


Multiple Pulse Amperometry—An Antifouling Approach for Nitrite Determination Using Carbon Fiber Microelectrodes

Abstract
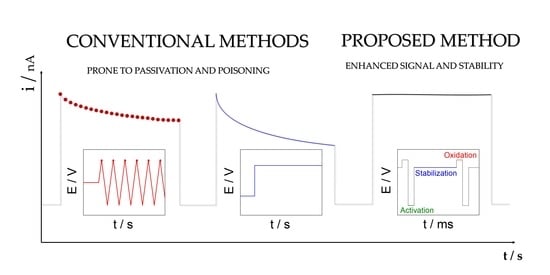
1. Introduction
2. Results and Discussion
2.1. Amperometric Reactivation Method Optimization
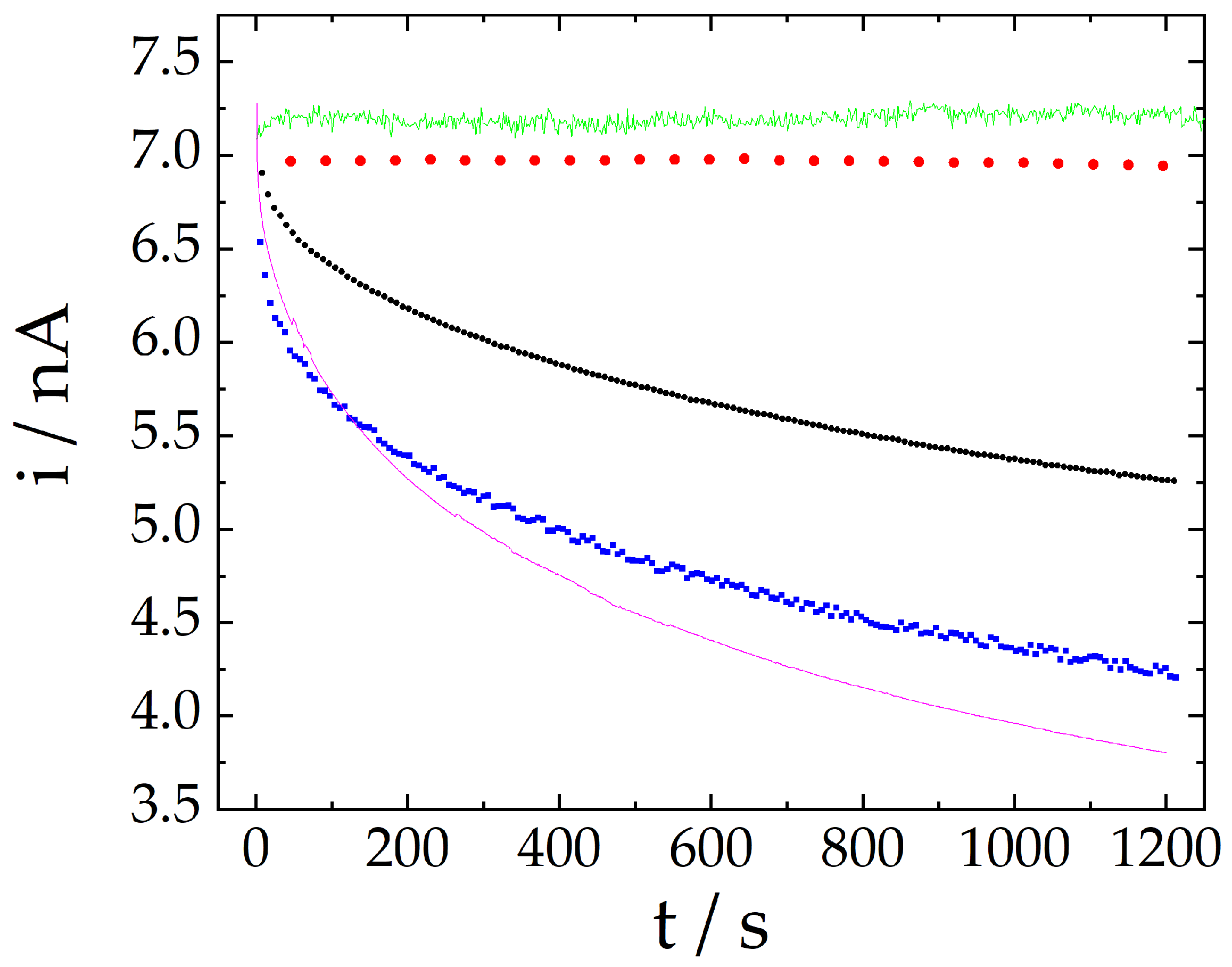
2.2. Signal Stability Comparison Using Different Techniques
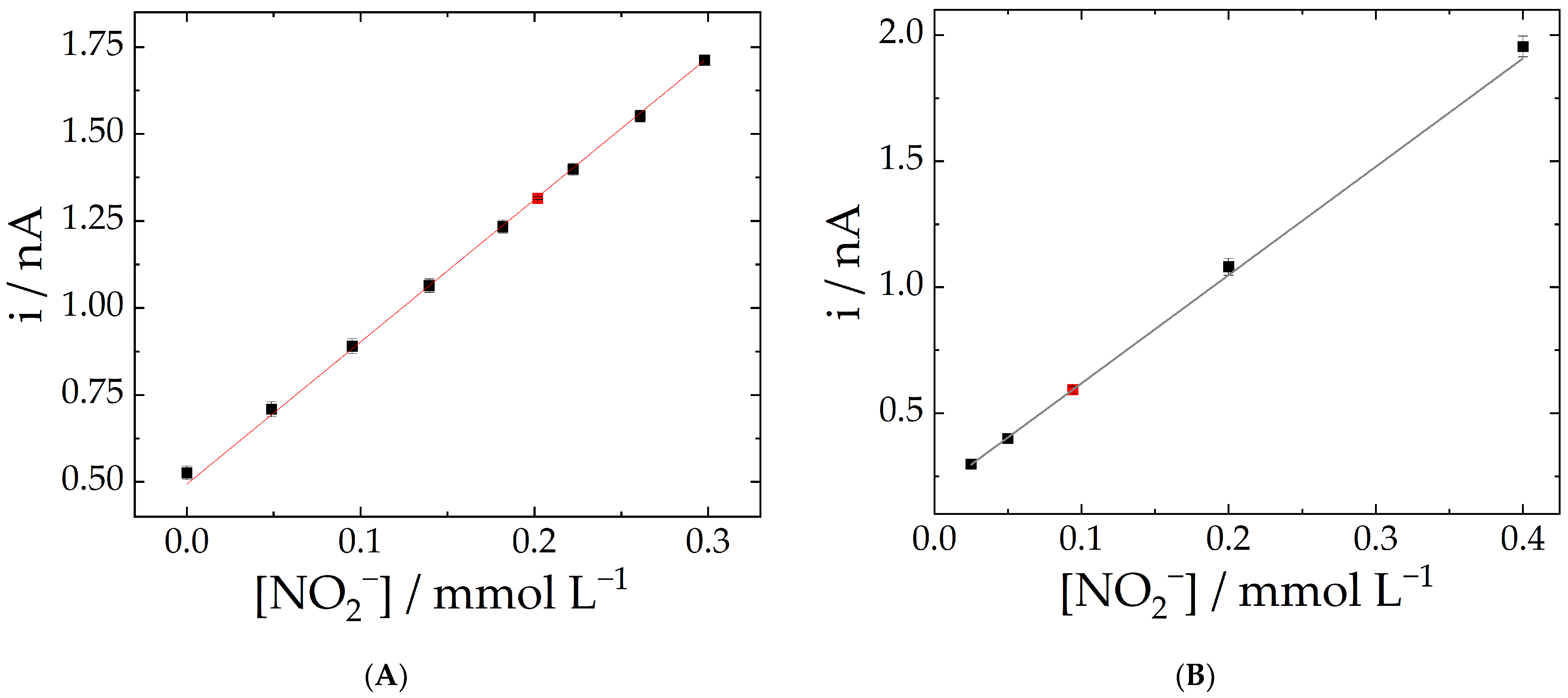
2.3. Analytical Parameters
3. Materials and Methods
3.1. Chemicals, Materials, and Samples
3.1.1. Synthetic Saliva Samples
3.1.2. Tap Water Samples
3.2. Electrodes and Instrumentation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Cvetković, D.; Živković, V.; Lukić, V.; Nikolić, S. Sodium nitrite food poisoning in one family. Forensic Sci. Med. Pathol. 2019, 15, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Chhem-Kieth, S.; Skou, P.B.; Lametsch, R.; Hansen, E.T.; Ruiz-Carrascal, J. Investigation of nitrite alternatives for the color stabilization of heme-iron hydrolysates. J. Food Sci. Technol. 2018, 55, 4287–4296. [Google Scholar] [CrossRef] [PubMed]
- Ferysiuk, K.; Wójciak, K.M. Reduction of Nitrite in Meat Products through the Application of Various Plant-Based Ingredi-ents. Antioxidants 2020, 9, 711. [Google Scholar] [CrossRef] [PubMed]
- Majoua, D.; Christieans, S. Mechanisms of the bactericidal effects of nitrate and nitrite in cured meats. Meat Sci. 2018, 145, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Bensid, A.; El Abed, N.; Houicher, A.; Regenstein, J.M.; Özogul, F. Antioxidant and antimicrobial preservatives: Properties, mechanism of action and applications in food—A review. Crit. Rev. Food Sci. Nutr. 2022, 62, 2985–3001. [Google Scholar] [CrossRef]
- Sharma, S.K.; Sharma, P.R.; Lin, S.; Chen, H.; Johnson, K.; Wang, R.; Borges, W.; Zhan, C.; Hsiao, B.S. Reinforcement of natural rubber latex using jute carboxycellulose nanofibers extracted using nitro-oxidation method. J. Nanomater. 2020, 10, 706. [Google Scholar] [CrossRef]
- Morway, A.J.; Knapp, C. Method for dispersing sodium nitrite in grease compositions. U.S. Patent US3078226A, 19 February 1963. [Google Scholar]
- Li, Q.; Liu, Y.; Li, S.; Guo, X.; Fu, Y.; He, N.; Ruan, G.; Wang, Q.; Gao, W.; Fang, L. Impact of nitrite exposure on oxidative stress and antioxidative-related genes responses in the gills of Procambarus clarkii. Fish Shellfish. Immunol. 2022, 131, 624–630. [Google Scholar] [CrossRef]
- Rodkey, F.L. A mechanism for the conversion of oxyhemoglobin to methemoglobin by nitrite. Clin. Chem. 1976, 22, 1986–1990. [Google Scholar] [CrossRef]
- IARC. Ingested Nitrate and Nitrite, and Cyanobacterial Peptide Toxins: IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 2010; Volume 94, pp. 314–320. Available online: https://publications.iarc.fr/Book-And-Report-Series/Iarc-Monographs-On-The-Identification-Of-Carcinogenic-Hazards-To-Humans/Ingested-Nitrate-And-Nitrite-And-Cyanobacterial-Peptide-Toxins-2010 (accessed on 23 November 2022).
- WHO. Health Criteria and Other Supporting Information: Guidelines for Drinking-Water Quality, 2nd ed.; WHO: Geneva, Switzerland, 1998; p. 11. Available online: https://apps.who.int/iris/bitstream/handle/10665/75380/WHO_SDE_WSH_04.03_56_eng.pdf (accessed on 23 November 2022).
- Code of Federal Regulations 21: Food for Human Consumption. Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=172.175 (accessed on 23 November 2022).
- Lucas, S.B.; Duarte, L.M.; Rezende, K.C.A.; Coltro, W.K.T. Nitrite Determination in Environmental Water Samples Using Microchip Electrophoresis Coupled with Amperometric Detection. Micromachines 2022, 13, 1736. [Google Scholar] [CrossRef]
- Cortés-Herrera, C.; Artavia, G.; Leiva, A.; Granados-Chinchilla, F. Liquid Chromatography Analysis of Common Nutri-tional Components, in Feed and Food. Foods 2019, 8, 1. [Google Scholar] [CrossRef]
- Wierzbicka, E. Novel methods of nitrate and nitrite determination—A review. J. Elem. 2020, 25, 97–106. [Google Scholar] [CrossRef]
- Amini, M.K.; Pourhossein, M.; Talebi, M. A chemiluminescence flow injection system for nitrite ion determination. JICS2 2005, 2, 305–314. [Google Scholar] [CrossRef]
- Singh, P.; Singh, M.K.; Beg, Y.R.; Nishad, G.R. A review on spectroscopic methods for determination of nitrite and nitrate in environmental samples. Talanta 2019, 191, 364–381. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Gonçalves, J.M.; Sukeri, A.; Araki, K.; Bertotti, M. Correlating surface growth of nanoporous gold with electro-deposition parameters to optimize amperometric sensing of nitrite. Sens. Actuators B Chem. 2018, 263, 237–247. [Google Scholar] [CrossRef]
- Stradiotto, N.R.; Yamanaka, H.; Zanoni, M.V.B. Electrochemical sensors: A powerful tool in analytical chemistry. J. Braz. Chem. Soc. 2003, 14, 159–173. [Google Scholar] [CrossRef]
- Salhi, O.; Ez-Zine, T.; Oularbi, L.; El Rhazi, M. Electrochemical Sensing of Nitrite Ions Using Modified Electrode by Poly 1,8-Diaminonaphthalene/Functionalized Multi-Walled Carbon Nanotubes. Front. Chem. 2022, 10, 870393. [Google Scholar] [CrossRef]
- Rocha, J.R.C.; Kosminsky, L.; Paixao, T.R.L.C.; Bertotti, M. Anodic oxidation of nitrite at a molybdenum oxide layer. Electroanal. Int. J. Devoted Fundam. Pract. Asp. Electroanal. 2001, 13, 155–160. [Google Scholar] [CrossRef]
- Etesami, M.; Chandran, N.; Hussin, M.H.; Ripin, A.; Adnan, R.; Sujari, A.N.A.; Mohamed, N. Electrooxidation of Nitrite Ions on Gold/Polyaniline/Carbon Paste Electrode. Int. J. Electrochem. Sci. 2016, 11, 8332–8345. [Google Scholar] [CrossRef]
- Bertotti, M.; Pletcher, D. A study of nitrite oxidation at platinum microelectrodes. J. Braz. Chem. Soc. 1997, 8, 391–395. [Google Scholar] [CrossRef]
- Palacios-Santander, J.; Terzi, F.; Zanardi, C.; Pigani, L.; Cubillana-Aguilera, L.M.; Naranjo-Rodriguez, I.; Seeber, R. Electro-catalytic and antifouling properties of CeO2-glassy carbon electrodes. J. Solid State Electrochem. 2016, 20, 3125–3131. [Google Scholar] [CrossRef]
- Nodehi, Z.; Ghaffarineja, A.; Rafati, A.A. Synergic and Antifouling Effect of ZnO on Ethanol Oxidation by Silver-Palladium Bimetallic Electrocatalyst. J. Electrochem. Soc. 2019, 166, 2556–2562. [Google Scholar] [CrossRef]
- Sudha, V.; Mothanty, S.A.; Thangamuthu, R. Facile synthesis of Co3O4 disordered circular sheets for selective electrochemi-cal determination of nitrite. New J. Chem. 2018, 42, 11869–11877. [Google Scholar] [CrossRef]
- Nitric Oxide Microsensors Instruction Manual, World Precision Instruments. Available online: https://www.wpiinc.com/media/wysiwyg/pdf/NO_Microsensors_IM.pdf (accessed on 23 November 2022).
- Fogg, A.; Chamsi, A. Oxidative flow injection amperometric determination of nitrite at an electrochemically pre-treated glassy carbon electrode. Analyst 1988, 113, 1723–1727. [Google Scholar] [CrossRef]
- Ramos, D.L.O.; Ribeiro, M.M.A.C.; Munoz, R.A.A.; Richter, E.M. Portable amperometric method for selective determination of caffeine in samples with the presence of interfering electroactive chemical species. J. Electroanal. Chem. 2022, 906, 111606. [Google Scholar] [CrossRef]
- dos Santos, W.T.P.; Almeida, E.N.; Ferreira, H.A.; Gimenes, D.; Richter, E.M. Simultaneous flow injection analysis of para-cetamol and ascorbic acid with multiple pulse amperometric detection. Electroanal. Int. J. Devoted Fundam. Pract. Asp. Electroanal. 2008, 20, 1878–1883. [Google Scholar] [CrossRef]
- Chaves, S.C.; Aguiar, P.N.; Torres, L.M.; Gil, E.S.; Luz, R.C.; Damos, F.S.; Munoz, R.A.; Richter, E.M.; dos Santos, W.T. Simultaneous determination of caffeine, ibuprofen, and paracetamol by flow-injection analysis with multiple-pulse am-perometric detection on boron-doped Diamond electrode. Electroanalysis 2015, 27, 2785–2791. [Google Scholar] [CrossRef]
- Baciu, A.; Ardelean, M.; Pop, A.; Pode, R.; Manea, F. Simultaneous Voltammetric/Amperometric Determination of Sulfide and Nitrite in Water at BDD Electrode. Sensors 2015, 15, 14526–14538. [Google Scholar] [CrossRef]
- Manea, F.; Remes, A.; Radovan, C.; Pode, R.; Picken, S.; Schoonman, J. Simultaneous electrochemical determination of ni-trate and nitrite in aqueous solution using Ag-doped zeolite-expanded graphite-epoxy electrode. Talanta 2010, 83, 66–71. [Google Scholar] [CrossRef]
- Piela, B.; Wrona, P.K. Oxidation of Nitrites on Solid Electrodes: I. Determination of the Reaction Mechanism on the Pure Electrode Surface. J. Electrochem. Soc. 2002, 142, 55–63. [Google Scholar] [CrossRef]
- Kozub, B.R.; Rees, N.V.; Compton, R.G. Electrochemical determination of nitrite at a bare glassy carbon electrode; why chemically modify electrodes? Sens. Actuators B Chem. 2010, 143, 539–546. [Google Scholar] [CrossRef]
- da Silva, S.M.; Mazo, L.H. Differential Pulse Voltammetric Determination of Nitrite with Gold Ultramicroelectrode. Electro-analysis 1998, 10, 1200–1203. [Google Scholar]
- da Rocha, J.R.C.; Angnes, L.; Bertotti, M.; Araki, K.; Toma, H.E. Amperometric detection of nitrite and nitrate at tetraruthe-nated porphyrin-modified electrodes in a continuous-flow assembly. Anal. Chim. Acta 2002, 452, 23–28. [Google Scholar] [CrossRef]
- Tau, P.; Nyokong, T. Electrocatalytic oxidation of nitrite by tetra-substituted oxotitanium(IV) phthalocyanines adsorbed or polymerized on glassy carbon electrode. J. Electroanal. Chem. 2007, 611, 10–18. [Google Scholar] [CrossRef]
- Zhang, Y.; Wen, F.; Tan, J.; Jiang, C.; Zhu, M.; Chen, Y.; Wang, H. Highly efficient electrocatalytic oxidation of nitrite by electrodeposition of Au nanoparticles on molybdenum sulfide and multi-walled carbon nanotubes. J. Electroanal. Chem. 2017, 786, 43–49. [Google Scholar] [CrossRef]
- Yin, H.; Zhang, Y.; Dong, H.; Liu, L.; Wang, X.; Zhang, Y.; Xu, M.; Zhou, Y. Self-Calibrating Electrochemical Sensors Based on Uniformly Dispersed Ag Nanoclusters in Nitrogen-Doped Carbon Sheets for Determination of Nitrite. ACS Appl. Nano Mater. 2022, 5, 9737–9746. [Google Scholar] [CrossRef]
- Ozarslan, M.; Can, D.B.; Avcioglu, N.H.; Çalişkan, S. Effect of different polishing techniques on surface properties and bacte-rial adhesion on resin ceramic CAD/CAM materials. Clin. Oral Investig. 2022, 26, 5289–5299. [Google Scholar] [CrossRef]
- Peña, R.C.; de Souza, A.P.R.; Bertotti, M. Determination of Fe(III) in wine samples using a ruthenium oxide hexacyanoferrate modified microelectrode. J. Electroanal. Chem. 2014, 731, 49–52. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Signal Change after 20 min |
|---|---|
| Amperometry | −47.7% |
| Differential pulse voltammetry | −35.5% |
| Cyclic voltammetry | −23.8% |
| Extended cyclic voltammetry | −0.35% |
| Proposed method | 0.86% |
| Nitrite Concentration/mmol L−1 | Mean/nA | Standard Deviation/nA | RSD |
|---|---|---|---|
| 0.25 | 1.334 | 0.004 | 0.4% |
| 0.40 | 1.579 | 0.003 | 0.2% |
| 0.80 | 2.611 | 0.004 | 0.2% |
| 2.00 | 5.89 | 0.01 | 0.2% |
| Material | Linear Range (µmol L−1) | LOD (µmol L−1) | Technique | Ref. |
|---|---|---|---|---|
| Bare CFE | 6–2000 | 5.5 | MPA | This work |
| Bare GCE | 50–4700 | 200 | CV | [35] |
| 20–6400 | 300 | DPV | ||
| 2.5–10 | 0.4 | Amp. | ||
| Bare Pt | 5–1000 | 2 | Amp. | [23] |
| Bare Au | 10–600 | 0.65 | DPV | [36] |
| CGE/Zn-TPPS | <1000 | 0.1 | FIA-Amp. | [37] |
| CGE/TiTaPc | <3500 | 1 | CV | [38] |
| Au/MWCNTs/MoS2 | 12–2100 | 4.0 | Amp. | [39] |
| Au/NPG | 1–1000 | 0.01 | Amp. | [18] |
| AgNC@NCS | 1–1400 | 0.38 | DPV | [40] |
| Sample | Fortification mg L−1 (µmol L−1) | Concentration Recovered mg L−1 (µmol L−1) | Deviation |
|---|---|---|---|
| Tap water | 4.6 (100) | 4.1 ± 0.1 (91 ± 3) | 8.9 % |
| Synthetic saliva | 9.6 (208) | 6.9 ± 0.2 (202.5 ± 0.8) | 2.6 % |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saraiva, D.P.M.; Braga, D.V.; Bossard, B.; Bertotti, M. Multiple Pulse Amperometry—An Antifouling Approach for Nitrite Determination Using Carbon Fiber Microelectrodes. Molecules 2023, 28, 387. https://doi.org/10.3390/molecules28010387
Saraiva DPM, Braga DV, Bossard B, Bertotti M. Multiple Pulse Amperometry—An Antifouling Approach for Nitrite Determination Using Carbon Fiber Microelectrodes. Molecules. 2023; 28(1):387. https://doi.org/10.3390/molecules28010387
Chicago/Turabian StyleSaraiva, Douglas P. M., Daniel V. Braga, Bruna Bossard, and Mauro Bertotti. 2023. "Multiple Pulse Amperometry—An Antifouling Approach for Nitrite Determination Using Carbon Fiber Microelectrodes" Molecules 28, no. 1: 387. https://doi.org/10.3390/molecules28010387
APA StyleSaraiva, D. P. M., Braga, D. V., Bossard, B., & Bertotti, M. (2023). Multiple Pulse Amperometry—An Antifouling Approach for Nitrite Determination Using Carbon Fiber Microelectrodes. Molecules, 28(1), 387. https://doi.org/10.3390/molecules28010387