
Virtual Screening for Identification of Dual Inhibitors against CDK4/6 and Aromatase Enzyme
 , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
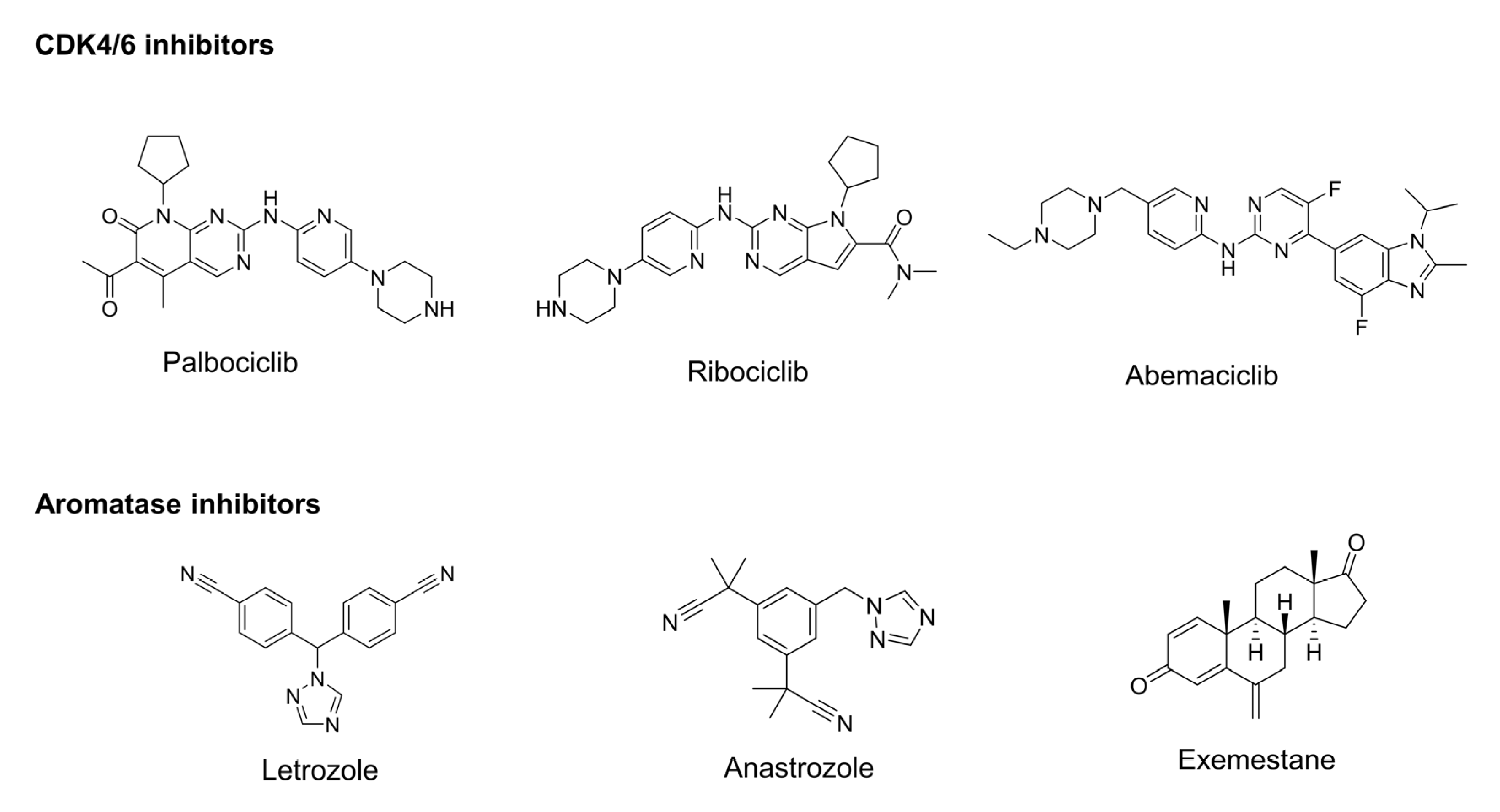
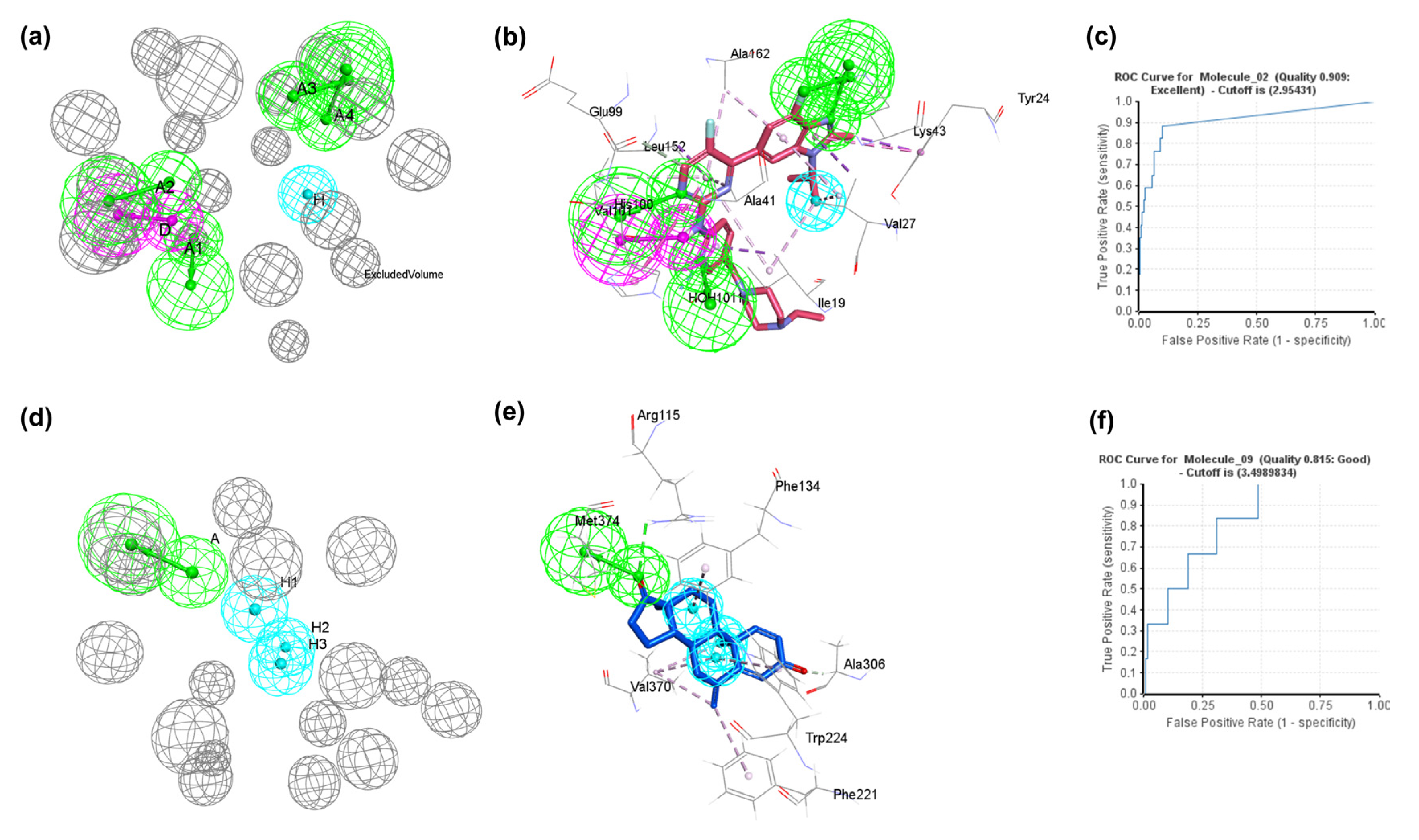
2.1. Development of Structure-Based Pharmacophore Model and Validation
2.2. Pharmacophore-Based Virtual Screening
2.3. Molecular Docking Analysis of Identified Potential Candidates
2.4. Binding Modes Analysis of Identified Candidates
2.4.1. Binding Mode of Candidate 1
2.4.2. Binding Mode of Candidate 2
2.4.3. Binding Mode of Candidate 3
2.4.4. Binding Mode of Candidate 4
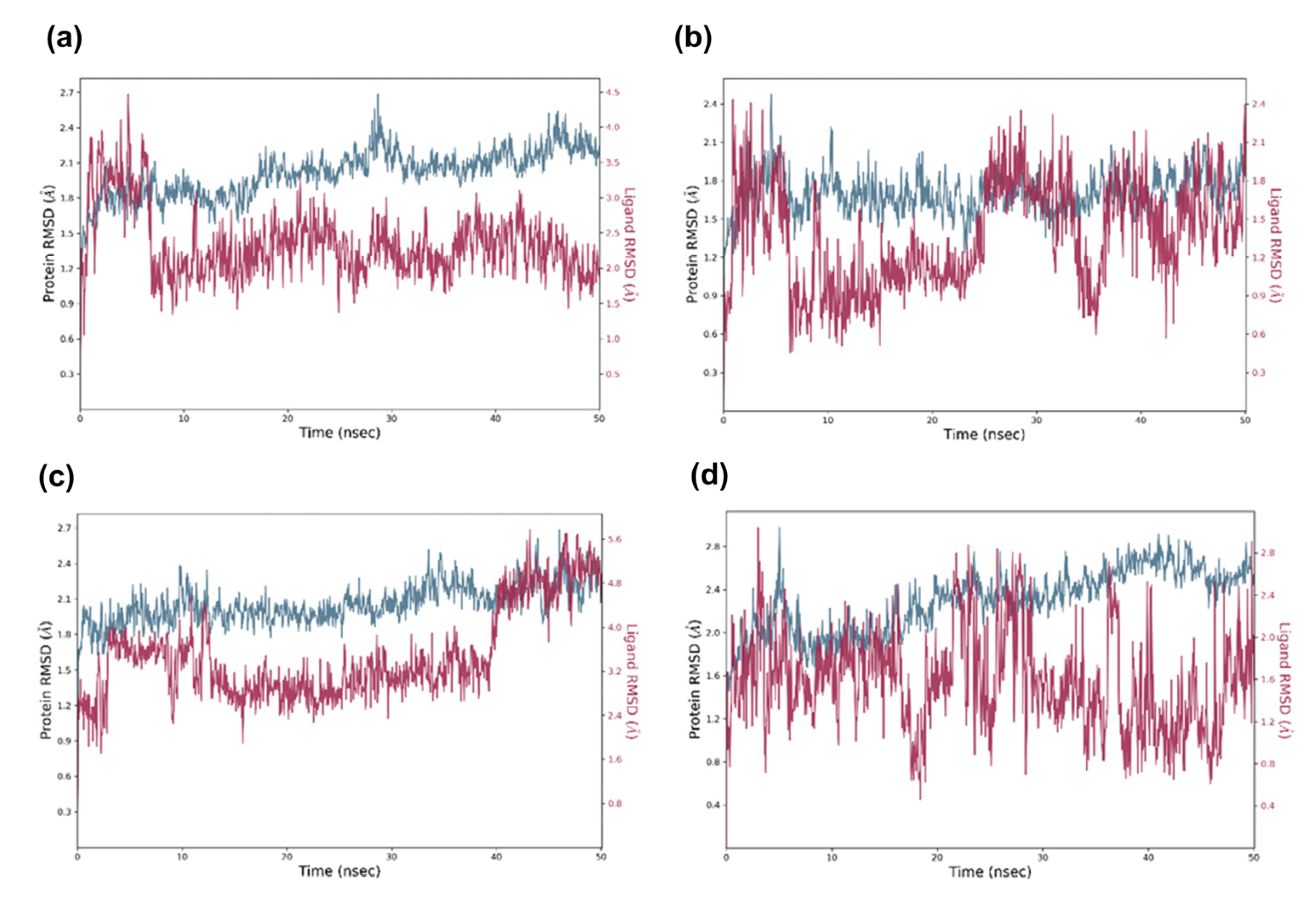
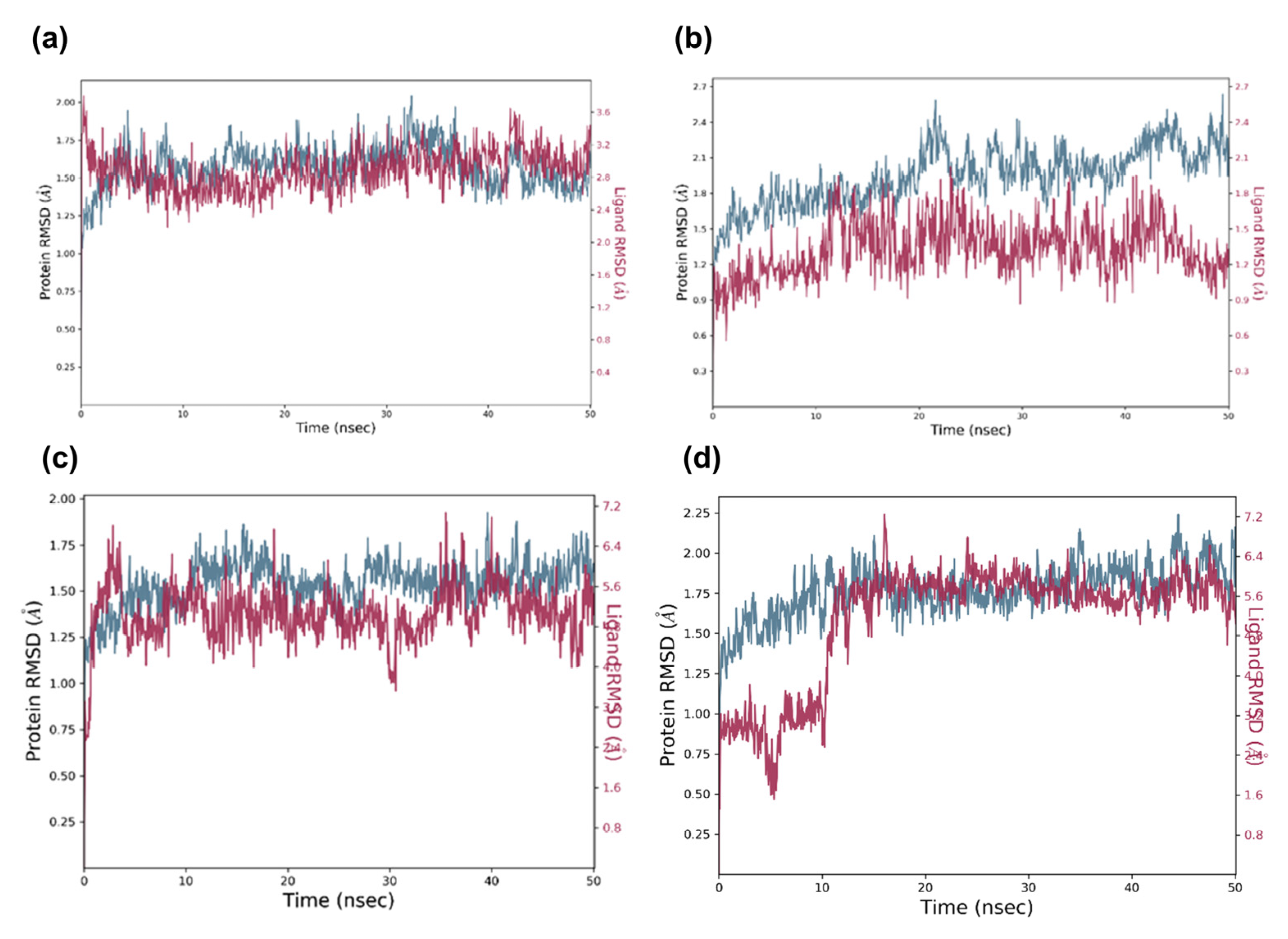
2.5. Molecular Dynamic Analysis of the Promising Candidates
2.6. Predicted ADMET Analysis
2.7. Molecular Orbital Properties
3. Materials and Methods
3.1. Preparation of Proteins
3.2. Generation and Validation of Structure-Based Pharmacophore Models
3.3. Pharmacophore-Based Virtual Screening
3.4. Molecular Docking
3.5. Molecular Dynamics (MD) Simulation
3.6. ADMET Prediction and Calculation of Molecular Orbitals
3.7. Density Functional Theory (DFT) Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Breast Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed on 12 January 2023).
- Omar, A.; Bakr, A.; Ibrahim, N. Female medical students’ awareness, attitudes, and knowledge about early detection of breast cancer in Syrian Private University, Syria. Heliyon 2020, 6, e03819. [Google Scholar] [CrossRef]
- Varlamis, I.; Apostolakis, I.; Sifaki-Pistolla, D.; Dey, N.; Georgoulias, V.; Lionis, C. Application of data mining techniques and data analysis methods to measure cancer morbidity and mortality data in a regional cancer registry: The case of the island of Crete, Greece. Comput. Methods Programs Biomed. 2017, 145, 73–83. [Google Scholar] [CrossRef]
- Cavalieri, E.; Chakravarti, D.; Guttenplan, J.; Hart, E.; Ingle, J.; Jankowiak, R.; Muti, P.; Rogan, E.; Russo, J.; Santen, R.; et al. Catechol estrogen quinones as initiators of breast and other human cancers: Implications for biomarkers of susceptibility and cancer prevention. Biochim. Biophys. Acta Rev. Cancer. 2006, 1766, 63–78. [Google Scholar]
- Samavat, H.; Kurzer, M.S. Estrogen metabolism and breast cancer. Cancer Lett. 2015, 356, 231–243. [Google Scholar] [CrossRef]
- Osborne, C.K.; Schiff, R. Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef]
- Saha Roy, S.; Vadlamudi, R.K. Role of Estrogen Receptor Signaling in Breast Cancer Metastasis. Int. J. Breast Cancer 2011, 2012, 654698. [Google Scholar] [CrossRef]
- García-Becerra, R.; Santos, N.; Díaz, L.; Camacho, J. Mechanisms of resistance to endocrine therapy in breast cancer: Focus on signaling pathways, miRNAs and genetically based resistance. Int. J. Mol Sci. 2013, 14, 108. [Google Scholar] [CrossRef]
- Szostakowska, M.; Trębińska-Stryjewska, A.; Grzybowska, E.A.; Fabisiewicz, A. Resistance to endocrine therapy in breast cancer: Molecular mechanisms and future goals. Breast Cancer Res Treat. 2019, 173, 489–497. [Google Scholar] [CrossRef]
- Tong, C.W.S.; Wu, M.; Cho, W.C.S.; To, K.K.W. Recent advances in the treatment of breast cancer. Front Oncol. 2018, 8, 227. [Google Scholar] [CrossRef]
- Sherr, C.J.; Beach, D.; Shapiro, G.I. Targeting CDK4 and CDK6: From discovery to therapy. Cancer Discov. 2016, 6, 353–367. [Google Scholar] [CrossRef]
- Sammons, S.L.; Topping, D.L.; Blackwell, K.L. HR+, HER2− Advanced Breast Cancer and CDK4/6 Inhibitors: Mode of Action, Clinical Activity, and Safety Profiles. Curr. Cancer Drug Targets 2017, 17, 637–649. [Google Scholar] [CrossRef] [PubMed]
- De Melo Gagliato, D.; C Buzaid, A.; Perez-Garcia, J.M.; Llombart, A.C.J. CDK4/6 Inhibitors in Hormone Receptor-Positive Metastatic Breast Cancer: Current Practice and Knowledge. Cancers 2020, 12, 2480. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Bartkova, J.; Bartek, J. Convergence of mitogenic signalling cascades from diverse classes of receptors at the cyclin D-cyclin-dependent kinase-pRb-controlled G1 checkpoint. Mol. Cell Biol. 1996, 16, 6917–6925. [Google Scholar] [CrossRef]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Zhang, S. Polypharmacology: Drug discovery for the future. Expert Rev. Clin. Pharmacol. 2013, 6, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Tomaselli, D.; Lucidi, A.; Rotili, D.M.A. Epigenetic Polypharmacology: A New Frontier for Epi-Drug Discovery. Med. Res. Rev. 2020, 40, 190–244. [Google Scholar] [CrossRef] [PubMed]
- Schuster, D. 3D pharmacophores as tools for activity profiling. Drug Discov. Today Technol. 2010, 7, e205–e211. [Google Scholar] [CrossRef]
- Wolber, G.; Seidel, T.; Bendix, F.; Langer, T. Molecule-pharmacophore superpositioning and pattern matching in computa-tional drug design. Drug Discov. Today 2008, 13, 23–29. [Google Scholar] [CrossRef]
- Lu, X.; Yang, H.; Chen, Y.; Li, Q.; He, S.Y.; Jiang, X.; Feng, F.; Qu, W.S.H. The Development of Pharmacophore Modeling: Generation and Recent Applications in Drug Discovery. Curr. Pharm. Des. 2018, 24, 3424–3439. [Google Scholar] [CrossRef]
- Chen, P.; Lee, N.V.; Hu, W.; Xu, M.; Ferre, R.A.; Lam, H.; Bergqvist, S.; Solowiej, J.; Diehl, W.; He, Y.A.; et al. Spectrum and degree of CDK drug interactions predicts clinical performance. Mol. Cancer Ther. 2016, 15, 2273–2281. [Google Scholar] [CrossRef]
- Ghosh, D.; Lo, J.; Morton, D.; Valette, D.; Xi, J.; Griswold, J.; Hubbell, S.; Egbuta, C.; Jiang, W.; An, J.D.H. Novel Aromatase Inhibitors by Structure-Guided Design Debashis. J. Med. Chem. 2012, 55, 8464–8476. [Google Scholar] [CrossRef] [PubMed]
- Desaphy, J.; Bret, G.; Rognan, D.; Kellenberger, E. Sc-PDB: A 3D-database of ligandable binding sites-10 years on. Nucleic Acids Res. 2015, 43, D399–D404. [Google Scholar] [CrossRef]
- Wermuth, C.G. Strategies in the Search for New Lead Compounds or Original Working Hypotheses. In The Practice of Medicinal Chemistry, 3rd ed.; Wermuth, C.G., Ed.; Academic Press: London, UK, 2008; pp. 125–143. [Google Scholar]
- Wood, P.M.; Woo, L.W.; Labrosse, J.R.; Trusselle, M.N.; Abbate, S.; Longhi, G.; Castiglioni, E.; Lebon, F.; Purohit, A.; Reed, M.J.; et al. Chiral aromatase and dual aromatase-steroid sulfatase inhibitors from the letrozole template: Synthesis, absolute configuration, and in vitro activity. J. Med. Chem. 2008, 51, 4226–4238. [Google Scholar] [CrossRef] [PubMed]
- Gitto, R.; Agnello, S.; Ferro, S.; De Luca, L.; Vullo, D.; Brynda, J.; Mader, P.; Supuran, C.T.; Chimirri, A. Identification of 3,4-dihydroisoquinoline-2(1H)-sulfonamides as potent carbonic anhydrase inhibitors: Synthesis, biological evaluation, and enzyme-ligand X-ray studies. J. Med. Chem. 2010, 53, 2401–2408. [Google Scholar] [CrossRef] [PubMed]
- Tsou, H.R.; Otteng, M.; Tran, T.; Floyd, M.B.; Reich, M.; Birnberg, G.; Kutterer, K.; Ayral-Kaloustian, S.; Ravi, M.; Nilakantan, R.; et al. 4-(Phenylaminomethylene)isoquinoline-1,3(2H,4H)-diones as potent and selective inhibitors of the cyclin-dependent kinase 4 (CDK4). J. Med. Chem. 2008, 51, 3507–3525. [Google Scholar] [CrossRef]
- Faheem; Karan Kumar, B.; Venkata Gowri Chandra Sekhar, K.; Chander, S.; Kunjiappan, S.; Murugesan, S. 1,2,3,4-Tetrahydroisoquinoline (THIQ) as privileged scaffold for anticancer de novo drug design. Expert Opin. Drug Discov. 2021, 16, 1119–1147. [Google Scholar] [CrossRef]
- Rampogu, S.; Park, C.; Ravinder, D.; Son, M.; Baek, A.; Zeb, A.; Bavi, A.; Kumar, R.; Lee, G.; Parate, S.; et al. Pharmacotherapeutics and molecular mechanism of phytochemicals in alleviating hormone-responsive breast cancer. Oxid Med. Cell Longev. 2019, 2019, 5189490. [Google Scholar] [CrossRef]
- Mandlik, V.; Bejugam, P.R.; Singh, S. Application of Artificial Neural Networks in Modern Drug Discovery. In Artificial Neural Network for Drug Design, Delivery and Disposition; Academic Press: Boca Raton, FL, USA, 2016; pp. 123–139. [Google Scholar]
- BIOVIA. Dassault System, QSAR, ADMET and Predictive Toxicology Package, 2019; Dassault System: San Diego, CA, USA, 2019. [Google Scholar]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Storelli, F.; Matthey, A.; Lenglet, S.; Thomas, A.; Desmeules, J.; Daali, Y. Impact of CYP2D6 Functional Allelic Variations on Phenoconversion and Drug-Drug Interactions. Clin. Pharmacol. Ther. 2018, 1, 148–157. [Google Scholar] [CrossRef]
- Wanat, K. Biological barriers, and the influence of protein binding on the passage of drugs across them. Mol. Biol. Rep. 2020, 47, 3221–3231. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Hoque, M.J.; Hossain, B. Molecular Docking, Pharmacokinetic, and DFT Calculation of Naproxen and its Degradants. Biomed. J. Sci. Tech. Res. 2018, 9, 7360–7365. [Google Scholar]
- Salihović, M.; Pazalja, M.; Špirtović Halilović, S.; Veljović, E.; Mahmutović-Dizdarević, I.; Roca, S.; Novakovic, I.; Tivunovic, S. Synthesis, characterization, antimicrobial activity and DFT study of some novel Schiff bases. J. Mol. Struct. 2021, 1241, 130670. [Google Scholar] [CrossRef]
- Venkatesh, G.; Govindaraju, M.; Vennila, P.; Kamal, C. Molecular structure, vibrational spectral assignments (FT-IR and FT-RAMAN), NMR, NBO, HOMO-LUMO and NLO properties of 2-nitroacetophenone based on DFT calculations. J. Theor. Comput. Chem. 2016, 15, 165007–165020. [Google Scholar] [CrossRef]
- BIOVIA. Dassault System, BIOVIA Discovery Studio Package, 2019; Dassault System: San Diego, CA, USA, 2019. [Google Scholar]
- Desmond Molecular Dynamics System; D. E. Shaw Research. Maestro-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2021. [Google Scholar]
- Kevin, J.; Bowers, E.C.; Huafeng, X.; Ron, O.D.; Michael, P.; Eastwood, B.A.; Gregersen, J.L.; Klepeis, I.K.; Mark, A.M.; Federico, D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, New York, NY, USA, 11–17 November 2006. [Google Scholar]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Radwan, H.A.; Ahmad, I.; Othman, I.M.M.; Gad-Elkareem, M.A.M.; Patel, H.; Aouadi, K.; Snoussi, M.; Kadri, A. Design, synthesis, in vitro anticancer and antimicrobial evaluation, SAR analysis, molecular docking and dynamic simulation of new pyrazoles, triazoles and pyridazines based isoxazole. J. Mol. Struct. 2022, 1264, 133312. [Google Scholar] [CrossRef]
- Shinoda, W.; Mikami, M. Rigid-body dynamics in the isothermal-isobaric ensemble: A test on the accuracy and computational efficiency. J. Comput. Chem. 2003, 24, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, K.; Grauffel, C.; Lim, C. How Molecular Size Impacts RMSD Applications in Molecular Dynamics Simulations. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef]
- Joshi, T.; Joshi, T.; Sharma, P.; Chandra, S.; Pande, V. Molecular docking and molecular dynamics simulation approach to screen natural compounds for inhibition of Xanthomonas oryzae pv. Oryzae by targeting peptide deformylase. J. Biomol. Struct. Dyn. 2021, 39, 823–840. [Google Scholar] [CrossRef]
- Sulpizi, M.; Folkers, G.; Rothlisberger, U.; Carloni, P.; Scapozza, L. Applications of density functional theory-based methods in medicinal chemistry. Quant. Struct. Relatsh. 2002, 21, 173–181. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacophore Model | No of Features | Feature Set | Selectivity Score | ROC | ||||
|---|---|---|---|---|---|---|---|---|
| CDK4/6 | Aromatase | CDK4/6 | Aromatase | CDK4/6 | Aromatase | CDK4/6 | Aromatase | |
| Pharmacophore_01 | 6 | 5 | AAAADHarom | AHHHH | 1.993 | 4.764 | 0.775 | 0.555 |
| Pharmacophore_02 | 6 | 5 | AAAADH | AHHHH | 1.993 | 4.764 | 0.909 | 0.546 |
| Pharmacophore_03 | 6 | 4 | AAAADH | AHHH | 1.993 | 6.347 | 0.790 | 0.600 |
| Pharmacophore_04 | 6 | 4 | AAAADHarom | AHHH | 1.993 | 6.347 | 0.643 | 0.570 |
| Pharmacophore_05 | 6 | 4 | AAAADH | AHHH | 1.993 | 6.347 | 0.903 | 0.679 |
| Pharmacophore_06 | 6 | 4 | AAAADH | AHHH | 1.993 | 6.347 | 0.654 | 0.704 |
| Pharmacophore_07 | 6 | 4 | AAAADHarom | AHHH | 1.993 | 6.347 | 0.792 | 0.593 |
| Pharmacophore_08 | 6 | 4 | AAAADH | AHHH | 1.993 | 6.347 | 0.904 | 0.582 |
| Pharmacophore_09 | 6 | 4 | AAAADH | AHHH | 1.993 | 6.347 | 0.774 | 0.815 |
| Pharmacophore_10 | 6 | 4 | AAAADHarom | AHHH | 1.993 | 6.347 | 0.625 | 0.715 |
| Sl.no | Compound | Compound ID | Fit Value | |
|---|---|---|---|---|
| Pharmacophore_02 Model | Pharmacophore_09 Model | |||
| 1 | Candidate 1 | ZINC77287236 | 0.748 | 1.887 |
| 2 | Candidate 2 | CHEMBL517070 | 3.538 | 3.064 |
| 3 | Candidate 3 | 51000421 | 2.642 | 3.515 |
| 4 | Candidate 4 | ZINC36924410 | 3.060 | 3.383 |
| Sl.no | Compound | 5L2S | 3S7S | ||
|---|---|---|---|---|---|
| CDOCKER Energy (kcal/mol) | CDOCKER Interaction Energy (kcal/mol) | CDOCKER Energy (kcal/mol) | CDOCKER Interaction Energy (kcal/mol) | ||
| 1 | Candidate1 | −13.04 | −56.37 | 3.892 | −56.27 |
| 2 | Candidate2 | −18.21 | −46.60 | −25.29 | −52.21 |
| 3 | Candidate3 | −31.82 | −35.28 | −28.57 | −32.98 |
| 4 | Candidate4 | −30.69 | −32.07 | −30.35 | −31.16 |
| 5 | Abemaciclib | −27.35 | −58.02 | - | - |
| 6 | Exemestane | - | - | −22.01 | −39.15 |
| Compound | Absorption | Solubility | a BBB | b PPB | c CYP2D6 | Hepatotoxicity | AMES Mutagenicity | AlogP98 | d PSA-2D |
|---|---|---|---|---|---|---|---|---|---|
| Candidate 1 | 0 | 3 | 4 | True | False | True | NM | 1.034 | 123.8 |
| Candidate 2 | 3 | 2 | 4 | False | False | True | NM | 0.434 | 185.8 |
| Candidate 3 | 0 | 3 | 3 | True | False | True | NM | 0.552 | 82.35 |
| Candidate 4 | 0 | 3 | 3 | True | False | True | NM | 0.175 | 82.35 |
| Sl.no | Compound | HUMO | LUMO | Energy Gap (ΔE) |
|---|---|---|---|---|
| 1 | Candidate 1 | −0.128 | −0.075 | 0.053 |
| 2 | Candidate 2 | −0.231 | −0.094 | 0.137 |
| 3 | Candidate 3 | −0.192 | −0.056 | 0.136 |
| 4 | Candidate 4 | −0.187 | −0.061 | 0.126 |
| ADMET Descriptor | Level | Description |
|---|---|---|
| Absorption | 0 | Good absorption |
| 1 | Moderate absorption | |
| 2 | Low absorption | |
| 3 | Very low absorption | |
| Solubility | 0 | Extremely low |
| 1 | Very low, but possible | |
| 2 | Yes, low | |
| 3 | Yes, good | |
| 4 | Yes, optimal | |
| 5 | No, too soluble | |
| 6 | Unknown | |
| BBB a | 0 | Very high |
| 1 | High | |
| 2 | Medium | |
| 3 | Low | |
| 4 | Undefined | |
| 5 | Unknown | |
| PPB b | 0 (False) | Binding is <90% |
| 1 (True) | Binding is ≥90% | |
| CYP2D6 c | 0 (False) | Non-inhibitor |
| 1 (True) | Inhibitor | |
| Hepatotoxicity | 0 (False) | Non-hepatotoxic |
| 1 (True) | Toxic | |
| AMES Mutagenicity | 0 (False) | Non-mutagen |
| 1 (True) | Mutagen |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adon, T.; Shanmugarajan, D.; Ather, H.; Ansari, S.M.A.; Hani, U.; Madhunapantula, S.V.; Honnavalli, Y.K. Virtual Screening for Identification of Dual Inhibitors against CDK4/6 and Aromatase Enzyme. Molecules 2023, 28, 2490. https://doi.org/10.3390/molecules28062490
Adon T, Shanmugarajan D, Ather H, Ansari SMA, Hani U, Madhunapantula SV, Honnavalli YK. Virtual Screening for Identification of Dual Inhibitors against CDK4/6 and Aromatase Enzyme. Molecules. 2023; 28(6):2490. https://doi.org/10.3390/molecules28062490
Chicago/Turabian StyleAdon, Tenzin, Dhivya Shanmugarajan, Hissana Ather, Shaik Mohammad Asif Ansari, Umme Hani, SubbaRao V. Madhunapantula, and Yogish Kumar Honnavalli. 2023. "Virtual Screening for Identification of Dual Inhibitors against CDK4/6 and Aromatase Enzyme" Molecules 28, no. 6: 2490. https://doi.org/10.3390/molecules28062490
APA StyleAdon, T., Shanmugarajan, D., Ather, H., Ansari, S. M. A., Hani, U., Madhunapantula, S. V., & Honnavalli, Y. K. (2023). Virtual Screening for Identification of Dual Inhibitors against CDK4/6 and Aromatase Enzyme. Molecules, 28(6), 2490. https://doi.org/10.3390/molecules28062490