6; 19F NMR (564 MHz, CDCl3) δ −80.3 (d, J = 25.9 Hz, 1F), −86.8 (d, J = 25.9 Hz, 1F); HRMS (ESI): calcd for C18H14F2N4O2SNa [M + Na]+: 411.0704, found: 411.0705.
(Z)-N-(3-(3,3-Difluoro-2-(4-(methylthio)phenyl)allyl)thiazolidin-2-ylidene)cyanamide (4gc). White solid, m.p. 133.1–134.7 °C, yield 58% (189.1 mg); 1H NMR (400 MHz, CDCl3) δ 7.25 (s, 4H), 4.48 (t, J = 2.0 Hz, 2H), 3.69 (t, J = 7.6 Hz, 2H), 3.21 (t, J = 7.6 Hz, 2H), 2.50 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 174.7, 155.0 (dd, 1JCF = 294.2, 290.9 Hz), 139.5, 128.3, 126.4, 126.1, 117.0, 88.2 (dd, 2JCF = 17.8, 15.2 Hz), 51.7, 43.1 (d, 3JCF = 2.6 Hz), 27.2, 15.3; 19F NMR (564 MHz, CDCl3) δ −84.9 (d, J = 30.4 Hz, 1F), −86.4 (d, J = 31.0 Hz, 1F); HRMS (EI): calcd for C14H13F2N3S2 [M]+: 325.0519, found: 325.0522.
(Z)-N-(3-(2-(4-(tert-Butyl)phenyl)-3,3-difluoroallyl)thiazolidin-2-ylidene)cyanamide (4hc). White solid, m.p. 133.5–135.0 °C, yield 55% (184.7 mg); 1H NMR (400 MHz, CDCl3) δ 7.34 (d, J = 7.6 Hz, 2H), 7.21 (d, J = 7.2 Hz, 2H), 4.43 (t, J = 2.0 Hz, 2H), 3.67–3.63 (m, 2H), 3.17–3.13 (m, 2H), 1.26 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 174.6, 155.1 (dd, 1JCF = 293.7, 289.5 Hz), 151.6, 128.5, 127.6 (t, 3JCF = 2.8 Hz), 126.8 (t, 4JCF = 3.0 Hz), 126.1, 125.9, 117.1, 88.3 (dd, 2JCF = 17.2, 15.0 Hz), 51.7, 43.3 (d, 3JCF = 2.4 Hz), 34.7, 31.2, 27.2; 19F NMR (564 MHz, CDCl3) δ −85.8 (d, J = 33.3 Hz, 1F), −87.8 (d, J = 34.4 Hz, 1F); HRMS (EI): calcd for C17H19F2N3S [M]+: 335.1268, found: 335.1263.
(Z)-N-(3-(2-(Benzo[d][1,3]dioxol-5-yl)-3,3-difluoroallyl)thiazolidin-2-ylidene)cyanamide (4xc). White solid, m.p. 123.5–124.9 °C, yield 68% (219.9 mg); 1H NMR (400 MHz, CDCl3) δ 6.82–6.76 (m, 3H), 5.99 (s, 2H), 4.43 (t, J = 2.0 Hz, 2H), 3.71 (t, J = 7.6 Hz, 2H), 3.24 (t, J = 7.6 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 174.6, 154.9 (dd, 1JCF = 292.5, 290.3 Hz), 148.1, 147.7, 123.4 (t, 3JCF = 3.0 Hz), 121.9 (t, 4JCF = 2.9 Hz), 117.1, 108.7, 108.5 (t, 4JCF = 3.3 Hz), 101.5, 88.3 (dd, 2JCF = 18.1, 16.0 Hz), 51.8, 43.6 (d, 3JCF = 2.8 Hz), 27.2; 19F NMR (564 MHz, CDCl3) δ −85.8 (d, J = 35.5 Hz, 1F), −88.1 (d, J = 35.0 Hz, 1F); HRMS (EI): calcd for C14H11F2N3O2S [M]+: 323.0540, found: 323.0543.
3.4. Procedure for the Synthesis of the Target Compound 6
To a glass tube charged with a stirring bar were added DBN (1.5 mmol, 1.5 equiv.), 2-bromo-3,3,3-trifluoroprop-1-ene 5 (1.0 mmol, 1.0 equiv.), 2-nitroimino-imidazolidine 2a (1.0 mmol, 1.0 equiv.), and CH3CN (3 mL). The reaction was stirred for 6 h under room temperature (monitored by TLC). After the completion of reaction, the reaction mixture was quenched with a saturated aqueous solution of NH4Cl (15 mL) and extracted with ethyl acetate (3 × 15 mL). The organic layer was separated and dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by column chromatography on silica gel using n-hexane/ethyl acetate (3/1) as eluent to afford the target compound 6.
(E)-N-(1-(2-Bromo-3,3,3-trifluoropropyl)imidazolidin-2-ylidene)nitramide (6). White solid, m.p. 114.5–115.3 °C, yield 63% (191.3 mg); 1H NMR (400 MHz, acetone-d6) δ 8.64 (s, 1H), 5.05–4.99 (m, 1H), 4.02–3.97 (m, 1H), 3.90–3.83 (m, 5H); 13C NMR (100 MHz, acetone-d6) δ 161.5, 123.9 (q, 1JCF = 275.8 Hz), 46.4, 45.8 (d, 3JCF = 1.8 Hz), 43.7 (q, 2JCF = 30.8 Hz), 41.9; 19F NMR (564 MHz, acetone-d6) δ −71.4 (d, J = 7.9 Hz, 3F); HRMS (EI): calcd for C6H8BrF3N4O2 [M]+: 303.9783, found: 303.9785.
4. Conclusions
In summary, two novel series of fluorinated analogues of neonicotinoids were synthesized. The hydroamination and defluorinative amination of α-(trifluoromethyl)styrenes can be controlled by the subtle choice of reaction conditions and nitrogen nucleophiles. The hydroamination of α-(trifluoromethyl)styrenes with 2-nitroimino-imidazolidine (2a), 2-(nitromethylene)imidazolidine (2b), 2-cyanoimino-thiazolidine (2c), and (E)-1-methyl-2-nitroguanidine (2d) proceeded efficiently in the presence of DBN and was completed at room temperature within 0.5–6 h, affording a number of structurally diverse β-trifluoromethyl-β-arylethyl analogues of neonicotinoids in moderate to good yields. The γ,γ-difluoro-β-arylallyl analogues of neonicotinoids were also successfully synthesized via defluorination of α-(trifluoromethyl)styrenes with 2-nitroimino-imidazolidine (2a) and 2-cyanoimino-thiazolidine (2c) using NaH as base at an elevated temperature together with a prolonged reaction time of 12 h. The preliminary insecticidal activity tests indicated that only compounds 3ta, 3tc, and 3ca displayed moderate insecticidal activity against cowpea aphids (Aphis craccivora). The mortalities of 3ta, 3tc, and 3ca were 55%, 42%, and 38% at 250 mg/L, respectively. These preliminary results further demonstrated that flexible linkage such as the methylene group (–CH2–) in imidacloprid, plays a key role in the insecticidal activity. Increasing the length of carbon chain might be unfavorable for retaining insecticidal activity. The insecticidal evaluation of the target compounds against other insects such as armyworm and carmine spider, is underway in our laboratory.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/molecules28083530/s1, 1H, 13C, 19F NMR and HRMS (EI/ESI) spectra of the target compounds.
Author Contributions
Conceptualization, S.C.; methodology, J.H.; synthesis of materials, J.H. and Y.D.; synthesis of chemical compounds, J.H. and Z.S.; data curation, J.H., Y.D., Y.L. and P.Z.; writing—original draft preparation, J.H.; writing—review and editing, S.C.; supervision, S.C.; project administration, S.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in this article.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Tomizawa, M.; Casida, J.E. Molecular recognition of neonicotinoid insecticides: The determinants of life or death. Acc. Chem. Res. 2009, 42, 260–269. [Google Scholar] [CrossRef]
- Tomizawa, M.; Casida, J.E. Neonicotinoid insecticides: Highlights of a symposium on strategic molecular designs. J. Agric. Food Chem. 2011, 59, 2883–2886. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, P.; Nauen, R. Neonicotinoids—From zero to hero in insecticide chemistry. Pest. Manag. Sci. 2008, 64, 1084–1098. [Google Scholar] [CrossRef]
- Ohno, I.; Tomizawa, M.; Durkin, K.A.; Naruse, Y.; Casida, J.E.; Kagabu, S. Molecular features of neonicotinoid pharmacophore variants interacting with the insect nicotinic receptor. Chem. Res. Toxicol. 2009, 22, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Kagabu, S. Discovery of imidacloprid and further developments from strategic molecular designs. J. Agric. Food Chem. 2011, 59, 2887–2896. [Google Scholar] [CrossRef]
- Shao, X.S.; Liu, Z.W.; Xu, X.Y.; Li, Z.; Qian, X.H. Overall status of neonicotinoid insecticides in China: Production, application and innovation. J. Pestic. Sci. 2013, 38, 1–9. [Google Scholar] [CrossRef]
- Maienfisch, P.; Huerlimann, H.; Rindlisbacher, A.; Gsell, L.; Dettwiler, H.; Haettenschwiler, J.; Sieger, E.; Walti, M. The discovery of thiamethoxam: A second-generation neonicotinoid. Pest. Manag. Sci. 2001, 57, 165–176. [Google Scholar] [CrossRef]
- Onozaki, Y.; Horikoshi, R.; Ohno, I.; Kitsuda, S.; Durkin, K.A.; Suzuki, T.; Asahara, C.; Hiroki, N.; Komabashiri, R.; Shimizu, R.; et al. Flupyrimin: A novel insecticide acting at the nicotinic acetylcholine receptors. J. Agric. Food Chem. 2017, 65, 7865–7873. [Google Scholar] [CrossRef]
- Buszewski, B.; Bukowska, M.; Ligor, M.; Staneczko-Baranowska, I. A holistic study of neonicotinoids neuroactive insecticides—Properties, applications, occurrence, and analysis. Environ. Sci. Pollut. Res. 2019, 26, 34723–34740. [Google Scholar] [CrossRef]
- Tian, Z.Z.; Shao, X.S.; Li, Z.; Qian, X.H.; Huang, Q.C. Synthesis, insecticidal activity, and QSAR of novel nitromethylene neonicotinoids with tetrahydropyridine fixed cis configuration and exo-ring ether modification. J. Agric. Food Chem. 2007, 55, 2288–2292. [Google Scholar] [CrossRef] [PubMed]
- Kagabu, S.; Nishimura, K.; Naruse, Y.; Ohno, I. Insecticidal and neuroblocking potencies of variants of the thiazolidine moiety of thiacloprid and quantitative relationship study for the key neonicotinoid pharmacophore. J. Pestic. Sci. 2008, 33, 58–66. [Google Scholar] [CrossRef]
- Krumlinde, P.; Bogár, K.; Bäckvall, J. Synthesis of a neonicotinoide pesticide derivative via chemoenzymatic dynamic kinetic resolution. J. Org. Chem. 2009, 74, 7407–7410. [Google Scholar] [CrossRef] [PubMed]
- Wakita, T.; Kinoshita, K.; Yamada, E.; Yasui, N.; Kawahara, N.; Naoi, A.; Nakaya, M.; Ebihara, K.; Matsuno, H.; Kodaka, K. The discovery of dinotefuran: A novel neonicotinoid. Pest. Manag. Sci. 2003, 59, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Ohno, I.; Tomizawa, M.; Durkin, K.A.; Casida, J.E.; Kagabu, S. Neonicotinoid substituents forming a water bridge at the nicotinic acetylcholine receptor. J. Agric. Food Chem. 2009, 57, 2436–2440. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.B.; Kumar, C.N.S.S.P.; Santhoshi, A.; Kumar, K.P.; Murthy, U.S.N.; Rao, V.J. Efficient synthesis of N-allylated 2-nitroiminoimidazolidine analogues from Baylis-Hillman bromides. Synth. Commun. 2017, 47, 131–136. [Google Scholar] [CrossRef]
- Luna-Hernández, S.A.; Bonilla-Landa, I.; Reyes-Luna, A.; Rodríguez-Hernández, A.; Cuapio-Muñoz, U.; Ibarra-Juárez, L.A.; Suarez-Mendez, G.; Barrera-Méndez, F.; Pérez-Landa, I.D.; Enríquez-Medrano, F.J.; et al. Synthesis and insecticidal evaluation of chiral neonicotinoids analogs: The laurel wilt case. Molecules 2021, 26, 4225. [Google Scholar] [CrossRef] [PubMed]
- Bonilla-Landa, I.; Cuapio-Muñoz, U.; Luna-Hernández, A.; Reyes-Luna, A.; Rodríguez-Hernández, A.; Ibarra-Juarez, A.; Suarez-Mendez, G.; Barrera-Méndez, F.; Caram-Salas, N.; Enríquez-Medrano, J.F.; et al. L-Proline as a valuable scaffold for the synthesis of novel enantiopure neonicotinoids analogs. J. Agric. Food Chem. 2021, 69, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Kagabu, S.; Aoki, E.; Ohno, I. Is pyridylmethyl group of imidacloprid replaceable with fluoroalkyl moiety as a hydrogen-bond acceptor. J. Pestic. Sci. 2007, 32, 128–130. [Google Scholar] [CrossRef]
- Ge, D.H.; Chu, X.Q. Multiple-fold C–F bond functionalization for the synthesis of (hetero)cyclic compounds: Fluorine as a detachable chemical handle. Org. Chem. Front. 2022, 9, 2013–2055. [Google Scholar] [CrossRef]
- Chen, S.J.; He, Z.Q.; Chen, G.S.; Zhao, C.; Chen, C.P.; Zhuang, Y.Y.; Chen, L.; Liu, Y.L. Synthesis of CF3-substituted alkylamines, 1,2-bisazoles, and 1,4,5,6-tetrahydro-1,2,4-triazines from newly designed tetrazole-activated trifluoromethyl alkenes. Org. Lett. 2022, 24, 9301–9305. [Google Scholar] [CrossRef]
- Li, W.Y.; Chen, X.F.; Zhou, L. Photocatalytic defluorinative three-component reaction of α-trifluoromethyl alkenes, alkenes, and sodium sulfinates: Synthesis of monofluorocyclopentenes. Org. Lett. 2022, 24, 5946–5950. [Google Scholar] [CrossRef] [PubMed]
- Claraz, A.; Allain, C.; Masson, G. Electroreductive cross-coupling of trifluoromethyl alkenes and redox active esters for the synthesis of gem-difluoroalkene. Chem. Eur. J. 2022, 28, e20210337. [Google Scholar] [CrossRef]
- Zhao, F.; Zhou, W.L.; Zuo, Z. Recent advances in the synthesis of difluorinated architectures from trifluoromethyl groups. Adv. Synth. Catal. 2022, 364, 234–267. [Google Scholar] [CrossRef]
- Ma, T.; Li, X.; Ping, Y.Y.; Kong, W.Q. Synthesis of gem-difluoroalkenes via Ni-catalyzed three-component defluorinative reductive cross-coupling of organohalides, alkenes and trifluoromethyl alkenes. Chin. J. Chem. 2022, 40, 2212–2218. [Google Scholar] [CrossRef]
- Wang, K.; Chen, J.C.; Liu, W.F.; Kong, W.Q. Nickel-catalyzed defluorinative asymmetric cyclization of fluoroalkyl-substituted 1,6-enynes for the synthesis of seletracetam. Angew. Chem. Int. Ed. 2022, 61, e202212664. [Google Scholar]
- Du, D.H.; Peng, H.; He, L.; Bai, S.P.; Li, Z.H.; Teng, H.L. Synthesis of remote fluoroalkenyl ketones by photo-induced ring-opening addition of cyclic alkoxy radicals to fluorinated alkenes. Org. Biomol. Chem. 2022, 20, 9313–9318. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.B.; Qiu, K.Y.; Guo, M. Recent advance in the C–F bond functionalization of trifluoromethyl-containing compounds. Org. Chem. Front. 2021, 8, 3915–3942. [Google Scholar] [CrossRef]
- Kim, H.; Jung, Y.L.; Cho, S.H. Defluorinative C−C bond-forming reaction of trifluoromethyl alkenes with gem-(diborylalkyl)lithiums. Org. Lett. 2022, 24, 2705–2710. [Google Scholar] [CrossRef]
- Qiu, J.; Wang, C.C.; Zhou, L.; Lou, Y.X.; Yang, K.; Song, Q.L. Ni-Catalyzed radical-promoted defluoroalkylborylation of trifluorom. Org. Lett. 2022, 24, 2446–2451. [Google Scholar] [CrossRef] [PubMed]
- Aelterman, M.; Biremond, T.; Jubault, P.; Poisson, T. Electrochemical synthesis of gem-difluoro- and γ-fluoro allyl boronates and silans. Chem. Eur. J. 2022, 28, e202202194. [Google Scholar] [CrossRef]
- Fan, P.; Zhang, C.; Lan, Y.; Lin, Z.Y.; Zhang, L.C.; Wang, C. Photocatalytic hydroacylation of trifluoromethyl alkenes. Chem. Commun. 2019, 55, 12691–12694. [Google Scholar] [CrossRef]
- Zhu, C.; Zhang, H.; Liu, Q.; Chen, K.; Liu, Z.Y.; Feng, C. Nickel-catalyzed anti-markovnikov hydroalkylation of trifluoromethylalkenes. ACS Catal. 2022, 12, 9410–9417. [Google Scholar] [CrossRef]
- Hu, M.; Tan, B.B.; Ge, S.Z. Enantioselective cobalt-catalyzed hydroboration of fluoroalkyl substituted alkenes to access chiral fluoroalkylboronates. J. Am. Chem. Soc. 2022, 144, 15333–15338. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Zhang, Y.Z.; Guo, Y.Y.; Liu, S.S.; Shen, X. Reductive quenching-initiated catalyst-controlled divergent alkylation of α-CF3-olefins. Chem. Catal. 2022, 2, 1380–1393. [Google Scholar] [CrossRef]
- Xie, Z.Z.; Zheng, Y.; Yuan, C.P.; Guan, J.P.; Ye, Z.P.; Xiao, J.A.; Xiang, H.Y.; Chen, K.; Chen, X.Q.; Yang, H. Photoredox-catalyzed deoxygenation of hexafluoroacetone hydrate enables hydroxypolyfluoroalkylation of alkenes. Angew. Chem. Int. Ed. 2022, e202211035. [Google Scholar]
- Deng, Y.P.; He, J.J.; Cao, S.; Qian, X.H. Advances in cycloaddition and hydroaddition reaction of α-(trifluoromethyl)styrenes without defluorination: An alternative approach to CF3-containing compounds. Chin. Chem. Lett. 2022, 33, 2363–2371. [Google Scholar] [CrossRef]
- Bégué, J.; Bonnet-Delpon, D.; Rock, M.H. Addition of organolithium reagents to α-(trifluoromethyl)styrene: Concise synthesis of functionalised gem-difluoroalkenes. J. Chem. Soc. Perkin Trans. 1996, 1, 1409–1413. [Google Scholar] [CrossRef]
- Zeng, H.; Cai, Y.Y.; Jiang, H.F.; Zhu, C.L. Two C(sp3)−F bond activation in a CF3 group: Ipso-defluorinative amination triggered 1,3-diamination of (trifluoromethyl)alkenes with indoles, carbazoles, pyrroles, and sulfonamides. Org. Lett. 2021, 23, 66–70. [Google Scholar] [CrossRef]
- Zeng, H.; Li, H.Y.; Li, C.X.; Jiang, H.F.; Zhu, C.L. Bond energy enabled amine distinguishing strategy: Chemo-, regioselective 1,3-diamination of (trifluoromethyl)alkenes with different amines by two C(sp3)–F bond cleavages. Org. Chem. Front. 2022, 9, 1383–1388. [Google Scholar] [CrossRef]
- He, J.J.; Liu, C.; Deng, Y.P.; Zeng, Q.D.; Zhang, Y.; Liu, Y.; Zheng, P.; Cao, S. DBN-Mediated addition reaction of α-(trifluoromethyl)styrenes with diazoles, triazoles, tetrazoles, and primary, secondary, and secondary cyclic amines. Org. Lett. 2022, 24, 2299–2304. [Google Scholar] [CrossRef]
- Chen, F.L.; Xu, X.F.; He, Y.L.; Huang, G.P.; Zhu, S.L. NiH-Catalyzed migratory defluorinative olefin cross-coupling: Trifluoromethyl-substituted alkenes as acceptor olefins to form gem-difluoroalkenes. Angew. Chem. Int. Ed. 2020, 59, 5398–5402. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Yang, J.D.; Chen, J.P. Chemoselective catalytic hydrodefluorination of trifluoromethylalkenes towards mono-/gem-difluoroalkenes under metal-free condition. Nat. Commun. 2021, 12, 2835. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Cao, X.F.; Chen, X.; Li, Z.; Xu, X.Y. The structure modification of seven-membered aza-brigded neonicotinoids in order to investigate their impact on honey bees. J. Chem. Res. 2021, 835–844. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).