
Pharmacophore-Based Virtual Screening and In-Silico Explorations of Biomolecules (Curcumin Derivatives) of Curcuma longa as Potential Lead Inhibitors of ERBB and VEGFR-2 for the Treatment of Colorectal Cancer
 ,
,  ,
,
Abstract
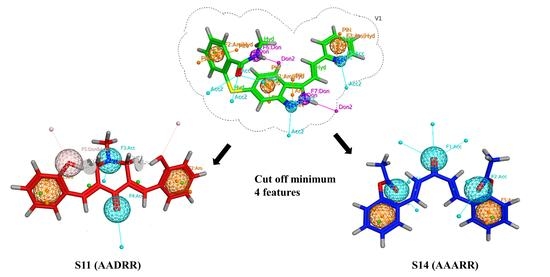
1. Introduction
2. Results and Discussion
2.1. Preparation of Chemical Database
2.2. Generation of Pharmacophore Model
2.3. Pharmacophore-Based Virtual Screening
2.4. Similarity Index
2.5. Density Function Theory (DFTs)
2.6. Filtration for Drug-Likeness and Virtual Screening
2.7. Molecular Docking Discussion
2.7.1. Binding Interactions of ERBB
2.7.2. Molecular Interactions with VEGFR2
2.8. Molecular Dynamics Simulations
2.8.1. MD Simulation Studies of VEGFR2 and Compound S14
2.8.2. MD Simulations Analysis of the ERBB–S14 Complex
2.8.3. The MMGBSA Free Energy Calculations
2.9. SeeSAR Analysis
3. Materials and Methods
3.1. Generation of Pharmacophore Model
3.2. Pharmacophore-Based Virtual Screening
3.3. Density Functional Theory (DFTs)
3.4. Filtration for Drug-Likeness and Virtual Screening
3.5. Molecular Docking Studies
3.6. Molecular Dynamics Simulation Studies
3.7. Compound Similarity Index
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zielińska, A.; Alves, H.; Marques, V.; Durazzo, A.; Lucarini, M.; Alves, T.F.; Morsink, M.; Willemen, N.; Eder, P.; Chaud, M.V.; et al. Properties, extraction methods, and delivery systems for curcumin as a natural source of beneficial health effects. Medicina 2020, 56, 336. [Google Scholar] [CrossRef]
- Baidoun, F.; Elshiwy, K.; Elkeraie, Y.; Merjaneh, Z.; Khoudari, G.; Sarmini, M.T.; Gad, M.; Al-Husseini, M.; Saad, A. Colorectal cancer epidemiology: Recent trends and impact on outcomes. Curr. Drug Targets 2021, 22, 998–1009. [Google Scholar] [PubMed]
- Rhee, P.; Joseph, B.; Pandit, V.; Aziz, H.; Vercruysse, G.; Kulvatunyou, M.; Friese, R.S. Increasing trauma deaths in the United States. Ann. Surg. 2014, 260, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Tannapfel, A.; Reinacher-Schick, A. Chemotherapy-Associated Hepatotoxicity in the Treatment of Colorectal Cancer (CRC). J. Gastroenterol. 2006, 46, 435–440. [Google Scholar]
- Joseph, D.A.; Meester, R.G.; Zauber, A.G.; Manninen, D.L.; Winges, L.; Dong, F.B.; Peaker, B.; van Ballegooijen, M. Colorectal cancer screening: Estimated future colonoscopy need and current volume and capacity. Cancer 2016, 122, 2479–2486. [Google Scholar] [CrossRef] [PubMed]
- Pikhurko, O.; Spencer, J.; Verbitsky, O. Decomposable graphs and definitions with no quantifier alternation. Eur. J. Comb. 2007, 28, 2264–2283. [Google Scholar] [CrossRef]
- Liszbinski, R.B.; Romagnoli, G.G.; Gorgulho, C.M.; Basso, C.R.; Pedrosa, V.A.; Kaneno, R. Anti-EGFR-coated gold nanoparticles in vitro carry 5-fluorouracil to colorectal cancer cells. Materials 2020, 13, 375. [Google Scholar] [CrossRef]
- Martinelli, E.; De Palma, R.; Orditura, M.; De Vita, F.; Ciardiello, F. Anti-epidermal growth factor receptor monoclonal antibodies in cancer therapy. Clin. Exp. Immunol. 2009, 158, 1–9. [Google Scholar] [CrossRef]
- Takahashi, S. Vascular endothelial growth factor (VEGF), VEGF receptors and their inhibitors for antiangiogenic tumor therapy. Biol. Pharm. Bull. 2011, 34, 1785–1788. [Google Scholar] [CrossRef]
- Oh, B.Y.; Lee, R.A.; Chung, S.S.; Kim, K.H. Epidermal growth factor receptor mutations in colorectal cancer patients. J. Korean Soc. Coloproctol. 2011, 27, 127. [Google Scholar] [CrossRef]
- Kanthan, R.; Senger, J.L.; Kanthan, S.C. Molecular events in primary and metastatic colorectal carcinoma: A review. Pathol. Res. Int. 2012, 2012, 597497. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, S.; Tolosano, E.; Munaron, L.; Genova, T. Targeting metabolism to counteract tumor angiogenesis: A review of patent literature. Recent Pat. Anticancer Drug Discov. 2018, 13, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Jonasch, E.; Boyle, S.; Carlo, M.I.; Manley, B.; Agarwal, N.; Alva, A.; Beckermann, K.; Choueiri, T.K.; Costello, B.A. NCCN guidelines insights: Kidney cancer, version 1.2021: Featured updates to the NCCN guidelines. J. Natl. Compr. Cancer Netw. 2020, 18, 1160–1170. [Google Scholar] [CrossRef] [PubMed]
- El-Metwally, S.A.; Abou-El-Regal, M.M.; Eissa, I.H.; Mehany, A.B.; Mahdy, H.A.; Elkady, H.; Elwan, A.; Elkaeed, E.B. Discovery of thieno [2, 3-d] pyrimidine-based derivatives as potent VEGFR-2 kinase inhibitors and anti-cancer agents. Bioorg. Med. Chem. 2021, 112, 104947. [Google Scholar] [CrossRef] [PubMed]
- Metibemu, D.S.; Akinloye, O.A.; Akamo, A.J.; Ojo, D.A.; Okeowo, O.T.; Omotuyi, O.I. Exploring receptor tyrosine kinases-inhibitors in Cancer treatments, Egyptian. J. Med. Hum. Genet. 2019, 20, 35. [Google Scholar] [CrossRef]
- Guo, M.; An, F.; Yu, H.; Wei, X.; Hong, M.; Lu, Y. Comparative effects of schisandrin, A.; B, and C on Propionibacterium acnes-induced, NLRP3 inflammasome activation-mediated IL-1β secretion and pyroptosis. Biomed. Pharmacother. 2017, 96, 129–136. [Google Scholar] [CrossRef]
- Gross-Goupil, M.; Françlois, L.; Quivy, A.; Ravaud, A. Axitinib: A review of its safety and efficacy in the treatment of adults with advanced renal cell carcinoma. Clin. Med. Insights Oncol. SAJE J. 2013, 7, S10594. [Google Scholar] [CrossRef]
- Tortora, G.; Ciardiello, F.; Gasparini, G. Combined targeting of EGFR-dependent and VEGF-dependent pathways: Rationale, preclinical studies and clinical applications. Nat. Clin. Pract. Oncol. 2008, 5, 521–530. [Google Scholar] [CrossRef]
- Adams, B.K.; Ferstl, E.M.; Davis, M.C.; Herold, M.; Kurtkaya, S.; Camalier, R.F.; Hollingshead, M.G.; Kaur, G.; Sausville, E.A.; Rickles, F.R.; et al. Synthesis and biological evaluation of novel curcumin analogs as anti-cancer and anti-angiogenesis agents. Bioorg. Med. Chem. 2004, 12, 3871–3883. [Google Scholar] [CrossRef]
- Can Baser, K. Biological and pharmacological activities of carvacrol and carvacrol bearing essential oils. Curr. Pharm. Des. 2008, 14, 3106–3119. [Google Scholar] [CrossRef]
- El-Azab, M.; Hishe, H.; Moustafa, Y.; El-Awady, E.S. Anti-angiogenic effect of resveratrol or curcumin in Ehrlich ascites carcinoma-bearing mice. Eur. J. Pharmacol. 2011, 652, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.C.; Arbab, A.S.; Jardim-Perassi, B.V.; Borin, T.F.; Varma, N.R.; Iskander, A.S.; Shankar, A.; Ali, M.M.; Zuccari, D.A. Effect of curcumin on pro-angiogenic factors in the xenograft model of breast cancer. Anticancer Agents Med. Chem. 2015, 15, 1285–1296. [Google Scholar] [CrossRef]
- Campos, D.; Ji, H. IMG2SMI: Translating Molecular Structure Images to Simplified Molecular-input Line-entry System. arXiv 2021, arXiv:2109.04202. [Google Scholar]
- Aziz, M.; Ejaz, S.A.; Rehman, H.M.; Alsubaie, A.S.; Mahmoud, K.H.; Siddique, F.; Al-Buriahi, M.S.; Alrowaili, Z.A. Identification of NEK7 inhibitors: Structure based virtual screening, molecular docking, density functional theory calculations and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2022, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Cousins, K. MOE (Molecular Operating Environment), 2016. Available online: https://www.chemcomp.com/Products.htm (accessed on 8 January 2023).
- Zhang, Y.; Zhang, T.J.; Tu, S.; Zhang, Z.H.; Meng, F.H. Identification of novel Src inhibitors: Pharmacophore-based virtual screening, molecular docking and molecular dynamics simulations. Molecules 2020, 25, 4094. [Google Scholar] [CrossRef]
- Frisch, M.E.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Goerigk, L.; Reimers, J.R. Efficient methods for the quantum chemical treatment of protein structures: The effects of London-dispersion and basis-set incompleteness on peptide and water-cluster geometries. J. Chem. Theory Comput. 2013, 9, 3240–3251. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, version 6.0.16; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Ahmed, A.; Saeed, A.; Ejaz, S.A.; Aziz, M.; Hashmi, M.Z.; Channar, P.A.; Abbas, Q.; Raza, H.; Shafiq, Z.; El-Seedi, H.R. Novel adamantyl clubbed iminothiazolidinones as promising elastase inhibitors: Design, synthesis, molecular docking, ADMET and DFT studies. RSC Adv. 2022, 12, 11974–11991. [Google Scholar] [CrossRef]
- Heinzerling, L.; Klein, R.; Rarey, M. Fast force field-based optimization of protein–ligand complexes with graphics processor. J. Comput. Chem. 2012, 33, 2554–2565. [Google Scholar] [CrossRef]
- Al-Hazmi, G.A.; Abou-Melha, K.S.; El-Metwaly, N.M.; Althagafi, I.; Shaaban, F.; Zaky, R. Green synthesis approach for Fe (III), Cu (II), Zn (II) and Ni (II)-Schiff base complexes, spectral, conformational, MOE-docking and biological studies. Appl. Organomet. Chem. 2020, 34, e5403. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing (SC’06), Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Barclay, P.L.; Zhang, D.Z. Periodic boundary conditions for arbitrary deformations in molecular dynamics simulations. J. Comput. Phys. 2021, 435, 110238. [Google Scholar] [CrossRef]
- Luty, B.A.; Davis, M.E.; Tironi, I.G.; Van Gunsteren, W.F. A comparison of particle-particle, particle-mesh and Ewald methods for calculating electrostatic interactions in periodic molecular systems. Mol. Simul. 1994, 14, 11–20. [Google Scholar] [CrossRef]
- Aziz, M.; Ejaz, S.A.; Tamam, N.; Siddique, F.; Riaz, N.; Qais, F.A.; Chtita, S.; Iqbal, J. Identification of potent inhibitors of NEK7 protein using a comprehensive computational approach. Sci. Rep. 2022, 12, 6404. [Google Scholar] [CrossRef] [PubMed]
- Bajusz, D.; Rácz, K.; Héberger, A. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Tresaugues, L.; Roos, A.; Arrowsmith, C.H.; Berglund, H.; Bountra, C.; Collins, R.; Edwards, A.M.; Flodin, S.; Flores, A.; Graslund, S.; et al. Crystal structure of VEGFR1 in complex with N-(4-Chlorophenyl)-2-((pyridin-4-ylmethyl) amino) benzamide. RCSB PDB 2009. [Google Scholar]
- Leppänen, V.M.; Tvorogov, D.; Kisko, K.; Prota, A.E.; Jeltsch, M.; Anisimov, A.; Markovic-Mueller, S.; Stuttfeld, E.; Goldie, K.N.; Ballmer-Hofer, K.; et al. Structural and mechanistic insights into VEGF receptor 3 ligand binding and activation. Proc. Natl. Acad. Sci. USA 2013, 110, 12960–12965. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IUPAC Formula | ERBB Binding Energies (kJ/mol) | VEGFR2 Binding Energies (kJ/mol) | VEGFR3 Binding Energies (kJ/mol) | VEGFR1 Binding Energies (kJ/mol) |
|---|---|---|---|---|---|
| S1 | 1E,6E)-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione | −27.65 | −33.4 | −22.8 | −34.16 |
| S2 | (2E,6E)-2,6-bis(4-hydroxy-3-methoxybenzylidene)cyclohexanone | −26.32 | −29.56 | −24.76 | −35.28 |
| S3 | (1E,4E)-1,5-bis(3,4-dimethoxyphenyl)penta-1,4-dien-3-one | 25.74 | −32.67 | −17.6 | −35.1 |
| S4 | (1E,4E)-1,5-bis(2-methoxyphenyl)penta-1,4-dien-3-one | −25.51 | −28.20 | −22.92 | −32.44 |
| S5 | (1E,4E)-1,5-bis(3-hydroxyphenyl)penta-1,4-dien-3-one | −23.79 | −26.9 | −22.68 | −34.96 |
| S6 | (1E,4E)-1,5-bis(2-fluorophenyl)penta-1,4-dien-3-one | −24.64 | −32.4 | −19.08 | −31.2 |
| S7 | (1E,4E)-1,5-bis(2-hydroxyphenyl)penta-1,4-dien-3-one | −26.54 | −37.5 | −21.72 | −30.5 |
| S8 | (1E,4E)-1,5-bis(2-hydroxyphenyl)penta-1,4-dien-3-one | −25.87 | −32.7 | −29.88 | −41.48 |
| S9 | (3E,5E)-3,5-bis(2-hydroxybenzylidene)dihydro-2H-pyran-4(3H)-one | −27.37 | −36.3 | −29.24 | −40.84 |
| S10 | (3E,5E)-3,5-bis(2-fluorobenzylidene)dihydro-2H-pyran-4(3H)-one | −25.87 | −37.2 | −22.36 | −15.2 |
| S11 | (3E,5E)-3,5-bis(2-hydroxybenzylidene)-1-methylpiperidin-4-one | −37.92 | −43.2 | −28.04 | −36.2 |
| S12 | (3E,5E)-3,5-bis(2-fluorobenzylidene)-1-(1-hydroxy-2-oxopropyl)piperidin-4-one | −31.68 | −38.78 | −26.1 | −32.68 |
| S13 | (1E,4E)-1,5-bis(4-hydroxyphenyl)penta-1,4-dien-3-one | −29.41 | −33.95 | −20.52 | −32.8 |
| S14 | ((1E,4E)-3-oxopenta-1,4-diene-1,5-diyl)bis(2,1-phenylene) diacetate | −38.5 | −46.5 | −22.24 | −35.2 |
| Irinotecan (reference) | 4,11-diethyl-4-hydroxy-3,14-dioxo-3,4,12,14-tetrahydro-1H-pyrano [3’,4’:6,7]indolizino [1,2-b]quinolin-9-yl [1,4’-bipiperidine]-1’-carboxylate | −28.4 | −40.24 | −38.32 | −41.21 |
| Compound | Tanimoto MACCS | Tanimoto Morgan | Dice MACCS | Dice Morgan |
|---|---|---|---|---|
| S11 | 0.300 | 0.271 | 0.461 | 0.426 |
| S14 | 0.215 | 0.251 | 0.354 | 0.401 |
| Axitinib | 1.000 | 1.000 | 1.000 | 1.000 |
| Compounds | Optimization Energy (Hatree) | Polarizability (α.u) | Dipole Moment (Debye) |
|---|---|---|---|
| S1 | −1256.586 | 257.490 | 6.373 |
| S2 | −1220.906 | 289.801 | 6.587 |
| S3 | −1182.989 | 285.627 | 4.918 |
| S4 | −955.207 | 233.085 | 3.816 |
| S5 | −877.013 | 204.518 | 4.692 |
| S6 | −924.814 | 193.712 | 2.963 |
| S7 | −877.024 | 215.917 | 1.545 |
| S8 | −993.128 | 239.791 | 1.551 |
| S9 | −1028.810 | 233.923 | 0.569 |
| S10 | −1076.612 | 222.898 | 4.450 |
| S11 | −1048.146 | 241.779 | 1.991 |
| S12 | −1322.551 | 259.230 | 4.842 |
| S13 | −877.021 | 228.199 | 5.811 |
| S14 | −1180.671 | 264.307 | 1.225 |
| Compound | Hardness (η) | Softness (S) | Electronegativity (X) | Chemical Potential (μ) | Electrophilicity Index (ω) |
|---|---|---|---|---|---|
| S1 | 0.071 | 0.285 | 1.138 | −1.138 | 9.107 |
| S2 | 0.069 | 0.276 | 1.105 | −1.105 | 8.838 |
| S3 | 0.064 | 0.256 | 1.023 | −1.023 | 8.186 |
| S4 | 0.072 | 0.289 | 1.156 | −1.156 | 9.248 |
| S5 | 0.073 | 0.290 | 1.160 | −1.160 | 9.282 |
| S6 | 0.076 | 0.303 | 1.212 | −1.212 | 9.696 |
| S7 | 0.071 | 7.04 | 0.142 | −0.142 | 0.142 |
| S8 | 0.070 | 7.14 | 0.130 | −0.130 | 0.121 |
| S9 | 0.070 | 7.14 | 0.140 | −0.140 | 0.140 |
| S10 | 0.075 | 6.67 | 0.155 | −0.155 | 0.160 |
| S11 | 0.068 | 7.35 | 0.132 | −0.132 | 0.128 |
| S12 | 0.067 | 7.46 | 0.147 | −0.147 | 0.161 |
| S13 | 0.069 | 7.25 | 0.142 | −0.142 | 0.146 |
| S14 | 0.073 | 6.90 | 0.155 | −0.155 | 0.165 |
| Codes | EHOMO (eV) | ELUMO (eV) | ∆Egap (eV) | Potential Ionization I(eV) | Affinity A(eV) | Electron Donating Power (ω−) | Electron Accepting Power (ω+) | Electrophilicity (Δω±) |
|---|---|---|---|---|---|---|---|---|
| S1 | −0.21 | −0.06 | 0.142 | 0.21 | 0.06 | 3.11 | 8.41 | 11.53 |
| S2 | −0.20 | −0.06 | 0.138 | 0.20 | 0.06 | 3.02 | 8.17 | 11.19 |
| S3 | −0.19 | −0.06 | 0.128 | 0.19 | 0.06 | 2.82 | 7.68 | 10.51 |
| S4 | −0.22 | −0.07 | 0.145 | 0.22 | 0.07 | 3.19 | 8.66 | 11.85 |
| S5 | −0.22 | −0.07 | 0.145 | 0.22 | 0.07 | 3.21 | 8.74 | 11.95 |
| S6 | −0.24 | −0.08 | 0.152 | 0.24 | 0.08 | 3.38 | 9.28 | 12.67 |
| S7 | −0.213 | −0.071 | 0.142 | 0.213 | 0.071 | 0.222 | 0.080 | 0.302 |
| S8 | −0.20 | −0.06 | 0.140 | 0.20 | 0.06 | 0.194 | 0.064 | 0.259 |
| S9 | −0.21 | −0.07 | 0.140 | 0.21 | 0.07 | 0.219 | 0.079 | 0.298 |
| S10 | −0.23 | −0.08 | 0.150 | 0.23 | 0.08 | 0.247 | 0.092 | 0.339 |
| Physicochemical Properties | ||||||||
|---|---|---|---|---|---|---|---|---|
| Molecular Weight | Density | nHA | nHD | TPSA | LogS | LogP | LogD | |
| S1 | 368.13 | 0.966 | 6 | 2 | 93.06 | −3.921 | 2.742 | 2.82 |
| S2 | 366.15 | 0.954 | 5 | 2 | 75.99 | −4.475 | 3.441 | 3.437 |
| S3 | 354.15 | 0.945 | 5 | 0 | 53.99 | −5.442 | 3.118 | 3.271 |
| S4 | 294.13 | 0.911 | 3 | 0 | 35.53 | −5.88 | 3.808 | 3.775 |
| S5 | 266.09 | 0.924 | 3 | 2 | 57.53 | −4.003 | 3.08 | 3.248 |
| S6 | 270.09 | 0.955 | 1 | 0 | 17.07 | −6.046 | 3.937 | 3.963 |
| S7 | 266.09 | 0.924 | 3 | 2 | 57.53 | −4.003 | 3.08 | 3.248 |
| S8 | 306.13 | 0.924 | 3 | 2 | 57.53 | −4.2 | 4.425 | 3.717 |
| S9 | 308.1 | 0.954 | 4 | 2 | 66.76 | −3.678 | 3.233 | 3.156 |
| S10 | 312.1 | 0.983 | 2 | 0 | 26.3 | −5.847 | 3.904 | 3.815 |
| S11 | 321.14 | 0.938 | 4 | 2 | 60.77 | −3.052 | 3.521 | 3.108 |
| S12 | 383.13 | 0.991 | 4 | 1 | 57.61 | −4.067 | 3.052 | 3.437 |
| S13 | 266.09 | 0.924 | 3 | 2 | 57.53 | −3.277 | 3.201 | 3.397 |
| S14 | 350.12 | 0.947 | 5 | 0 | 69.67 | −5.518 | 3.123 | 2.829 |
| Absorption and Distribution Properties | ||||||||
|---|---|---|---|---|---|---|---|---|
| Volume of Distribution (vd) | Human Intestinal Absorption (hia) | Caco-2 Permeability | Blood Brain Barrier (bbb) and Blood-Placenta Barrier (bpb) | Plasma Protein Binding (ppb) | pgp-Inhibitor | p-Glycoprotein Substrate (pgp-Substrate) | MDCK Permeability | |
| S1 | 2.52 | 0.008 | −4.668 | 0.218 | 99.08% | 0.087 | 0.001 | 1.3 × 10−5 |
| S2 | 2.442 | 0.006 | −4.584 | 0.176 | 99.53% | 0.008 | 0.001 | 1.3 × 10−5 |
| S3 | 2.398 | 0.006 | −4.604 | 0.185 | 99.73% | 0.057 | 0.003 | 1.2 × 10−5 |
| S4 | 2.505 | 0.006 | −4.589 | 0.165 | 99.52% | 0.069 | 0.002 | 1.2 × 10−5 |
| S5 | 2.243 | 0.006 | −4.581 | 0.187 | 96.94% | 0.004 | 0.004 | 1.3 × 10−5 |
| S6 | 2.155 | 0.005 | −4.513 | 0.172 | 98.12% | 0.045 | 0.004 | 1.2 × 10−5 |
| S7 | 2.179 | 0.005 | −4.508 | 0.161 | 100% | 0.056 | 0.005 | 1.2 × 10−5 |
| S8 | 2.817 | 0.005 | −4.548 | 0.159 | 99.99% | 0.007 | 0.002 | 1.3 × 10−5 |
| S9 | 2.418 | 0.006 | −4.611 | 0.16 | 100% | 0.015 | 0.008 | 1.2 × 10−5 |
| S10 | 1.701 | 0.005 | −4.557 | 0.13 | 100% | 0.33 | 0.015 | 1.1 × 10−5 |
| S11 | 2.964 | 0.005 | −4.539 | 0.167 | 99.86% | 0.057 | 0.001 | 1.3 × 10−5 |
| S12 | 2.846 | 0.006 | −4.542 | 0.166 | 99.94% | 0.341 | 0.002 | 1.2 × 10−5 |
| S13 | 2.629 | 0.005 | −4.513 | 0.077 | 99.89% | 0.717 | 0.001 | 1.3 × 10−5 |
| S14 | 2.854 | 0.006 | −4.539 | 0.545 | 100% | 0.627 | 0.001 | 1.2 × 10−5 |
| Metabolism | Excretion | ||||||
|---|---|---|---|---|---|---|---|
| Codes | CYP1A2 Inhibitor | CYP2C19 Inhibitor | CYP2C9 Inhibitor | CYP2D6 Inhibitor | CYP3A4 Inhibitor | CL (mL/min) | T1/2 (Hours) |
| S1 | 0.593 | 0.287 | 0.661 | 0.037 | 0.674 | 13.839 | 0.948 |
| S2 | 0.851 | 0.823 | 0.689 | 0.421 | 0.6 | 7.792 | 0.89 |
| S3 | 0.511 | 0.478 | 0.148 | 0.009 | 0.474 | 10.232 | 0.785 |
| S4 | 0.978 | 0.904 | 0.626 | 0.256 | 0.883 | 7.828 | 0.431 |
| S5 | 0.987 | 0.421 | 0.447 | 0.64 | 0.939 | 12.32 | 0.912 |
| S6 | 0.98 | 0.742 | 0.512 | 0.366 | 0.254 | 5.519 | 0.106 |
| S7 | 0.987 | 0.421 | 0.447 | 0.64 | 0.939 | 12.32 | 0.912 |
| S8 | 0.963 | 0.958 | 0.894 | 0.947 | 0.477 | 5.072 | 0.657 |
| S9 | 0.945 | 0.884 | 0.867 | 0.554 | 0.282 | 6.894 | 0.835 |
| S10 | 0.919 | 0.946 | 0.837 | 0.03 | 0.055 | 6.738 | 0.056 |
| S11 | 0.703 | 0.741 | 0.433 | 0.944 | 0.062 | 17.699 | 0.799 |
| S12 | 0.435 | 0.868 | 0.759 | 0.159 | 0.054 | 12.063 | 0.169 |
| S13 | 0.829 | 0.636 | 0.432 | 0.195 | 0.558 | 13.675 | 0.937 |
| S14 | 0.976 | 0.94 | 0.857 | 0.498 | 0.206 | 1.413 | 0.813 |
| Medicinal Properties | Toxicity | ||||||
|---|---|---|---|---|---|---|---|
| Synthetic Accessibility Score | Lipinski Rule | AMES Toxicity | Carcinogenicity | Eye Corrosion | Eye Irritation | Respiratory Toxicity | |
| S1 | 2.426 | Accepted | 0.234 | 0.706 | 0.007 | 0.792 | 0.951 |
| S2 | 2.408 | Accepted | 0.103 | 0.78 | 0.004 | 0.303 | 0.905 |
| S3 | 2.095 | Accepted | 0.19 | 0.828 | 0.02 | 0.404 | 0.516 |
| S4 | 2.048 | Accepted | 0.187 | 0.636 | 0.581 | 0.988 | 0.688 |
| S5 | 2.269 | Accepted | 0.083 | 0.517 | 0.767 | 0.99 | 0.917 |
| S6 | 2.143 | Accepted | 0.049 | 0.708 | 0.651 | 0.98 | 0.622 |
| S7 | 2.269 | Accepted | 0.083 | 0.517 | 0.767 | 0.99 | 0.917 |
| S8 | 2.394 | Accepted | 0.638 | 0.347 | 0.011 | 0.952 | 0.514 |
| S9 | 2.605 | Accepted | 0.913 | 0.272 | 0.013 | 0.936 | 0.633 |
| S10 | 2.512 | Accepted | 0.911 | 0.887 | 0.004 | 0.216 | 0.666 |
| S11 | 2.555 | Accepted | 0.724 | 0.239 | 0.004 | 0.018 | 0.861 |
| S12 | 3.264 | Accepted | 0.55 | 0.608 | 0.003 | 0.008 | 0.713 |
| S13 | 2.148 | Accepted | 0.131 | 0.463 | 0.051 | 0.978 | 0.947 |
| S14 | 2.345 | Accepted | 0.632 | 0.44 | 0.857 | 0.988 | 0.801 |
| TOX21 Pathway | ||||
|---|---|---|---|---|
| Compound | NR-AR | NR-AR-LBD | NR-ER | Antioxidant Response Element |
| S1 | 0.807 | 0.787 | 0.6 | 0.891 |
| S2 | 0.772 | 0.121 | 0.926 | 0.977 |
| S3 | 0.381 | 0.906 | 0.892 | 0.91 |
| S4 | 0.429 | 0.954 | 0.964 | 0.933 |
| S5 | 0.484 | 0.973 | 0.975 | 0.967 |
| S6 | 0.005 | 0.935 | 0.625 | 0.84 |
| S7 | 0.484 | 0.973 | 0.975 | 0.967 |
| S8 | 0.515 | 0.439 | 0.932 | 0.982 |
| S9 | 0.141 | 0.636 | 0.908 | 0.974 |
| S10 | 0.001 | 0.925 | 0.447 | 0.956 |
| S11 | 0.073 | 0.049 | 0.422 | 0.969 |
| S12 | 0.002 | 0.685 | 0.113 | 0.953 |
| S13 | 0.788 | 0.9 | 0.981 | 0.971 |
| S14 | 0.103 | 0.963 | 0.751 | 0.921 |
| Complex | ΔGbind (kcal/mol) | ΔE H-bond (kcal/mol) | ΔE vdW (kcal/mol) | ΔE coulomb (kcal/mol) |
|---|---|---|---|---|
| VEGFR2-S14 | −65.4 | −2.1 | −44.21 | −10.76 |
| ERBB–S14 | −45.32 | −1.4 | −28.54 | −8.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ejaz, S.A.; Aziz, M.; Fawzy Ramadan, M.; Fayyaz, A.; Bilal, M.S. Pharmacophore-Based Virtual Screening and In-Silico Explorations of Biomolecules (Curcumin Derivatives) of Curcuma longa as Potential Lead Inhibitors of ERBB and VEGFR-2 for the Treatment of Colorectal Cancer. Molecules 2023, 28, 4044. https://doi.org/10.3390/molecules28104044
Ejaz SA, Aziz M, Fawzy Ramadan M, Fayyaz A, Bilal MS. Pharmacophore-Based Virtual Screening and In-Silico Explorations of Biomolecules (Curcumin Derivatives) of Curcuma longa as Potential Lead Inhibitors of ERBB and VEGFR-2 for the Treatment of Colorectal Cancer. Molecules. 2023; 28(10):4044. https://doi.org/10.3390/molecules28104044
Chicago/Turabian StyleEjaz, Syeda Abida, Mubashir Aziz, Mohamed Fawzy Ramadan, Ammara Fayyaz, and Muhammad Sajjad Bilal. 2023. "Pharmacophore-Based Virtual Screening and In-Silico Explorations of Biomolecules (Curcumin Derivatives) of Curcuma longa as Potential Lead Inhibitors of ERBB and VEGFR-2 for the Treatment of Colorectal Cancer" Molecules 28, no. 10: 4044. https://doi.org/10.3390/molecules28104044
APA StyleEjaz, S. A., Aziz, M., Fawzy Ramadan, M., Fayyaz, A., & Bilal, M. S. (2023). Pharmacophore-Based Virtual Screening and In-Silico Explorations of Biomolecules (Curcumin Derivatives) of Curcuma longa as Potential Lead Inhibitors of ERBB and VEGFR-2 for the Treatment of Colorectal Cancer. Molecules, 28(10), 4044. https://doi.org/10.3390/molecules28104044