3.2. Experimental Procedures
Dimethyl 2-(3-((N-benzyl-4-methylphenyl)sulfonamido)prop-2-yn-1-yl)-2-(2-bromoallyl)-malonate (11). To a stirred solution of alkyne 16 (50 mg, 0.17 mmol, 1.0 eq.) in acetone (0.34 mL) at room temperature was added AgNO3 (2.9 mg, 10 mol%). After stirring for 5 min, N-bromosuccinimide (34 mg, 0.19 mmol, 1.1 eq.) was added and the resulting mixture was stirred for a further 4 h at rt. The reaction mixture was concentrated, then pentane was added, and the suspension was filtered through cotton wool to remove the white precipitate. The resulting solution was concentrated to obtain the corresponding bromoalkyne 17 (58 mg, 0.16 mmol), which was of sufficient purity to be used without further purification. Note 1: Because of similar Rf values of reactant and product, the reaction was monitored by NMR aliquot (conversion of singlet at 5.84 to 5.80 ppm). Note 2: If acetone is not evaporated completely before trituration with pentane, some succinimide will dissolve and contaminate the product. Note 3: The product was used immediately in the next step due to its tendency to decompose on storage.
To a mixture of N-tosylbenzylamine (245 mg, 0.938 mmol, 1.0 eq.), K3PO4 (398 mg, 1.88 mmol, 2.0 eq.), CuSO4 (23 mg, 0.094 mmol, 0.1 eq.) and 1,10-phenanthroline (34 mg, 0.187 mmol, 0.2 eq.) in a vial was added a solution of bromoalkyne 17 (380 mg, 1.03 mmol, 1.1 eq.) in toluene (1 mL). The vial was sealed and heated in an oil bath at 75 °C for 4 days. The reaction mixture was cooled to room temperature, diluted with EtOAc, filtered through Celite and concentrated. The product was purified via flash chromatography (pentane/EtOAc 7:3) to afford 11 (481 mg, 0.877 mmol, 93%) as a pale green gel. 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 8.0 Hz, 2HTs), 7.33 (d, J = 8.0 Hz, 2HTs), 7.30–7.27 (m, 3H), 7.25–7.21 (m, 2H), 5.45 (s, 2H), 4.41 (s, 2H), 3.67 (s, 6H), 3.07 (s, 2H), 2.91 (s, 2H), 2.45 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 169.8, 144.9, 135.1, 134.8, 130.1, 128.9, 128.9, 128.6, 127.9, 126.3, 123.2, 76.9, 65.6, 56.5, 55.6, 53.8, 53.3, 43.1, 22.0. HRMS (ESI) calc. for C25H26BrNO6S [M+H]+ 548.0737, found 548.0734.
Dimethyl 7-((N-benzyl-4-methylphenyl)sulfonamido)-1,3,4,5-tetrahydro-2H-indene-2,2-dicarboxylate (12). A vial loaded with potassium vinyltrifluoroborate (3.7 mg, 0.027 mmol, 1.5 eq.) was taken into the glovebox, and a solution of bromoynamide 11 (10.0 mg, 0.018 mmol, 1.0 eq. in 100 µL) and Pd(PPh3)4 (2.1 mg, 0.002 mmol, 10 mol% in 300 µL) in previously degassed THF were added. The vial was sealed, removed from the glovebox, and then a degassed solution of K2CO3 in water (7.6 mg, 0.055 mmol, 3.0 eq. in 50 µL) was quickly added under an inverted cone of nitrogen. The mixture was heated at 80 °C for 10 h. The mixture was cooled to room temperature and directly purified via flash chromatography (pentane/EtOAc 7:3) to afford 12 (8.0 mg, 0.016 mmol, 89%). 1H NMR (400 MHz, acetone) δ 7.74 (d, J = 8.1 Hz, 2H), 7.44 (d, J = 8.1 Hz, 2H), 7.36–7.15 (m, 5H), 5.20 (t, J = 4.6 Hz, 1H), 4.46 (s, 2H), 3.66 (s, 6H), 2.91 (s, 2H), 2.87 (s, 2H), 2.46 (s, 3H), 2.22–2.13 (m, 2H), 2.03 (d, J = 9.8 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 172.4, 143.5, 136.2, 136.1, 136.0, 134.8, 131.3, 129.5, 129.3, 128.4, 128.0, 127.8, 125.1, 54.7, 52.9, 43.6, 39.8, 29.8, 23.7, 22.5, 21.7. HRMS (ESI) calc. for C27H29NO6S [M+Na]+ 518.1608, found 548.1609.
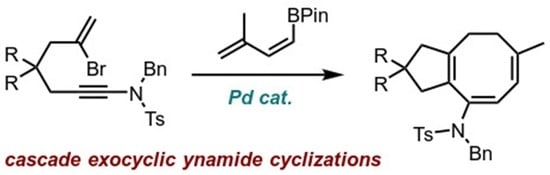
Dimethyl (6Z,8E)-9-((N-benzyl-4-methylphenyl)sulfonamido)-6-methyl-1,3,4,5-tetra-hydro-2H-cyclopenta[8]annulene-2,2-dicarboxylate (13). To a vial loaded with bromoynamide 11 (34 mg, 0.062 mmol, 1.0 eq.) and (Z)-boronate ester 23 (18 mg, 0.093 mmol, 1.5 eq.) was added dry and degassed THF (1.3 mL, 50 mM), degassed K2CO3 solution in water (0.13 mL, 19 mg/mL, 0.19 mmol, 3.0 eq.) and Pd(PPh3)4 (7.2 mg, 0.006 mmol, 10 mol%). The mixture was further degassed by bubbling N2, then sealed and heated overnight at 80 °C. After cooling to rt, water and EtOAc were added, and the organic layer was separated and washed with brine, dried over MgSO4 and concentrated. Purification via flash chromatography (pentane/EtOAc 7:3) afforded 13 (32 mg, 0.060 mmol, 96%) of the desired product. 1H NMR (400 MHz, CDCl3) δ 7.76 (d, J = 8.3 Hz, 2H), 7.37–7.13 (m, 7H), 5.77 (dd, J = 5.1, 1.5 Hz, 1H), 5.51 (d, J = 4.5 Hz, 1H), 4.55 (s, 2H), 3.71 (s, 6H), 2.91 (s, 2H), 2.61 (s, 2H), 2.44 (s, 3H), 2.02–1.96 (m, 2H), 1.84–1.78 (m, 2H), 1.65 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 172.4, 143.4, 143.2, 143.0, 137.8, 136.7, 131.7, 129.6, 129.2, 128.6, 128.5, 128.3, 127.8, 127.6, 120.1, 56.9, 52.9, 52.7, 46.0, 43.4, 30.5, 27.8, 25.4, 21.7. HRMS (ESI) calc. for C30H33NO6S [M+H]+ 536.2100, found 536.2101.
Dimethyl 2-(2-bromoallyl)-2-(prop-2-yn-1-yl)malonate (16). To a stirred suspension of pentane-washed NaH (162 mg pre-wash weight, 4.04 mmol, 1.5 eq., 60% in oil) in dry THF (12 mL) at room temperature under N
2 was slowly added
29 (676 mg, 2.69 mmol, 1.0 eq.), and the mixture was stirred for 30 min. Propargyl bromide (160 µL, 5.38 mmol, 2.0 eq., 80% wt. in toluene) was slowly added and the reaction mixture was stirred overnight. Diethyl ether and NH
4Cl (sat., aq.) were added and the organic layer was separated, washed with brine, dried over MgSO
4 and concentrated. Purification via flash chromatography (pentane/EtOAc 9:1) afforded
16 (490 mg, 1.69 mmol, 63%) as a colourless oil.
1H NMR (400 MHz, CDCl
3) δ 5.84 (br s, 1H), 5.63 (d,
J = 1.6 Hz, 1H), 3.77 (s, 6H), 3.31 (br s, 2H), 2.93 (d,
J = 2.7 Hz, 2H), 2.05 (t,
J = 2.7 Hz, 1H). Data in agreement with literature values [
15].
(Z)-4,4,5,5-Tetramethyl-2-(3-methylbuta-1,3-dien-1-yl)-1,3,2-dioxaborolane (23). A protocol for
trans-selective alkyne hydroboration developed by Miyaura and co-workers [
25] was applied. To a flame dried Schlenk flask was added anhydrous cyclohexane (27 mL, 0.25 M). The flask was taken into the glovebox and [Rh(cod)Cl]
2 (66 mg, 0.13 mmol, 1.5 mol%) and
i-Pr
3P (86 mg, 0.54 mmol, 6.0 mol%) were added. The flask capped with a septum and removed from the glovebox, then Et
3N (6.26 mL, 44.9 mmol, 5.0 eq.) and HBPin (1.15 g, 8.99 mmol, 1.0 eq.) were added and the mixture was stirred at room temperature for 30 min. 2-Methyl-1-buten-3-yne** (0.71 g, 10.8 mmol, 1.2 eq.) was added and the mixture was stirred for 3–4 h at room temperature (it could be also left overnight). The reaction was quenched by addition of MeOH (~10 mL) and evaporated (150 mbar, 30 °C). Purification via flash chromatography (pentane/EtOAc 98:2) afforded
23 (850 mg, 5.29 mmol, 49%) as a ~1:1
Z/
E mixture that was separated by reversed phase HPLC (Gemini-NX C18 5 µm, 21×150 mm, H
2O/MeCN 20:80, 9 mL/min, 6.6 min
Z-
23, 7.0 min
E-
23) to afford pure
Z isomer.
1H NMR (500 MHz, CDCl
3) δ 6.73 (d,
J = 14.9 Hz, 1H), 5.36 (d,
J = 14.9 Hz, 1H), 5.07–5.04 (m, 2H), 1.96 (s, 3H), 1.29 (s, 12H).
13C NMR (126 MHz, CDCl
3) δ 148.6, 144.0, 119.2, 83.7, 24.9, 20.5. The alkene carbon atom bonded to boron was not observed due to quadrupolar relaxation.
E isomer
1H NMR (400 MHz, CDCl
3) δ 7.11 (d,
J = 18.2 Hz, 1H), 5.56 (d,
J = 18.2 Hz, 1H), 5.16 (s, 2H), 1.85 (s, 3H), 1.29 (s, 12H). **2-Methyl-1-buten-3-yne (b.p. 35 °C) could be synthesized from 2-methylbut-3-yn-2-ol according to a literature procedure [
24], or purchased directly from a commercial source.
Dimethyl (3aR,7aR)-7-((N-benzyl-4-methylphenyl)sulfonamido)-3a,7a-dihydroxy-1,3,3a,4,5,7a-hexahydro-2H-indene-2,2-dicarboxylate (25). To a solution of diene
12 (10.0 mg, 0.020 mmol) in acetone (0.30 mL) was added NMO·H
2O (6.2 mg, 0.040 mmol, 2.0 eq.) and solution of K
2OsO
2(OH)
4 (0.7 mg, 0.002 mmol, 10 mol%) in water (0.10 mL). The reaction mixture was stirred for 4 h at room temperature. Aqueous Na
2S
2O
3 and EtOAc were added, and the organic layer was washed with sat. NaHCO
3, brine, dried (MgSO
4) and concentrated. Purification via flash chromatography (pentane/EtOAc 1:1) afforded
25 (3.2 mg, 0.006 mmol, 30%).
1H NMR (400 MHz, CDCl
3) δ 7.69 (d,
J = 8.3 Hz, 1H), 7.33–7.19 (m, 2H+5H), 5.27 (dd,
J = 5.0, 3.0 Hz, 1H), 4.57 (d,
J = 14.1 Hz, 1H), 4.41 (d,
J = 14.1 Hz, 1H), 3.74 (s, 3H), 3.63 (s, 3H), 2.64 (d,
J = 14.4 Hz, 1H), 2.50–2.44 (m, 1H), 2.44 (s, 3H), 2.31–2.19 (m, 1H +1H), 2.06–1.97 (m, 1H) 1.95–1.86 (m, 1H), 1.82–1.74 (m, 1H), 1.72–1.60 (m, 1H).
13C NMR (126 MHz, CDCl
3) δ 172.8, 144.2, 137.5, 135.7, 135.4, 134.1, 129.9, 129.6, 128.6, 128.6, 128.3, 79.5, 56.7, 55.7, 53.2, 53.1, 44.6, 44.5, 30.5, 22.7, 21.7.
HRMS (ESI) calc. for C
27H
31NO
8S [M+Na]
+ 552.1663, found 552.1659.
Dimethyl (3aR,7aR)-7-((N-benzyl-4-methylphenyl)sulfonamido)-4,5-dihydro-1H-3a,7a-epoxyindene-2,2(3H)-dicarboxylate (26). To a solution of diene 12 (10.0 mg, 0.020 mmol) in CH2Cl2 (0.40 mL) was added NaHCO3 (2.0 mg, 0.024 mmol, 1.2 eq.) and m-CPBA (70%, 5.5 mg, 0.022 mmol, 1.1 eq.) and the mixture was stirred for 30 min at rt. The colour turned from red to yellow upon addition of m-CPBA. The reaction was diluted with CH2Cl2, and the organic layer was washed with aqueous NaHCO3, Na2S2O3 and brine, dried (MgSO4) and concentrated. Purification via flash chromatography (pentane/EtOAc 7:3 to 1:1) afforded 26 (6.1 mg, 0.012 mmol, 59%) of the epoxide product. 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 8.3 Hz, 1H), 7.35–7.21 (m, 2H+5H), 5.29 (t, J = 4.9 Hz, 1H), 4.65 (d, J = 13.5 Hz, 1H), 4.26 (d, J = 13.5 Hz, 1H), 3.70 (s, 3H), 3.66 (s, 3H), 3.02 (d, J = 14.2 Hz, 1H), 2.96 (d, J = 14.2 Hz, 1H), 2.44 (s, 3H), 2.19–1.96 (m, 5H), 1.48 (dt, J = 14.0, 9.1 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 171.4, 171.3, 143.8, 135.8, 135.6, 134.8, 131.1, 129.8, 129.6, 128.5, 128.1, 128.1, 68.8, 63.5, 56.6, 55.2, 53.1, 53.0, 38.9, 36.3, 22.1, 21.9, 21.7. HRMS (ESI) calc. for C27H29NO7S [M+Na]+ 534.1557, found 534.1550.
Dimethyl (6R,7S,E)-9-((N-benzyl-4-methylphenyl)sulfonamido)-6,7-dihydroxy-6-methyl-1,3,4,5,6,7-hexahydro-2H-cyclopenta[8]annulene-2,2-dicarboxylate (27). To a solution of triene 14 (10.0 mg, 0.019 mmol) in acetone (0.30 mL) was added NMO.H2O (3.4 mg, 0.040 mmol, 2.0 eq.) and a solution of K2OsO2(OH)4 (0.7 mg, 0.002 mmol, 10 mol%) in water (0.10 mL). The reaction mixture was stirred at rt, 3 h. Aqueous Na2S2O3 and EtOAc were added, and the organic layer was washed with sat. NaHCO3, brine, dried (MgSO4) and concentrated. Purification via flash chromatography (pentane/EtOAc 1:1) afforded 27 (7.5 mg, 0.013 mmol, 71%). 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 8.2 Hz, 1HTs), 7.35–7.14 (m, 2H+5H), 5.41 (d, J = 7.6 Hz, 1H), 4.95 (d, J = 14.4 Hz, 1H), 4.15 (d, J = 14.4 Hz, 1H), 3.93 (d, J = 7.6 Hz, 1H), 3.75 (s, 3H), 3.67 (s, 3H), 3.01–2.91 (m, +1H), 2.76 (d, J = 17.1 Hz, 1H), 2.44 (s, 3H), 2.39–2.31 (m, 1H), 2.07–1.90 (m, 2H), 1.25–1.20 (m, 2H), 1.19 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 172.8, 144.2, 137.5, 135.7, 135.4, 134.1, 129.9, 129.6, 128.6, 128.6, 128.3, 79.5, 56.7, 55.7, 53.2, 53.1, 44.6, 44.5, 30.5, 22.7, 21.7. HRMS (ESI) calc. for C27H31NO8S [M+Na]+ 552.1663, found 552.1659.
Dimethyl (1aR,8aS,E)-7-((N-benzyl-4-methylphenyl)sulfonamido)-1a-methyl-1a,2, 3,4,6,8a-hexahydro-5H-cyclopenta[5,6]cycloocta[1,2-b]oxirene-5,5-dicarboxylate (28). To a solution of triene 14 (20 mg, 0.037 mmol) in CH2Cl2 (0.5 mL) was added NaHCO3 (5 mg, 0.056 mmol, 1.5 eq.) and m-CPBA (70%, 10 mg, 0.041 mmol, 1.1 eq.). The mixture was stirred overnight at rt, diluted with CH2Cl2, and washed with aqueous Na2S2O3 (aq., sat.), NaHCO3 (aq., sat.), and brine, dried (MgSO4) and concentrated. Purification via flash chromatography (pentane/EtOAc 7:3) afforded 28 (8.7 mg, 0.016 mmol, 42%). 1H NMR (400 MHz, CDCl3) δ 7.76 (d, J = 8.2 Hz, 2H), 7.32 (d, J = 8.2 Hz, 2H), 7.25–7.18 (m, 3H), 7.14–7.07 (m, 2H), 5.54 (s, 1H), 4.86 (d, J = 15.0 Hz, 1H), 4.18 (d, J = 15.0 Hz, 1H), 3.76 (s, 3H), 3.70 (s, 3H), 3.24 (s, 1H), 2.98–2.84 (m, 2H), 2.81 (d, J = 16.0 Hz, 1H), 2.56 (d, J = 16.0 Hz, 1H), 2.45 (s, 3H), 1.89–1.84 (m, 2H), 1.49 (dt, J = 13.8, 4.4 Hz, 1H), 1.13 (s, 3H), 1.06–0.96 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 172.3, 172.1, 143.9, 137.6, 136.3, 129.8, 128.7, 128.5, 127.9, 127.8, 127.7, 124.8, 61.0, 59.9, 57.2, 53.1, 53.0, 51.8, 47.2, 43.8, 29.8, 27.3, 21.7. HRMS (ESI) calc. for C30H33NO7S [M+H]+ 552.2050, found 552.2052.
Dimethyl 2-(2-bromoallyl)malonate (29). To a stirred suspension of pentane-washed NaH (1.09 g pre-wash weight, 27.2 mmol, 1.2 eq., 60% in oil) in dry THF (68 mL) at room temperature under N
2 was slowly added dimethyl malonate (2.61 mL, 22.7 mmol, 1.0 eq.), and the mixture was stirred for 30 min. 2,3-Dibromopropene (2.22 mL, 22.7 mmol, 1.0 eq. neat) was slowly added and the reaction mixture was stirred overnight. Diethyl ether and NH
4Cl (sat., aq.) were added. The organic layer was separated, washed with brine, dried over MgSO
4 and concentrated. The product was distilled under reduced pressure (0.5 mbar) and three fractions were collected. The first fraction (45 °C) contained unreacted dimethyl malonate, the second (65–90 °C) contained pure monoallylated product and the third fraction (100–110 °C) contained mostly diallylated product (these products were very hard to separate by chromatography). The desired product
29 was thus obtained (3.49 g, 13.9 mmol, 61%) as a colourless liquid.
1H NMR (400 MHz, CDCl
3) δ 5.69 (s, 1H), 5.47 (s, 1H
9), 3.82 (t,
J = 7.5 Hz, 1H), 3.75 (s, 6H), 3.02 (d,
J = 7.5 Hz, 2H). Data in agreement with literature values [
28].
Dimethyl 2-(2-bromoallyl)-2-(3-oxopropyl)malonate (30). A modified literature protocol for acrolein conjugate addition was applied [
29]. To a mixture of acrolein (120 µL, 1.78 mmol, 1.0 eq.) and bromoallyl malonate
29 (448 mg, 1.78 mmol, 1.0 eq.) in MeOH (5.2 mL) at room temperature was slowly added NaOMe (25% wt solution in MeOH, 82 µL, 0.36 mmol, 0.2 eq.). The resulting mixture was stirred for 4 h, then the solvent was evaporated under reduced pressure. The residue was dissolved in Et
2O and washed with water, brine, dried over MgSO
4, filtered and concentrated under reduced pressure. The residue was purified via flash column chromatography (pentane/EtOAc 7:3) to give
30 (259 mg, 0.84 mmol, 47%) as a viscous liquid.
1H NMR (400 MHz, CDCl
3) δ 9.73 (t,
J = 1.3 Hz, 1H), 5.67 (dt,
J = 1.8, 0.9 Hz, 1H), 5.60 (d,
J = 1.8 Hz, 1H), 3.74 (s, 6H), 3.18 (d,
J = 0.9 Hz, 2H), 2.52–2.46 (m, 2H), 2.37–2.27 (m, 2H).
13C NMR (101 MHz, CDCl
3) δ 200.3, 170.5, 126.6, 122.3, 56.3, 52.8, 44.0, 39.1, 24.4.
(E)-N-Benzyl-N-(1,2-dichlorovinyl)-4-methylbenzenesulfonamide (32). Prepared according to a literature procedure [
26]. To a stirred suspension of
N-tosylbenzylamine (1.50 g, 5.74 mmol, 1.0 eq.) and powdered Cs
2CO
3 (2.81 g, 8.61 mmol, 1.5 eq.) in DMF (4.3 mL), at 50 °C was added trichloroethylene (0.57 mL, 6.31 mmol, 1.1 eq.) dropwise over 10 min. The resulting mixture was stirred at 50 °C until the reaction reached completion as judged by TLC (~1–2 h). The mixture was cooled to room temperature, then partitioned between EtOAc and H
2O. The organic layer was separated and further washed with water (×2) and brine. The organic layer was then dried (MgSO
4), filtered and concentrated. Recrystallisation from EtOAc (10–20 mL) afforded
32 (1.50 g, 4.20 mmol, 73%) as white crystals.
1H NMR (400 MHz, CDCl
3) δ 7.86 (d,
J = 8.3 Hz, 2H), 7.36 (d,
J = 8.3 Hz, 2H), 7.34–7.27 (m, 5H), 6.27 (s, 1H), 5.08–3.69 (very br s, 2H) 2.47 (s, 3H).
13C NMR (101 MHz, CDCl
3) δ 144.7, 135.2, 133.4, 129.8, 129.4, 128.5, 128.5, 128.4, 121.7, 77.2, 51.8, 21.7. Data in agreement with literature values [
26].
Dimethyl 2-(5-((N-benzyl-4-methylphenyl)sulfonamido)-3-((tert-butyldimethylsilyl)oxy)-pent-4-yn-1-yl)-2-(2-bromoallyl)malonate (34). Synthesized according to the protocol for ynamide synthesis developed by Anderson et al. [
26]. To an oven dried, argon flushed flask was added 1,2-dichloroenamide
32 (116 mg, 0.33 mmol, 2.5 eq.) and anhydrous THF (1.1 mL), and the mixture was cooled to −78 °C whilst stirring. A solution of phenyllithium (1.9 M solution in dibutyl ether, 0.34 mL, 5.0 eq.) was then added dropwise, and the mixture was left to stir at −78 °C for 15 min. After complete conversion to the lithiated ynamide (as confirmed by TLC consumption of the dichloroenamide), a solution of the aldehyde
30 (40 mg, 0.13 mmol, 1.0 eq.) in anhydrous THF (0.6 mL) was added at −78 °C and the mixture was stirred for 1 h. The reaction was quenched with NH
4Cl (at −78 °C, sat., aq.), then warmed to room temperature and extracted with Et
2O. The organic extract was washed with brine, dried (MgSO
4) and concentrated. The crude product was used directly in the next step.
To a stirred solution of the crude alcohol in DMF (1 mL) at room temperature was added imidazole (14 mg, 0.19 mmol, 1.5 eq.) and TBSCl (20 mg, 0.13 mmol, 1.0 eq.), and the mixture was stirred for 1 h until complete (as monitored by TLC). Water and Et2O were added, then the organic layer was separated and washed with brine, dried (MgSO4) and concentrated. Since the ynamide impurity (resulting from protonation of excess lithiated ynamide from previous step) has a similar Rf value to the product, it was removed by washing the crude product with pentane several times (until the yellow colour faded). A solid, white impurity that is not soluble in pentane remains as a precipitate. The pentane washes were concentrated, and the residue was purified via flash chromatography (pentane/EtOAc 85:15) to afforded 34 (28 mg, 0.04 mmol, 30% over two steps) as an oil. 1H NMR (400 MHz, d6-acetone) δ 7.84 (d, J = 8.3 Hz, 2H), 7.49 (d, J = 8.3 Hz, 2H), 7.40–7.24 (m, 5H), 5.81–5.69 (m, 1H), 5.56 (d, J = 1. Hz, 1H), 4.54 (s, 1H), 4.52 (s, 1H), 4.50–4.46 (m, 1H), 3.72 (s, 3H), 3.71 (s, 3H), 3.13 (s, 2H), 2.48 (s, 3H), 2.15–2.09 (m, 2H), 1.54–1.42 (m, 2H), 0.83 (s, 9H), 0.01 (s, 3H), −0.04 (s, 3H). 13C NMR (101 MHz, d6-acetone) δ 170.4, 144.9, 135.1, 134.9, 130.0, 128.7, 128.5, 128.2, 127.7, 126.9, 122.1, 78.2, 72.0, 62.5, 56.4, 55.4, 52.1, 52.1, 42.9, 33.3, 27.2, 25.3, 20.7, 17.8, −5.3, −5.9. HRMS (ESI) calc. for C33H44BrNO7SSi [M+Na]+ 728.1683, found 728.1679.
Dimethyl 8-((N-benzyl-4-methylphenyl)sulfonamido)-6-((tert-butyldimethylslyl)-oxy)bicyclo[5.2.0]nona-1,7-diene-3,3-dicarboxylate (37) and
dimethyl 2-(5-((N-benzyl-4-methylphenyl)sulfonamido)-3-((tert-butyldimethylsilyl)oxy)pent-4-yn-1-yl)-2-(2-methylenebut-3-en-1-yl)malonate (38). To a vial loaded with bromoynamide
34 (11 mg, 0.016 mmol, 1.0 eq.) and potassium vinyltrifluoroborate
20 (3.1 mg, 0.023 mmol, 1.5 eq.) was added dry and degassed THF (0.40 mL), degassed K
2CO
3 solution in water (6.4 mg in 40 µL water, 0.047 mmol, 3.0 eq.) and Pd(PPh
3)
4 (1.8 mg, 0.002 mmol, 10 mol%). The mixture was further degassed by bubbling N
2, then sealed and heated overnight at 80 °C. After cooling to rt, water and EtOAc were added, the organic layer was separated and washed with brine, dried over MgSO
4 and concentrated. Purification via flash chromatography (pentane/EtOAc 7:3) afforded undesired products
37 (2.8 mg, 0.005 mmol, 28%) and 38 (3.1 mg, 0.005 mmol, 30%). 37:
1H NMR (400 MHz, Acetone) δ 7.76 (d,
J = 8.4 Hz, 2H), 7.52–7.38 (m, 2H), 7.40–7.24 (m, 5H), 5.14 (d,
J = 16.8 Hz, 1H), 5.00 (s, 1H), 4.79 (d,
J = 16.8 Hz, 1H), 4.28 (br s, 1H), 3.68 (s, 3H), 3.65 (s, 3H), 3.23–3.18 (m, 2H), 2.44 (s, 3H), 2.44–2.40 (m, 1H), 2.08–2.02 (m, 1H), 1.82–1.71 (m, 1H), 1.68–1.58 (m, 1H), 0.79 (s, 9H), −0.00 (s, 3H), −0.02 (s, 3H). 38:
1H NMR (400 MHz, CDCl
3) δ 7.74 (d,
J = 8.3 Hz, 2H), 7.34–7.27 (m, 5H+2H), 6.25 (dd,
J = 17.5, 10.9 Hz, 1H), 5.20 (d,
J = 17.5 Hz, 1H), 5.14 (s, 1H), 5.00 (d,
J = 10.9 Hz, 1H), 4.94 (s, 1H), 4.51 (d,
J = 14.0 Hz, 1H), 4.43 (d,
J = 14.0 Hz, 1H), 4.33 (t,
J = 6.1 Hz, 1H), 3.68 (s, 3H), 3.67 (s, 3H), 2.82 (s, 2H), 2.44 (s, 3H), 2.07–1.85 (m, 2H), 1.54–1.39 (m, 2H), 0.81 (s, 9H), −0.04 (s, 3H), −0.08 (s, 3H). Further structural assignment is provided in the
Supporting information.
N-Benzyl-N-(4-((3S,3aR,6aR)-3a-(2-bromoallyl)-2,2-dimethyl-5-oxohexahydrofuro[3,2-b]furan-3-yl)-3-hydroxybut-1-yn-1-yl)-4-methylbenzenesulfonamide (39). To an oven dried, argon flushed flask was added 1,2-dichloroenamide
32 (18 mg, 0.051 mmol, 3.0 eq.) and anhydrous THF (0.5 mL), and the mixture was cooled to −78 °C. A solution of phenyllithium (1.9 M solution in dibutyl ether, 25 µL, 2.8 eq.) was then added dropwise, and the mixture was left to stir at −78 °C for 15 min. After almost complete conversion to the lithiated ynamide (the 1,2-dichloroenamide is in excess, so a small amount remains), the aldehyde 6 (5.4 mg, 0.017 mmol) in anhydrous THF (0.5 mL) was added at −78 °C and stirred for 1 h. The reaction was quenched by the addition of NH
4Cl (at −78 °C, sat., aq.), then warmed to room temperature and extracted with Et
2O. The organic extract was washed with brine, dried (MgSO
4) and concentrated. The product was purified via flash chromatography to afford
39 (5.1 mg, 0.008 mmol, 50%) as a 2:1 mixture of diastereomeric alcohols.
1H NMR (500 MHz, CDCl
3) δ 7.78–7.74 (m, 2H), 7.39–7.25 (m, 2H+5H), 5.77–5.63 (m, 2H), 4.83–4.79 (m, 1H), 4.58–4.54 (m, 1H), 4.51–4.43 (m, 2H), 3.37–3.25 (m, 1H), 2.93–2.82 (m, 1H), 2.71–2.59 (m, 1H), 2.59–2.52 (m, 1H), 2.46 (s, 3H), 2.29 (dd,
J = 9.6, 3.9 Hz, 1H), 1.94 (d,
J = 5.6 Hz, 1H), 1.74–1.67 (m, 1H), 1.46–1.38 (m, 1H), 1.13 (s, 3H), 1.02 (s, 3H).
HRMS (ESI) calc. for C
29H
32BrNO
6S [M+H]
+ 602.1207, found 602.1207. Due to insufficient material, the product structure was assigned using
1H NMR,
1H–
1H COSY and
13C edited-HSQC (see the
Supplementary Materials).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
