
Identification of Some Glutamic Acid Derivatives with Biological Potential by Computational Methods
 , , and
, , and 
Abstract
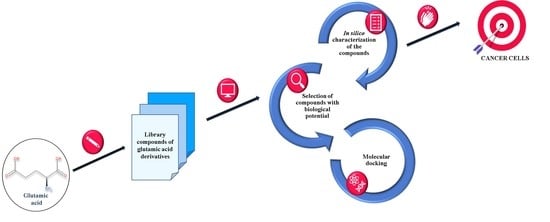
1. Introduction
2. Results and Discussion
2.1. Algorithm for Designing Glutamic Acid Derivatives and Studies Underlying Their Development
2.2. The Elimination of Reactive and Toxic Compounds
- Step 1. In the first stage, compounds belonging to at least two toxicity classes are eliminated, as the risk of them causing severe adverse reactions is high.
- Step 2. This step involves the removal of compounds that do not follow Lipinski and Veber’s rules, and which have a CNS MPO score less than 4, as well as compounds with low solubility and/or an inhibitory effect on cytochrome P450 and/or gp-P enzymes.
- Step 3. Compounds with medium toxicity, which fall into Class III (Cramer rules) and are positive for at least one toxicity criterion, are eliminated if the overall drug-likeness score does not exceed 0.90.
- Step 4. Compounds that have violated all Ghose’s rule criteria (four out of four) and belong to Cramer class III or II or overlap with the violation of at least one Muegge rule are eliminated.
- Step 5. Compounds that have violated at least three Ghose criteria and at least two Muegge rules and belong to Cramer class III are eliminated.
- Step 6. Removal of Cramer Class III compounds that violate at least one Ghose and Muegge rule, having an SA score below 2.
- Step 7. Elimination of Class III Cramer compounds that violate at least one Ghose and Muegge rule, regardless of the SA score achieved.
- Step 8. Removal of compounds that violate at least one Ghose and Muegge rule with a low GI absorption value.
- Step 9. Compounds that violate at least one Ghose and Muegge rule with an SA score below 4, regardless of Cramer toxicity class, are eliminated.
- Step 10. Elimination of Cramer Class III compounds that violate at least two Muegge criteria and have an SA score below 3 and/or overall drug-likeness score below 0.5.
2.3. Characterisation of the “Lead” Compounds
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| ADME | Absorption, Distribution, Metabolism and Excretion |
| HAA | Heavy aromatic atoms |
| BBB | Blood–Brain Barrier |
| BD | Bioavailability |
| CLC-Pred | Cell Line Cytotoxicity Predictor |
| CNS MPO | Central Nervous System Multiparameter Optimisation |
| ESOL | Estimating Aqueous Solubility Directly from Molecular Structure |
| GS | Glutamine synthetase |
| GSH | Glutathione |
| GPCR | G-protein coupled receptor |
| GPL | General Public License |
| GUSAR | General Unrestricted Structure–Activity Relationships |
| HA | Heavy atoms |
| HLB | Hydrophilic Lipophilic Balance |
| Ki | Inhibition constant |
| LD50 | Lethal dose 50 |
| MR | Molar refractivity |
| PDB | Protein Data Bank |
| P-gp | P-glycoprotein |
| pI | Isoelectric point |
| QSAR | Quantitative Structure–Activity Relationships |
| QSPR | Quantitative Structure–Property Relationships |
| RB | Rotatable bonds |
| SA | Synthetic accessibility score |
| SLC25 | The solute carrier family 25 |
| SN1 | Nucleophilic substitution type 1 |
| SN2 | Aliphatic nucleophilic substitution type 2 |
| SOMP | Site of Metabolism Prediction |
| TPSA | Total polar surface area |
| TTC | Threshold of Toxicological Concern |
References
- Ageing as an Important Risk Factor for Cancer|Anticancer Research. Available online: https://ar.iiarjournals.org/content/36/10/5009.long (accessed on 25 January 2023).
- Solary, E.; Abou-Zeid, N.; Calvo, F. Ageing and Cancer: A Research Gap to Fill. Mol. Oncol. 2022, 16, 3220–3237. [Google Scholar] [CrossRef]
- Risk Factors: Age—NCI. Available online: https://www.cancer.gov/about-cancer/causes-prevention/risk/age (accessed on 25 January 2023).
- American Cancer Society. Global Cancer Facts & Figures, 4th ed.; American Cancer Society: Atlanta, GA, USA, 2018; Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/global-cancer-facts-and-figures/global-cancer-facts-and-figures-4th-edition.pdf (accessed on 13 January 2023).
- Lieu, E.L.; Nguyen, T.; Rhyne, S.; Kim, J. Amino Acids in Cancer. Exp. Mol. Med. 2020, 52, 15–30. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Li, J.; Chen, H.; He, X.; Zhang, H.; Liang, H.; Lu, J. Biosynthetic Energy Cost for Amino Acids Decreases in Cancer Evolution. Nat. Commun. 2018, 9, 4124. [Google Scholar] [CrossRef]
- Nguyen, T.; Kirsch, B.J.; Asaka, R.; Nabi, K.; Quinones, A.; Tan, J.; Antonio, M.J.; Camelo, F.; Li, T.; Nguyen, S.; et al. Uncovering the Role of N-Acetyl-Aspartyl-Glutamate as a Glutamate Reservoir in Cancer. Cell Rep. 2019, 27, 491–501.e6. [Google Scholar] [CrossRef]
- Palmieri, E.M.; Menga, A.; Martín-Pérez, R.; Quinto, A.; Riera-Domingo, C.; De Tullio, G.; Hooper, D.C.; Lamers, W.H.; Ghesquière, B.; McVicar, D.W.; et al. Pharmacologic or Genetic Targeting of Glutamine Synthetase Skews Macrophages toward an M1-like Phenotype and Inhibits Tumor Metastasis. Cell Rep. 2017, 20, 1654–1666. [Google Scholar] [CrossRef]
- Jiang, J.; Srivastava, S.; Zhang, J. Starve Cancer Cells of Glutamine: Break the Spell or Make a Hungry Monster? Cancers 2019, 11, 804. [Google Scholar] [CrossRef]
- Natarajan, S.K.; Venneti, S. Glutamine Metabolism in Brain Tumors. Cancers 2019, 11, 1628. [Google Scholar] [CrossRef]
- Boysen, G.; Jamshidi-Parsian, A.; Davis, M.A.; Siegel, E.R.; Simecka, C.M.; Kore, R.A.; Dings, R.P.M.; Griffin, R.J. Glutaminase Inhibitor CB-839 Increases Radiation Sensitivity of Lung Tumor Cells and Human Lung Tumor Xenografts in Mice. Int. J. Radiat. Biol. 2019, 95, 436–442. [Google Scholar] [CrossRef]
- Walker, M.C.; van der Donk, W.A. The Many Roles of Glutamate in Metabolism. J. Ind. Microbiol. Biotechnol. 2016, 43, 419–430. [Google Scholar] [CrossRef]
- Jin, H.; Wang, S.; Zaal, E.A.; Wang, C.; Wu, H.; Bosma, A.; Jochems, F.; Isima, N.; Jin, G.; Lieftink, C.; et al. A Powerful Drug Combination Strategy Targeting Glutamine Addiction for the Treatment of Human Liver Cancer. eLife 2020, 9, e56749. [Google Scholar] [CrossRef]
- Bertero, T.; Oldham, W.M.; Grasset, E.M.; Bourget, I.; Boulter, E.; Pisano, S.; Hofman, P.; Bellvert, F.; Meneguzzi, G.; Bulavin, D.V.; et al. Tumor-Stroma Mechanics Coordinate Amino Acid Availability to Sustain Tumor Growth and Malignancy. Cell Metab. 2019, 29, 124–140.e10. [Google Scholar] [CrossRef]
- Bott, A.; Maimouni, S.; Zong, W.-X. The Pleiotropic Effects of Glutamine Metabolism in Cancer. Cancers 2019, 11, 770. [Google Scholar] [CrossRef]
- Edwards, D.N.; Ngwa, V.M.; Raybuck, A.L.; Wang, S.; Hwang, Y.; Kim, L.C.; Cho, S.H.; Paik, Y.; Wang, Q.; Zhang, S.; et al. Selective Glutamine Metabolism Inhibition in Tumor Cells Improves Antitumor T Lymphocyte Activity in Triple-Negative Breast Cancer. J. Clin. Investig. 2021, 131, e140100. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, F.; Fan, N.; Zhou, C.; Li, D.; Macvicar, T.; Dong, Q.; Bruns, C.J.; Zhao, Y. Targeting Glutaminolysis: New Perspectives to Understand Cancer Development and Novel Strategies for Potential Target Therapies. Front. Oncol. 2020, 10, 589508. [Google Scholar] [CrossRef]
- Shen, Y.-A.; Chen, C.-L.; Huang, Y.-H.; Evans, E.E.; Cheng, C.-C.; Chuang, Y.-J.; Zhang, C.; Le, A. Inhibition of Glutaminolysis in Combination with Other Therapies to Improve Cancer Treatment. Curr. Opin. Chem. Biol. 2021, 62, 64–81. [Google Scholar] [CrossRef]
- Wu, C.; Chen, L.; Jin, S.; Li, H. Glutaminase Inhibitors: A Patent Review. Expert Opin. Ther. Pat. 2018, 28, 823–835. [Google Scholar] [CrossRef]
- Hanaford, A.R.; Alt, J.; Rais, R.; Wang, S.Z.; Kaur, H.; Thorek, D.L.J.; Eberhart, C.G.; Slusher, B.S.; Martin, A.M.; Raabe, E.H. Orally Bioavailable Glutamine Antagonist Prodrug JHU-083 Penetrates Mouse Brain and Suppresses the Growth of MYC-Driven Medulloblastoma. Transl. Oncol. 2019, 12, 1314–1322. [Google Scholar] [CrossRef]
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J.; Coffey, R.J.; Johnson, M.O.; Rathmell, J.C.; et al. Pharmacological Blockade of ASCT2-Dependent Glutamine Transport Leads to Antitumor Efficacy in Preclinical Models. Nat. Med. 2018, 24, 194–202. [Google Scholar] [CrossRef]
- Iovino, L.; Tremblay, M.E.; Civiero, L. Glutamate-Induced Excitotoxicity in Parkinson’s Disease: The Role of Glial Cells. J. Pharmacol. Sci. 2020, 144, 151–164. [Google Scholar] [CrossRef]
- Crupi, R.; Impellizzeri, D.; Cuzzocrea, S. Role of Metabotropic Glutamate Receptors in Neurological Disorders. Front. Mol. Neurosci. 2019, 12, 20. [Google Scholar] [CrossRef]
- Montero Domínguez, P.A.; Garduño Juárez, R.; Mares Sámano, S. Computer Simulation Studies of a Kainate (GluK1) Receptor with Two Glutamate Analogues. CyS 2019, 23, 313–324. [Google Scholar] [CrossRef]
- Esslinger, C.S.; Cybulski, K.A.; Rhoderick, J.F. Nγ-Aryl Glutamine Analogues as Probes of the ASCT2 Neutral Amino Acid Transporter Binding Site. Bioorg. Med. Chem. 2005, 13, 1111–1118. [Google Scholar] [CrossRef]
- Dubey, S.K.; Sharma, A.K.; Narain, U.; Misra, K.; Pati, U. Design, Synthesis and Characterization of Some Bioactive Conjugates of Curcumin with Glycine, Glutamic Acid, Valine and Demethylenated Piperic Acid and Study of Their Antimicrobial and Antiproliferative Properties. Eur. J. Med. Chem. 2008, 43, 1837–1846. [Google Scholar] [CrossRef]
- Pagire, S.H.; Lee, E.; Pagire, H.S.; Bae, E.J.; Ryu, S.J.; Lee, D.; Kim, M.H.; Kim, G.R.; Hwang, K.-S.; Ahn, S.; et al. Design, Synthesis and Biological Evaluation of Glutamic Acid Derivatives as Anti-Oxidant and Anti-Inflammatory Agents. Bioorg. Med. Chem. Lett. 2018, 28, 529–532. [Google Scholar] [CrossRef]
- Harvey, R.A. Lippincott Ilustrated Reviews-Pharmacology, Ediţia a 5-a, Ed. Medicală Callisto, Bucureşti, 2013, 496, 499–501, 507–508, 5th ed.; Medical Callisto: Bucharest, Romania, 2013. [Google Scholar]
- Swinney, D.C. Annual Reports in Medicinal Chemistry, Chapter 18, Molecular Mechanism of Action (MMoA) in Drug Discovery; Academic Press: Cambridge, MA, USA, 2011; Volume 46. [Google Scholar]
- Semenyuta, I.; Kovalishyn, V.; Tanchuk, V.; Pilyo, S.; Zyabrev, V.; Blagodatnyy, V.; Trokhimenko, O.; Brovarets, V.; Metelytsia, L. 1,3-Oxazole Derivatives as Potential Anticancer Agents: Computer Modeling and Experimental Study. Comput. Biol. Chem. 2016, 65, 8–15. [Google Scholar] [CrossRef]
- Sanderson, B.J.S.; Shield, A.J. Mutagenic Damage to Mammalian Cells by Therapeutic Alkylating Agents. Mutat. Res./Fundam. Mol. Mech. Mutagen. 1996, 355, 41–57. [Google Scholar] [CrossRef]
- Puyo, S.; Montaudon, D.; Pourquier, P. From Old Alkylating Agents to New Minor Groove Binders. Crit. Rev. Oncol./Hematol. 2014, 89, 43–61. [Google Scholar] [CrossRef]
- Nikolova, T.; Roos, W.P.; Krämer, O.H.; Strik, H.M.; Kaina, B. Chloroethylating Nitrosoureas in Cancer Therapy: DNA Damage, Repair and Cell Death Signaling. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2017, 1868, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, S.; Yamamoto, K. Mechanism of Site-Specific DNA Damage Induced by Methylhydrazines in the Presence of Copper(II) or Manganese(III). Biochemistry 1991, 30, 3069–3075. [Google Scholar] [CrossRef]
- Moreira-Pais, A.; Ferreira, R.; Gil da Costa, R. Platinum-Induced Muscle Wasting in Cancer Chemotherapy: Mechanisms and Potential Targets for Therapeutic Intervention. Life Sci. 2018, 208, 1–9. [Google Scholar] [CrossRef]
- Anh, D.T.; Hai, P.-T.; Huong, L.-T.-T.; Park, E.J.; Jun, H.W.; Kang, J.S.; Kwon, J.-H.; Dung, D.T.M.; Anh, V.T.; Hue, V.T.M.; et al. Exploration of Certain 1,3-Oxazole- and 1,3-Thiazole-Based Hydroxamic Acids as Histone Deacetylase Inhibitors and Antitumor Agents. Bioorg. Chem. 2020, 101, 103988. [Google Scholar] [CrossRef] [PubMed]
- de Ruijter, A.J.M.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B.P. Histone Deacetylases (HDACs): Characterization of the Classical HDAC Family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC Family: What Are the Cancer Relevant Targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef]
- Licciardi, P.; Ververis, K.; Hiong, A.; Karagiannis, T. Histone Deacetylase Inhibitors (HDACIs): Multitargeted Anticancer Agents. Biol. Targets Ther. 2013, 7, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Fritzer-Szekeres, M.; Grusch, M.; Luxbacher, C.; Horvath, S.; Krupitza, G.; Elford, H.L.; Szekeres, T. Trimidox, an Inhibitor of Ribonucleotide Reductase, Induces Apoptosis and Activates Caspases in HL-60 Promyelocytic Leukemia Cells. Exp. Hematol. 2000, 28, 924–930. [Google Scholar] [CrossRef]
- Aye, Y.; Li, M.; Long, M.J.C.; Weiss, R.S. Ribonucleotide Reductase and Cancer: Biological Mechanisms and Targeted Therapies. Oncogene 2015, 34, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Liu, X.; Zhu, L.; Yen, Y. Targeting Ribonucleotide Reductase for Cancer Therapy. Expert Opin. Ther. Targets 2013, 17, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Suárez, I.; Bodega, G.; Fernández, B. Glutamine Synthetase in Brain: Effect of Ammonia. Neurochem. Int. 2002, 41, 123–142. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Marin-Valencia, I.; Yang, C.; Mashimo, T.; Cho, S.; Baek, H.; Yang, X.-L.; Rajagopalan, K.N.; Maddie, M.; Vemireddy, V.; Zhao, Z.; et al. Analysis of Tumor Metabolism Reveals Mitochondrial Glucose Oxidation in Genetically Diverse Human Glioblastomas in the Mouse Brain In Vivo. Cell Metab. 2012, 15, 827–837. [Google Scholar] [CrossRef]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.Ø.; Weinstock, A.; Wagner, A.; et al. Glutamine Synthetase Activity Fuels Nucleotide Biosynthesis and Supports Growth of Glutamine-Restricted Glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Wasfy, R.E.; Eldeen, A.A.S. Roles of Combined Glypican-3 and Glutamine Synthetase in Differential Diagnosis of Hepatocellular Lesions. Asian Pac. J. Cancer Prev. 2015, 16, 4769–4775. [Google Scholar] [CrossRef] [PubMed]
- Kung, H.-N.; Marks, J.R.; Chi, J.-T. Glutamine Synthetase Is a Genetic Determinant of Cell Type–Specific Glutamine Independence in Breast Epithelia. PLoS Genet. 2011, 7, e1002229. [Google Scholar] [CrossRef]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Wang, Y.; Zhang, Z.; Lu, J.; Wu, Z.; Shan, Q.; Sun, C.; Wu, D.; Li, M.; Sheng, N.; et al. High Expression of Glutamate-ammonia Ligase Is Associated with Unfavorable Prognosis in Patients with Ovarian Cancer. J. Cell. Biochem. 2018, 119, 6008–6015. [Google Scholar] [CrossRef]
- Furusawa, A.; Miyamoto, M.; Takano, M.; Tsuda, H.; Song, Y.S.; Aoki, D.; Miyasaka, N.; Inazawa, J.; Inoue, J. Ovarian Cancer Therapeutic Potential of Glutamine Depletion Based on GS Expression. Carcinogenesis 2018, 39, 758–766. [Google Scholar] [CrossRef]
- Yuneva, M.O.; Fan, T.W.M.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Matés, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The Metabolic Profile of Tumors Depends on Both the Responsible Genetic Lesion and Tissue Type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef]
- Bott, A.J.; Shen, J.; Tonelli, C.; Zhan, L.; Sivaram, N.; Jiang, Y.-P.; Yu, X.; Bhatt, V.; Chiles, E.; Zhong, H.; et al. Glutamine Anabolism Plays a Critical Role in Pancreatic Cancer by Coupling Carbon and Nitrogen Metabolism. Cell Rep. 2019, 29, 1287–1298.e6. [Google Scholar] [CrossRef]
- Yang, L.; Achreja, A.; Yeung, T.-L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; Kunji, E.R.S. The SLC25 Mitochondrial Carrier Family: Structure and Mechanism. Trends Biochem. Sci. 2020, 45, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Dituri, F.; Convertini, P.; Santarsiero, A.; Palmieri, F.; Todisco, S.; Mancarella, S.; Giannelli, G.; Iacobazzi, V. Epigenetic Upregulation and Functional Role of the Mitochondrial Aspartate/Glutamate Carrier Isoform 1 in Hepatocellular Carcinoma. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2019, 1865, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Qian, Y.; Li, X.; Xu, J.; Kang, W.; Tong, J.H.; To, K.-F.; Jin, Y.; Li, W.; Chen, H.; et al. SLC25A22 Promotes Proliferation and Survival of Colorectal Cancer Cells With KRAS Mutations and Xenograft Tumor Progression in Mice via Intracellular Synthesis of Aspartate. Gastroenterology 2016, 151, 945–960.e6. [Google Scholar] [CrossRef]
- Chen, M.-W.; Wu, X. SLC25A22 Promotes Proliferation and Metastasis of Osteosarcoma Cells via the PTEN Signaling Pathway. Technol. Cancer Res. Treat. 2018, 17, 153303381881114. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Liang, H.; Fu, X.; Wu, P.; Wang, C.; Chen, H.; Zheng, B.; Zhang, J.; Hu, S.; Zeng, R.; et al. SLC25A22 Promotes Proliferation and Metastasis by Activating MAPK/ERK Pathway in Gallbladder Cancer. Cancer Cell Int. 2019, 19, 33. [Google Scholar] [CrossRef]
- Ali, I.; Wani, W.A.; Haque, A.; Saleem, K. Glutamic Acid and Its Derivatives: Candidates for Rational Design of Anticancer Drugs. Future Med. Chem. 2013, 5, 961–978. [Google Scholar] [CrossRef]
- Valente, S.; Mai, A. Small-Molecule Inhibitors of Histone Deacetylase for the Treatment of Cancer and Non-Cancer Diseases: A Patent Review (2011–2013). Expert Opin. Ther. Pat. 2014, 24, 401–415. [Google Scholar] [CrossRef]
- Nudelman, A.; Levovich, I.; Cutts, S.M.; Phillips, D.R.; Rephaeli, A. The Role of Intracellularly Released Formaldehyde and Butyric Acid in the Anticancer Activity of Acyloxyalkyl Esters. J. Med. Chem. 2005, 48, 1042–1054. [Google Scholar] [CrossRef]
- Nudelman, A.; Ruse, M.; Aviram, A.; Rabizadeh, E.; Shaklai, M.; Zimrah, Y.; Rephaeli, A. Novel Anticancer Prodrugs of Butyric Acid. 2. J. Med. Chem. 1992, 35, 687–694. [Google Scholar] [CrossRef]
- Zwergel, C.; Valente, S.; Jacob, C.; Mai, A. Emerging Approaches for Histone Deacetylase Inhibitor Drug Discovery. Expert Opin. Drug Discov. 2015, 10, 599–613. [Google Scholar] [CrossRef]
- Berlicki, L. Inhibitors of Glutamine Synthetase and Their Potential Application in Medicine. Mini Rev. Med. Chem. 2008, 8, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Berlicki, Ł.; Kafarski, P. The Use of Molecular Modelling for Comparison of Three Possible Modes of Action of Herbicidally Active Derivatives of Aminomethylenebisphosphonic Acid. Pestic. Biochem. Physiol. 2002, 73, 94–103. [Google Scholar] [CrossRef]
- Sinden, S.L.; Durbin, R.D. Glutamine Synthetase Inhibition: Possible Mode of Action of Wildfire Toxin from Pseudomonas Tabaci. Nature 1968, 219, 379–380. [Google Scholar] [CrossRef] [PubMed]
- Durbin, R.D.; Uchytil, T.F. The Role of Zinc in Regulating Tabtoxin Production. Experientia 1983, 41, 136–137. [Google Scholar] [CrossRef]
- Langston-Unkefer, P.L.; Macy, P.A.; Durbin, R.D. Inactivation of Glutamine Synthetase by Tabtoxinine-Beta-Lactam: Effects of Substrates and PH. Plant Physiol. 1984, 76, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Lücking, U. Neglected Sulfur(vi) Pharmacophores in Drug Discovery: Exploration of Novel Chemical Space by the Interplay of Drug Design and Method Development. Org. Chem. Front. 2019, 6, 1319–1324. [Google Scholar] [CrossRef]
- Roy, M.; Liang, L.; Xiao, X.; Feng, P.; Ye, M.; Liu, J. Lycorine: A Prospective Natural Lead for Anticancer Drug Discovery. Biomed. Pharmacother. 2018, 107, 615–624. [Google Scholar] [CrossRef]
- Kasai, T.; Nishitoba, T.; Shiroshita, Y.; Sakamura, S. Several 4-Substituted Glutamic Acid Derivatives and Small Peptides in Some Liliaceae Plants. Agric. Biol. Chem. 1984, 48, 2271–2278. [Google Scholar] [CrossRef]
- Alkadi, H.; Khubeiz, M.J.; Jbeily, R. Colchicine: A Review on Chemical Structure and Clinical Usage. Infect. Disord.-Drug Targets 2018, 18, 105–121. [Google Scholar] [CrossRef]
- Dudkiewicz, I.; Brosh, T.; Perelman, M.; Salai, M. Colchicine Inhibits Fracture Union and Reduces Bone Strength—In Vivo Study. J. Orthop. Res. 2005, 23, 877–881. [Google Scholar] [CrossRef]
- Zhang, T.; Chen, W.; Jiang, X.; Liu, L.; Wei, K.; Du, H.; Wang, H.; Li, J. Anticancer Effects and Underlying Mechanism of Colchicine on Human Gastric Cancer Cell Lines in Vitro and in Vivo. Biosci. Rep. 2019, 39, BSR20181802. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.K.; Mukherjee, D.; Maji, A.K.; Rai, S.; Heinrich, M. The Sacred Lotus (Nelumbo nucifera)—Phytochemical and Therapeutic Profile. J. Pharm. Pharmacol. 2009, 61, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-M.; Kao, C.-L.; Wu, H.-M.; Li, W.-J.; Huang, C.-T.; Li, H.-T.; Chen, C.-Y. Antioxidant and Anticancer Aporphine Alkaloids from the Leaves of Nelumbo nucifera Gaertn. cv. Rosa-Plena. Molecules 2014, 19, 17829–17838. [Google Scholar] [CrossRef] [PubMed]
- Chaichompoo, W.; Chokchaisiri, R.; Apiratikul, N.; Chairoungdua, A.; Yingyongnarongkul, B.; Chunglok, W.; Tocharus, C.; Suksamrarn, A. Cytotoxic Alkaloids against Human Colon Adenocarcinoma Cell Line (HT-29) from the Seed Embryos of Nelumbo nucifera. Med. Chem. Res. 2018, 27, 939–943. [Google Scholar] [CrossRef]
- Asokan, S.M.; Mariappan, R.; Muthusamy, S.; Velmurugan, B.K. Pharmacological Benefits of Neferine—A Comprehensive Review. Life Sci. 2018, 199, 60–70. [Google Scholar] [CrossRef]
- Kadioglu, O.; Law, B.Y.K.; Mok, S.W.F.; Xu, S.-W.; Efferth, T.; Wong, V.K.W. Mode of Action Analyses of Neferine, a Bisbenzylisoquinoline Alkaloid of Lotus (Nelumbo nucifera) against Multidrug-Resistant Tumor Cells. Front. Pharmacol. 2017, 8, 238. [Google Scholar] [CrossRef]
- Law, B.Y.K.; Michelangeli, F.; Qu, Y.Q.; Xu, S.-W.; Han, Y.; Mok, S.W.F.; Dias, I.R.D.S.R.; Javed, M.-H.; Chan, W.-K.; Xue, W.-W.; et al. Neferine Induces Autophagy-Dependent Cell Death in Apoptosis-Resistant Cancers via Ryanodine Receptor and Ca2+-Dependent Mechanism. Sci. Rep. 2019, 9, 20034. [Google Scholar] [CrossRef]
- Poornima, P.; Kumar, V.B.; Weng, C.F.; Padma, V.V. Doxorubicin Induced Apoptosis Was Potentiated by Neferine in Human Lung Adenocarcima, A549 Cells. Food Chem. Toxicol. 2014, 68, 87–98. [Google Scholar] [CrossRef]
- Sivalingam, K.S.; Paramasivan, P.; Weng, C.F.; Viswanadha, V.P. Neferine Potentiates the Antitumor Effect of Cisplatin in Human Lung Adenocarcinoma Cells Via a Mitochondria-Mediated Apoptosis Pathway. J. Cell. Biochem. 2017, 118, 2865–2876. [Google Scholar] [CrossRef]
- Manogaran, P.; Beeraka, N.M.; Huang, C.-Y.; Vijaya Padma, V. Neferine and Isoliensinine Enhance ‘Intracellular Uptake of Cisplatin’ and Induce ‘ROS-Mediated Apoptosis’ in Colorectal Cancer Cells—A Comparative Study. Food Chem. Toxicol. 2019, 132, 110652. [Google Scholar] [CrossRef]
- Liu, W.; Yi, D.-D.; Guo, J.-L.; Xiang, Z.-X.; Deng, L.-F.; He, L. Nuciferine, Extracted from Nelumbo Nucifera Gaertn, Inhibits Tumor-Promoting Effect of Nicotine Involving Wnt/β-Catenin Signaling in Non-Small Cell Lung Cancer. J. Ethnopharmacol. 2015, 165, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Lamoral-Theys, D.; Decaestecker, C.; Mathieu, V.; Dubois, J.; Kornienko, A.; Kiss, R.; Evidente, A.; Pottier, L. Lycorine and Its Derivatives for Anticancer Drug Design. Mini-Rev. Med. Chem. 2010, 10, 41–50. [Google Scholar] [CrossRef] [PubMed]
- McNulty, J.; Nair, J.J.; Bastida, J.; Pandey, S.; Griffin, C. Structure-Activity Studies on the Lycorine Pharmacophore: A Potent Inducer of Apoptosis in Human Leukemia Cells. Phytochemistry 2009, 70, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Hao, B.; Shen, S.-F.; Zhao, Q.-J. Cytotoxic and Antimalarial Amaryllidaceae Alkaloids from the Bulbs of Lycoris Radiata. Molecules 2013, 18, 2458–2468. [Google Scholar] [CrossRef] [PubMed]
- AquaSol: Predict Aqueous Solublity of Small Molecules Using UG-RNN Ensembles (Version 2.0). Available online: http://cdb.ics.uci.edu/cgibin/tools/AquaSolWeb.py (accessed on 21 January 2023).
- Chemicalize—Instant Cheminformatics Solutions. Available online: https://chemicalize.com (accessed on 20 January 2023).
- SwissADME. Available online: http://www.swissadme.ch/ (accessed on 17 January 2023).
- Toxtree—Toxtree—Toxic Hazard Estimation by Decision Tree Approach (Version 3.1.0.1851). Available online: https://toxtree.sourceforge.net/ (accessed on 26 January 2023).
- Molecular Properties Prediction—Osiris Property Explorer. Available online: https://www.organic-chemistry.org/prog/peo/ (accessed on 13 February 2023).
- Kerns, E.H.; Di, L. Drug-like Properties: Concepts, Structure Design and Methods: From ADME to Toxicity Optimization; Academic Press: Amsterdam, The Netherlands; Boston, MA, USA, 2008; ISBN 978-0-12-369520-8. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Drug Likeness Tool (DruLiTo 1) (Version 1.0.). Available online: http://www.niper.gov.in/pi_dev_tools/DruLiToWeb/DruLiTo_index.html (accessed on 1 February 2023).
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Khan, T.; Lawrence, A.J.; Azad, I.; Raza, S.; Joshi, S.; Khan, A.R. Computational Drug Designing and Prediction Of Important Parameters Using in Silico Methods- A Review. Curr. Comput.-Aided Drug Des. 2019, 15, 384–397. [Google Scholar] [CrossRef]
- Adki, K.M.; Murugesan, S.; Kulkarni, Y.A. In Silico and In Vivo Toxicological Evaluation of Paeonol. Chem. Biodivers. 2020, 17, e2000422. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Teague, S.J.; Davis, A.M.; Leeson, P.D.; Oprea, T. The Design of Leadlike Combinatorial Libraries. Angew. Chem. Int. Ed. 1999, 38, 3743–3748. [Google Scholar] [CrossRef]
- Marvinsketch (Version 5.0). Available online: https://chemaxon.com/marvin (accessed on 19 January 2023).
- Baba, Y.; Isomura, T.; Kashima, H. Wisdom of Crowds for Synthetic Accessibility Evaluation. J. Mol. Graph. Model. 2018, 80, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Baber, J.; Feher, M. Predicting Synthetic Accessibility: Application in Drug Discovery and Development. Mini-Rev. Med. Chem. 2004, 4, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Schuffenhauer, A. Estimation of Synthetic Accessibility Score of Drug-like Molecules Based on Molecular Complexity and Fragment Contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Fukunishi, Y.; Kurosawa, T.; Mikami, Y.; Nakamura, H. Prediction of Synthetic Accessibility Based on Commercially Available Compound Databases. J. Chem. Inf. Model. 2014, 54, 3259–3267. [Google Scholar] [CrossRef]
- Patlewicz, G.; Jeliazkova, N.; Safford, R.J.; Worth, A.P.; Aleksiev, B. An Evaluation of the Implementation of the Cramer Classification Scheme in the Toxtree Software. SAR QSAR Environ. Res. 2008, 19, 495–524. [Google Scholar] [CrossRef]
- Dolan, D.G.; Naumann, B.D.; Sargent, E.V.; Maier, A.; Dourson, M. Application of the Threshold of Toxicological Concern Concept to Pharmaceutical Manufacturing Operations. Regul. Toxicol. Pharmacol. 2005, 43, 1–9. [Google Scholar] [CrossRef]
- Renwick, A. Structure-Based Thresholds of Toxicological Concern—Guidance for Application to Substances Present at Low Levels in the Diet. Toxicol. Appl. Pharmacol. 2005, 207, 585–591. [Google Scholar] [CrossRef]
- Verhaar, H.J.M.; van Leeuwen, C.J.; Hermens, J.L.M. Classifying Environmental Pollutants. Chemosphere 1992, 25, 471–491. [Google Scholar] [CrossRef]
- Verhaar, H.J.M.; Solbé, J.; Speksnijder, J.; van Leeuwen, C.J.; Hermens, J.L.M. Classifying Environmental Pollutants: Part 3. External Validation of the Classification System. Chemosphere 2000, 40, 875–883. [Google Scholar] [CrossRef] [PubMed]
- von der Ohe, P.C.; Kühne, R.; Ebert, R.-U.; Altenburger, R.; Liess, M.; Schüürmann, G. Structural AlertsA New Classification Model to Discriminate Excess Toxicity from Narcotic Effect Levels of Organic Compounds in the Acute Daphnid Assay. Chem. Res. Toxicol. 2005, 18, 536–555. [Google Scholar] [CrossRef] [PubMed]
- Ellison, C.M.; Madden, J.C.; Cronin, M.T.D.; Enoch, S.J. Investigation of the Verhaar Scheme for Predicting Acute Aquatic Toxicity: Improving Predictions Obtained from Toxtree Ver. 2.6. Chemosphere 2015, 139, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Enoch, S.J.; Hewitt, M.; Cronin, M.T.D.; Azam, S.; Madden, J.C. Classification of Chemicals According to Mechanism of Aquatic Toxicity: An Evaluation of the Implementation of the Verhaar Scheme in Toxtree. Chemosphere 2008, 73, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Krishna, K.A.; Goel, S.; Krishna, G. SAR Genotoxicity and Tumorigenicity Predictions for 2-MI and 4-MI Using Multiple SAR Software. Toxicol. Mech. Methods 2014, 24, 284–293. [Google Scholar] [CrossRef]
- Zeiger, E. The Test That Changed the World: The Ames Test and the Regulation of Chemicals. Mutat. Res./Genet. Toxicol. Environ. Mutagen. 2019, 841, 43–48. [Google Scholar] [CrossRef]
- McCann, J.; Ames, B.N. Detection of Carcinogens as Mutagens in the Salmonella/Microsome Test: Assay of 300 Chemicals: Discussion. Proc. Natl. Acad. Sci. USA 1976, 73, 950–954. [Google Scholar] [CrossRef]
- Clayden, J.; Greeves, N.; Warren, S. Organic Chemistry; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Saini, N.; Sterling, J.F.; Sakofsky, C.J.; Giacobone, C.K.; Klimczak, L.J.; Burkholder, A.B.; Malc, E.P.; Mieczkowski, P.A.; Gordenin, D.A. Mutation Signatures Specific to DNA Alkylating Agents in Yeast and Cancers. Nucleic Acids Res. 2020, 48, 3692–3707. [Google Scholar] [CrossRef]
- Uddin, N.; Rashid, F.; Ali, S.; Tirmizi, S.A.; Ahmad, I.; Zaib, S.; Zubair, M.; Diaconescu, P.L.; Tahir, M.N.; Iqbal, J.; et al. Synthesis, Characterization, and Anticancer Activity of Schiff Bases. J. Biomol. Struct. Dyn. 2020, 38, 3246–3259. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef]
- Szakács, G.; Váradi, A.; Özvegy-Laczka, C.; Sarkadi, B. The Role of ABC Transporters in Drug Absorption, Distribution, Metabolism, Excretion and Toxicity (ADME–Tox). Drug Discov. Today 2008, 13, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Martin, Y.C. A Bioavailability Score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef] [PubMed]
- Olsen, L.; Montefiori, M.; Tran, K.P.; Jørgensen, F.S. SMARTCyp 3.0: Enhanced Cytochrome P450 Site-of-Metabolism Prediction Server. Bioinformatics 2019, 35, 3174–3175. [Google Scholar] [CrossRef] [PubMed]
- Montanari, F.; Ecker, G.F. Prediction of Drug–ABC-Transporter Interaction—Recent Advances and Future Challenges. Adv. Drug Deliv. Rev. 2015, 86, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Kirchmair, J.; Göller, A.H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I.D.; Glen, R.C.; Schneider, G. Predicting Drug Metabolism: Experiment and/or Computation? Nat. Rev. Drug Discov. 2015, 14, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Veith, H.; Southall, N.; Huang, R.; James, T.; Fayne, D.; Artemenko, N.; Shen, M.; Inglese, J.; Austin, C.P.; Lloyd, D.G.; et al. Comprehensive Characterization of Cytochrome P450 Isozyme Selectivity across Chemical Libraries. Nat. Biotechnol. 2009, 27, 1050–1055. [Google Scholar] [CrossRef]
- SMARTCyp (Version 3.0). Available online: https://smartcyp.sund.ku.dk/mol_to_som (accessed on 3 February 2023).
- Prediction of Site of Metabolism (Version 2.0). Available online: http://www.way2drug.com/SOMP/ (accessed on 4 February 2023).
- SwissTargetPrediction. Available online: http://www.swisstargetprediction.ch/ (accessed on 8 February 2023).
- Calculation of Molecular Properties and Bioactivity Score. Available online: https://www.molinspiration.com/cgi-bin/properties (accessed on 5 February 2023).
- Lagunin, A.A.; Dubovskaja, V.I.; Rudik, A.V.; Pogodin, P.V.; Druzhilovskiy, D.S.; Gloriozova, T.A.; Filimonov, D.A.; Sastry, N.G.; Poroikov, V.V. CLC-Pred: A Freely Available Web-Service for in Silico Prediction of Human Cell Line Cytotoxicity for Drug-like Compounds. PLoS ONE 2018, 13, e0191838. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for Rigid and Symmetric Docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef]
- Way2Drug—Main (Version 2.0). Available online: http://www.way2drug.com/PASSOnline/index.php (accessed on 10 February 2023).
- Lagunin, A.; Zakharov, A.; Filimonov, D.; Poroikov, V. QSAR Modelling of Rat Acute Toxicity on the Basis of PASS Prediction. Mol. Inf. 2011, 30, 241–250. [Google Scholar] [CrossRef]
- Glutamine Synthetase. Available online: https://chem.libretexts.org/Courses/Saint_Marys_College_Notre_Dame_IN/CHEM_342%3A_Bio-inorganic_Chemistry/Readings/Metals_in_Biological_Systems_(Saint_Mary’s_College)/Glutamine_Synthetase (accessed on 16 January 2023).
- Eisenberg, D.; Gill, H.S.; Pfluegl, G.M.U.; Rotstein, S.H. Structure–Function Relationships of Glutamine Synthetases. Biochim. Biophys. Acta (BBA)—Protein Struct. Mol. Enzymol. 2000, 1477, 122–145. [Google Scholar] [CrossRef]
- Liaw, S.-H.; Kuo, I.; Eisenberg, D. Discovery of the Ammonium Substrate Site on Glutamine Synthetase, A Third Cation Binding Site. Protein Sci. 1995, 4, 2358–2365. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Cryst. Sect. D Biol. Cryst. 2002, 58, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Liaw, S.-H.; Jun, G.; Eisenberg, D. Interactions of Nucleotides with Fully Unadenylylated Glutamine Synthetase from Salmonella Typhimurium. Biochemistry 1994, 33, 11184–11188. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera?A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Huang, C.C.; Ferrin, T.E. Tools for Integrated Sequence-Structure Analysis with UCSF Chimera. BMC Bioinform. 2006, 7, 339. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a Protein-Small Molecule Docking Web Service Based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef]
- Bentley, H.R.; McDermott, E.E.; Moran, T.; Pace, J.; Whitehead, J.K. Action of Nitrogen Trichloride on Certain Proteins I. Isolation and Identification of the Toxic Factor. Proc. R. Soc. Lond. B 1950, 137, 402–417. [Google Scholar] [CrossRef]
- Zoete, V.; Daina, A.; Bovigny, C.; Michielin, O. SwissSimilarity: A Web Tool for Low to Ultra High Throughput Ligand-Based Virtual Screening. J. Chem. Inf. Model. 2016, 56, 1399–1404. [Google Scholar] [CrossRef]
- Gill, H.S.; Eisenberg, D. The Crystal Structure of Phosphinothricin in the Active Site of Glutamine Synthetase Illuminates the Mechanism of Enzymatic Inhibition. Biochemistry 2001, 40, 1903–1912. [Google Scholar] [CrossRef]
- SwissDock—The Online Docking Web Server of the Swiss Institute of Bioinformatics—TargetProteins. Available online: http://www.swissdock.ch/target_proteins (accessed on 11 February 2023).
- Grasso, G.; Di Gregorio, A.; Mavkov, B.; Piga, D.; Labate, G.F.D.; Danani, A.; Deriu, M.A. Fragmented Blind Docking: A Novel Protein–Ligand Binding Prediction Protocol. J. Biomol. Struct. Dyn. 2021, 40, 1–10. [Google Scholar] [CrossRef]
- Bitencourt-Ferreira, G.; de Azevedo, W.F. Docking with SwissDock. In Docking Screens for Drug Discovery; de Azevedo, W.F., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 2053, pp. 189–202. ISBN 978-1-4939-9751-0. [Google Scholar]
- Schneidman-Duhovny, D.; Inbar, Y.; Polak, V.; Shatsky, M.; Halperin, I.; Benyamini, H.; Barzilai, A.; Dror, O.; Haspel, N.; Nussinov, R.; et al. Taking Geometry to Its Edge: Fast Unbound Rigid (and Hinge-Bent) Docking. Proteins 2003, 52, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Duhovny, D.; Nussinov, R.; Wolfson, H.J. Efficient Unbound Docking of Rigid Molecules. In Algorithms in Bioinformatics; Guigó, R., Gusfield, D., Eds.; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2002; Volume 2452, pp. 185–200. ISBN 978-3-540-44211-0. [Google Scholar]
- AutoDock Vina (Version 1.1.2). Available online: https://vina.scripps.edu/ (accessed on 12 February 2023).
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern Web App for 3D Visualization and Analysis of Large Biomolecular Structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, A.; Zoete, V.; Michielin, O. Blind Docking of 260 Protein-Ligand Complexes with EADock 2.0. J. Comput. Chem. 2009, 30, 2021–2030. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. EADock: Docking of Small Molecules into Protein Active Sites with a Multiobjective Evolutionary Optimization. Proteins 2007, 67, 1010–1025. [Google Scholar] [CrossRef]
- Choudhary, D.K.; Kumar, M.; Prasad, R.; Kumar, V. Silico Approach for Sustainable Agriculture; Springer: Singapore, 2018; ISBN 9789811303463. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Fang, X.; Lu, Y.; Yang, C.-Y.; Wang, S. The PDBbind Database: Methodologies and Updates. J. Med. Chem. 2005, 48, 4111–4119. [Google Scholar] [CrossRef]
- Jain, A.N. Scoring Noncovalent Protein-Ligand Interactions: A Continuous Differentiable Function Tuned to Compute Binding Affinities. J. Comput.-Aided Mol. Des. 1996, 10, 427–440. [Google Scholar] [CrossRef]
- Bursulaya, B.D.; Totrov, M.; Abagyan, R.; Brooks, C.L., III. Comparative Study of Several Algorithms for Flexible Ligand Docking. J. Comput.-Aided Mol. Des. 2003, 17, 755–763. [Google Scholar] [CrossRef]
- López-Camacho, E.; García-Godoy, M.J.; García-Nieto, J.; Nebro, A.J.; Aldana-Montes, J.F. A New Multi-Objective Approach for Molecular Docking Based on RMSD and Binding Energy. In Algorithms for Computational Biology; Botón-Fernández, M., Martín-Vide, C., Santander-Jiménez, S., Vega-Rodríguez, M.A., Eds.; Lecture Notes in Computer Science; Springer International Publishing: Cham, Switzerland, 2016; Volume 9702, pp. 65–77. ISBN 978-3-319-38826-7. [Google Scholar]
- Moldovean, S.N.; Timaru, D.-G.; Chiş, V. All-Atom Molecular Dynamics Investigations on the Interactions between D2 Subunit Dopamine Receptors and Three 11C-Labeled Radiopharmaceutical Ligands. Int. J. Mol. Sci. 2022, 23, 2005. [Google Scholar] [CrossRef]
- Systèmes, D. BIOVIA Draw for Academics (Version 21.1.0.2363). Available online: https://discover.3ds.com/biovia-draw-academic (accessed on 12 January 2023).
- Delaney, J.S. ESOL: Estimating Aqueous Solubility Directly from Molecular Structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Ali, J.; Camilleri, P.; Brown, M.B.; Hutt, A.J.; Kirton, S.B. Revisiting the General Solubility Equation: In Silico Prediction of Aqueous Solubility Incorporating the Effect of Topographical Polar Surface Area. J. Chem. Inf. Model. 2012, 52, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Arnott, J.A.; Planey, S.L. The Influence of Lipophilicity in Drug Discovery and Design. Expert Opin. Drug Discov. 2012, 7, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Mannhold, R.; Poda, G.I.; Ostermann, C.; Tetko, I.V. Calculation of Molecular Lipophilicity: State-of-the-Art and Comparison of LogP Methods on More than 96,000 Compounds. J. Pharm. Sci. 2009, 98, 861–893. [Google Scholar] [CrossRef]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of Octanol−Water Partition Coefficients by Guiding an Additive Model with Knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef] [PubMed]
- Wildman, S.A.; Crippen, G.M. Prediction of Physicochemical Parameters by Atomic Contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Moriguchi, I.; Hirono, S.; Liu, Q.; Nakagome, I.; Matsushita, Y. Simple Method of Calculating Octanol/Water Partition Coefficient. Chem. Pharm. Bull. 1992, 40, 127–130. [Google Scholar] [CrossRef]
- Moriguchi, I.; Hirono, S.; Nakagome, I.; Hirano, H. Comparison of Reliability of Log P Values for Drugs Calculated by Several Methods. Chem. Pharm. Bull. 1994, 42, 976–978. [Google Scholar] [CrossRef]
- Gusar—Create QSAR/QSPR Models on the Basis of the Appropriate Training Sets. Available online: http://www.way2drug.com/gusar/acutoxpredict.html (accessed on 29 January 2023).
- PatchDock Server: An Automatic Server for Molecular Docking (Version Beta 1.3). Available online: https://bioinfo3d.cs.tau.ac.il/PatchDock/php.php (accessed on 11 February 2023).
- Download UCSF Chimera (Version 1.15). Available online: https://www.cgl.ucsf.edu/chimera/download.html (accessed on 12 February 2023).
- SwissSimilarity. Available online: http://www.swisssimilarity.ch/ (accessed on 13 February 2023).
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 17 January 2023).
- ProteinsPlus. Available online: https://proteins.plus/ (accessed on 20 April 2023).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Group | Subgroup | |||
|---|---|---|---|---|---|
| 1 | Compounds resulting from reactions at the carboxyl group | A | Esters | a | - |
| B | Amides | a | - | ||
| C | Acid chlorides | a | - | ||
| D | Anhydrides | a | - | ||
| 2 | Compounds resulting from reactions at the amino group | A | Amides | a | - |
| B | Alkylated glutamic acid derivatives | a | Azotyperites | ||
| C | Alcohols resulting from diazotisation | a | - | ||
| 3 | Heterocyclic derivatives | A | Thiazole derivatives | a | Simple |
| b | With cyclic anhydride | ||||
| B | 1,3 Oxazole derivatives | a | Simple | ||
| b | With cyclic anhydride | ||||
| 4 | Other derivatives and their potential mechanism of action | A | Alkylating agents | a | Azotyperites |
| b | Nitrosoureas | ||||
| c | Methylhydrazine | ||||
| d | Alkyl sulphonates | ||||
| e | Platinum complexes | ||||
| B | Histone deacetylase inhibitors | a | - | ||
| C | Ribonucleotide reductase inhibitors | a | Hydroxyurea derivatives | ||
| b | Cyclic compounds (based on the structure of Trimidox) | ||||
| D | Inhibitors of glutamate synthetase and/or SLC25A mitochondrial transporters | a | Methionine–sulfoximine analogues | ||
| b | Phosphinothricin analogues | ||||
| c | Biphosphonates | ||||
| d | Various inhibitors starting from different structures:
| ||||
| 5 | Natural substances with proven anti-cancer effects (Table S11; Supplementary Materials) conjugated with glutamic acid molecules | A | Colchicine derivatives | a | Spindle inhibitors |
| B | Neferine derivatives | a | - | ||
| C | 7-Hydroxycinuciferine derivatives | a | - | ||
| D | Lycorine derivatives | a | - | ||
| E | Derivatives of 5,6-dehydrolycorine | a | - | ||
| Drug-Likeness Rules | ||||
|---|---|---|---|---|
| Lipinski | Ghose | Veber | Egan | Muegge |
| MW ≤ 500 Da MlogP ≤ 4.15 N or O ≤ 10 NH or OH ≤ 5 | 160 ≤ MW ≤ 480 Da −0.4 ≤ WlogP ≤ 5.6 40 ≤ MR ≤ 130 20 ≤ atoms ≤ 70 | RB ≤ 10 TPSA ≤ 140 | WlogP ≤ 5.88 TPSA ≤ 131.6 | 200 ≤ MW ≤ 600 Da −2 ≤ XlogP ≤ 5 TPSA ≤ 150 No. of rings ≤ 7 No. of carbon atoms > 4 No. of heteroatoms > 1 No. of RB ≤ 15 H-bond acceptors ≤ 10 H-bond donors ≤ 5 |
| No. | ID Code | Chemical Structure | Geometric Isomers | Isomerism | Conformations | ||
|---|---|---|---|---|---|---|---|
| Asymmetric Atoms | Chiral Centres | Tautomers | Stereoisomers | Emin (kcal/mol) | |||
| 1 | 1Aa7 |  | 1 | 1 | 4 | 2 | 10.66 |
| 2 | 1Aa8 |  | 1 | 1 | 2 | 2 | 10.79 |
| 3 | 2Ba2 |  | 1 | 1 | 18 | 2 | 12.11 |
| 4 | 2Ba5 |  | 1 | 1 | 4 | 2 | 26.45 |
| 5 | 2Ba6 |  | 1 | 1 | 4 | 2 | 25.4 |
| 6 | 3Aa3 |  | 1 | 1 | 16 | 2 | 31.59 |
| 7 | 3Aa5 |  | 1 | 1 | 16 | 2 | 31.56 |
| 8 | 4Da11 |  | 3 | 3 | 46 | 8 | 73.63 |
| 9 | 4Db6 |  | 2 | 2 | 30 | 4 | 62.49 |
| No. | ID Code | Primary Sites of Metabolism | Secondary Sites of Metabolism | Tertiary Sites of Metabolism | Quaternary Sites of Metabolism |
|---|---|---|---|---|---|
| 1 | 1Aa7 | N-dealkylation | Amine hydroxylation | Aliphatic hydroxylation | O-dealkylation |
| 2 | 1Aa8 | N-dealkylation | Amine hydroxylation | Aliphatic hydroxylation | O-dealkylation |
| 3 | 2Ba2 | N-dealkylation | N-oxidation | N-dealkylation | Aliphatic hydroxylation |
| 4 | 2Ba5 | N-dealkylation | N-dealkylation | N-oxidation | Aliphatic hydroxylation |
| 5 | 2Ba6 | N-dealkylation | None | N-dealkylation | N-oxidation |
| 6 | 3Aa3 | N-dealkylation | Amine hydroxylation | Aromatic hydroxylation | Aliphatic hydroxylation |
| 7 | 3Aa5 | N-dealkylation | Amine hydroxylation | Aliphatic hydroxylation | Aromatic hydroxylation |
| 8 | 4Da11 | N-dealkylation | None | Amine hydroxylation | Aliphatic hydroxylation |
| 9 | 4Db6 | N-dealkylation | None | Amine hydroxylation | Aliphatic hydroxylation |
| No. | ID Code | 3A4 | 2D6 | 2C9 | |||
|---|---|---|---|---|---|---|---|
| The Most Reactive Atom | Score | The Most Reactive Atom | Score | The Most Reactive Atom | Score | ||
| 1 | 1Aa7 | C8 | 34.7 | C1 | 93.7 | C2 | 86.3 |
| 2 | 1Aa8 | C6 | 36.7 | C1 | 107.1 | C2 | 86.4 |
| 3 | 2Ba2 | C1 | 30.9 | C1 | 85.8 | C1 | 50.7 |
| 4 | 2Ba5 | C2 | 33.2 | C2 | 93.7 | C2 | 57.8 |
| 5 | 2Ba6 | C4 | 32.2 | C4 | 92.7 | C4 | 56.8 |
| 6 | 3Aa3 | C2 | 34.7 | C10 | 74.6 | C10 | 67.9 |
| 7 | 3Aa5 | C2 | 36.8 | C13 | 88 | C13 | 67.9 |
| 8 | 4Da11 | C7 | 35.3 | N3 | 85.8 | N3 | 64.1 |
| 9 | 4Db6 | C7 | 34.5 | C7 | 100.1 | C7 | 75.2 |
| No. | ID Code | GPCR Ligand | Ion Channel Modulator | Kinase Inhibitor | Nuclear Receptor ligand | Protease Inhibitor | Enzyme Inhibitor |
|---|---|---|---|---|---|---|---|
| 1 | 1Aa7 | −0.42 | 0.17 | −1.01 | −0.86 | −0.2 | 0.09 |
| 2 | 1Aa8 | −0.41 | 0.13 | −1 | −0.84 | −0.21 | 0.09 |
| 3 | 2Ba2 | −0.11 | 0.18 | −1.01 | −0.9 | −0.28 | 0.19 |
| 4 | 2Ba5 | −0.02 | 0.11 | −0.89 | −0.55 | −0.15 | 0.14 |
| 5 | 2Ba6 | −0.02 | 0.15 | −0.96 | −0.68 | −0.24 | 0.11 |
| 6 | 3Aa3 | −0.1 | 0.23 * | −0.38 | −0.94 | 0.27 * | 0.7 ** |
| 7 | 3Aa5 | −0.21 | 0 | −0.28 | −0.67 | 0.33 * | 0.43 * |
| 8 | 4Da11 | −0.69 | −0.26 | −1.36 | −0.93 | −0.44 | 0.27 * |
| 9 | 4Db6 | 0.12 | 0.83 ** | −0.65 | −1.06 | 0.67 ** | 0.87 ** |
| No. | ID Code | Target | Common Name | Uniprot ID | Target Class | Probability |
|---|---|---|---|---|---|---|
| 1 | 1Aa7 | Kynureninase | KYNU | Q16719 | Enzyme | 0.141787 |
| 2 | 1Aa8 | Aminopeptidase A | ENPEP | Q07075 | Protease | 0.125076 |
| Kynurenine 3-monooxygenase | KMO | O15229 | Oxidoreductase | 0.125076 | ||
| Glutamate receptor ionotropic, AMPA 1 | GRIA1 | P42261 | Ligand-gated ion channel | 0.125076 | ||
| 3 | 2Ba2 | Metabotropic glutamate receptor 3 | GRM3 | Q14832 | Family C G protein-coupled receptor | 0.150098 |
| Metabotropic glutamate receptor 6 | GRM6 | O15303 | Family C G protein-coupled receptor | 0.150098 | ||
| Metabotropic glutamate receptor 2 | GRM2 | Q14416 | Family C G protein-coupled receptor | 0.150098 | ||
| 4 | 2Ba5 | Glutamate receptor ionotropic kainate 1 | GRIK1 | P39086 | Ligand-gated ion channel | 0.031227 |
| Glutamate receptor ionotropic AMPA 1 | GRIA1 | P42261 | Ligand-gated ion channel | 0.031227 | ||
| Adenosine A3 receptor | ADORA3 | P0DMS8 | Family A G protein-coupled receptor | 0.031227 | ||
| 5 | 2Ba6 | Glutamate receptor ionotropic kainate 1 | GRIK1 | P39086 | Ligand-gated ion channel | 0.08057 |
| Glutamate receptor ionotropic AMPA 1 | GRIA1 | P42261 | Ligand-gated ion channel | 0.08057 | ||
| Adenosine A3 receptor | ADORA3 | P0DMS8 | Family A G protein-coupled receptor | 0.08057 | ||
| 6 | 3Aa3 | Kynurenine 3-monooxygenase | KMO | O15229 | Oxidoreductase | 0.04147 |
| Kynureninase | KYNU | Q16719 | Enzyme | 0.04147 | ||
| 7 | 3Aa5 | Caspase-3 | CASP3 | P42574 | Protease | 0.031227 |
| Lysine-specific demethylase 2A | KDM2A | Q9Y2K7 | Eraser | 0.031227 | ||
| Histone lysine demethylase PHF8 | PHF8 | Q9UPP1 | Eraser | 0.031227 | ||
| 8 | 4Da11 | Fructose-1,6-bisphosphatase | FBP1 | P09467 | Enzyme | 0.053518 |
| G protein-coupled receptor 44 | PTGDR2 | Q9Y5Y4 | Family A G protein-coupled receptor | 0.053518 | ||
| 9 | 4Db6 | Glutamate receptor ionotropic kainate 1 | GRIK1 | P39086 | Ligand-gated ion channel | 0.08057 |
| Glutamate receptor ionotropic AMPA 1 | GRIA1 | P42261 | Ligand-gated ion channel | 0.08057 | ||
| Glutamate receptor ionotropic kainate 5 | GRIK5 | Q16478 | Ligand-gated ion channel | 0.08057 |
| No. | ID Code | Pa | Pi | Cell Line | Cell Line (Full Name) | Tissue | Tumour Type |
|---|---|---|---|---|---|---|---|
| 1 | 1Aa7 | 0.694 | 0.004 | NCI-H1299 | Non-small cell lung carcinoma | Lung | Carcinoma |
| 2 | 1Aa8 | 0.541 | 0.004 | NCI-H1299 | Non-small cell lung carcinoma | Lung | Carcinoma |
| 3 | 2Ba2 | 0.458 | 0.023 | MDA-MB-453 | Breast adenocarcinoma | Breast | Adenocarcinoma |
| 4 | 2Ba5 | 0.451 | 0.008 | Jurkat | Acute leukaemia T-cells | Blood | Leukaemia |
| 5 | 2Ba6 | 0.438 | 0.039 | MDA-MB-453 | Breast adenocarcinoma | Breast | Adenocarcinoma |
| 6 | 3Aa3 | 0.717 | 0.004 | DMS-114 | Lung carcinoma | Lung | Carcinoma |
| 0.527 | 0.005 | RKO | Colon carcinoma | Colon | Carcinoma | ||
| 7 | 3Aa5 | 0.728 | 0.004 | DMS-114 | Lung carcinoma | Lung | Carcinoma |
| 0.543 | 0.005 | RKO | Colon carcinoma | Colon | Carcinoma | ||
| 8 | 4Da11 | 0.595 | 0.01 | DMS-114 | Lung carcinoma | Lung | Carcinoma |
| 9 | 4Db6 | 0.657 | 0.012 | HCT-116 | Colon carcinoma | Colon | Carcinoma |
| No. | ID Code | Mechanism of Action | Toxic Effects | ||||
|---|---|---|---|---|---|---|---|
| Pa | Pi | Activity | Pa | Pi | Activity | ||
| 1 | 1Aa7 | 0.965 | 0.001 | Arginine 2-monooxygenase inhibitor | 0.982 | 0.004 | Respiratory toxicity |
| 0.962 | 0.002 | Protein-disulphide reductase (GSH) inhibitor | 0.952 | 0.004 | Euphoria | ||
| 0.961 | 0.002 | Methylenetetrahydrofolate reductase (NADPH) inhibitor | 0.904 | 0.008 | Weakness | ||
| 0.952 | 0.001 | Levanase inhibitor | 0.892 | 0.007 | Pure red cell aplasia | ||
| 0.951 | 0.002 | Acylcarnitine hydrolase inhibitor | 0.885 | 0.007 | Muscle weakness | ||
| 2 | 1Aa8 | 0.969 | 0.001 | Protein-disulphide reductase (GSH) inhibitor | 0.976 | 0.005 | Toxic, respiratory failure |
| 0.961 | 0.002 | Methylenetetrahydrofolate reductase (NADPH) inhibitor | 0.932 | 0.005 | Euphoria | ||
| 0.956 | 0.001 | Arginine 2-monooxygenase inhibitor | 0.900 | 0.004 | Apnoea | ||
| 0.953 | 0.001 | Levanase inhibitor | 0.900 | 0.008 | Weakness | ||
| 0.949 | 0.001 | Aspartate kinase inhibitor | 0.871 | 0.009 | Neurotoxic | ||
| 3 | 2Ba2 | 0.956 | 0.001 | Methylamine-glutamate N-methyltransferase inhibitor | 0.925 | 0.006 | Euphoria |
| 0.952 | 0.002 | Acylcarnitine hydrolase inhibitor | 0.919 | 0.015 | Toxic, respiratory failure | ||
| 0.915 | 0.003 | NADPH peroxidase inhibitor | 0.870 | 0.011 | Pure red cell aplasia | ||
| 0.906 | 0.004 | Anaphylatoxin receptor antagonist | 0.860 | 0.003 | Skin irritation, corrosive | ||
| 0.906 | 0.006 | Methylenetetrahydrofolate reductase (NADPH) inhibitor | 0.851 | 0.019 | Shivering | ||
| 4 | 2Ba5 | 0.945 | 0.002 | Acylcarnitine hydrolase inhibitor | 0.958 | 0.009 | Toxic, respiratory failure |
| 0.941 | 0.001 | Methylamine-glutamate N-methyltransferase inhibitor | 0.935 | 0.005 | Euphoria | ||
| 0.920 | 0.002 | Dimethylargininase inhibitor | 0.920 | 0.004 | Pure red cell aplasia | ||
| 0.909 | 0.002 | Aminoacylase inhibitor | 0.901 | 0.006 | Shivering | ||
| 0.905 | 0.004 | Gluconate 2-dehydrogenase (acceptor) inhibitor | 0.888 | 0.003 | Skin irritation, corrosive | ||
| 5 | 2Ba6 | 0.946 | 0.002 | Acylcarnitine hydrolase inhibitor | 0.962 | 0.009 | Toxic, respiratory failure |
| 0.943 | 0.001 | Methylamine-glutamate N-methyltransferase inhibitor | 0.954 | 0.004 | Euphoria | ||
| 0.900 | 0.001 | Flavin-containing monooxygenase inhibitor | 0.918 | 0.002 | Skin irritation, corrosive | ||
| 0.889 | 0.007 | Phobic disorders treatment | 0.894 | 0.007 | Pure red cell aplasia | ||
| 0.884 | 0.003 | Dimethylargininase inhibitor | 0.876 | 0.006 | Postural (orthostatic) hypotension | ||
| 6 | 3Aa3 | 0.866 | 0.003 | Glutamine-phenylpyruvate transaminase inhibitor | 0.766 | 0.020 | Respiratory failure |
| 0.853 | 0.005 | Monodehydroascorbate reductase (NADH) inhibitor | 0.731 | 0.035 | Ulcer, aphthous | ||
| 0.800 | 0.009 | Arginine 2-monooxygenase inhibitor | 0.686 | 0.009 | Anaemia, sideroblastic | ||
| 0.803 | 0.018 | Methylenetetrahydrofolate reductase (NADPH) inhibitor | 0.707 | 0.041 | Pure red cell aplasia | ||
| 0.793 | 0.013 | NADPH peroxidase inhibitor | 0.667 | 0.033 | Stomatitis | ||
| 7 | 3Aa5 | 0.797 | 0.014 | Acylcarnitine hydrolase inhibitor | 0.764 | 0.022 | Stomatitis |
| 0.787 | 0.005 | Glutamine-phenylpyruvate transaminase inhibitor | 0.719 | 0.026 | Respiratory failure | ||
| 0.794 | 0.019 | Methylenetetrahydrofolate reductase (NADPH) inhibitor | 0.702 | 0.020 | Asthma | ||
| 0.734 | 0.002 | Pyrimidine-deoxynucleoside 2′-dioxygenase inhibitor | 0.689 | 0.015 | Respiratory impairment | ||
| 0.736 | 0.021 | NADPH peroxidase inhibitor | 0.655 | 0.020 | Haematuria | ||
| 8 | 4Da11 | 0.932 | 0.004 | Angiogenesis inhibitor | 0.496 | 0.074 | Haematemesis |
| 0.930 | 0.004 | Anti-inflammatory | 0.439 | 0.038 | Thrombocytopoiesis inhibitor | ||
| 0.923 | 0.004 | Glutamate-5-semialdehyde dehydrogenase inhibitor | 0.436 | 0.078 | Interstitial nephritis | ||
| 0.869 | 0.001 | CDK1/cyclin B inhibitor | 0.463 | 0.109 | Occult bleeding | ||
| 0.865 | 0.002 | Macular degeneration treatment | 0.450 | 0.105 | Nephritis | ||
| 9 | 4Db6 | 0.957 | 0.002 | Glutamate-5-semialdehyde dehydrogenase inhibitor | 0.651 | 0.023 | Ototoxicity |
| 0.952 | 0.000 | Sphingosine 1-phosphate receptor 5 antagonist | 0.520 | 0.069 | Bronchoconstriction | ||
| 0.793 | 0.002 | GABA C receptor antagonist | 0.343 | 0.158 | Sneezing | ||
| 0.782 | 0.003 | Ornithine cyclodeaminase inhibitor | 0.280 | 0.097 | Demyelination | ||
| 0.701 | 0.003 | Bone formation stimulant | 0.319 | 0.159 | Fibrosis, interstitial | ||
| No. | ID Code | Rat IP LD50 (mg/kg) | Rat IV LD50 (mg/kg) | Rat Oral LD50 (mg/kg) | Rat SC LD50 (mg/kg) |
|---|---|---|---|---|---|
| 1 | 1Aa7 | 2593.000 in AD | 1256.000 in AD | 5859.000 in AD | 6254.000 in AD |
| 2 | 1Aa8 | 3059.000 in AD | 1268.000 in AD | 4228.000 in AD | 4014.000 in AD |
| 3 | 2Ba2 | 1069.000 in AD | 1017.000 in AD | 1978.000 in AD | 1027.000 in AD |
| 4 | 2Ba5 | 436.000 in AD | 865.000 in AD | 1861.000 in AD | 1026.000 out of AD |
| 5 | 2Ba6 | 375.200 in AD | 613.100 in AD | 1198.000 in AD | 505.500 in AD |
| 6 | 3Aa3 | 418.900 in AD | 643.600 in AD | 3172.000 in AD | 2290.000 in AD |
| 7 | 3Aa5 | 585.600 in AD | 464.800 in AD | 2623.000 out of AD | 1923.000 in AD |
| 8 | 4Da11 | 551.700 out of AD | 580.800 in AD | 3362.000 in AD | 298.500 in AD |
| 9 | 4Db6 | 298.100 out of AD | 180.400 in AD | 1456.000 out of AD | 76.460 in AD |
| No. | ID Code | Rat IP LD50 Classification | Rat IV LD50 Classification | Rat Oral LD50 Classification | Rat SC LD50 Classification |
|---|---|---|---|---|---|
| 1 | 1Aa7 | Non-Toxic in AD | Non-Toxic in AD | Non-Toxic in AD | Non-Toxic in AD |
| 2 | 1Aa8 | Non-Toxic in AD | Non-Toxic in AD | Class 5 in AD | Non-Toxic in AD |
| 3 | 2Ba2 | Class 5 in AD | Non-Toxic in AD | Class 4 in AD | Class 5 in AD |
| 4 | 2Ba5 | Class 4 in AD | Non-Toxic in AD | Class 4 in AD | Class 5 out of AD |
| 5 | 2Ba6 | Class 4 in AD | Class 5 in AD | Class 4 in AD | Class 4 in AD |
| 6 | 3Aa3 | Class 4 in AD | Class 5 in AD | Class 5 in AD | Class 5 in AD |
| 7 | 3Aa5 | Class 5 in AD | Class 5 in AD | Class 5 out of AD | Class 5 in AD |
| 8 | 4Da11 | Class 5 out of AD | Class 5 in AD | Class 5 in AD | Class 4 in AD |
| 9 | 4Db6 | Class 4 out of AD | Class 4 in AD | Class 4 out of AD | Class 3 in AD |
| Step | Time (fs) | Potential Energy (J) | Kinetic Energy (J) |
|---|---|---|---|
| 0 | 0.0 | 341.630730 | 86.279577 |
| 100 | 0.1 | 335.578989 | 92.292502 |
| 200 | 0.2 | 351.037095 | 77.385248 |
| 300 | 0.3 | 333.800802 | 94.478719 |
| 400 | 0.4 | 353.520040 | 74.902008 |
| 500 | 0.5 | 363.225563 | 65.233746 |
| 600 | 0.6 | 365.055252 | 63.321001 |
| 700 | 0.7 | 359.244207 | 69.127817 |
| 800 | 0.8 | 333.010201 | 95.326086 |
| 900 | 0.9 | 336.650457 | 91.597157 |
| 1000 | 1 | 362.614614 | 65.517278 |
| No. | Score | Interface Area | Coordinates |
|---|---|---|---|
| 1 | 2900 | 318.4 | −1.34; −0.09; 1.38; −23.80; −22.56; −39.82 |
| 2 | 2858 | 318.3 | 1.02; 0.08; 0.88; −48.08; 21.92; −56.70 |
| 3 | 2834 | 310.4 | −1.95; 0.16; −1.70; −74.21; −8.23; −46.64 |
| 4 | 2830 | 306 | 1.32; 0.01; −2.83; −44.00; −28.47; −52.58 |
| 5 | 2814 | 318.5 | −1.33; −0.27; 1.59; −27.20; −37.47; −60.51 |
| 6 | 2792 | 316.6 | −1.14; −0.17; −1.48; −66.81; 35.58; −67.43 |
| 7 | 2792 | 308.1 | −1.78; −0.03; 2.84; −41.83; 28.83; −45.53 |
| 8 | 2790 | 301.5 | −1.64; 0.44; 1.33; −71.19; −15.76; −81.43 |
| 9 | 2786 | 310.8 | 2.09; −0.02; −1.89; −16.57; 19.50; −51.15 |
| 10 | 2786 | 296.7 | −2.08; 0.27; −1.76; −57.26; −20.74; −68.35 |
| Cluster | ΔG (kcal/mol) | FullFitness (kcal/mol) | Ki |
|---|---|---|---|
| 1 | −8.1 | −2139.9 | 11.264 × 10−7 |
| 6 | −7.6 | −2137.1 | 22.904 × 10−7 |
| 33 | −6.8 | −2126.6 | 94.047 × 10−7 |
| Mode | Affinity (kcal/mol) | Dist. from RMSD L. B | Dist. from RMSD U. B |
|---|---|---|---|
| 1 | −6.3 | 0 | 0 |
| 2 | −6 | 1.805 | 4.092 |
| 3 | −5.7 | 2.296 | 2.819 |
| 4 | −5.3 | 4.405 | 5.497 |
| 5 | −5.3 | 9.763 | 11.591 |
| 6 | −5.3 | 2.674 | 3.792 |
| 7 | −5.3 | 3.029 | 5.193 |
| 8 | −5.2 | 2.142 | 2.893 |
| 9 | −5.1 | 2.042 | 2.793 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moldovan, O.-L.; Sandulea, A.; Lungu, I.-A.; Gâz, Ș.A.; Rusu, A. Identification of Some Glutamic Acid Derivatives with Biological Potential by Computational Methods. Molecules 2023, 28, 4123. https://doi.org/10.3390/molecules28104123
Moldovan O-L, Sandulea A, Lungu I-A, Gâz ȘA, Rusu A. Identification of Some Glutamic Acid Derivatives with Biological Potential by Computational Methods. Molecules. 2023; 28(10):4123. https://doi.org/10.3390/molecules28104123
Chicago/Turabian StyleMoldovan, Octavia-Laura, Alexandra Sandulea, Ioana-Andreea Lungu, Șerban Andrei Gâz, and Aura Rusu. 2023. "Identification of Some Glutamic Acid Derivatives with Biological Potential by Computational Methods" Molecules 28, no. 10: 4123. https://doi.org/10.3390/molecules28104123
APA StyleMoldovan, O.-L., Sandulea, A., Lungu, I.-A., Gâz, Ș. A., & Rusu, A. (2023). Identification of Some Glutamic Acid Derivatives with Biological Potential by Computational Methods. Molecules, 28(10), 4123. https://doi.org/10.3390/molecules28104123