

Synergistic Catalysis in Heterobimetallic Complexes for Homogeneous Carbon Dioxide Hydrogenation
 , ,
, ,  , and
, and 
Abstract
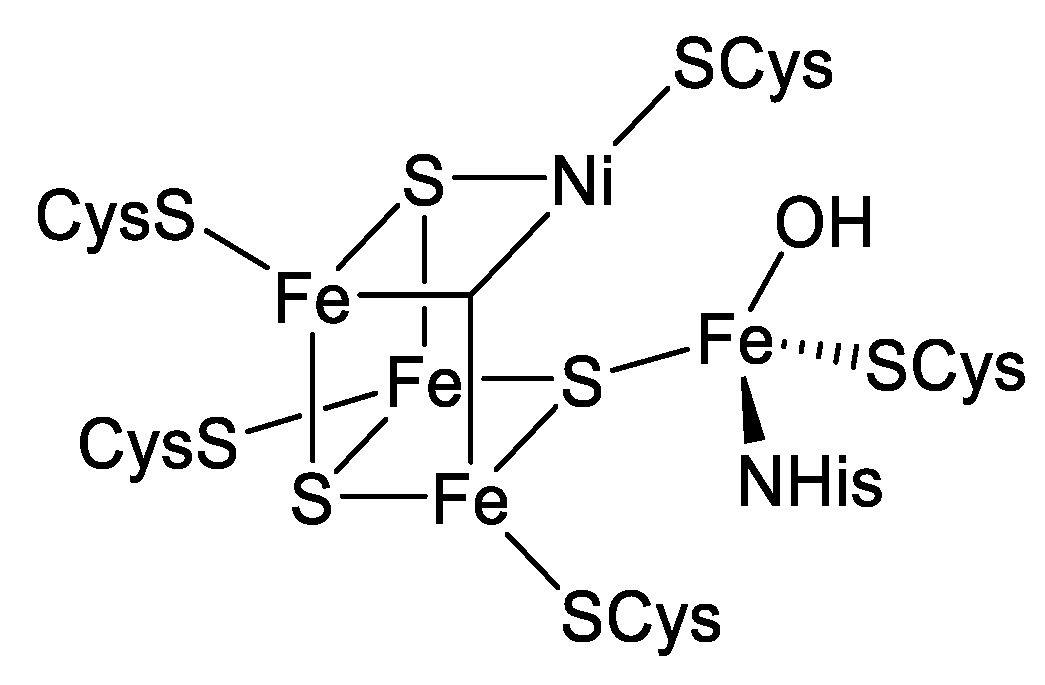
1. Introduction
2. Results and Discussion
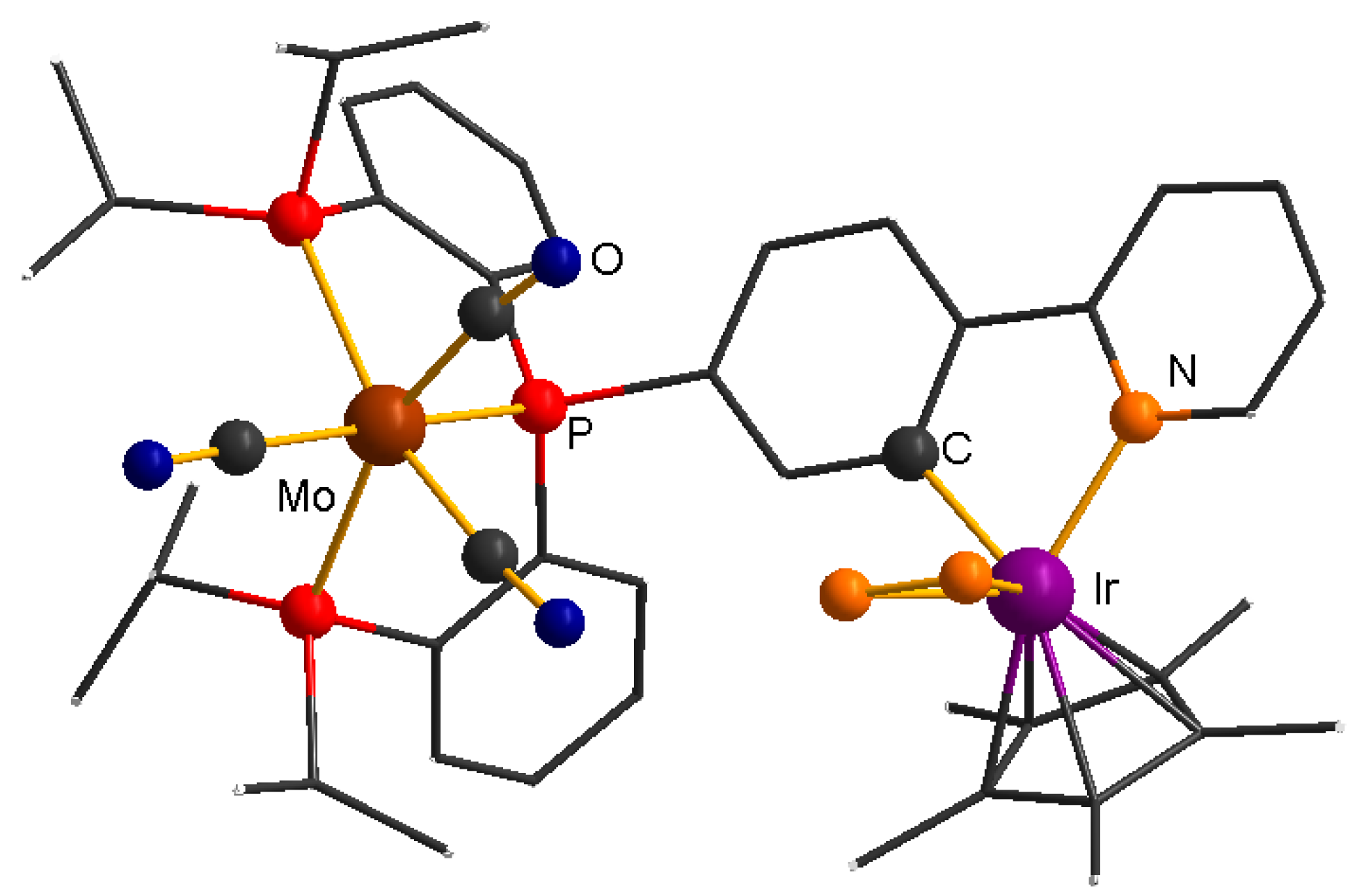
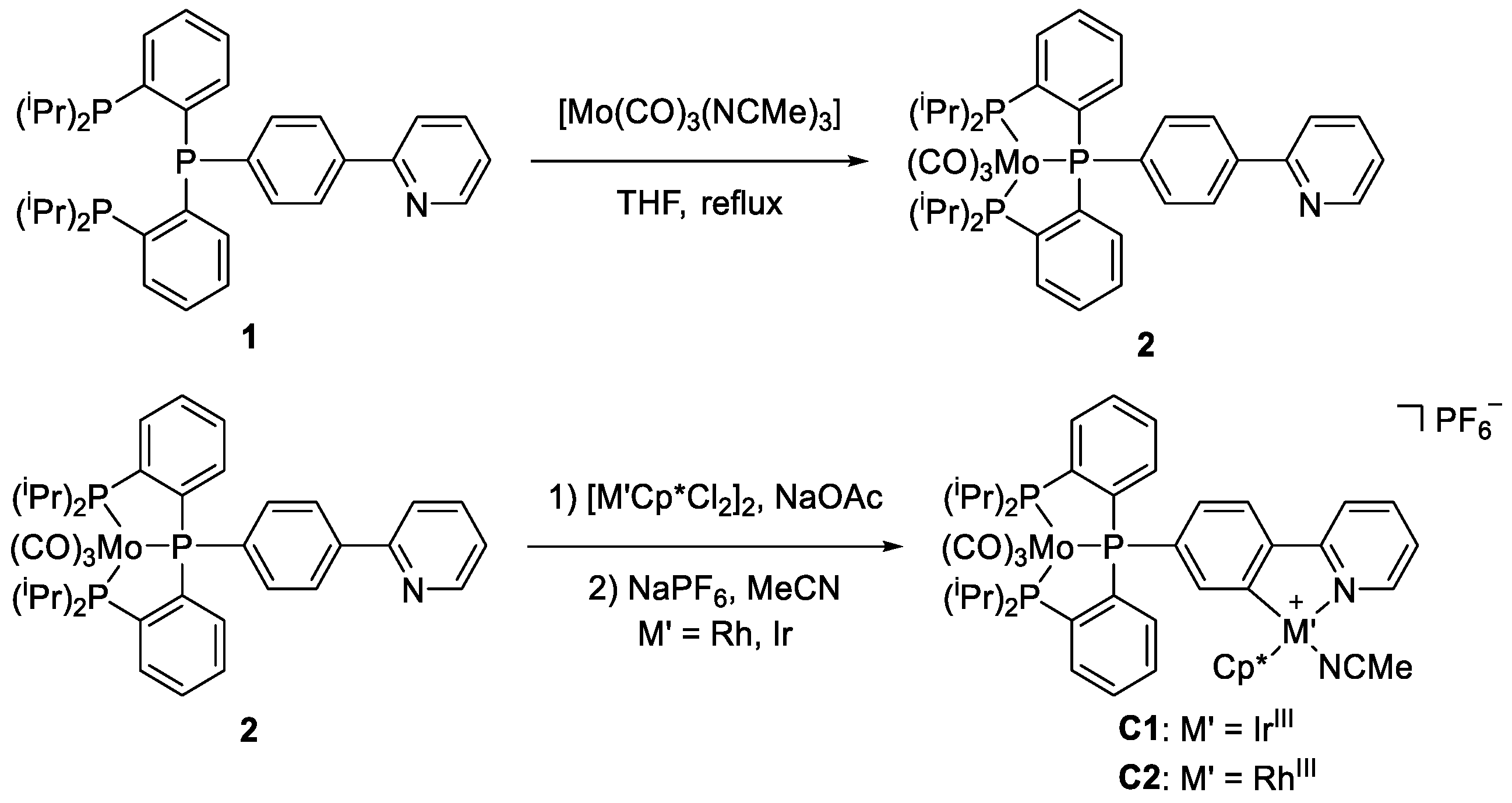
2.1. Complex Synthesis
2.2. Catalysis
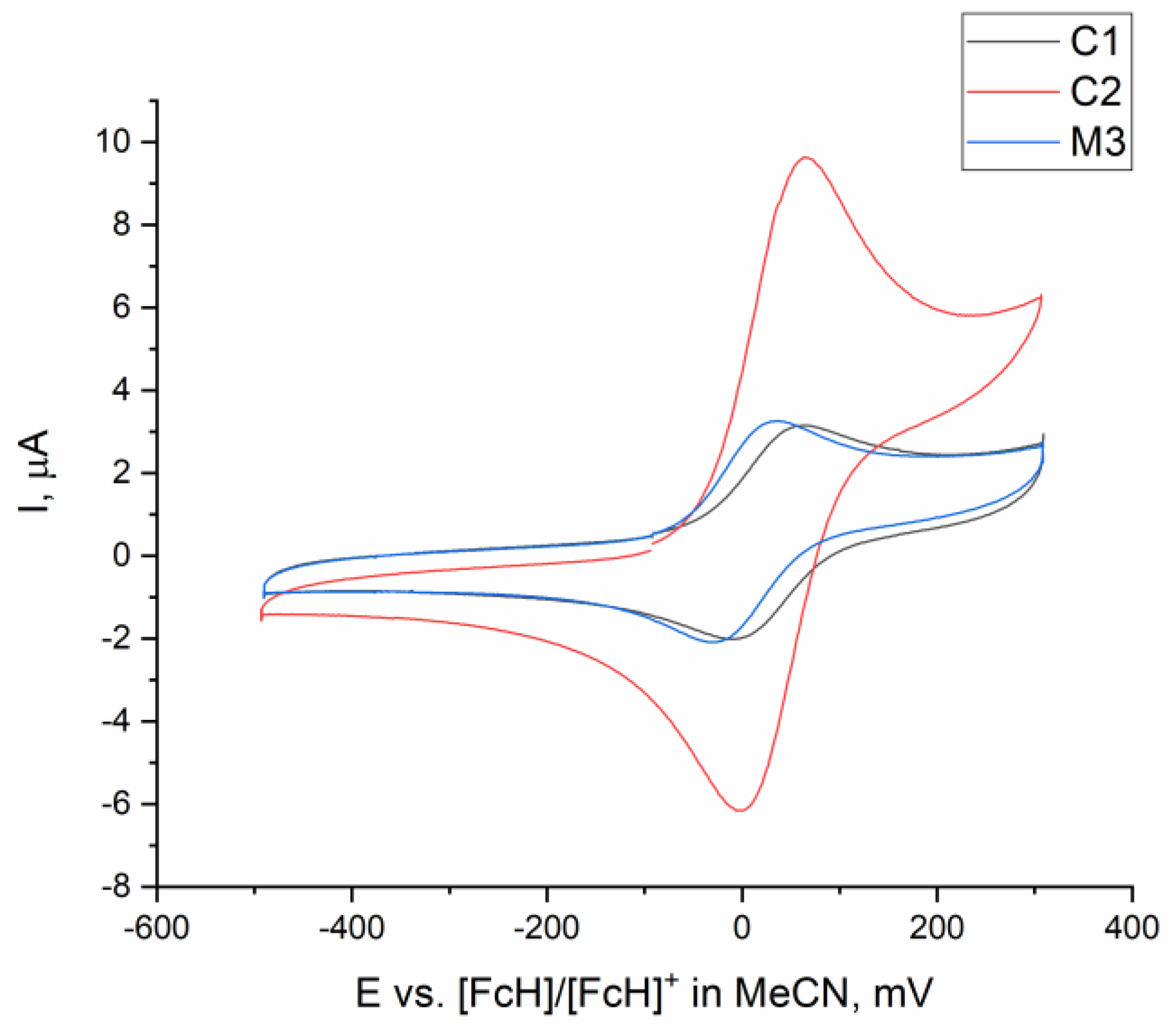
2.3. Comparing Heterobimetallic and Monometallic Systems
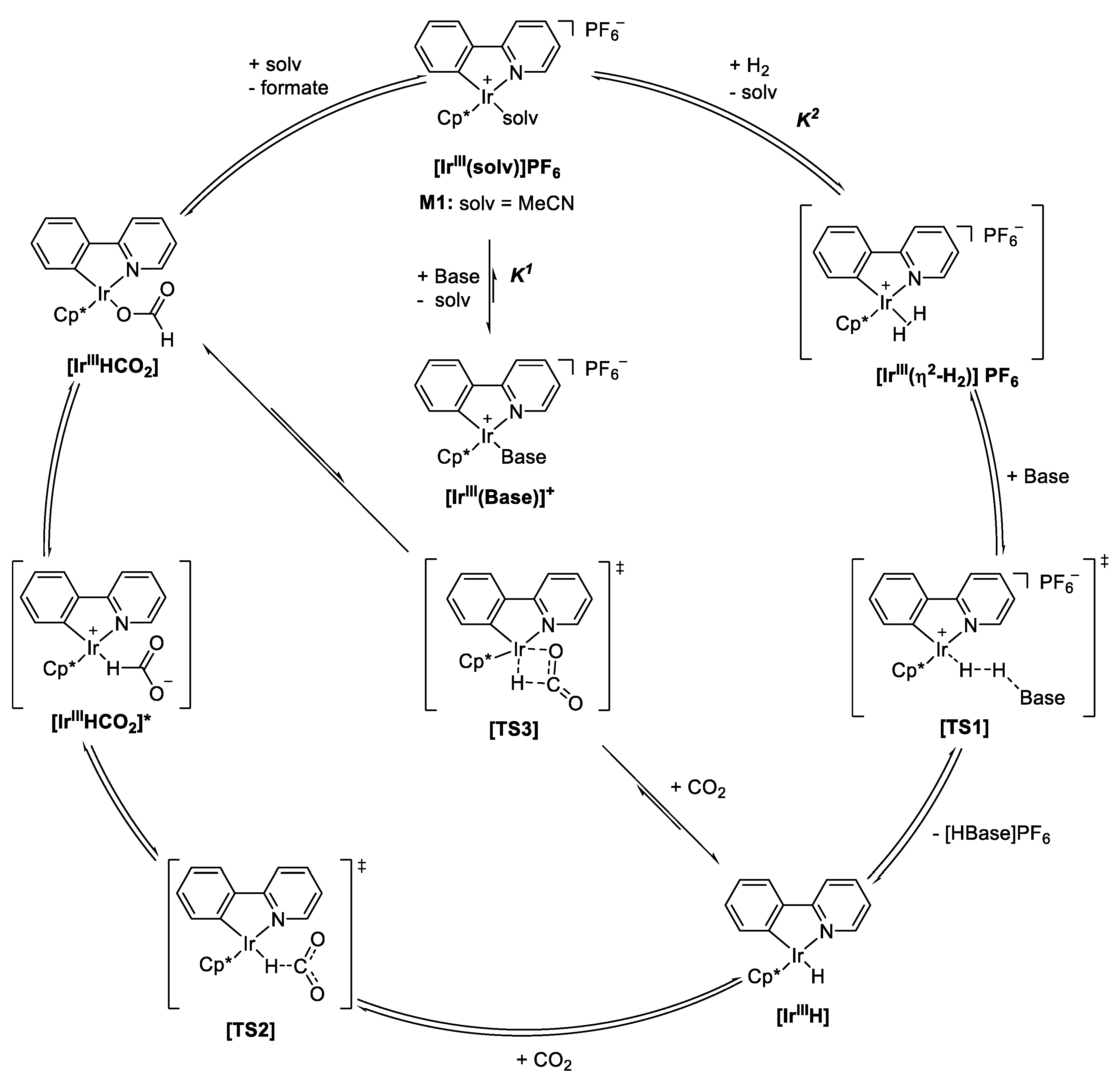
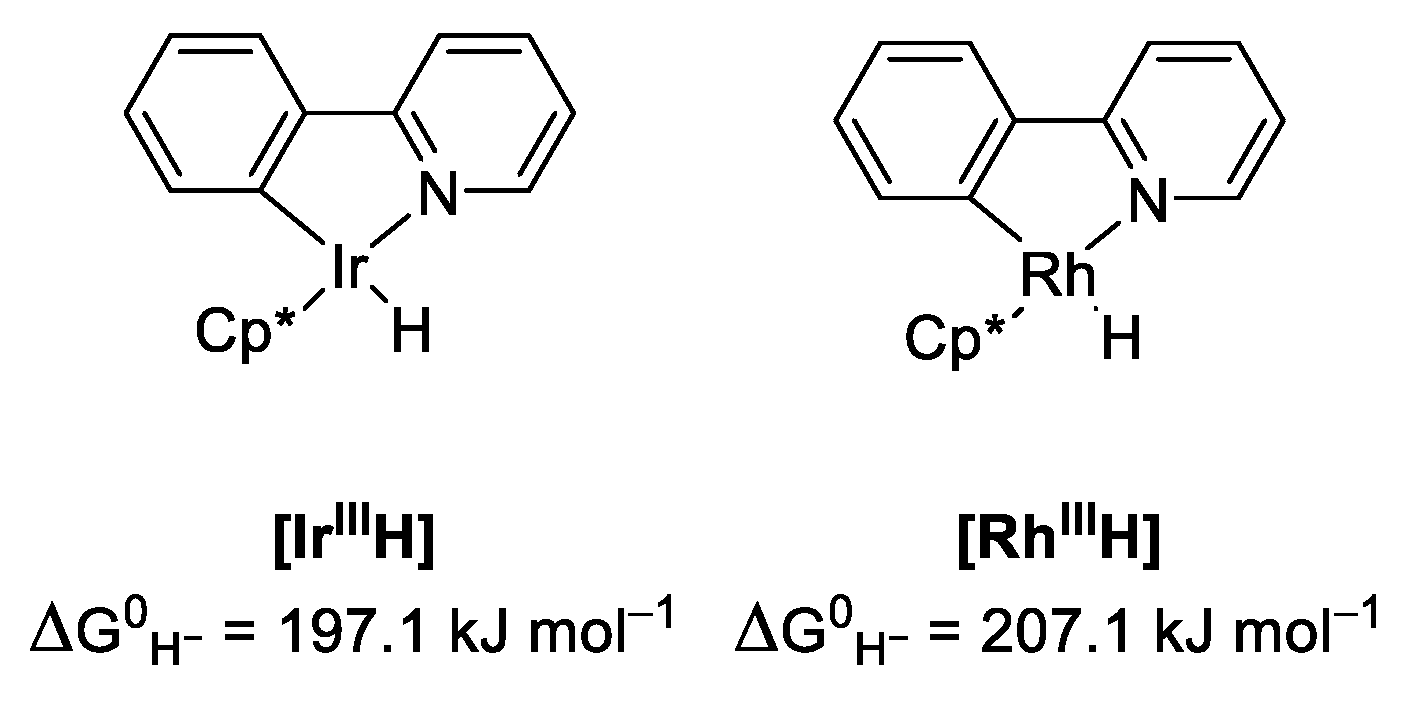
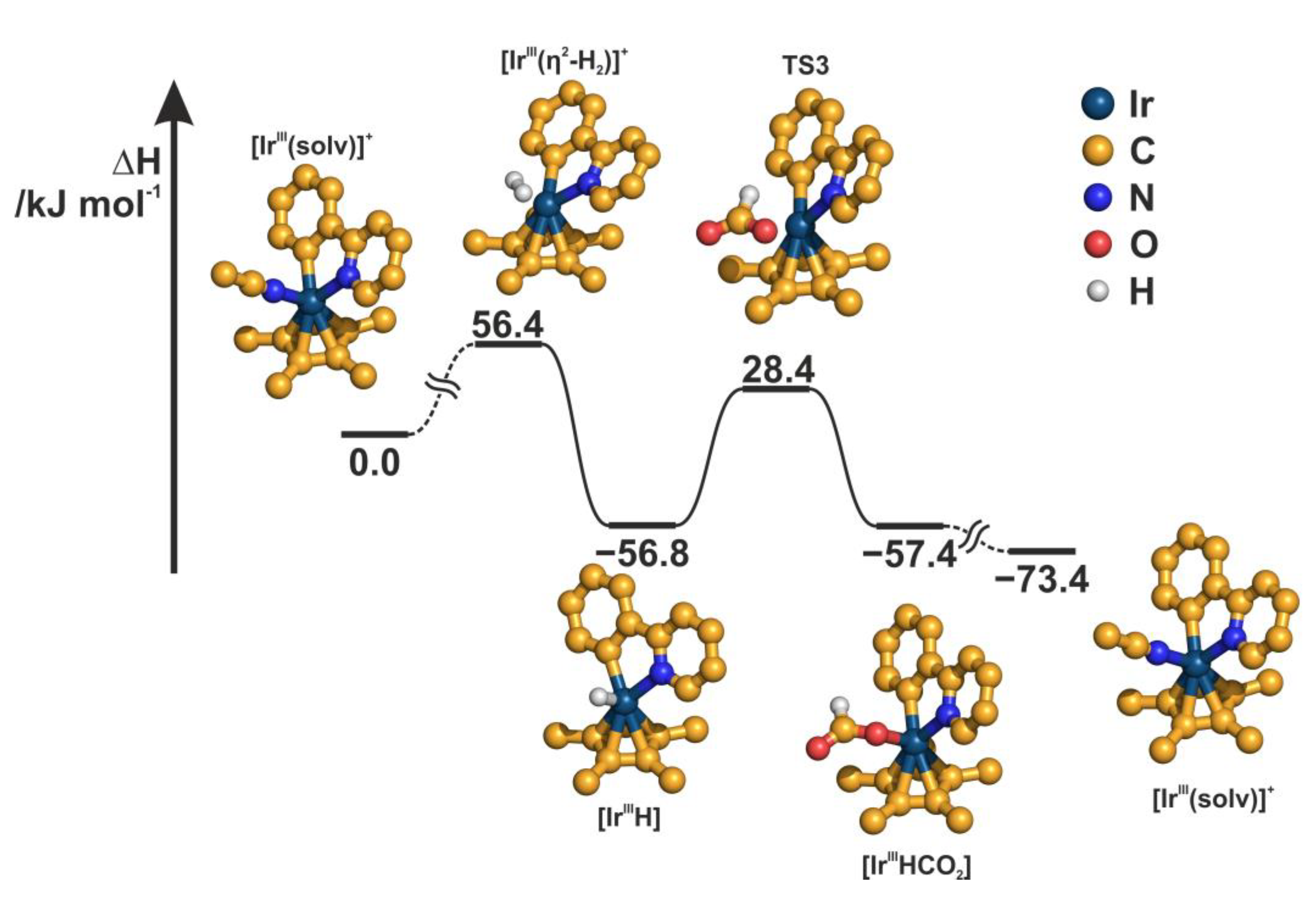
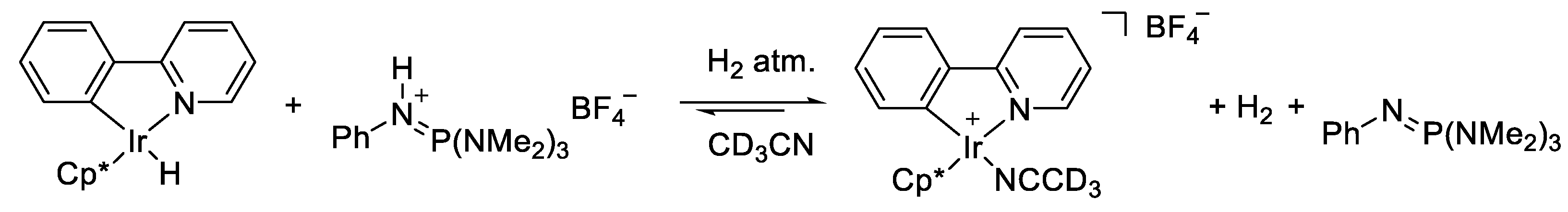
2.4. Understanding the CO2 Hydrogenation Using M1 as Catalyst

2.5. Identifying the Synergistic Interaction
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rockström, J.; Steffen, W.; Noone, K.; Persson, A.; Chapin, F.S.; Lambin, E.F.; Lenton, T.M.; Scheffer, M.; Folke, C.; Schellnhuber, H.J.; et al. A safe operating space for humanity. Nature 2009, 461, 472–475. [Google Scholar] [CrossRef]
- Beller, M.; Bolm, C. Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals, 2nd ed.; Wiley VCH: Weinheim, Germany, 2004; ISBN 9783527306138. [Google Scholar]
- Steinhagen, H.; Helmchen, G. Asymmetric Two-Center Catalysis—Learning from Nature. Angew. Chem. Int. Ed. 1996, 35, 2339–2342. [Google Scholar] [CrossRef]
- Steinhagen, H.; Helmchen, G. Asymmetrische Zweizentren-Katalyse—Von der Natur lernen. Angew. Chem. 1996, 108, 2489–2492. [Google Scholar] [CrossRef]
- Appel, A.M.; Bercaw, J.E.; Bocarsly, A.B.; Dobbek, H.; DuBois, D.L.; Dupuis, M.; Ferry, J.G.; Fujita, E.; Hille, R.; Kenis, P.J.A.; et al. Frontiers, opportunities, and challenges in biochemical and chemical catalysis of CO2 fixation. Chem. Rev. 2013, 113, 6621–6658. [Google Scholar] [CrossRef] [PubMed]
- Can, M.; Armstrong, F.A.; Ragsdale, S.W. Structure, function, and mechanism of the nickel metalloenzymes, CO dehydrogenase, and acetyl-CoA synthase. Chem. Rev. 2014, 114, 4149–4174. [Google Scholar] [CrossRef] [PubMed]
- Buchwalter, P.; Rosé, J.; Braunstein, P. Multimetallic catalysis based on heterometallic complexes and clusters. Chem. Rev. 2015, 115, 28–126. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, A.; Singh, A.; Kamboj, R.C. Heterobimetallic Complexes as Promising Catalysts. Chem. Sci. Rev. Lett. 2016, 5, 170–192. [Google Scholar]
- Cooper, B.G.; Napoline, J.W.; Thomas, C.M. Catalytic Applications of Early/Late Heterobimetallic Complexes. Catal. Rev. 2012, 54, 1–40. [Google Scholar] [CrossRef]
- Kalck, P. Homo-and Heterobimetallic Complexes in Catalysis; Springer International Publishing: Basel, Switzerland, 2016; ISBN 978-3-319-34182-8. [Google Scholar]
- Mankad, N.P. Catalysis with Multinuclear Complexes. In Non-Noble Metal Catalysis: Molecular Approaches and Reactions; Moret, M.-E., Gebbink, R.J.M., Eds.; Wiley-VCH: Weinheim, Germany, 2019; pp. 49–68. ISBN 9783527699087. [Google Scholar]
- Bratko, I.; Gómez, M. Polymetallic complexes linked to a single-frame ligand: Cooperative effects in catalysis. Dalton Trans. 2013, 42, 10664–10681. [Google Scholar] [CrossRef] [PubMed]
- Mata, J.A.; Hahn, F.E.; Peris, E. Heterometallic complexes, tandem catalysis and catalytic cooperativity. Chem. Sci. 2014, 5, 1723–1732. [Google Scholar] [CrossRef]
- Bitzer, M.J.; Kühn, F.E.; Baratta, W. Tandem Suzuki–Miyaura/transfer hydrogenation reaction catalyzed by a Pd–Ru complex bearing an anionic dicarbene. J. Catal. 2016, 338, 222–226. [Google Scholar] [CrossRef]
- Gu, S.; Xu, D.; Chen, W. Heterobimetallic complexes containing an N-heterocyclic carbene based multidentate ligand and catalyzed tandem click/Sonogashira reactions. Dalton Trans. 2011, 40, 1576–1583. [Google Scholar] [CrossRef]
- Sabater, S.; Mata, J.A.; Peris, E. Heterobimetallic Iridium–Ruthenium Assemblies through an Ambidentate Triazole-Diylidene Ligand: Electrochemical Properties and Catalytic Behavior in a Cascade Reaction. Organometallics 2012, 31, 6450–6456. [Google Scholar] [CrossRef]
- Wei, L.; Zhu, Q.; Xu, S.-M.; Chang, X.; Wang, C.-J. Stereodivergent Synthesis of α,α-Disubstituted α-Amino Acids via Synergistic Cu/Ir Catalysis. J. Am. Chem. Soc. 2018, 140, 1508–1513. [Google Scholar] [CrossRef] [PubMed]
- Zanardi, A.; Mata, J.A.; Peris, E. Well-defined Ir/Pd complexes with a triazolyl-diylidene bridge as catalysts for multiple tandem reactions. J. Am. Chem. Soc. 2009, 131, 14531–14537. [Google Scholar] [CrossRef]
- Zanardi, A.; Mata, J.A.; Peris, E. An Ir-Pt catalyst for the multistep preparation of functionalized indoles from the reaction of amino alcohols and alkynyl alcohols. Chemistry 2010, 16, 13109–13115. [Google Scholar] [CrossRef] [PubMed]
- Brereton, K.R.; Pitman, C.L.; Cundari, T.R.; Miller, A.J.M. Solvent-Dependent Thermochemistry of an Iridium/Ruthenium H2 Evolution Catalyst. Inorg. Chem. 2016, 55, 12042–12051. [Google Scholar] [CrossRef] [PubMed]
- Garden, J.A.; Saini, P.K.; Williams, C.K. Greater than the Sum of Its Parts: A Heterodinuclear Polymerization Catalyst. J. Am. Chem. Soc. 2015, 137, 15078–15081. [Google Scholar] [CrossRef]
- Trott, G.; Garden, J.A.; Williams, C.K. Heterodinuclear zinc and magnesium catalysts for epoxide/CO2 ring opening copolymerizations. Chem. Sci. 2019, 10, 4618–4627. [Google Scholar] [CrossRef] [PubMed]
- Deacy, A.C.; Durr, C.B.; Garden, J.A.; White, A.J.P.; Williams, C.K. Groups 1, 2 and Zn(II) Heterodinuclear Catalysts for Epoxide/CO2 Ring-Opening Copolymerization. Inorg. Chem. 2018, 57, 15575–15583. [Google Scholar] [CrossRef]
- Hong, D.; Shimoyama, Y.; Ohgomori, Y.; Kanega, R.; Kotani, H.; Ishizuka, T.; Kon, Y.; Himeda, Y.; Kojima, T. Cooperative Effects of Heterodinuclear IrIII-MII Complexes on Catalytic H2 Evolution from Formic Acid Dehydrogenation in Water. Inorg. Chem. 2020, 59, 11976–11985. [Google Scholar] [CrossRef]
- Inoue, Y.; Izumida, H.; Sasaki, Y.; Hashimoto, H. Catalytic Fixation of Carbon Dioxide to Formic Acid by Transition-Metal Complexes under Mild Conditions. Chem. Lett. 1976, 5, 863–864. [Google Scholar] [CrossRef]
- Yu, X.; Pickup, P.G. Recent advances in direct formic acid fuel cells (DFAFC). J. Power Sources 2008, 182, 124–132. [Google Scholar] [CrossRef]
- Klankermayer, J.; Wesselbaum, S.; Beydoun, K.; Leitner, W. Selective Catalytic Synthesis Using the Combination of Carbon Dioxide and Hydrogen: Catalytic Chess at the Interface of Energy and Chemistry. Angew. Chem. Int. Ed. 2016, 55, 7296–7343. [Google Scholar] [CrossRef]
- Klankermayer, J.; Wesselbaum, S.; Beydoun, K.; Leitner, W. Selektive katalytische Synthesen mit Kohlendioxid und Wasserstoff: Katalyse-Schach an der Nahtstelle zwischen Energie und Chemie. Angew. Chem. 2016, 128, 7416–7467. [Google Scholar] [CrossRef]
- Dong, K.; Razzaq, R.; Hu, Y.; Ding, K. Homogeneous Reduction of Carbon Dioxide with Hydrogen. Top. Curr. Chem. 2017, 375, 23. [Google Scholar] [CrossRef]
- Wang, W.-H.; Himeda, Y.; Muckerman, J.T.; Manbeck, G.F.; Fujita, E. CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction. Chem. Rev. 2015, 115, 12936–12973. [Google Scholar] [CrossRef]
- Cammarota, R.C.; Vollmer, M.V.; Xie, J.; Ye, J.; Linehan, J.C.; Burgess, S.A.; Appel, A.M.; Gagliardi, L.; Lu, C.C. A Bimetallic Nickel-Gallium Complex Catalyzes CO2 Hydrogenation via the Intermediacy of an Anionic d10 Nickel Hydride. J. Am. Chem. Soc. 2017, 139, 14244–14250. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Cammarota, R.C.; Xie, J.; Vollmer, M.V.; Truhlar, D.G.; Cramer, C.J.; Lu, C.C.; Gagliardi, L. Rationalizing the Reactivity of Bimetallic Molecular Catalysts for CO2 Hydrogenation. ACS Catal. 2018, 8, 4955–4968. [Google Scholar] [CrossRef]
- Vollmer, M.V.; Ye, J.; Linehan, J.C.; Graziano, B.J.; Preston, A.; Wiedner, E.S.; Lu, C.C. Cobalt-Group 13 Complexes Catalyze CO2 Hydrogenation via a Co(−I)/Co(I) Redox Cycle. ACS Catal. 2020, 10, 2459–2470. [Google Scholar] [CrossRef]
- Tshepelevitsh, S.; Kütt, A.; Lõkov, M.; Kaljurand, I.; Saame, J.; Heering, A.; Plieger, P.G.; Vianello, R.; Leito, I. On the Basicity of Organic Bases in Different Media. Eur. J. Org. Chem. 2019, 2019, 6735–6748. [Google Scholar] [CrossRef]
- Matsubara, Y.; Grills, D.C.; Koide, Y. Thermodynamic Cycles Relevant to Hydrogenation of CO2 to Formic Acid in Water and Acetonitrile. Chem. Lett. 2019, 48, 627–629. [Google Scholar] [CrossRef]
- Hansen, P.E.; Lund, T.; Krake, J.; Spanget-Larsen, J.; Hvidt, S. A Reinvestigation of the Ionic Liquid Diisopropylethylammonium Formate by NMR and DFT Methods. J. Phys Chem. B 2016, 120, 11279–11286. [Google Scholar] [CrossRef] [PubMed]
- Corre, Y.; Iali, W.; Hamdaoui, M.; Trivelli, X.; Djukic, J.-P.; Agbossou-Niedercorn, F.; Michon, C. Efficient hydrosilylation of imines using catalysts based on iridium(III) metallacycles. Catal. Sci. Technol. 2015, 5, 1452–1458. [Google Scholar] [CrossRef]
- Hamdaoui, M.; Desrousseaux, C.; Habbita, H.; Djukic, J.-P. Iridacycles as Catalysts for the Autotandem Conversion of Nitriles into Amines by Hydrosilylation: Experimental Investigation and Scope. Organometallics 2017, 36, 4864–4882. [Google Scholar] [CrossRef]
- Hong, D.; Ohgomori, Y.; Shimoyama, Y.; Kotani, H.; Ishizuka, T.; Kon, Y.; Kojima, T. Mechanistic Insight into Synergistic Catalysis of Olefin Hydrogenation by a Hetero-Dinuclear RuII-CoII Complex with Adjacent Reaction Sites. Inorg. Chem. 2019, 58, 11284–11288. [Google Scholar] [CrossRef]
- van den Beuken, E.K.; Feringa, B.L. Bimetallic catalysis by late transition metal complexes. Tetrahedron 1998, 54, 12985–13011. [Google Scholar] [CrossRef]
- Kim, S.; Loose, F.; Bezdek, M.J.; Wang, X.; Chirik, P.J. Hydrogenation of N-Heteroarenes Using Rhodium Precatalysts: Reductive Elimination Leads to Formation of Multimetallic Clusters. J. Am. Chem. Soc. 2019, 141, 17900–17908. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Norton, J.R. Kinetics and thermodynamics of H-/H•/H+ transfer from a rhodium(III) hydride. J. Am. Chem. Soc. 2014, 136, 5938–5948. [Google Scholar] [CrossRef]
- Huber, K.-P.; Herzberg, G. Molecular Spectra and Molecular Structure: IV. Constants of Diatomic Molecules; Springer US; Imprint; Springer: Boston, MA, USA, 1979; ISBN 978-1-4757-0963-6. [Google Scholar]
- DuBois, D.L.; Berning, D.E. Hydricity of transition-metal hydrides and its role in CO2 reduction. Appl. Organometal. Chem. 2000, 14, 860–862. [Google Scholar] [CrossRef]
- Hu, Y.; Li, L.; Shaw, A.P.; Norton, J.R.; Sattler, W.; Rong, Y. Synthesis, Electrochemistry, and Reactivity of New Iridium(III) and Rhodium(III) Hydrides. Organometallics 2012, 31, 5058–5064. [Google Scholar] [CrossRef]
- Evans, M.G.; Polanyi, M. Inertia and driving force of chemical reactions. Trans. Faraday Soc. 1938, 34, 11. [Google Scholar] [CrossRef]
- Alvarez, R.; Carmona, E.; Marin, J.M.; Poveda, M.L.; Gutierrez-Puebla, E.; Monge, A. Carbon dioxide chemistry. Synthesis, properties, and structural characterization of stable bis(carbon dioxide) adducts of molybdenum. J. Am. Chem. Soc. 1986, 108, 2286–2294. [Google Scholar] [CrossRef]
- Bernskoetter, W.H.; Tyler, B.T. Kinetics and Mechanism of Molybdenum-Mediated Acrylate Formation from Carbon Dioxide and Ethylene. Organometallics 2011, 30, 520–527. [Google Scholar] [CrossRef]
- Carmona, E.; Munoz, M.A.; Perez, P.J.; Poveda, M.L. Rotational isomerism in bis(carbon dioxide) complexes of molybdenum generated by conrotatory motion of the CO2 ligands. Organometallics 1990, 9, 1337–1339. [Google Scholar] [CrossRef]
- Hanna, B.S.; MacIntosh, A.D.; Ahn, S.; Tyler, B.T.; Palmore, G.T.R.; Williard, P.G.; Bernskoetter, W.H. Ancillary Ligand Effects on Carbon Dioxide-Ethylene Coupling at Zerovalent Molybdenum. Organometallics 2014, 33, 3425–3432. [Google Scholar] [CrossRef]
- Zhang, Y.; Hanna, B.S.; Dineen, A.; Williard, P.G.; Bernskoetter, W.H. Functionalization of Carbon Dioxide with Ethylene at Molybdenum Hydride Complexes. Organometallics 2013, 32, 3969–3979. [Google Scholar] [CrossRef]
- Ziegler, W.; Nicholas, K.M. Photochemistry of (Me3P)4Mo(η-CO2)2: Deoxygenation of coordinated carbon dioxide and phosphine oxidation. J. Organomet. Chem. 1992, 423, C35–C37. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A Gen. Phys. 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Bellan, J.; Marre, M.R.; Sanchez, M.; Wolf, R. Nouveaux ylures du phosphore a partir e tris(amino)iminophosphanes et d’esters acetyleniques. etude structurale par RMN de 13C. Phosphorus Sulfur Silicon Relat. Elem. 1981, 12, 11–18. [Google Scholar] [CrossRef]
- Buscagan, T.M.; Oyala, P.H.; Peters, J.C. N2 -to-NH3 Conversion by a triphos-Iron Catalyst and Enhanced Turnover under Photolysis. Angew. Chem. Int. Ed. 2017, 56, 6921–6926. [Google Scholar] [CrossRef] [PubMed]
- Mankad, N.P.; Rivard, E.; Harkins, S.B.; Peters, J.C. Structural snapshots of a flexible Cu2P2 core that accommodates the oxidation states CuICuI, Cu1.5Cu1.5, and CuIICuII. J. Am. Chem. Soc. 2005, 127, 16032–16033. [Google Scholar] [CrossRef]
- Li, L.; Brennessel, W.W.; Jones, W.D. C−H Activation of Phenyl Imines and 2-Phenylpyridines with [Cp*MCl2]2 (M = Ir, Rh): Regioselectivity, Kinetics, and Mechanism. Organometallics 2009, 28, 3492–3500. [Google Scholar] [CrossRef]
- Kang, J.W.; Moseley, K.; Maitlis, P.M. Pentamethylcyclopentadienylrhodium and -iridium halides. I. Synthesis and properties. J. Am. Chem. Soc. 1969, 91, 5970–5977. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; Cabral De Menezes, S.M.; Goodfellow, R.; Granger, P. NMR nomenclature: Nuclear spin properties and conventions for chemical shifts (IUPAC recommendations 2001). Concepts Magn. Reson. 2002, 14, 326–346. [Google Scholar] [CrossRef]
- Clark, R.C.; Reid, J.S. The analytical calculation of absorption in multifaceted crystals. Acta. Crystallogr. A 1995, 51, 887–897. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction. CrysAlisPro Software Systems, Rigaku Corporation, Oxford, UK 201-2020. Available online: https://www.rigaku.com/products/crystallography/crysalis (accessed on 17 February 2023).
- Sheldrick, G.M. SHELXT - integrated space-group and crystal-structure determination. Acta. Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Gritzner, G.; Kuta, J. Recommendations on reporting electrode potentials in nonaqueous solvents (Recommendations 1983). Pure Appl. Chem. 1984, 56, 461–466. [Google Scholar] [CrossRef]
- Noviandri, I.; Brown, K.N.; Fleming, D.S.; Gulyas, P.T.; Lay, P.A. The Decamethylferrocenium/Decamethylferrocene Redox Couple: A Superior Redox Standard to the Ferrocenium/Ferrocene Redox Couple for Studying Solvent Effects on the Thermodynamics of Electron Transfer. J Phys Chem B 1999, 103, 6713–6722. [Google Scholar] [CrossRef]
- Burchat, A.F.; Chong, J.; Nielsen, N. Titration of alkyllithiums with a simple reagent to a blue endpoint. J. Organomet. Chem. 1997, 542, 281–283. [Google Scholar] [CrossRef]
- Krasovskiy, A.; Knochel, P. Convenient Titration Method for Organometallic Zinc, Magnesium, and Lanthanide Reagents. Synthesis 2006, 2006, 890–891. [Google Scholar] [CrossRef]
- Albinati, A.; Bakhmutov, V.I.; Caulton, K.G.; Clot, E.; Eckert, J.; Eisenstein, O.; Gusev, D.G.; Grushin, V.V.; Hauger, B.E. Reaction of molecular hydrogen (H2) with chlorohydridoiridium phosphines IrHCl2P2 (P = PiPr3 or PtBu2Ph): Stereoelectronic control of the stability of molecular H2 transition metal complexes. J. Am. Chem. Soc. 1993, 115, 7300–7312. [Google Scholar] [CrossRef]
- Desrosiers, P.J.; Cai, L.; Lin, Z.; Richards, R.; Halpern, J. Assessment of the "T1 criterion" for distinguishing between classical and nonclassical transition-metal hydrides: Hydride relaxation rates in tris(triarylphosphine)osmium tetrahydrides and related polyhydrides. J. Am. Chem. Soc. 1991, 113, 4173–4184. [Google Scholar] [CrossRef]
- Kubas, G.J. Dihydrogen complexes as prototypes for the coordination chemistry of saturated molecules. Proc. Natl. Acad. Sci. USA 2007, 104, 6901–6907. [Google Scholar] [CrossRef]
- Kubas, G.J. Activation of dihydrogen and coordination of molecular H2 on transition metals. J. Organomet. Chem. 2014, 751, 33–49. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. C 2015, 71, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta. Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
| Entry | Catalyst | Catalyst Loading (mol%) (b) | Base | TON (c) | Yield (%) (d) |
| 1 | C1 | 0.6 | DBU | 128 | 77 |
| 2 | M1 | 0.6 | DBU | 23 | 14 |
| 3 | M3 | 0.6 | DBU | traces | <1 |
| 4 | M1 + M3 (e) | 0.6 | DBU | 34 | 21 |
| 5 | C2 | 0.6 | DBU | 17 | 10 |
| 6 | M2 | 0.6 | DBU | 5 | 3 |
| 7 | M2 + M3 (e) | 0.6 | DBU | 3 | 2 |
| 8 | C1 | 0.6 | TMG | 168 | Quant. |
| 9 | C1 | 0.6 | NEt3 | 45 | 27 |
| 10 | C1 | 0.6 | - | - | 0 |
| 11 | C1 (f) | 0.6 | DBU | 12 | 7 |
| Intermediate. | Base | MeCN (M1) | DMSO | H2O | MeOH |
|---|---|---|---|---|---|
| [IrIII(solv)]+ | DBU | 0.0 | 0.0 | 0.0 | 0.0 |
| NEt3 | 0.0 | 0.0 | 0.0 | 0.0 | |
| [IrIII(base)]+ | DBU | −66.7 | −34.2 | −86.2 | −84.0 |
| NEt3 | −9.0 | 22.5 | −28.7 | −26.2 | |
| [IrIII(η2-H2)]+ | DBU | 56.4 | 87.7 | 36.3 | 39.3 |
| NEt3 | 56.4 | 87.7 | 36.3 | 39.3 | |
| [IrIIIH] | DBU | −56.8 | −25.8 | −77.6 | −73.8 |
| NEt3 | −9.0 | 21.7 | −30.4 | −25.8 | |
| [TS3] | DBU | 28.4 | 59.1 | 7.8 | 11.6 |
| NEt3 | 76.3 | 106.7 | 55.0 | 59.6 | |
| [IrIIIHCO2] | DBU | −57.4 | −26.6 | −78.7 | −74.3 |
| NEt3 | −9.6 | 21.0 | −31.5 | −26.3 | |
| [IrIII(solv)]+ (a) | DBU | −73.4 | −73.3 | −73.3 | −73.4 |
| NEt3 | −59.6 | −59.7 | −59.7 | −59.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fickenscher, Z.B.G.; Lönnecke, P.; Müller, A.K.; Hollóczki, O.; Kirchner, B.; Hey-Hawkins, E. Synergistic Catalysis in Heterobimetallic Complexes for Homogeneous Carbon Dioxide Hydrogenation. Molecules 2023, 28, 2574. https://doi.org/10.3390/molecules28062574
Fickenscher ZBG, Lönnecke P, Müller AK, Hollóczki O, Kirchner B, Hey-Hawkins E. Synergistic Catalysis in Heterobimetallic Complexes for Homogeneous Carbon Dioxide Hydrogenation. Molecules. 2023; 28(6):2574. https://doi.org/10.3390/molecules28062574
Chicago/Turabian StyleFickenscher, Zeno B. G., Peter Lönnecke, Anna K. Müller, Oldamur Hollóczki, Barbara Kirchner, and Evamarie Hey-Hawkins. 2023. "Synergistic Catalysis in Heterobimetallic Complexes for Homogeneous Carbon Dioxide Hydrogenation" Molecules 28, no. 6: 2574. https://doi.org/10.3390/molecules28062574
APA StyleFickenscher, Z. B. G., Lönnecke, P., Müller, A. K., Hollóczki, O., Kirchner, B., & Hey-Hawkins, E. (2023). Synergistic Catalysis in Heterobimetallic Complexes for Homogeneous Carbon Dioxide Hydrogenation. Molecules, 28(6), 2574. https://doi.org/10.3390/molecules28062574