

Palladium-Catalyzed N-Alkenylation of N-Aryl Phosphoramidates with Alkenes
Abstract
1. Introduction
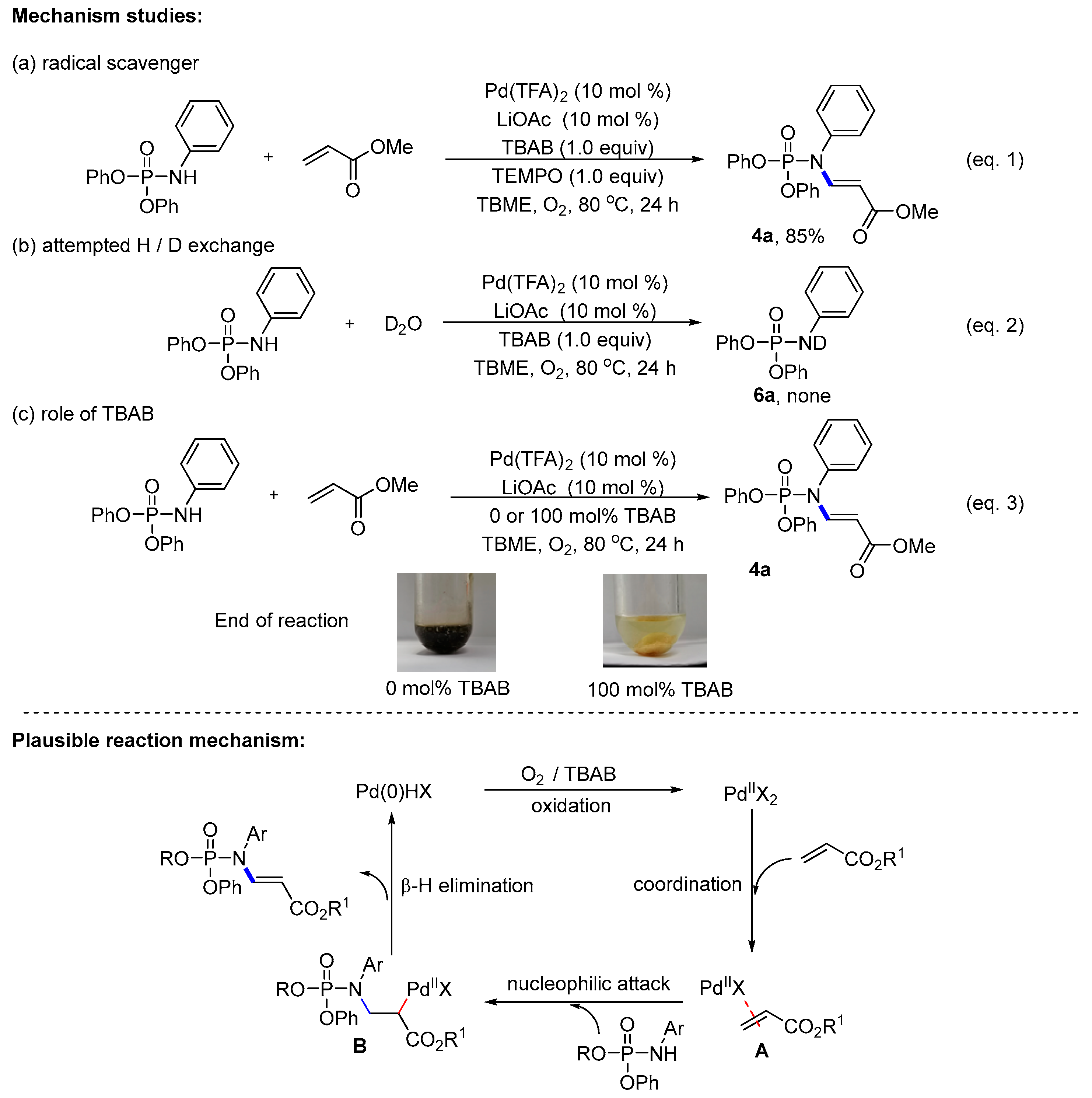
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Procedure of Palladium-Catalyzed N-Alkenylation of Phosphoramidates with Alkenes
3.3. Characterization of Products in Details
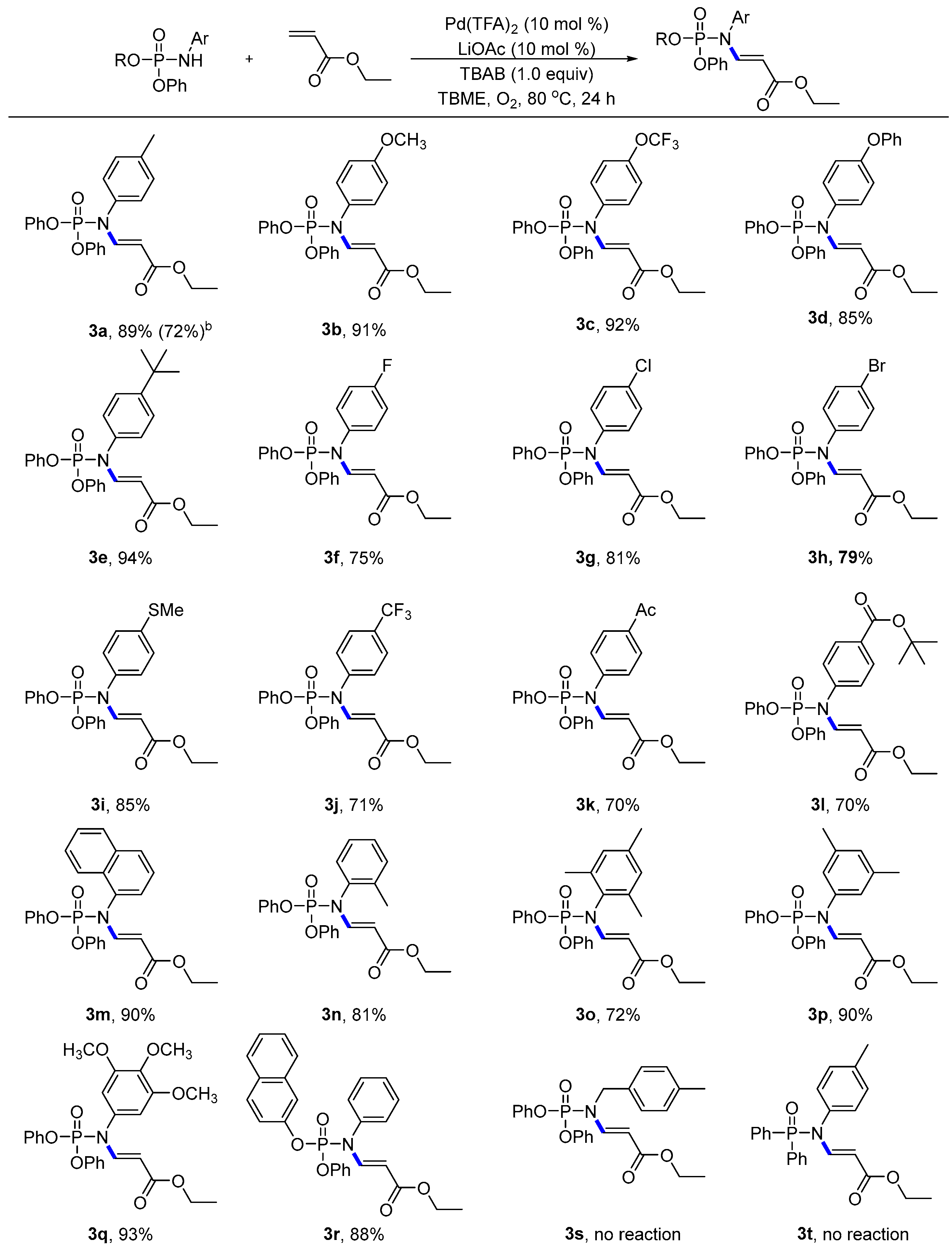
- Compound: ethyl (E)-3-((diphenoxyphosphoryl)(p-tolyl)amino)acrylate (3a): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (77.8 mg, 89% yield). 1H NMR (400 MHz, CDCl3): δ 8.31 (dd, J = 13.7, 7.8 Hz, 1H), 7.36 (t, J = 7.8 Hz, 4H), 7.23 (d, J = 7.7 Hz, 8H), 7.01 (d, J = 7.9 Hz, 2H), 4.79 (d, J = 13.7 Hz, 1H), 4.17 (q, J = 7.1 Hz, 2H), 2.40 (s, 3H), 1.27 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 167.43, 150.26 (d, J = 6.7 Hz), 147.20 (d, J = 9.3 Hz), 139.10, 134.02 (d, J = 2.3 Hz), 130.65, 130.00, 128.57 (d, J = 2.5 Hz), 125.67, 120.10 (d, J = 5.1 Hz), 100.42 (d, J = 9.7 Hz), 60.00, 21.23, 14.43. 31P NMR (162 MHz, CDCl3) δ-9.54; HRMS (ESI): calculated for C24H25NO5P [M + H]+ 438.14649, found 438.14714.
- Compound: ethyl (E)-3-((diphenoxyphosphoryl)(4-methoxyphenyl)amino)acrylate (3b): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (82.5 mg, 91% yield). 1H NMR (400 MHz, CDCl3): δ 8.20 (dd, J = 13.6, 7.5 Hz, 1H), 7.29 (t, J = 7.9 Hz, 4H), 7.14 (t, J = 8.1 Hz, 6H), 6.95 (d, J = 8.8 Hz, 2H), 6.85 (d, J = 8.9 Hz, 2H), 4.68 (dd, J = 13.7, 1.1 Hz, 1H), 4.09 (q, J = 7.2 Hz, 2H), 3.76 (s, 3H), 1.19 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 167.45, 159.81, 150.25 (d, J = 6.7 Hz), 147.43 (d, J = 9.8 Hz), 130.00, 129.95 (d, J = 2.6 Hz), 129.04 (d, J = 2.3 Hz), 125.67, 120.07 (d, J = 5.2 Hz), 115.17, 100.37 (d, J = 9.8 Hz), 60.02, 55.56, 14.42. 31P NMR (162 MHz, CDCl3) δ-9.47; HRMS (ESI): calculated for C24H25O6NP [M + H]+ 454.14140, found 454.14206.
- Compound: ethyl (E)-3-((diphenoxyphosphoryl)(4-(trifluoromethoxy)phenyl)amino)acrylate (3c): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (93.3 mg, 92% yield). 1H NMR (400 MHz, CDCl3): δ 8.26 (dd, J = 13.8, 7.7 Hz, 1H), 7.41–7.38 (m, 4H), 7.28–7.22 (m, 8H), 7.16–7.14 (m, 2H), 4.78 (d, J = 13.8 Hz, 1H), 4.20 (q, J = 7.1 Hz, 2H), 1.29 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 167.10, 150.04 (d, J = 6.9 Hz), 149.25, 146.47 (d, J = 8.5 Hz), 135.25 (d, J = 2.9 Hz), 130.50 (d, J = 2.8 Hz), 130.11, 125.91, 122.32, 120.4 (q, J = 257.0 Hz), 120.05 (d, J = 5.1 Hz), 100.97 (d, J = 9.3 Hz), 60.22, 14.39. 31P NMR (162 MHz, CDCl3) δ-9.96; 19F NMR (375 MHz, CDCl3) δ-57.85; HRMS (ESI): calculated for C24H22O6NF3P [M + H]+ 508.11313, found 508.11384.
- Compound: ethyl (E)-3-((diphenoxyphosphoryl)(4-phenoxyphenyl)amino)acrylate (3d): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (87.6 mg, 85% yield). 1H NMR (400 MHz, CDCl3): δ 8.30 (dd, J = 13.7, 7.6 Hz, 1H), 7.44–7.37 (m, 6H), 7.26–7.19 (m, 7H), 7.12–7.01 (m, 6H), 4.84 (d, J = 13.7 Hz, 1H), 4.21 (q, J = 7.1 Hz, 2H), 1.30 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 167.36, 158.15, 156.09, 150.21 (d, J = 6.7 Hz), 147.15 (d, J = 9.4 Hz), 131.07 (d, J = 2.4 Hz), 130.24 (d, J = 2.5 Hz), 125.76, 124.34, 120.09 (d, J = 5.1 Hz), 119.90, 119.20, 100.59 (d, J = 9.5 Hz), 60.11, 14.46. 31P NMR (162 MHz, CDCl3) δ-9.57; HRMS (ESI): calculated for C29H27NO6P [M + H]+ 516.15705, found 516.15730.
- Compound: ethyl (E)-3-((4-(tert-butyl)phenyl)(diphenoxyphosphoryl)amino)acrylate (3e): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a white solid (90.1 mg, 94% yield), Mp = 67–68 °C. 1H NMR (400 MHz, CDCl3): δ 8.29 (dd, J = 13.6, 7.8 Hz, 1H), 7.44 (d, J = 8.6 Hz, 2H), 7.38 (t, J = 7.8 Hz, 4H), 7.25–7.22 (m, 6H), 7.06–7.03 (m, 2H), 4.81 (d, J = 13.7 Hz, 1H), 4.48–4.02 (m, 12H), 1.37 (s, 9H), 1.29 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 167.47, 152.12, 150.25 (d, J = 6.8 Hz), 147.19 (d, J = 9.1 Hz), 133.91 (d, J = 2.3 Hz), 129.99, 128.22 (d, J = 2.5 Hz), 126.95, 125.68, 120.14 (d, J = 5.1 Hz), 100.39 (d, J = 9.8 Hz), 60.03, 34.80, 31.37, 14.45. 31P NMR (162 MHz, CDCl3) δ-9.47; HRMS (ESI): calculated for C27H31NO5P [M + H]+ 480.19344, found 480.19401.
- Compound: ethyl (E)-3-((diphenoxyphosphoryl)(4-fluorophenyl)amino)acrylate (3f): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (66.1 mg, 75% yield). 1H NMR (400 MHz, CDCl3): δ 8.27 (dd, J = 13.7, 7.6 Hz, 1H), 7.41–7.37 (m, 4H), 7.27–7.21 (m, 6H), 7.15–7.07 (m, 4H), 4.75 (d, J = 13.7 Hz, 1H), 4.19 (q, J = 7.1 Hz, 2H), 1.28 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 167.20, 162.53 (d, J = 249.6 Hz), 150.11 (d, J = 6.8 Hz), 146.84 (d, J = 9.1 Hz), 132.64, 130.74 (dd, J = 8.8, 2.6 Hz), 130.07, 125.83, 120.04 (d, J = 5.1 Hz), 117.05 (d, J = 23.0 Hz), 100.76 (d, J = 9.5 Hz), 60.14, 14.40. 31P NMR (162 MHz, CDCl3) δ-9.82; 19F NMR (375 MHz, CDCl3) δ-111.60; HRMS (ESI): calculated for C23H22O5NFP [M + H]+ 442.12141, found 442.12214.
- Compound: ethyl (E)-3-((4-chlorophenyl)(diphenoxyphosphoryl)amino)acrylate (3g): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (74.0 mg, 81% yield). 1H NMR (400 MHz, CDCl3): δ 8.25 (dd, J = 13.7, 7.7 Hz, 1H), 7.43–7.37 (m, 6H), 7.28–7.21 (m, 6H), 7.07–7.04 (m, 2H), 4.77 (d, J = 13.7 Hz, 1H), 4.19 (q, J = 7.1 Hz, 2H), 1.29 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 167.15, 150.07 (d, J = 6.8 Hz), 146.54 (d, J = 8.9 Hz), 135.21 (d, J = 29.0 Hz), 130.32, 130.26 (d, J = 2.5 Hz), 130.09, 125.87, 100.90 (d, J = 9.4 Hz), 60.17, 14.40. 31P NMR (162 MHz, CDCl3) δ-10.00; HRMS (ESI): calculated for C23H22NO5ClP [M + H]+ 458.09186, found 458.09268.
- Compound: ethyl (E)-3-((4-bromophenyl)(diphenoxyphosphoryl)amino)acrylate (3h): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (79.2 mg, 79% yield). 1H NMR (400 MHz, CDCl3): δ 8.25 (dd, J = 13.7, 7.7 Hz, 1H), 7.58 (d, J = 8.6 Hz, 2H), 7.39 (t, J = 7.9 Hz, 4H), 7.27–7.22 (m, 6H), 7.00–6.98 (m, 2H), 4.77 (d, J = 13.7 Hz, 1H), 4.19 (q, J = 7.1 Hz, 2H), 1.29 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 167.12, 150.06 (d, J = 6.8 Hz), 146.46 (d, J = 8.6 Hz), 135.91 (d, J = 2.8 Hz), 133.33, 130.58 (d, J = 2.6 Hz), 130.10, 125.88, 123.13, 120.05 (d, J = 5.2 Hz), 100.93 (d, J = 9.5 Hz), 60.18, 14.41. 31P NMR (162 MHz, CDCl3) δ-10.10; HRMS (ESI): calculated for C23H22NO5BrP [M + H]+ 502.04135, found 502.04192.
- Compound: ethyl (E)-3-((diphenoxyphosphoryl)(4-(methylthio)phenyl)amino)acrylate (3i): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (79.7 mg, 85% yield). 1H NMR (400 MHz, CDCl3): δ 8.28 (dd, J = 13.7, 7.6 Hz, 1H), 7.40–7.36 (m, 4H), 7.30–7.22 (m, 8H), 7.04–7.02 (m, 2H), 4.79 (d, J = 13.7 Hz, 1H), 4.18 (q, J = 7.1 Hz, 2H), 2.53 (s, 3H), 1.28 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 167.33, 150.18 (d, J = 6.8 Hz), 146.98 (d, J = 9.3 Hz), 140.30, 133.35, 130.04, 129.16 (d, J = 2.7 Hz), 127.31, 125.75, 120.08 (d, J = 5.2 Hz), 100.61 (d, J = 9.6 Hz), 60.08, 15.51, 14.42. 31P NMR (162 MHz, CDCl3) δ-9.72; HRMS (ESI): calculated for C24H25O5NPS [M + H]+ 470.11856, found 470.11920.
- Compound: ethyl (E)-3-((diphenoxyphosphoryl)(4-(trifluoromethyl)phenyl)amino)acrylate (3j): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (69.7 mg, 71% yield). 1H NMR (400 MHz, CDCl3): δ 8.25 (dd, J = 15.3, 7.8 Hz, 1H), 7.71 (d, J = 8.3 Hz, 1H), 7.57 (d, J = 8.5 Hz, 1H), 7.42–7.37 (m, 4H), 7.27–7.21 (m, 7H), 6.99 (d, J = 8.5 Hz, 1H), 4.77 (d, J = 13.7 Hz, 1H), 4.18 (q, J = 7.1 Hz, 2H), 1.29 (t, J = 6.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 167.01, 149.97 (d, J = 7.0 Hz), 146.46 (d, J = 9.1 Hz), 146.08 (d, J = 8.0 Hz), 140.32, 133.33, 130.58, 130.12 (d, J = 5.4 Hz), 129.45 (d, J = 2.5 Hz), 127.25, 126.00, 125.87, 120.08 (d, J = 5.1 Hz), 101.24 (d, J = 9.4 Hz), 60.28, 14.38. 31P NMR (162 MHz, CDCl3) δ-10.18; 19F NMR (375 MHz, CDCl3) δ-62.71; HRMS (ESI): calculated for C24H22NO5F3P [M + H]+ 492.11822, found 492.11904.
- Compound: ethyl (E)-3-((4-acetylphenyl)(diphenoxyphosphoryl)amino)acrylate (3k): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (65.1 mg, 70% yield). 1H NMR (400 MHz, CDCl3): δ 8.24 (dd, J = 13.8, 8.0 Hz, 1H), 8.03 (d, J = 8.4 Hz, 2H), 7.39 (t, J = 7.8 Hz, 4H), 7.28–7.21 (m, 8H), 4.78 (d, J = 13.8 Hz, 1H), 4.19 (q, J = 7.1 Hz, 2H), 2.65 (s, 3H), 1.28 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 196.93, 167.06, 149.98, 146.11 (d, J = 8.0 Hz), 141.26, 137.29, 130.11, 130.06, 129.14 (d, J = 2.6 Hz), 125.93, 120.08 (d, J = 5.1 Hz), 101.19 (d, J = 9.3 Hz), 60.21, 26.77, 14.39. 31P NMR (162 MHz, CDCl3) δ-10.23; HRMS (ESI): calculated for C26H29NO5P [M + H]+ 466.17779, found 466.17825.
- Compound: tert-butyl(E)-4-((diphenoxyphosphoryl)(3-ethoxy-3-oxoprop-1-en-1-yl)amino)benzoate (3l): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (73.2 mg, 70% yield). 1H NMR (400 MHz, CDCl3): δ 8.26 (dd, J = 13.8, 7.9 Hz, 1H), 8.07 (d, J = 8.4 Hz, 2H), 7.39 (t, J = 7.8 Hz, 4H), 7.27–7.18 (m, 8H), 4.77 (d, J = 13.7 Hz, 1H), 4.18 (q, J = 7.1 Hz, 2H), 1.64 (s, 9H), 1.27 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 167.12, 164.70, 150.07 (d, J = 6.6 Hz), 146.36 (d, J = 8.5 Hz), 140.62, 132.64, 131.22, 130.08, 128.75 (d, J = 2.7 Hz), 120.06 (d, J = 5.2 Hz), 101.01 (d, J = 9.5 Hz), 81.74, 60.13, 28.21, 14.39. 31P NMR (162 MHz, CDCl3) δ-10.21; HRMS (ESI): calculated for C28H31NO7P [M + H]+ 524.18327, found 524.18371.
- Compound: ethyl (E)-3-((diphenoxyphosphoryl)(naphthalen-1-yl)amino)acrylate (3m): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (85.1 mg, 90% yield). 1H NMR (400 MHz, CDCl3): δ 8.51 (dd, J = 13.6, 7.4 Hz, 1H), 7.96–7.91 (m, 2H), 7.73 (d, J = 8.5 Hz, 1H), 7.53–7.49 (m, 2H), 7.42–7.37 (m, 4H), 7.34–7.31 (m, 2H), 7.29–7.24 (m, 3H), 7.19–7.15 (m, 1H), 7.07–7.04 (m, 2H), 4.63 (d, J = 13.7 Hz, 1H), 4.20–4.12 (m, 2H), 1.24 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 167.34, 150.52 (d, J = 7.1 Hz), 150.25 (d, J = 7.0 Hz), 146.38 (d, J = 8.8 Hz), 134.87, 133.08, 130.14, 129.92 (d, J = 2.1 Hz), 129.85, 128.57, 127.29, 127.10 (d, J = 2.7 Hz), 126.77, 123.09, 120.23 (d, J = 2.4 Hz), 120.18 (d, J = 2.1 Hz), 101.28 (d, J = 9.9 Hz), 60.07, 14.38. 31P NMR (162 MHz, CDCl3) δ-9.31; HRMS (ESI): calculated for C27H25NO5P [M + H]+ 474.14649, found 474.14661.
- Compound: ethyl (E)-3-((diphenoxyphosphoryl)(o-tolyl)amino)acrylate (3n): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (70.8 mg, 81% yield). 1H NMR (400 MHz, CDCl3): δ 8.28 (dd, J = 13.6, 7.5 Hz, 1H), 7.39–7.30 (m, 6H), 7.28–7.21 (m, 5H), 7.16–7.13 (m, 2H), 7.08–7.06(m, 1H), 4.67 (d, J = 13.6 Hz, 1H), 4.19 (qd, J = 7.1, 1.3 Hz, 2H), 2.14 (s, 3H), 1.28 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 167.45, 150.32 (dd, J = 21.5, 7.2 Hz), 145.56 (d, J = 8.7 Hz), 137.16 (d, J = 3.3 Hz), 135.26 (d, J = 2.3 Hz), 131.89, 129.99 (d, J = 10.8 Hz), 129.17 (d, J = 34.9 Hz), 127.58, 125.75 (d, J = 5.7 Hz), 120.28 (d, J = 4.8 Hz), 120.16 (d, J = 5.0 Hz), 100.14 (d, J = 10.0 Hz), 60.06, 17.52, 14.42. 31P NMR (162 MHz, CDCl3) δ-9.53; HRMS (ESI): calculated for C24H25NO5P [M + H]+ 438.14649, found 438.14709.
- Compound: ethyl (E)-3-((diphenoxyphosphoryl)(mesityl)amino)acrylate (3o): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (66.9 mg, 72% yield). 1H NMR (400 MHz, CDCl3): δ 8.25 (dd, J = 13.5, 7.3 Hz, 1H), 7.36–7.32 (m, 4H), 7.24–7.19 (m, 6H), 6.95 (s, 2H), 4.75 (d, J = 13.4 Hz, 1H), 4.22 (q, J = 7.1 Hz, 2H), 2.32 (s, 3H), 2.13 (s, 6H), 1.31 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 167.74, 150.52 (d, J = 7.7 Hz), 144.67 (d, J = 8.5 Hz), 138.72, 136.77 (d, J = 2.5 Hz), 131.66 (d, J = 2.9 Hz), 130.12, 129.89, 125.68, 120.45 (d, J = 4.8 Hz), 99.32 (d, J = 9.8 Hz), 60.06, 21.02, 18.04, 14.42. 31P NMR (162 MHz, CDCl3) δ-9.34; HRMS (ESI): calculated for C26H29NO5P [M + H]+ 466.17779, found 466.17815.
- Compound: ethyl (E)-3-((3,5-dimethylphenyl)(diphenoxyphosphoryl)amino)acrylate (3p): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a white solid (81.2 mg, 90% yield), Mp = 69–70 °C. 1H NMR (400 MHz, CDCl3): δ 8.39 (dd, J = 13.7, 7.7 Hz, 1H), 7.52–7.48 (m, 4H), 7.37–7.34 (m, 6H), 7.15 (s, 1H), 6.79 (s, 2H), 4.90 (d, J = 13.6 Hz, 1H), 4.30 (q, J = 7.0 Hz, 2H), 2.41 (s, 4H), 1.40 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 167.49, 150.31 (d, J = 6.6 Hz), 147.15 (d, J = 9.0 Hz), 139.71, 136.40 (d, J = 2.3 Hz), 130.68, 129.96, 126.33 (d, J = 2.7 Hz), 125.66, 120.14 (d, J = 5.3 Hz), 100.41 (d, J = 9.8 Hz), 60.00, 21.22, 14.44. 31P NMR (162 MHz, CDCl3) δ-9.60; HRMS (ESI): calculated for C25H27NO5P [M + H]+ 452.16214, found 452.16280.
- Compound: ethyl (E)-3-((diphenoxyphosphoryl)(3,4,5-trimethoxyphenyl)amino)acrylate (3q): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (95.4 mg, 93% yield). 1H NMR (400 MHz, CDCl3): δ 8.25 (dd, J = 13.6, 7.6 Hz, 1H), 7.40–7.36 (m, 4H), 7.27–7.21 (m, 6H), 6.20 (s, 2H), 4.84 (d, J = 13.6 Hz, 1H), 4.19 (q, J = 7.1 Hz, 2H), 3.87 (s, 3H), 3.67 (s, 6H), 1.29 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 167.37, 153.90, 150.24 (d, J = 6.6 Hz), 146.91 (d, J = 9.0 Hz), 138.29, 132.00 (d, J = 2.6 Hz), 130.04, 125.72, 120.04 (d, J = 5.3 Hz), 105.79 (d, J = 2.5 Hz), 100.67 (d, J = 9.5 Hz), 60.96, 60.12, 56.05, 14.43. 31P NMR (162 MHz, CDCl3) δ-9.99; HRMS (ESI): calculated for C26H29NO8P [M + H]+ 514.16253, found 514.16286.
- Compound: ethyl (E)-3-(((naphthalen-2-yloxy)(phenoxy)phosphoryl)(phenyl)amino)acrylate (3r): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (83.3 mg, 88% yield). 1H NMR (400 MHz, CDCl3): δ 8.37 (dd, J = 13.7, 7.8 Hz, 1H), 7.87–7.82 (m, 3H), 7.74–7.73 (m, 1H), 7.58–7.43 (m, 5H), 7.41–7.37 (m, 2H), 7.34–7.30 (m, 1H), 7.27–7.23 (m, 3H), 7.19–7.16 (m, 2H), 4.81 (d, J = 14.5 Hz, 1H), 4.20 (q, J = 7.1 Hz, 2H), 1.29 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 167.37, 150.27 (d, J = 7.0 Hz), 147.82 (d, J = 7.0 Hz), 147.01 (d, J = 9.0 Hz), 136.82, 133.86, 131.21, 130.23, 130.06, 129.11, 128.93 (d, J = 2.7 Hz), 127.79 (d, J = 9.9 Hz), 127.04, 125.93, 125.77, 120.13 (d, J = 5.2 Hz), 119.77 (d, J = 5.6 Hz), 116.77 (d, J = 5.0 Hz), 100.70 (d, J = 9.8 Hz), 60.08, 14.45. 31P NMR (162 MHz, CDCl3) δ-9.59; HRMS (ESI): calculated for C27H25NO5P [M + H]+ 474.14649, found 474.14693.
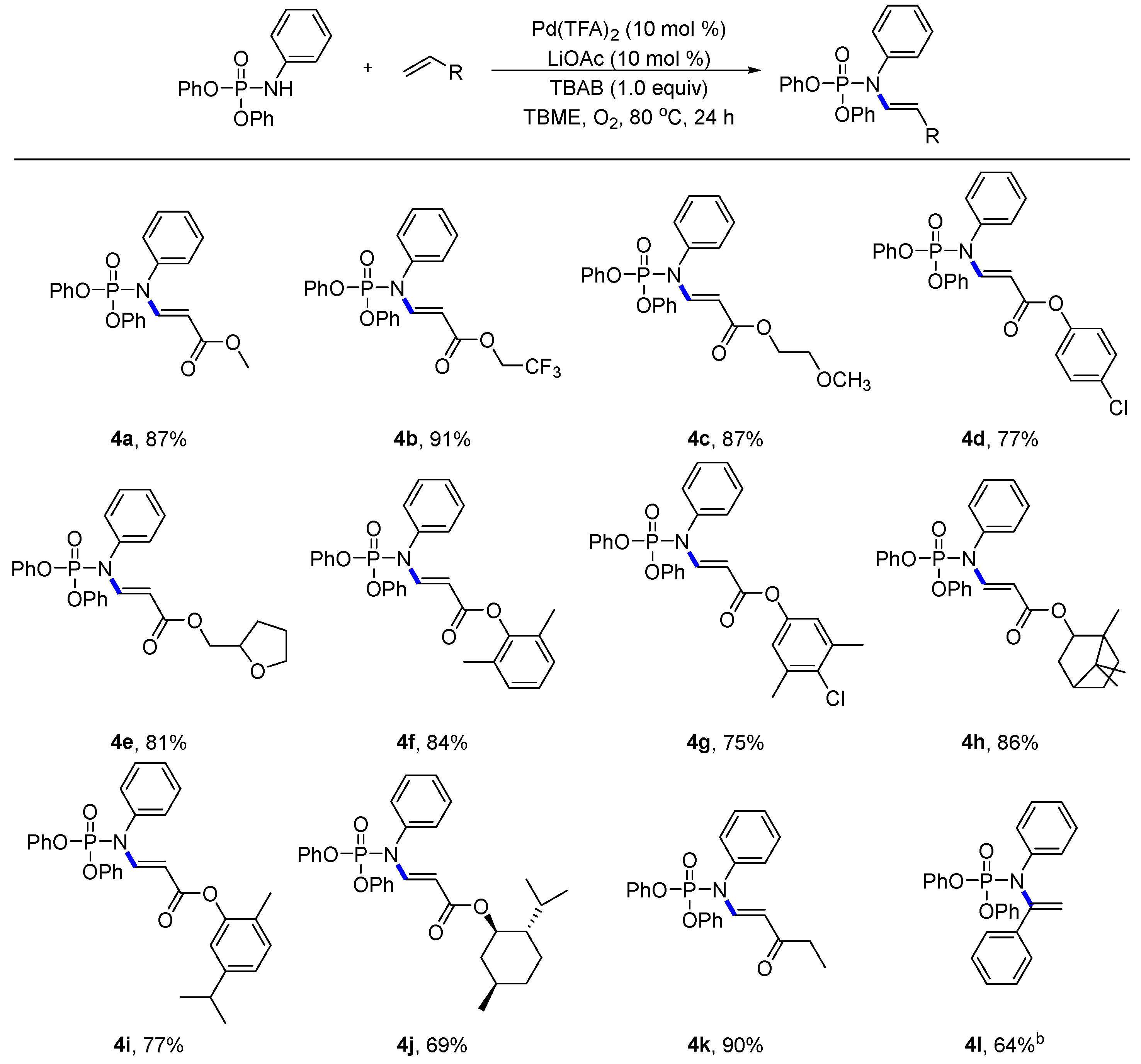
- Compound: methyl (E)-3-((diphenoxyphosphoryl)(phenyl)amino)acrylate (4a): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (71.2 mg, 87% yield). 1H NMR (400 MHz, CDCl3): δ 8.30 (dd, J = 13.7, 7.9 Hz, 1H), 7.46–7.43 (m, 3H), 7.41–7.37 (m, 4H), 7.27–7.21 (m, 6H), 7.15–7.12 (m, 2H), 4.78 (d, J = 13.7 Hz, 1H), 3.72 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 167.77, 150.20 (d, J = 6.7 Hz), 147.21 (d, J = 9.0 Hz), 136.75, 130.02, 129.07, 128.84 (d, J = 2.5 Hz), 125.72, 120.08 (d, J = 5.2 Hz), 100.15 (d, J = 9.7 Hz), 51.31. 31P NMR (162 MHz, CDCl3) δ-9.77; HRMS (ESI): calculated for C22H21NO5P [M + H]+ 410.11519, found 410.11543.
- Compound: 2,2,2-trifluoroethyl (E)-3-((diphenoxyphosphoryl)(phenyl)amino)acrylate (4b): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a white solid (86.8 mg, 91% yield), Mp = 84–85 °C. 1H NMR (400 MHz, CDCl3): δ 8.39 (dd, J = 13.7, 8.1 Hz, 1H), 7.49–7.47 (m, 3H), 7.39 (t, J = 7.8 Hz, 4H), 7.27–7.20 (m, 6H), 7.17–7.15(m, 2H), 4.80 (d, J = 13.7 Hz, 1H), 4.51 (q, J = 8.5 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ 165.63, 150.13 (d, J = 6.8 Hz), 149.15 (d, J = 9.4 Hz), 136.46, 130.16, 130.06, 129.82, 129.33, 128.72, 125.83, 120.02 (d, J = 5.1 Hz), 98.01 (d, J = 9.8 Hz), 59.76 (t, J = 36.3 Hz). 31P NMR (162 MHz, CDCl3) δ-10.16; 19F NMR (375 MHz, CDCl3) δ-73.73 (3F); HRMS (ESI): calculated for C23H20NO5F3P [M + H]+ 478.10257, found 478.10314.
- Compound: 2-methoxyethyl (E)-3-((diphenoxyphosphoryl)(phenyl)amino)acrylate (4c): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a white solid (78.8 mg, 87% yield), Mp = 46–47 °C. 1H NMR (400 MHz, CDCl3): δ 8.32 (dd, J = 13.7, 7.8 Hz, 1H), 7.44–7.42 (m, 3H), 7.40–7.36 (m, 4H), 7.26–7.20 (m, 6H), 7.13–7.11 (m, 2H), 4.84 (dd, J = 13.7, 1.1 Hz, 1H), 4.29–4.26 (m, 2H), 3.62–3.60 (m, 2H), 3.39 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 167.41, 150.19 (d, J = 6.7 Hz), 147.45 (d, J = 9.0 Hz), 136.77, 130.01, 129.84, 129.07, 128.87 (d, J = 2.7 Hz), 125.72, 120.09 (d, J = 5.1 Hz), 100.12 (d, J = 9.8 Hz), 70.67, 63.12, 59.04.
- 31P NMR (162 MHz, CDCl3) δ-9.79; HRMS (ESI): calculated for C24H25NO6P [M + H]+ 454.14140, found 454.14180.
- Compound: 4-chlorophenyl (E)-3-((diphenoxyphosphoryl)(phenyl)amino)acrylate (4d): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a white solid (77.8 mg, 77% yield), Mp = 123–124 °C. 1H NMR (400 MHz, CDCl3): δ 8.51 (dd, J = 13.7, 8.0 Hz, 1H), 7.52–7.49 (m, 3H), 7.42–7.35 (m, 6H), 7.28–7.23 (m, 8H), 7.08–7.06 (m, 2H), 4.94 (d, J = 13.7 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 165.58, 150.17 (d, J = 6.8 Hz), 149.34, 149.13 (d, J = 9.3 Hz), 136.54, 130.91, 130.20, 130.10, 129.44, 129.38, 128.80 (d, J = 2.6 Hz), 125.87, 123.20, 120.04 (d, J = 5.1 Hz), 99.06 (d, J = 9.9 Hz). 31P NMR (162 MHz, CDCl3) δ-10.10; HRMS (ESI): calculated for C27H22NO5ClP [M + H]+ 506.09186, found 506.09252.
- Compound: (tetrahydrofuran-2-yl)methyl (E)-3-((diphenoxyphosphoryl)(phenyl)amino)acrylate (4e): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a white solid (77.6 mg, 81% yield), Mp = 64–65 °C. 1H NMR (400 MHz, CDCl3): δ 8.32 (dd, J = 13.7, 7.8 Hz, 1H), 7.45–7.41 (m, 3H), 7.39–7.30 (m, 5H), 7.24–7.21 (m, 5H), 7.13–7.10 (m, 2H), 4.84 (d, J = 13.7 Hz, 1H), 4.23 (dd, J = 11.3, 3.3 Hz, 1H), 4.13 (qd, J = 7.1, 3.3 Hz, 1H), 4.02 (dd, J = 11.3, 7.3 Hz, 1H), 3.89 (dt, J = 8.5, 6.7 Hz, 1H), 3.82–3.77 (m, 1H), 2.07–1.98 (m, 1H), 1.97–1.86 (m, 2H), 1.64–1.56 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 167.37, 150.19 (d, J = 6.7 Hz), 147.40 (d, J = 8.9 Hz), 136.75, 130.01, 129.81, 129.48, 129.06, 128.86 (d, J = 2.6 Hz), 125.72, 125.42, 122.57, 120.47 (d, J = 4.8 Hz), 120.09 (d, J = 5.1 Hz), 100.16 (d, J = 9.8 Hz), 76.70, 68.44, 66.23, 28.02, 25.66. 31P NMR (162 MHz, CDCl3) δ-9.77; HRMS (ESI): calculated for C26H27NO6P [M + H]+ 480.15705, found 480.15766.
- Compound: 2,6-dimethylphenyl (E)-3-((diphenoxyphosphoryl)(phenyl)amino)acrylate (4f): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a white solid (83.8 mg, 84% yield), Mp = 121–122 °C. 1H NMR (400 MHz, CDCl3): δ 8.52 (dd, J = 13.7, 8.0 Hz, 1H), 7.53–7.48 (m, 3H), 7.42–7.38 (m, 4H), 7.28–7.23 (m, 8H), 7.09–7.07 (m, 3H), 4.99 (d, J = 13.0 Hz, 1H), 2.18 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 165.03, 150.21 (d, J = 7.0 Hz), 148.50 (d, J = 9.3 Hz), 148.20, 136.65, 130.52, 130.16, 130.05, 129.26, 128.88 (d, J = 2.4 Hz), 128.52, 125.81, 125.71, 120.09 (d, J = 5.1 Hz), 99.04 (d, J = 9.9 Hz), 16.45. 31P NMR (162 MHz, CDCl3) δ-9.97; HRMS (ESI): calculated for C29H27NO5P [M + H]+ 500.16214, found 500.16265.
- Compound: 4-chloro-3,5-dimethylphenyl (E)-3-((diphenoxyphosphoryl)(phenyl)amino)acrylate (4g): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (79.9 mg, 75% yield). 1H NMR (400 MHz, CDCl3): δ 8.50 (dd, J = 13.6, 8.0 Hz, 1H), 7.51–7.49 (m, 3H), 7.40 (t, J = 7.8 Hz, 4H), 7.29–7.21 (m, 8H), 6.87 (s, 2H), 4.94 (d, J = 13.6 Hz, 1H), 2.40 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 165.90, 150.19 (d, J = 6.9 Hz), 148.89 (d, J = 9.2 Hz), 148.45, 137.39, 136.58, 131.51, 130.19, 130.09, 129.34, 128.82 (d, J = 2.6 Hz), 125.85, 121.64, 120.06 (d, J = 5.2 Hz), 115.43, 99.31 (d, J = 9.8 Hz), 20.91. 31P NMR (162 MHz, CDCl3) δ-10.06; HRMS (ESI): calculated for C29H26NO5ClP [M + H]+ 534.12316, found 534.12377.
- Compound: 1,7,7-trimethylbicyclo[2.2.1]heptan-2-yl (E)-3-((diphenoxyphosphoryl)(phenyl)amino)acrylate (4h): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (91.4 mg, 86% yield). 1H NMR (400 MHz, CDCl3): δ 8.22 (dd, J = 13.6, 8.0 Hz, 1H), 7.46–7.44 (m, 3H), 7.40–7.36 (m, 4H), 7.26–7.21 (m, 6H), 7.15–7.12 (m, 2H), 4.75–4.71 (m, 2H), 1.84–1.70 (m, 6H), 1.67 (s, 1H), 1.04 (s, 3H), 0.88–0.87 (m, 6H). 13C NMR (100 MHz, CDCl3): δ 166.86, 150.21(d, J = 7.0 Hz), 146.49 (d, J = 8.9 Hz), 136.79, 129.42 (d, J = 116.1 Hz), 128.99, 128.86, 125.69, 120.09 (d, J = 5.2 Hz), 100.98 (d, J = 9.6 Hz), 80.62, 48.87, 46.99, 45.11, 38.91, 33.80, 27.11, 20.18, 20.09, 11.61. 31P NMR (162 MHz, CDCl3) δ-9.70; HRMS (ESI): calculated for C31H35NO5P [M + H]+ 532.22474, found 532.22549.
- Compound: 5-isopropyl-2-methylphenyl (E)-3-((diphenoxyphosphoryl)(phenyl)amino)acrylate (4i): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (81.2 mg, 77% yield). 1H NMR (400 MHz, CDCl3): δ 8.52 (dd, J = 13.7, 8.0 Hz, 1H), 7.54–7.49 (m, 3H), 7.43–7.39 (m, 4H), 7.30–7.24 (m, 8H), 7.18 (d, J = 7.8 Hz, 1H), 7.05 (dd, J = 7.8, 1.9 Hz, 1H), 6.92 (d, J = 1.9 Hz, 1H), 4.99 (d, J = 13.7 Hz, 1H), 2.92 (p, J = 6.9 Hz, 1H), 2.18 (s, 3H), 1.28 (d, J = 6.9 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ 165.63, 150.23 (d, J = 6.9 Hz), 149.36, 148.53 (d, J = 9.1 Hz), 147.99, 136.69, 130.85, 130.16, 130.07, 129.27, 128.89 (d, J = 2.5 Hz), 127.59, 125.81, 124.04, 120.10 (d, J = 5.1 Hz), 120.03, 99.44 (d, J = 9.7 Hz), 33.63, 24.00, 15.95. 31P NMR (162 MHz, CDCl3) δ-9.95; HRMS (ESI): calculated for C31H31NO5P [M + H]+ 528.19344, found 528.19397.
- Compound: (1R,2S,5R)-2-isopropyl-5-methylcyclohexyl (E)-3-((diphenoxyphosphoryl)(phenyl)amino)acrylate (4j): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (73.6 mg, 69% yield). 1H NMR (400 MHz, CDCl3): δ 8.26 (dd, J = 13.7, 8.0 Hz, 1H), 7.46–7.44 (m, 3H), 7.40–7.36 (m, 4H), 7.26–7.21 (m, 6H), 7.17–7.15 (m, 2H), 4.78–4.74 (m, 2H), 2.05–2.00 (m, 1H), 1.92–1.85 (m, 1H), 1.73–1.67 (m, 3H), 1.43–1.36 (m, 1H), 0.98–0.91 (m, 9H), 0.80 (d, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 166.96, 150.24 (d, J = 6.8 Hz), 146.70 (d, J = 8.7 Hz), 136.83, 129.99, 129.71, 129.00, 128.94 (d, J = 2.5 Hz), 125.68, 120.10 (d, J = 5.1 Hz), 100.93 (d, J = 9.9 Hz), 73.67, 47.15, 41.17, 34.35, 31.45, 26.27, 23.53, 22.10, 20.85, 16.45. 31P NMR (162 MHz, CDCl3) δ-9.72; HRMS (ESI): calculated for C31H37NO5P [M + H]+ 534.24039, found 534.24108.
- Compound: diphenyl (E)-(3-oxopent-1-en-1-yl)(phenyl)phosphoramidate (4k): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (73.3 mg, 90% yield). 1H NMR (400 MHz, CDCl3): δ 8.24 (dd, J = 13.9, 7.9 Hz, 1H), 7.48–7.44 (m, 3H), 7.39–7.36 (m, 4H), 7.26–7.20 (m, 6H), 7.16–7.13 (m, 2H), 5.15 (d, J = 13.9 Hz, 1H), 2.49 (q, J = 7.3 Hz, 2H), 1.09 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 199.93, 150.19 (d, J = 6.9 Hz), 145.99 (d, J = 8.8 Hz), 136.75, 130.02, 129.10, 128.84 (d, J = 2.5 Hz), 125.75, 120.06 (d, J = 5.2 Hz), 109.71 (d, J = 8.5 Hz), 33.76, 8.51. 31P NMR (162 MHz, CDCl3) δ-9.60; HRMS (ESI): calculated for C23H23NO4P [M + H]+ 408.13592, found 408.13654.
- Compound: diphenyl (E)-phenyl(styryl)phosphoramidate (4l): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a white solid (54.7 mg, 64% yield), Mp = 87–88 °C. 1H NMR (400 MHz, CDCl3): δ 7.51–7.49 (m, 2H), 7.44–7.42 (m, 2H), 7.36–7.25 (m, 9H), 7.23–7.19 (m, 5H), 7.14–7.11 (m, 1H), 5.75 (s, 1H), 5.62 (s, 1H). 13C NMR (100 MHz, CDCl3): δ 150.91 (d, J = 7.1 Hz), 146.37, 141.52, 137.13, 129.84, 129.02, 128.54, 128.34, 128.15, 127.09, 125.35, 124.98 (d, J = 3.6 Hz), 120.63 (d, J = 5.0 Hz), 113.51. 31P NMR (162 MHz, CDCl3) δ-7.65; HRMS (ESI): calculated for C26H23NO3P [M + H]+ 428.14101, found 428.14153.
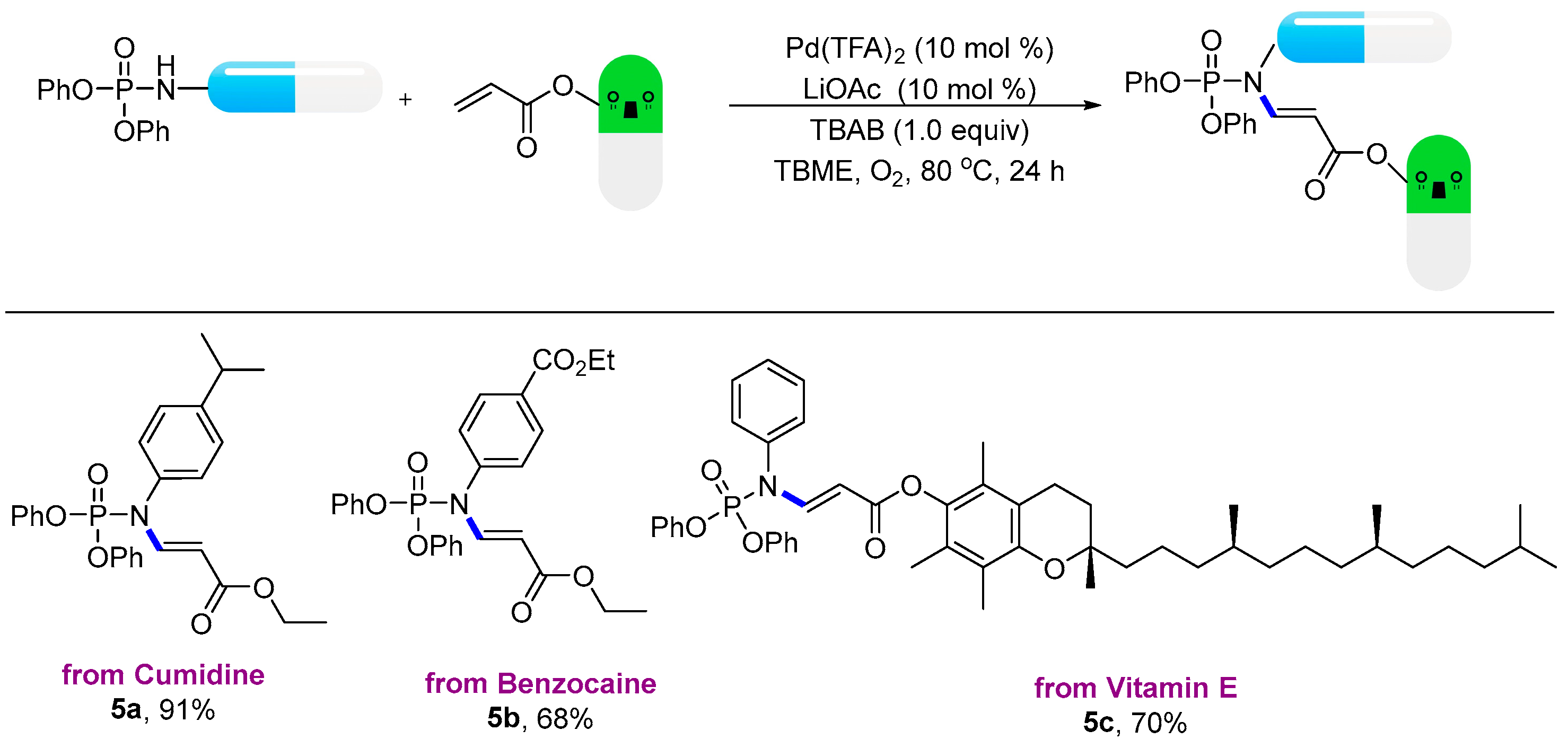
- Compound: ethyl (E)-3-((diphenoxyphosphoryl)(4-isopropylphenyl)amino)acrylate (5a): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (84.6 mg, 91% yield). 1H NMR (400 MHz, CDCl3): δ 8.29 (dd, J = 13.6, 7.8 Hz, 1H), 7.39–7.35 (m, 4H), 7.30–7.27 (m, 2H), 7.25–7.21 (m, 6H), 7.05–7.03 (m, 2H), 4.80 (d, J = 14.2 Hz, 1H), 4.18 (q, J = 7.1 Hz, 2H), 2.96 (p, J = 6.9 Hz, 1H), 1.30–1.29 (m, 6H). 13C NMR (100 MHz, CDCl3): δ 167.46, 150.26 (d, J = 6.7 Hz), 149.84, 147.22 (d, J = 9.2 Hz), 134.19 (d, J = 2.3 Hz), 129.99, 128.58 (d, J = 2.6 Hz), 128.01, 125.67, 120.12 (d, J = 5.2 Hz), 100.40 (d, J = 9.5 Hz), 60.02, 33.90, 23.97, 14.45. 31P NMR (162 MHz, CDCl3) δ-9.50; HRMS (ESI): calculated for C26H29NO5P [M + H]+ 466.17779, found 466.17812.
- Compound: ethyl (E)-4-((diphenoxyphosphoryl)(3-ethoxy-3-oxoprop-1-en-1-yl)amino)benzoate (5b): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (67.3 mg, 68% yield). 1H NMR (400 MHz, CDCl3): δ 8.26 (dd, J = 13.8, 7.9 Hz, 1H), 8.13 (d, J = 8.2 Hz, 2H), 7.39 (t, J = 7.8 Hz, 4H), 7.30–7.19 (m, 8H), 4.77 (d, J = 13.8 Hz, 1H), 4.43 (q, J = 7.2 Hz, 2H), 4.18 (q, J = 7.1 Hz, 2H), 1.44 (t, J = 7.2 Hz, 3H), 1.28 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 167.09, 165.59, 150.05 (d, J = 6.7 Hz), 146.25 (d, J = 8.4 Hz), 141.02, 131.34, 131.09, 130.09, 128.89, 125.88, 120.06 (d, J = 5.1 Hz), 101.09 (d, J = 9.4 Hz), 61.45, 60.17, 14.38, 14.36. 31P NMR (162 MHz, CDCl3) δ-10.25; HRMS (ESI): calculated for C26H27NO7P [M + H]+ 496.15197, found 496.15246.
- Compound: (S)-2,5,7,8-tetramethyl-2-((4S,8S)-4,8,12-trimethyltridecyl)chroman-6-yl (E)-3-((diphenoxyphosphoryl)(phenyl)amino)acrylate (5c): Following the general procedure, using (petroleum ether:EtOAc = 9:1) as the eluant afforded a colourless liquid (113.0 mg, 70% yield). 1H NMR (400 MHz, CDCl3): δ 8.50 (dd, J = 13.8, 7.9 Hz, 1H), 7.51–7.49 (m, 3H), 7.42–7.38 (m, 4H), 7.28–7.23 (m, 8H), 5.01 (d, J = 13.8 Hz, 1H), 2.62 (t, J = 6.8 Hz, 1H), 2.12 (s, 3H), 2.05 (s, 3H), 2.00 (s, 3H), 1.81 (dq, J = 20.7, 6.8 Hz, 2H), 1.61–1.53 (m, 4H), 1.46–1.25 (m, 14H), 1.20–1.14 (m, 3H), 1.15–1.09 (m, 3H), 0.92–0.88 (m, 12H). 13C NMR (100 MHz, CDCl3): δ 165.92, 150.25, 149.36, 148.13, 140.40, 136.74, 130.12, 130.03, 129.18, 128.91, 127.07, 125.78, 125.29, 122.96, 120.12 (d, J = 5.2 Hz), 117.33, 99.50 (d, J = 9.1 Hz), 75.05, 39.44, 37.52, 37.36, 32.86, 32.79, 28.05, 24.87, 24.52, 22.80, 22.70, 21.11, 20.66, 19.83, 19.73, 13.10, 12.26, 11.90. 31P NMR (162 MHz, CDCl3) δ-9.89; HRMS (ESI): calculated for C50H67NO6P [M + H]+ 808.47005, found 808.47048.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Zhu, Y.-Y.; Niu, Y.; Niu, Y.-N.; Yang, S.-D. Recent Advances in the Synthesis and Applications of Phosphoramides. Org. Biomol. Chem. 2021, 19, 10296–10313. [Google Scholar] [CrossRef]
- Oliveira, F.M.; Barbosa, L.C.A.; Ismailc, F.M.D. The Diverse Pharmacology and Medicinal Chemistry of Phosphoramidates A Review. RSC Adv. 2014, 4, 18998–19012. [Google Scholar] [CrossRef]
- Liu, Z.; Klapars, A.; Simmons, B.; Bellomo, A.; Kalinin, A.; Weisel, M.; Hill, J.; Silverman, S.M. Development and Implementation of an Aluminum-Promoted Phosphorylation in the Uprifosbuvir Manufacturing Route. Org. Process Res. Dev. 2021, 25, 661–667. [Google Scholar] [CrossRef]
- Mehellou, Y.; Rattan, H.S.; Balzarini, J. The ProTide Prodrug Technology: From the Concept to the Clinic. J. Med. Chem. 2018, 61, 2211–2226. [Google Scholar] [CrossRef]
- Holshue, M.L.; DeBolt, C.; Lindquist, S.; Lofy, K.H.; Wiesman, J.; Bruce, H.; Spitters, C.; Ericson, K.; Wilkerson, S.; Tural, A.; et al. First Case of 2019 Novel Coronavirus in the United States. N. Engl. J. Med. 2020, 382, 929–936. [Google Scholar] [CrossRef]
- Raia, A.; Yadav, L.D.S. Strategic Applications of Baylis-Hillman Adducts to General Syntheses of 3-Nitroazetidines. Org. Biomol. Chem. 2011, 9, 8058–8061. [Google Scholar] [CrossRef]
- Yin, G.; Zhu, Y.; Zhang, L.; Lu, P.; Wang, Y. Preparation of Allenephosphoramide and Its Utility in the Preparation of 4,9-Dihydro-2H-benzo[f]isoindoles. Org. Lett. 2011, 13, 940–943. [Google Scholar] [CrossRef]
- Shinde, A.H.; Thomas, A.A.; Mague, J.T.; Sathyamoorthi, S. Highly Regio- and Diastereoselective Tethered Aza-Wacker Cyclizations of Alkenyl Phosphoramidates. J. Org. Chem. 2021, 86, 14732–14758. [Google Scholar] [CrossRef]
- DeKorver, K.A.; Walton, M.C.; North, T.D.; Hsung, R.P. Introducing a New Class of N-Phosphoryl Ynamides via Cu(I)-Catalyzed Amidations of Alkynyl Bromides. Org. Lett. 2011, 13, 4862–4865. [Google Scholar] [CrossRef]
- Jillella, R.; Raju, S.; Hsiao, H.-C.; Hsu, D.-S.; Chuang, S.-C. Pd-Catalyzed Redox-Neutral C-N Coupling Reaction of Iminoquinones with Electron-Deficient Alkenes without External Oxidants Access of Tertiary (E)-Enamines and Application to the Synthesis of Indoles and Quinolin-4-ones. Org. Lett. 2020, 22, 6252–6256. [Google Scholar] [CrossRef]
- Pratap, K.; Kumar, A. Palladium-Catalyzed Intermolecular Dehydrogenative Carboamination of Alkenes with Amines and N-Substituted Isatin. Org. Lett. 2018, 20, 7451–7454. [Google Scholar] [CrossRef]
- Ji, X.; Huang, H.; Wu, W.; Li, X.; Jiang, H. Palladium-Catalyzed Oxidative Coupling of Aromatic Primary Amines and Alkenes under Molecular Oxygen Stereoselective Assembly of (Z)-Enamines. J. Org. Chem. 2013, 78, 11155–11162. [Google Scholar] [CrossRef]
- Mizuta, Y.; Yasuda, K.; Obora, Y. Palladium-Catalyzed Z-Selective Oxidative Amination of ortho-Substituted Primary Anilines with Olefins under an Open Air Atmosphere. J. Org. Chem. 2013, 78, 6332–6337. [Google Scholar] [CrossRef]
- Chary, B.C.; Kim, S.; Park, Y.; Kim, J.; Lee, P.H. Palladium-Catalyzed C-H Arylation Using Phosphoramidate as a Directing Group at Room Temperature. Org. Lett. 2013, 15, 2692–2695. [Google Scholar] [CrossRef]
- Wu, G.-J.; Han, F.-S.; Zhao, Y.-L. Palladacycles Derived from Arylphosphinamides for Mild Suzuki-Miyaura Cross-Couplings. RSC Adv. 2015, 5, 69776–69781. [Google Scholar] [CrossRef]
- Garcia, P.; Lau, Y.Y.; Perry, M.R.; Schafer, L.L. Phosphoramidate Tantalum Complexes for Room-Temperature C-H Functionalization: Hydroaminoalkylation Catalysis. Angew. Chem. Int. Ed. 2013, 52, 9144–9148. [Google Scholar] [CrossRef]
- Jiao, L.-Y.; Ferreira, A.V.; Oestreich, M. Phosphinic Amide as Directing Group Enabling Palladium(II)-Catalyzed ortho C-H Alkenylation of Anilines without and with Alkylation at the Nitrogen Atom. Chem. Asian J. 2016, 11, 367–370. [Google Scholar] [CrossRef]
- Park, S.; Seo, B.; Shin, S.; Son, J.-Y.; Lee, P.H. Rhodium-Catalyzed Oxidative Coupling through C-H Activation and Annulation Directed by Phosphonamide and Phosphinamide Groups. Chem. Commun. 2013, 49, 8671–8673. [Google Scholar] [CrossRef]
- Shi, S.; Ma, Y.; Zhou, J.; Li, J.; Chen, L.; Wu, G. Copper-Catalyzed Oxidative Thioamination of Maleimides with Amines and Bunte Salts. Org. Lett. 2020, 22, 1863–1867. [Google Scholar] [CrossRef]
- Gao, X.; Tang, L.; Huang, L.; Huang, Z.-S.; Ma, Y.; Wu, G. Oxidative Aminoarylselenation of Maleimides via Copper-Catalyzed Four-Component Cross-Coupling. Org. Lett. 2019, 21, 745–748. [Google Scholar] [CrossRef]
- Wu, G.; Su, W. Regio- and Stereoselective Direct N-Alkenylation of Indoles via Pd-Catalyzed Aerobic Oxidation. Org. Lett. 2013, 15, 5278–5281. [Google Scholar] [CrossRef]
- Bhatt, S.; Wang, Y.-N.; Pham, H.T.P.; Hull, K.L. Palladium-Catalyzed Oxidative Amination of α-Olefins with Indoles. Org. Lett. 2022, 24, 5746–5750. [Google Scholar] [CrossRef]
- Timokhin, V.I.; Stahl, S.S. Brønsted Base-Modulated Regioselectivity in the Aerobic Oxidative Amination of Styrene Catalyzed by Palladium. J. Am. Chem. Soc. 2005, 127, 17888–17893. [Google Scholar] [CrossRef]
- Geng, M.; Kuang, J.; Miao, M.; Ma, Y. Cu(ii)-Catalyzed Domino Construction of Spironaphthalenones by Dearomatization of β-Naphthols and using N,N-Dimethylaminoethanol as a C1 Synthon. Org. Biomol. Chem. 2023, 21, 3101–3104. [Google Scholar] [CrossRef]
- Fang, J.; Pan, Z.; Liu, T.; Rao, Y.; Jiang, H.; Ma, Y. I2-Mediated Coupling of Quinazolinone Enamines with 2-Aminopyridines: A New Strategy to Access Spiroquinazolinones. Org. Biomol. Chem. 2023, 21, 2355–2360. [Google Scholar] [CrossRef]
- Reetz, M.T.; de Vries, J.G. Ligand-Free Heck Reactions Using Low Pd-Loading. Chem. Commun. 2004, 35, 1559–1563. [Google Scholar] [CrossRef]
- de Vries, A.H.M.; Parlevliet, F.J.; Schmieder-van de Vondervoort, L.; Mommers, J.H.M.; Henderickx, H.J.W.; Walet, M.A.M.; de Vries, J.G. A Practical Recycle of a Ligand-Free Palladium Catalyst for Heck Reactions. Adv. Synth. Catal. 2002, 344, 996–1002. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Catalyst | Additive | Solvent | Yield (%) b |
| 1 | Pd(OAc)2 | Cu(OAc)2 | DME | 0 |
| 2 | Pd(OAc)2 | CuCl2 | DME | 0 |
| 3 | Pd(OAc)2 | BQ | DME | 0 |
| 4 | Pd(OAc)2 | pyridine | DME | 0 |
| 5 | Pd(OAc)2 | LiBr | DME | 0 |
| 6 | Pd(OAc)2 | TBAB | DME | 65 |
| 7 | Pd(OAc)2 | TBAF | DME | 0 |
| 8 | Pd(OAc)2 | TBAC | DME | 0 |
| 9 | Pd(OAc)2 | TBAI | DME | 0 |
| 10 | Pd(OAc)2 | TBAB | TBME | 80 |
| 11 c | Pd(OAc)2 | TBAB | TBME | 11 |
| 12 d | Pd(OAc)2 | TBAB | TBME | 14 |
| 13 | Pd(TFA)2 | TBAB | TBME | 89 |
| 14 e | Pd(TFA)2 | TBAB | TBME | 7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.-A.; Wu, G.; Li, J. Palladium-Catalyzed N-Alkenylation of N-Aryl Phosphoramidates with Alkenes. Molecules 2023, 28, 4466. https://doi.org/10.3390/molecules28114466
Li Y-A, Wu G, Li J. Palladium-Catalyzed N-Alkenylation of N-Aryl Phosphoramidates with Alkenes. Molecules. 2023; 28(11):4466. https://doi.org/10.3390/molecules28114466
Chicago/Turabian StyleLi, Yu-An, Ge Wu, and Jia Li. 2023. "Palladium-Catalyzed N-Alkenylation of N-Aryl Phosphoramidates with Alkenes" Molecules 28, no. 11: 4466. https://doi.org/10.3390/molecules28114466
APA StyleLi, Y.-A., Wu, G., & Li, J. (2023). Palladium-Catalyzed N-Alkenylation of N-Aryl Phosphoramidates with Alkenes. Molecules, 28(11), 4466. https://doi.org/10.3390/molecules28114466