

Novel 7-Deazapurine Incorporating Isatin Hybrid Compounds as Protein Kinase Inhibitors: Design, Synthesis, In Silico Studies, and Antiproliferative Evaluation

Abstract
1. Introduction
2. Results and Discussion
2.1. Design
2.2. Chemistry
2.3. Biological Evaluation
2.3.1. In Vitro Cytotoxic Assay against HepG2, MCF-7, MDA-MB-231, and HeLa Cell Lines
2.3.2. Kinase Inhibition Assay over CDK2, EGFR, HER2, and VEGFR2
2.3.3. Cell Cycle Analysis
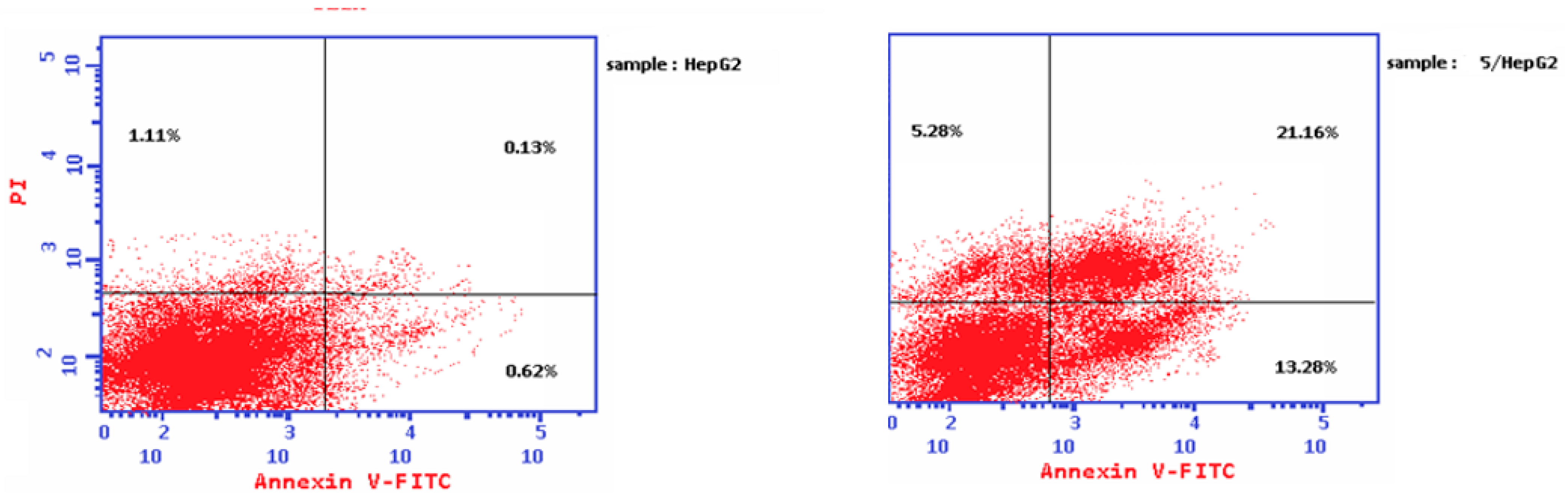
2.3.4. Apoptosis Analysis
2.3.5. Caspase 3, Caspase 9, BAX, and Bcl-2 Level Protein Assays
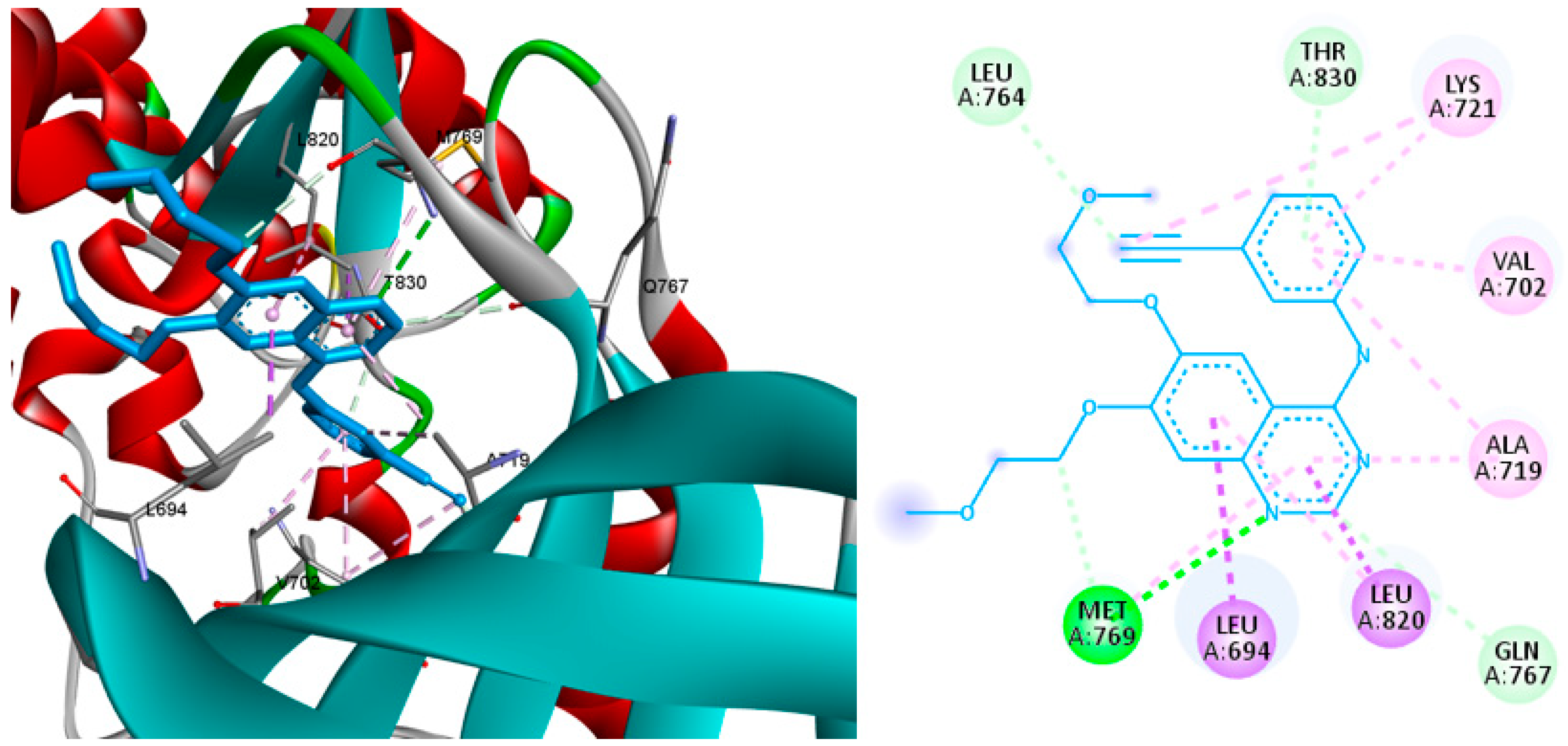
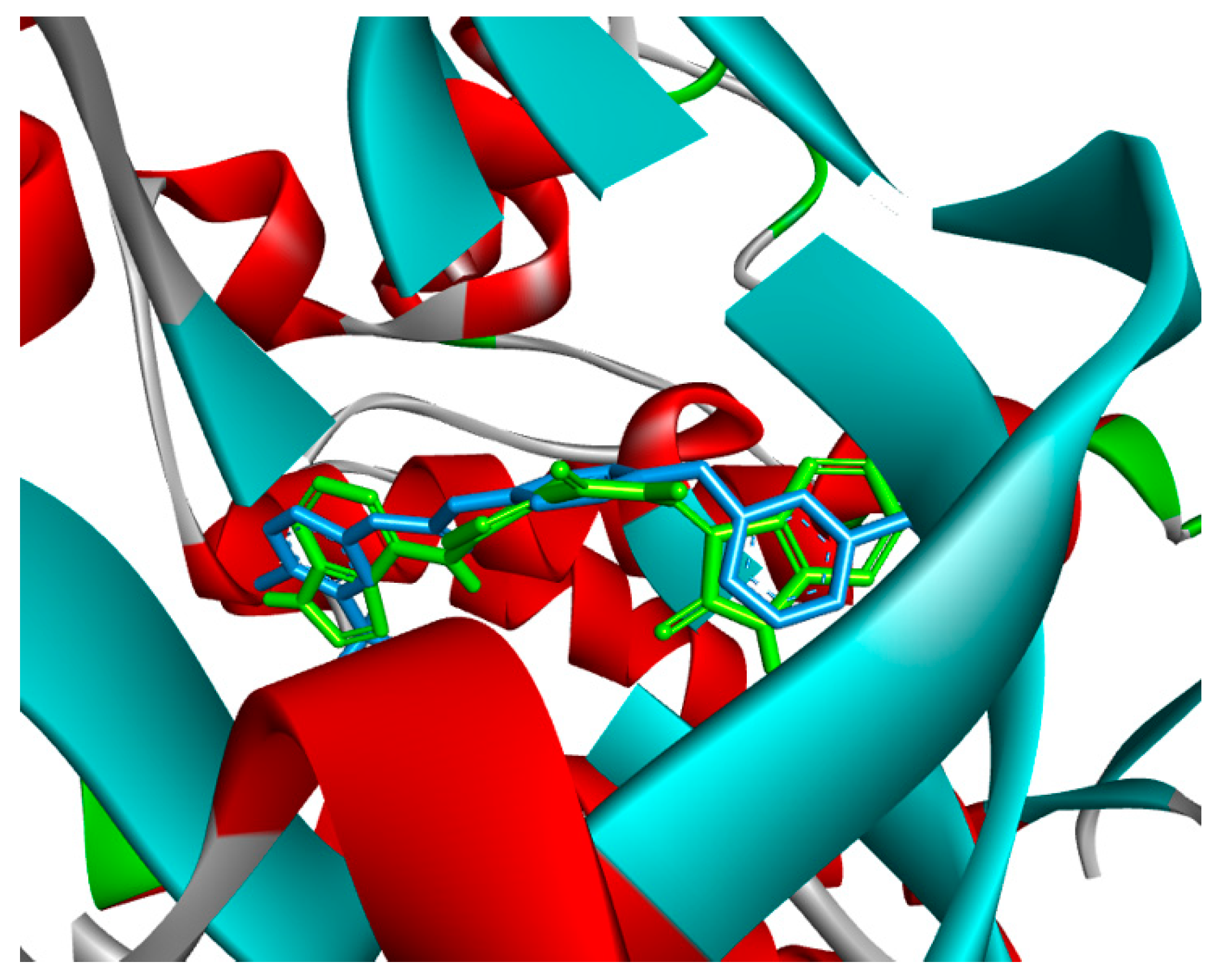
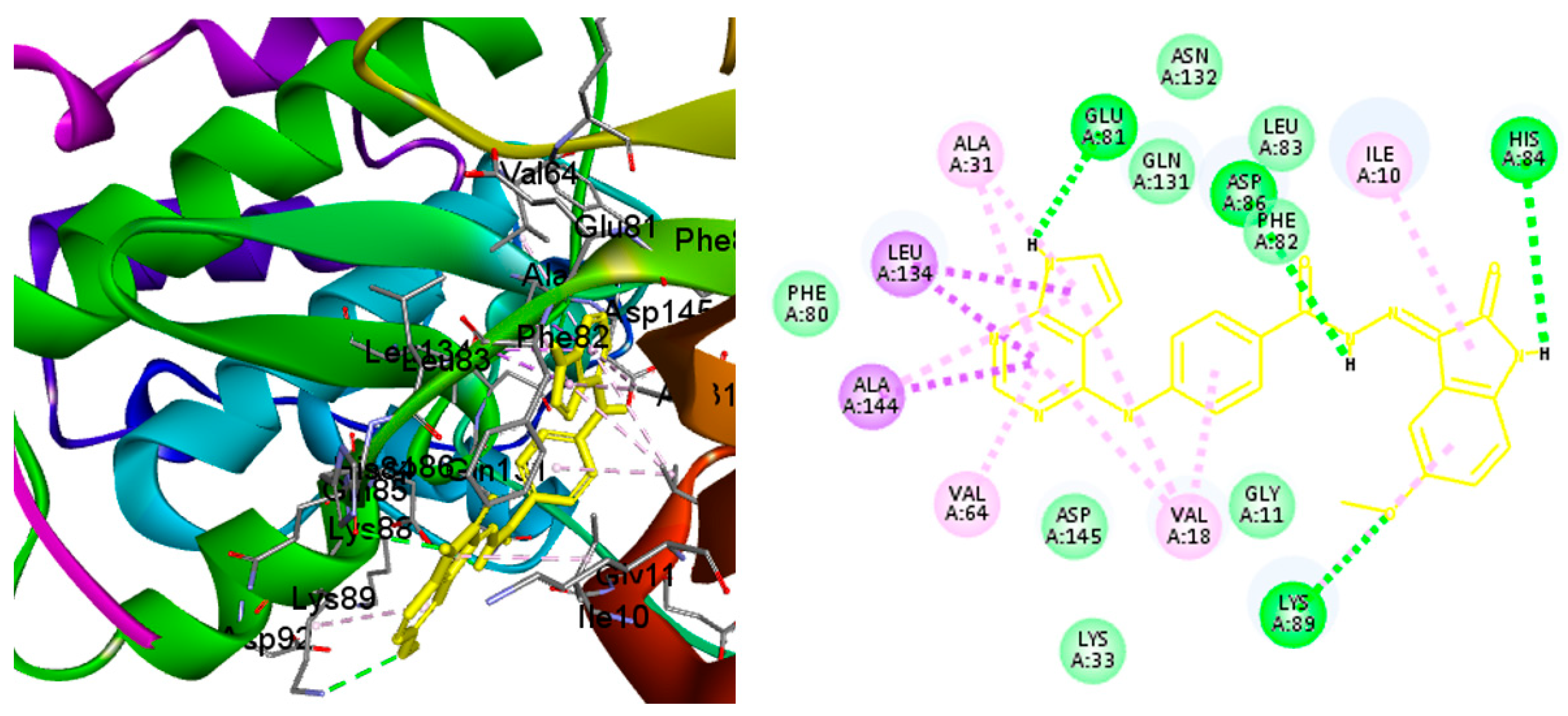
2.4. Molecular Docking
3. Material and Methods
3.1. Chemistry
3.1.1. Preparation of Compounds 1–5
3.1.2. Preparation of Ethyl 4-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)benzoate (7)
3.1.3. Preparation of 4-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)benzohydrazide (8)
3.2. HPLC Analysis
3.3. Biological Evaluation
3.3.1. In Vitro Cytotoxic Assay against HepG2, MCF-7, MDA-MB-231, and HeLa Cell Lines
3.3.2. Kinase Inhibition Assay over CDK2, EGFR, HER2, and VEGFR2
3.3.3. Cell Cycle Analysis
3.3.4. Apoptosis Analysis
3.3.5. Caspase 3, Caspase 9, BAX, and Bcl-2 Level Protein Assays
3.4. Molecular Docking
3.5. ADMET Profile Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Saraon, P.; Pathmanathan, S.; Snider, J.; Lyakisheva, A.; Wong, V.; Stagljar, I. Receptor Tyrosine Kinases and Cancer: Oncogenic Mechanisms and Therapeutic Approaches. Oncogene 2021, 40, 4079–4093. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kaur, N.; Sahu, S.; Sharma, V.; Kumar, D.; Sharma, A.; Wadhwa, P. Role of Tyrosine Kinases and Their Inhibitors in Cancer Therapy: A Comprehensive Review. Curr. Med. Chem. 2022, 30, 1464–1481. [Google Scholar] [CrossRef]
- Ayala-Aguilera, C.C.; Valero, T.; Lorente-Macías, Á.; Baillache, D.J.; Croke, S.; Unciti-Broceta, A. Small Molecule Kinase Inhibitor Drugs (1995–2021): Medical Indication, Pharmacology, and Synthesis. J. Med. Chem. 2022, 65, 1047–1131. [Google Scholar] [CrossRef]
- Sun, D.; Zhao, Y.; Zhang, S.; Zhang, L.; Liu, B.; Ouyang, L. Dual-Target Kinase Drug Design: Current Strategies and Future Directions in Cancer Therapy. Eur. J. Med. Chem. 2020, 188, 112025. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Classification of Small Molecule Protein Kinase Inhibitors Based upon the Structures of Their Drug-Enzyme Complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef]
- Roskoski, R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2021 Update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef] [PubMed]
- Levitzki, A. Tyrosine Kinases as Targets for Cancer Therapy. Eur. J. Cancer 2002, 38 (Suppl. 5), S11–S18. [Google Scholar] [CrossRef]
- Hussaarts, K.G.A.M.; Veerman, G.D.M.; Jansman, F.G.A.; van Gelder, T.; Mathijssen, R.H.J.; van Leeuwen, R.W.F. Clinically Relevant Drug Interactions with Multikinase Inhibitors: A Review. Ther. Adv. Med. Oncol. 2019, 11, 1758835918818347. [Google Scholar] [CrossRef]
- Hanks, S.K.; Quinn, A.M.; Hunter, T. The Protein Kinase Family: Conserved Features and Deduced Phylogeny of the Catalytic Domains. Science 1988, 241, 42–52. [Google Scholar] [CrossRef]
- Ur Rashid, H.; Ahmad, N.; Abdalla, M.; Khan, K.; Martines, M.A.U.; Shabana, S. Molecular Docking and Dynamic Simulations of Cefixime, Etoposide and Nebrodenside A against the Pathogenic Proteins of SARS-CoV-2. J. Mol. Struct. 2022, 1247, 131296. [Google Scholar] [CrossRef]
- Chacia, T. Analysis of Benzodiazepines by High Performance Liquid Chromatography. In Contributions to Forensic Toxicology, Proceedings of the 31st International Meeting of The International Association of Forensic Toxicologists (TIAFT), Leipzig, Germany, August 1993; MOLINApress: Leipzig, Germany, 1994; pp. 133–137. [Google Scholar]
- Alanazi, M.M.; Elkady, H.; Alsaif, N.A.; Obaidullah, A.J.; Alkahtani, H.M.; Alanazi, M.M.; Alharbi, M.A.; Eissa, I.H.; Dahab, M.A. New Quinoxaline-Based VEGFR-2 Inhibitors: Design, Synthesis, and Antiproliferative Evaluation with in Silico Docking, ADMET, Toxicity, and DFT Studies. RSC Adv. 2021, 11, 30315–30328. [Google Scholar] [CrossRef]
- Swanton, C.; Futreal, A.; Eisen, T.; Engelman, J.; Johnson, D.; Haber, D.; Lynch, T.; Johnson, B.; Heymach, J. Her2-Targeted Therapies in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2006, 12, 4377s–4383s. [Google Scholar] [CrossRef]
- Sadek, M.M.; Serrya, R.A.; Kafafy, A.H.N.; Ahmed, M.; Wang, F.; Abouzid, K.A.M. Discovery of New HER2/EGFR Dual Kinase Inhibitors Based on the Anilinoquinazoline Scaffold as Potential Anti-Cancer Agents. J. Enzyme Inhib. Med. Chem. 2014, 29, 215–222. [Google Scholar] [CrossRef]
- Al-Sanea, M.M.; Obaidullah, A.J.; Shaker, M.E.; Chilingaryan, G.; Alanazi, M.M.; Alsaif, N.A.; Alkahtani, H.M.; Alsubaie, S.A.; Abdelgawad, M.A. A New CDK2 Inhibitor with 3-Hydrazonoindolin-2-One Scaffold Endowed with Anti-Breast Cancer Activity: Design, Synthesis, Biological Evaluation, and In Silico Insights. Molecules 2021, 26, 412. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M. Cyclin-Dependent Kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, A.S.; Mirgany, T.O.; Alsfouk, A.A.; Alsaif, N.A.; Alanazi, M.M. Antiproliferative Activity, Multikinase Inhibition, Apoptosis- Inducing Effects and Molecular Docking of Novel Isatin–Purine Hybrids. Medicina 2023, 59, 610. [Google Scholar] [CrossRef]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the Translation Machinery in Cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef]
- Alanazi, A.S.; Mirgany, T.O.; Alsaif, N.A.; Alsfouk, A.A.; Alanazi, M.M. Design, Synthesis, Antitumor Evaluation, and Molecular Docking of Novel Pyrrolo[2,3-d]Pyrimidine as Multi-Kinase Inhibitors. Saudi Pharm. J. 2023, 31, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Alsaif, N.A.; Elwan, A.; Alanazi, M.M.; Obaidullah, A.J.; Alanazi, W.A.; Alasmari, A.F.; Albassam, H.; Mahdy, H.A.; Taghour, M.S. Design, Synthesis and Molecular Docking of New [1,2,4] Triazolo[4,3-a]Quinoxaline Derivatives as Anticancer Agents Targeting VEGFR-2 Kinase. Mol. Divers. 2022, 26, 1915–1932. [Google Scholar] [CrossRef]
- Alsaif, N.A.; Taghour, M.S.; Alanazi, M.M.; Obaidullah, A.J.; Alanazi, W.A.; Alasmari, A.; Albassam, H.; Dahab, M.A.; Mahdy, H.A. Identification of New [1,2,4]Triazolo[4,3-a]Quinoxalines as Potent VEGFR-2 Tyrosine Kinase Inhibitors: Design, Synthesis, Anticancer Evaluation, and in Silico Studies. Bioorg. Med. Chem. 2021, 46, 116384. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Alaa, E.; Alsaif, N.A.; Obaidullah, A.J.; Alkahtani, H.M.; Al-Mehizia, A.A.; Alsubaie, S.M.; Taghour, M.S.; Eissa, I.H. Discovery of New 3-Methylquinoxalines as Potential Anti-Cancer Agents and Apoptosis Inducers Targeting VEGFR-2: Design, Synthesis, and in Silico Studies. J. Enzyme Inhib. Med. Chem. 2021, 36, 1732–1750. [Google Scholar] [CrossRef]
- Alsaif, N.A.; Taghour, M.S.; Alanazi, M.M.; Obaidullah, A.J.; Al-Mehizia, A.A.; Alanazi, M.M.; Aldawas, S.; Elwan, A.; Elkady, H. Discovery of New VEGFR-2 Inhibitors Based on Bis([1, 2, 4]Triazolo)[4,3-a:3’,4’-c]Quinoxaline Derivatives as Anticancer Agents and Apoptosis Inducers. J. Enzyme Inhib. Med. Chem. 2021, 36, 1093. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Thabrew, M.I.; Hughes, R.D.; Mcfarlane, I.G. Screening of Hepatoprotective Plant Components Using a HepG2 Cell Cytotoxicity Assay. J. Pharm. Pharmacol. 2011, 49, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Abou-Seri, S.M.; Eldehna, W.M.; Ali, M.M.; Abou El Ella, D.A. 1-Piperazinylphthalazines as Potential VEGFR-2 Inhibitors and Anticancer Agents: Synthesis and in Vitro Biological Evaluation. Eur. J. Med. Chem. 2016, 107, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Suresh, P.S.; Mullangi, R.; Srinivas, N.R. Quantitation of VEGFR2 (Vascular Endothelial Growth Factor Receptor) Inhibitors-Review of Assay Methodologies and Perspectives. Biomed. Chromatogr. 2014, 29, 803–834. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; Hassan, G.S.; Al-Rashood, S.T.; Al-Warhi, T.; Altyar, A.E.; Alkahtani, H.M.; Almehizia, A.A.; Abdel-Aziz, H.A. Synthesis and in Vitro Anticancer Activity of Certain Novel 1-(2-Methyl-6-Arylpyridin-3-Yl)-3-Phenylureas as Apoptosis-Inducing Agents. J. Enzyme Inhib. Med. Chem. 2019, 34, 322–332. [Google Scholar] [CrossRef]
- Sabt, A.; Abdelhafez, O.M.; El-Haggar, R.S.; Madkour, H.M.F.; Eldehna, W.M.; El-Khrisy, E.E.D.A.M.; Abdel-Rahman, M.A.; Rashed, L.A. Novel Coumarin-6-Sulfonamides as Apoptotic Anti-Proliferative Agents: Synthesis, in Vitro Biological Evaluation, and QSAR Studies. J. Enzyme Inhib. Med. Chem. 2018, 33, 1095–1107. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Aldawas, S.; Alsaif, N.A. Design, Synthesis, and Biological Evaluation of 2-Mercaptobenzoxazole Derivatives as Potential Multi-Kinase Inhibitors. Pharmaceuticals 2023, 16, 97. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | 1 | 2 | 3 | 4 | 5 | Sunitinib | |
|---|---|---|---|---|---|---|---|
| Compound | |||||||
| Molecular properties | |||||||
| Molecular weight | 397.398 | 431.843 | 415.388 | 411.425 | 427.424 | 398.482 | |
| LogP | 2.7877 | 3.4411 | 2.9268 | 3.0961 | 2.7963 | 3.33494 | |
| H-acceptor | 6 | 6 | 6 | 6 | 7 | 3 | |
| H-donors | 4 | 4 | 4 | 4 | 4 | 3 | |
| Surface area | 169.803 | 180.106 | 173.968 | 176.168 | 181.281 | 169.730 | |
| Absorption | |||||||
| Water solubility | −3.118 | −3.122 | −3.117 | −3.127 | −3.116 | −4.338 | |
| Intestinal absorption (human) | 79.999% | 81.175% | 78.38% | 80.467% | 76.475% | 94.534% | |
| Skin permeability | −2.735 | −2.735 | −2.735 | −2.735 | −2.735 | −2.79 | |
| Distribution | |||||||
| BBB permeability | −1.325 | −1.507 | −1.486 | −1.354 | −1.476 | −1.122 | |
| CNS permeability | −2.474 | −2.367 | −2.541 | −2.407 | −2.667 | −2.601 | |
| Metabolism | |||||||
| CYP1A2 inhibitor | YES | YES | YES | YES | YES | NO | |
| CYP2C19 inhibitor | YES | YES | YES | YES | YES | NO | |
| CYP2C9 inhibitor | YES | YES | YES | YES | YES | NO | |
| CYP2D6 inhibitor | NO | NO | NO | NO | NO | YES | |
| CYP3A4 inhibitor | YES | YES | YES | YES | YES | YES | |
| Excretion | |||||||
| Total clearance | −0.022 | −0.281 | 0.08 | −0.018 | 0.186 | 1.055 | |
| Renal OCT2 substrate | NO | NO | NO | NO | NO | NO | |
| Toxicity | |||||||
| Max. tolerated dose (human) | 0.587 | 0.626 | 0.621 | 0.616 | 0.635 | 0.223 | |
| Oral rate acute toxicity (LD50) | 2.837 | 2.528 | 2.773 | 2.854 | 2.776 | 2.387 | |
| Oral rate chronic toxicity (LOAEL) | 2.651 | 2.528 | 2.363 | 2.481 | 2.314 | 1.662 | |
| Hepatotoxicity | YES | YES | YES | YES | YES | YES | |
| Skin sensitization | NO | NO | NO | NO | NO | NO | |
| Compound | R | In Vitro Cytotoxicity IC50 (µM) | |||
|---|---|---|---|---|---|
| HepG2 | MCF-7 | MDA-MB-231 | HeLa | ||
| 1 | H | 25.78 ± 1.9 | 18.49 ± 1.5 | 16.03 ± 1.3 | 12.39 ± 0.9 |
| 2 | Cl | 67.42 ± 3.6 | 72.51 ± 3.7 | 51.29 ± 2.9 | 45.40 ± 2.6 |
| 3 | F | 59.31 ± 3.3 | 63.68 ± 3.4 | 42.85 ± 2.6 | 39.15 ± 2.4 |
| 4 | CH3 | 92.67 ± 4.5 | 80.23 ± 4.2 | 73.22 ± 3.3 | 75.36 ± 3.5 |
| 5 | OCH3 | 6.11 ± 0.4 | 5.93 ± 0.3 | 2.48 ± 0.1 | 1.98 ± 0.1 |
| DOXORUBICIN | - | 4.50 ± 0.2 | 4.17 ± 0.2 | 3.18 ± 0.1 | 5.57 ± 0.4 |
| Sunitinib | - | 6.82 ± 0.5 | 5.19 ± 0.4 | 8.41 ± 0.7 | 7.48 ± 0.6 |
| IC50 (μM) | CDK2 | EGFR | HER2 | VEGFR2 |
|---|---|---|---|---|
| Compound 5 | 0.131 ± 0.007 | 0.103 ± 0.006 | 0.081 ± 0.002 | 0.178 ± 0.009 |
| Reference | * 0.063 ± 0.003 | ** 0.041 ± 0.003 | *** 0.06 ± 0.006 | **** 0.045 ± 0.002 |
| Cell Line | Cell Cycle Distribution (%) | |||
|---|---|---|---|---|
| %G0-G1 | %S | %G2/M | %Pre-G1 | |
| Compound 5 | 49.02 | 44.93 | 6.05 | 39.72 |
| Cont. HepG2 | 43.97 | 41.12 | 14.91 | 1.86 |
| Sample | Apoptosis | Necrosis | ||
|---|---|---|---|---|
| Viable | Early | Late | ||
| Compound 5/HepG2 | 39.72 | 13.28 | 21.16 | 5.28 |
| Cont. HepG2 | 1.86 | 0.62 | 0.13 | 1.11 |
| Sample | Gene Expression Fold Change | |||
|---|---|---|---|---|
| Caspase 3 | Caspase 9 | Bax | Bcl-2 | |
| Compound 5/HepG2 | 7.984 | 4.853 | 6.13 | 0.184 |
| Cont. HepG2 | 1 | 1 | 1 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alanazi, M.M.; Alanazi, A.S. Novel 7-Deazapurine Incorporating Isatin Hybrid Compounds as Protein Kinase Inhibitors: Design, Synthesis, In Silico Studies, and Antiproliferative Evaluation. Molecules 2023, 28, 5869. https://doi.org/10.3390/molecules28155869
Alanazi MM, Alanazi AS. Novel 7-Deazapurine Incorporating Isatin Hybrid Compounds as Protein Kinase Inhibitors: Design, Synthesis, In Silico Studies, and Antiproliferative Evaluation. Molecules. 2023; 28(15):5869. https://doi.org/10.3390/molecules28155869
Chicago/Turabian StyleAlanazi, Mohammed M., and Ashwag S. Alanazi. 2023. "Novel 7-Deazapurine Incorporating Isatin Hybrid Compounds as Protein Kinase Inhibitors: Design, Synthesis, In Silico Studies, and Antiproliferative Evaluation" Molecules 28, no. 15: 5869. https://doi.org/10.3390/molecules28155869
APA StyleAlanazi, M. M., & Alanazi, A. S. (2023). Novel 7-Deazapurine Incorporating Isatin Hybrid Compounds as Protein Kinase Inhibitors: Design, Synthesis, In Silico Studies, and Antiproliferative Evaluation. Molecules, 28(15), 5869. https://doi.org/10.3390/molecules28155869