Abstract
There has been a great focus on halogen-bonding as a unique interaction between electron-deficient halogen atoms with Lewis basic moieties. Although the application of halogen-bonded atoms in organic chemistry has been eagerly researched in these decades, the development of chiral molecules with halogen-bonding functionalities and their utilization in asymmetric catalysis are still in the\ir infancy. We have previously developed chiral halonium salts with amide functionalities, which behaved as excellent catalysts albeit in only two reactions due to the lack of substrate activation abilities. In this manuscript, we have developed chiral halonium salts with an N-nitrosamine moiety and applied them to the Mannich reaction of isatin-derived ketimines with malonic esters. The study focused on our novel bromonium salt catalyst which provided the corresponding products in high yields with up to 80% ee. DFT calculations of the chiral catalyst structure suggested that the high asymmetric induction abilities of this catalyst are due to the Lewis basic role of the N-nitrosamine part. To the best of our knowledge, this is the first catalytic application of N-nitrosamines.
1. Introduction
Halogen-bonding (XB), which is the non-covalent interaction formed between electron-deficient halogen atoms and Lewis basic moieties, has been focused on in the wide field of chemistry due to its unique power [1,2,3,4]. The key feature of XB is its relatively strong yet soft character, providing higher directionality compared to hydrogen-bonding. In organic chemistry, XB has been employed for molecule recognition and for the activation of electron-rich substrates in several reactions [5,6]. In the past decade, XB has been utilized as a key interaction in metal catalysis and organocatalysis, demonstrating its characteristic reactivities and selectivities in various reactions [7,8,9,10,11,12,13,14,15,16,17]. In 2022, Fiksdahl, Erdelyi and co-workers reported the cyclopropanation of propargyl acetate with styrene derivatives under gold catalysis [8]. In their reaction, the addition of a halogen bond donor as a co-catalyst drastically increased the reaction rate.
In asymmetric catalysis, the application of XB has been limited [18,19,20,21,22,23,24]. In 2014, Tan and co-workers reported the alkylation reaction of sulfenes with alkyl halides catalyzed by an ammonium salt with halogen atom, to form the corresponding products with up to 96% ee [18]. In their report, the iodine atom played a crucial role and switching to other halogen atoms provided decreased enantioselectivities. In 2018, Arai and co-workers developed the cinchona-derived Brønsted base catalyst with the halogen-bonding functionality of the perfluoro iodophenyl group, which effectively catalyzed the Mannich reaction of malononitrile with isatin-derived ketimines to afford products in high yields with up to 97% ee (Figure 1) [20]. Finally, in 2021, Huber and co-workers developed the bulky bis(2-iodoimidazolium) based chiral molecules, which catalyzed the Mukaiyama–aldol reaction of silyl ketene acetals with ketones, to provide the chiral products with up to 33% ee (Figure 1) [23].
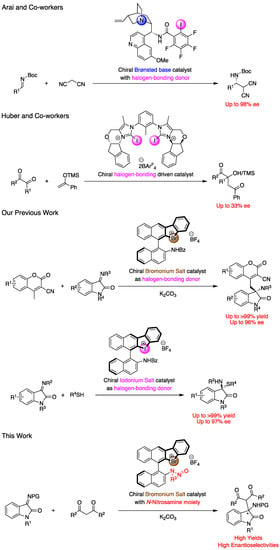
Figure 1.
Examples of chiral halogen-bonding catalysts.
Hypervalent halogen reagents, especially hypervalent iodine reagent, have been researched as highly reactive chemical species which act as substrates and as catalysts in oxidation reactions [25,26,27,28,29,30,31]. Although the iodonium and bromonium salts, which are kinds of hypervalent halogen compounds, have been found to work as XB catalysts [32,33,34,35,36,37], findings on their chiral equivalents to be used for asymmetric catalysis have been quite limited thus far [38,39,40]. In 2015, Liu, Han, and co-workers reported the syntheses of iodonium salts with a chiral phosphate counter anion, and their application as asymmetric XB catalysts in a three-component Mannich reaction to form the desired products with up to 7% ee [38]. Recently, we have developed chiral binaphthyl-based cyclic iodonium and bromonium salts with amide functionalities, which efficiently catalyzed the vinylogous Mannich reaction of cyanomethyl coumarins with imines, as well as the thiol addition reaction to isatin-derived ketimines in high yields with up to 96% and 97% ee, respectively (Figure 1) [39,40]. Although our chiral halonium salts worked as excellent catalysts, they could not be applied to other kinds of reactions due to the difficulties in introducing other functionalities except the amide group. Compounds with the N-nitrosamine moiety have been utilized as precursors of azomethine imines [41], as directing groups in C–H functionalization by transition metal catalysis [42,43,44], and as active substrates in several reactions [45]. Therefore, this study aimed to develop chiral bromonium salts with N-nitrosamine functionalities, which behaved as a stronger Lewis basic part than previous amide group, and explore their applications in the enantioselective Mannich reaction of malonic esters with ketimines. To the best of our knowledge, this is the first example of the successful enantioselective catalytic applications of N-nitrosamines.
2. Results and Discussion
2.1. Synthesis of Catalysts
The chiral binathyl-based halonium salts were synthesized as follows (Scheme 1). Commercial (R)-BINAM was chosen as a starting material, which was converted to 3-iodo-BINAM 1 according to the reported procedure [46]. The acylation of 1 was conducted with 1.1 equivalents of an acetic anhydride under acidic condition to obtain amide 2 in 90% yield with almost perfect chemoselectivity. The 2-halophenyl group was introduced to 2 through the Suzuki coupling reaction with 1.1 equivalents of 2-halophenylboronic acid under palladium catalysis to form the intermediate, and its amide parts were reduced into amine functionalities by the treatment with 5.0 equivalents of borane in THF to give 3. Finally, the primary and the secondary amino groups of 3 were converted to the diazonium salt and the N-nitrosamine moiety, respectively, by the treatments with 8.0 equivalents of tert-butyl nitrite and 4.0 equivalents of tetrafluoroboric acid, which were then cyclized through the pyrolysis of diazonium moieties in chloroform at 60 °C to form the desired halonium salts 4 in low to moderate yields; 4d was synthesized through the counteranion exchange of 4a by treatment with 10 equivalents of NaCl in 25% yield.
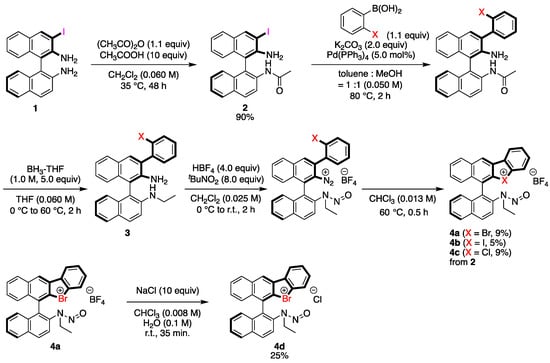
Scheme 1.
Synthesis of chiral halonium salts with N-nitrosamine moiety.
2.2. Application of Chiral Catalysts
The synthesized chiral halonium salts with N-nitrosamine moieties were applied to enantioselective reactions (Scheme 2). We chose the Mannich reaction of malonic esters to ketimines as a model reaction, as it affords valuable chiral amines in an enantioselective fashion [47,48]. The catalyst screenings were conducted in this reaction. First, the employed halogen atoms were screened, and the bromonium salt 4a was observed to have provided the corresponding products in the highest yield with enantioselectivity. This result revealed that the N-nitrosamine moiety could be included to the chiral catalyst structure as a Lewis basic part, which demonstrated the high asymmetric induction abilities. The iodonium salt 4b also worked as an asymmetric catalyst in the present reaction, but decreased enantioselectivity, and the chloronium salt 4c catalyzed the reaction only in racemic form. These reasons can be explained by the strength of the halogen bond of the halogen atoms. The bromonium salt interacted with the substrate in its bidentate form, composed of both halonium salt and N-nitrosamine moiety, to construct an efficient asymmetric environment around the prochiral carbon center, and form the products in good enantioselectivities. Conversely, the iodonium salt bears a strong halogen bond donor character, which enabled the intramolecular interaction with N-nitrosamine part to form the monodentate catalyst and it mediated the reaction in low and reversed enantioselectivity. The chloronium salt weakly coordinated to the imine substrate, and the background reaction went faster to form the product in racemic form. To investigate the importance of the halogen bond in the present catalysis, the bromonium salt with chloride anion 4d was synthesized and applied, which afforded the corresponding product in decreased enantioselectivity. The chloride anion is known to make a halogen bond with halogen atoms such as bromonium moieties, which weaken the halogen bond of the catalyst with a substrate. This result suggests the importance of the halogen bond for the enantioinduction to the products in the present reaction.
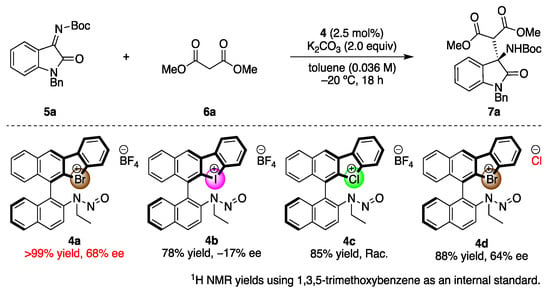
Scheme 2.
Screening of the chiral halonium salt catalysts.
After the optimal catalyst was established, the reaction conditions were optimized (Table 1). Reaction in non-polar solvents such as toluene, THF, and dichloromethane provided the products in excellent yields with high enantioselectivities (Entries 1–3); however, the employment of polar solvents (diethyl ether and acetonitrile) gave diminished enantioselectivities (Entries 4 and 5). These results suggest the importance of the Lewis basic interaction of the N-nitrosamine moiety in the product’s enantioselectivity, which was disturbed by the hydrogen bond of the polar solvents. Subsequently, the catalyst loading, and the reaction temperatures were optimized, and the reaction in toluene at −20 °C with 2.5 mol% of 4a was found to be optimal. The lower catalyst loading caused the shortage of the chiral catalyst due to its decomposition under basic condition, which relatively accelerated the background reaction, and the racemic product was obtained. On the other hands, the higher catalyst concentration perhaps increased the intermolecular interaction of 4a, which formed the oligomeric catalyst species, and it worked as an inferior asymmetric catalyst, and the enantioselectivity of the product decreased [40].

Table 1.
Reaction condition optimizations.
After obtaining the optimal conditions, the substrate scope for the study transformation was conducted (Figure 2). Most of the reactions were conducted with excess longer time because the products had very similar RF values to substrates on TLC, which made it difficult to judge the completion of reactions. The scopes for imines and N-protecting group of benzyl and phenyl moieties tolerated the reaction well, and products 7a and 7c were isolated in high yields with high ee, while the methyl protected product 7b was obtained with a lower ee of 31%. The aromatic substituents of the imines were investigated, and the corresponding products 7d–7h were obtained in high yields with high ee’s, irrespective of their steric and electronic natures except for 4-chlorinated 7d, which was obtained in a racemic form. N-Benzyloxycarbonyl- (Cbz) protected imine 5i could also be applied well to the present reaction to form 7i in good enantioselectivity. Next, the scopes for nucleophiles were conducted. Scopes for malonic ester derivatives revealed that the enantioselectivities of the products were highly affected by the bulkiness of the nucleophiles, and a sterically less-hindered dimethyl malonate provided product 7a with a high ee. However, the sterically hindered dibenzyl malonate and diethyl malonate gave 7j and 7k with only 28% ee and 3% ee, respectively, and the more hindered di-tert-butyl malonate did not provide any products. Other active methylene compounds were also employed as pre-nucleophiles. The reaction outcomes had almost same trend to that of the malonic esters, as the sterically less-hindered acetylacetone provided 7m in 51% yield with 61% ee, and the hindered dibenzoyl methane and tertiary nucleophile gave poor results (7n and 7o, respectively). Nitrocyclohexane was also applied as a pre-nucleophile, but no reaction was observed. The bulky nucleophiles were interacted with chiral catalyst weaker than that of less-bulky one, which decreased the Si-face selectivity in the enantioselectivity determining step. The absolute configurations of the 7b and 7m were determined according to the HPLC retention times of the reported values, and the other configurations were determined according to their structures [47,48].
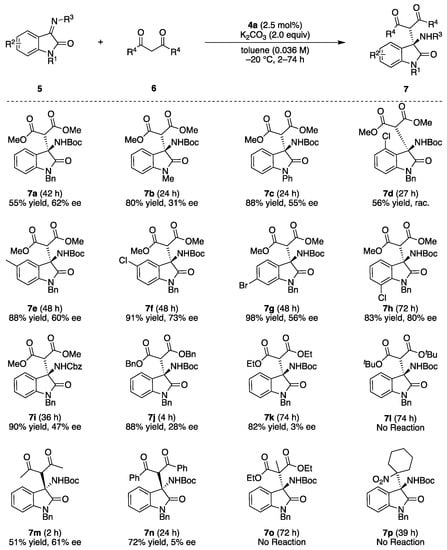
Figure 2.
Substrate scopes. Isolated product yields are shown.
Next, to verify the asymmetric induction mechanism of the present reaction, the interactions between the chiral halonium salt with an imine 5a or a dimethyl malonate (6a) were investigated by 1H NMR titration experiments (Figure 3 and Figure 4) [49]. Due to the instability of 4a under a basic condition, these titration experiments were conducted in the absence of potassium carbonate. When 4a was mixed with different equivalents of imine 5a, the 1H NMR chemical shifts of H1, H2 or H3 and H4 were significantly shifted as increasing the amount of 5a (Figure 4, left). On the other hand, when 4a was mixed with dimethyl malonate, the 1H NMR chemical shifts of only H1 and H4 were shifted (Figure 4, right). These observations suggested that catalyst 4a had interaction with both an imine substrate and a dimethyl malonate. To get the insight into a more detailed activation pattern, binding constants (K) and Gibbs free energies (ΔG) were determined by Bindfit program (Figure 5) [50,51]. Because the protons for H2 or H3 were rather overlapped, the binding constants were calculated only using H1 and H4 protons. The spots in Figure 5 shows the absolute values for chemical shift differences of 4a by mixing with 5a (left) or 6a (right). In both cases, the chemical shifts were highly affected by the quantity of additives, although, their binding constants were significantly different from each other. When the binding constants for the cases of mixing 4a with 5a were calculated, the K values were almost 0 for both H1 and H4. On the other hands, the K values for mixing 4a with 6a were 0.69 and 1.53 M−1 for H1 and H4, respectively. These results suggested that 4a did not interact with 5a but only 6a. Furthermore, the titration curve indicated that both H1 and H4 of 4a were affected by the binding with 6a, which located close to the bromonium salt moiety and the N-nitrosamine part, respectively. These observations suggests that 4a binds and activates dimethyl malonate with both a bromonium and a N-nitrosamine part even under the neutral condition.
Figure 3.
Structure of 4a and 8.
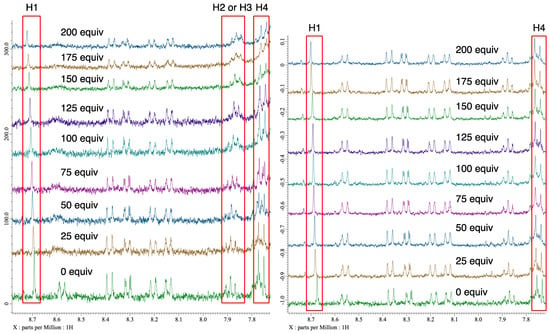
Figure 4.
1H NMR charts (in CDCl3) of 4a with different equivalents of 5a (left), and 6a (right).
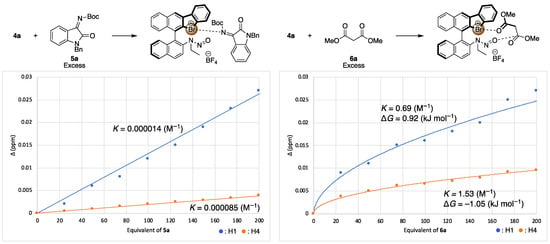
Figure 5.
1H NMR titration experiments (in CDCl3) of 4a with 5a (left), and 6a (right). The spots show the chemical shift differences from additive-free values at various equivalent of additives. Blue spot means the chemical shifts differences for H1, and the orange spot means that of H4 (Figure 4). The binding constants K were calculated by Bindfit.
Finally, to check the reaction acceleration ability of our chiral catalyst, the reaction conversions were monitored by 1H NMR (Figure 6). When the reaction of 5a with 6a was conducted under the optimized condition without catalyst, the reaction was very slow and considerable amount of 5a was remained even after 180 min (black line). On the other hands, when the same reaction was carried out in the presence of 2.5 mol% of 4a, the reaction was drastically accelerated and most of 5a was consumed after 60 min (blue line). Interestingly, the reaction was more efficiently catalyzed by the achiral bromonium salt 8, which gave the almost complete conversion of 5a only within 10 min (orange line). These results suggest that 4a has lower catalytic activity than 8 due to its bulkiness and the half number of sigma holes, which can activate a substrate.
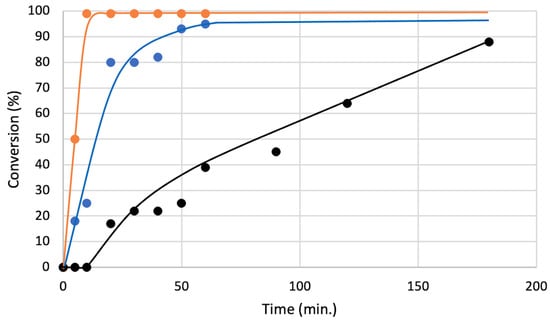
Figure 6.
Conversions of 5a in an optimal condition (Table 1, Entry 1) with different catalyst (black: without catalyst, blue: with 4a (2.5 mol%), orange: with 8 (2.5 mol%)) monitored by 1H NMR.
From mechanistic studies, the plausible reaction mechanism for the present transformation was proposed (Figure 7). First, the proton of the active methylene moiety was deprotonated by potassium carbonate to form an ionic intermediate. Then, the counteranion of the latter was exchanged by the chiral halonium salt catalyst to form chiral intermediate II, which worked as a nucleophile with imines to form III. Finally, protonation of III provided desired product 7 together with the regeneration of the catalyst. In this catalytic cycle, the step from intermediate II to III is the enantioselectivity determining step. In this step, the plausible transition structure is constructed according to the 1H NMR titration experiment results of mixing the chiral catalyst with an imine or a methyl malonate. The halonium salt activated an enolate anion of the active methylene species-derived molecule by halogen-bonding, and the N-nitrosamine part recognized another carbonyl carbon atom in the same molecule. The structure of 4a was optimized at the M06-2X/6-31G(d) level of theory in gas phase within Gaussian 16 [52], with structures generated by GaussView 6 [53]. The Br···O distance of the optimized structure was 3.11 Å, which suggested the independently existence of both the bromonium salt and the N-nitrosamine part [1].
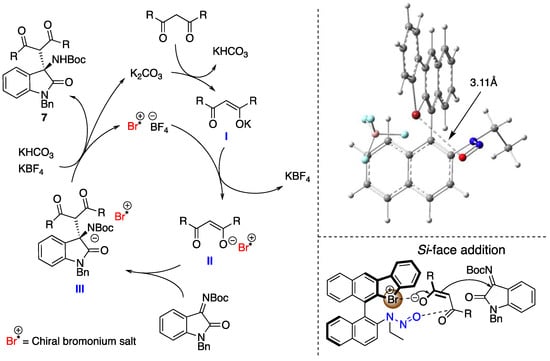
Figure 7.
Plausible reaction mechanism (left), the DFT-calculated optimized structure of 4a (M06-2X/6-31G(d)) generated by GaussView 6 (right, upper), and the plausible transition state structure (right, lower). The Br···O distance of the calculated structure is 3.11 Å.
3. Materials and Methods
3.1. General Information
1H-, 13C-NMR spectra were recorded with Bruker AVANCE III-400M (1H-NMR 400 MHz, 13C-NMR 100 MHz, 19F-NMR 376 MHz). 1H-NMR spectra are reported as follows: chemical shift in ppm (δ) relative to the chemical shift of CHCl3 at 7.26 ppm or tetramethylsilane at 0 ppm, integration, multiplicities (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), and coupling constants (Hz). 13C-NMR spectra reported in ppm (δ) relative to the central line of triplet for CDCl3 at 77 ppm. CF3CO2H used as external standards for 19F. ESI-MS spectra were obtained with Thermo Fisher, Exactive. FT-IR spectra were recorded on a JASCO FT-IR system (FT/IR-460 Plus). Mp was measured with AS ONE ATM-02. Column chromatography on SiO2 and neutral SiO2 was performed with Kanto Silica Gel 60 (40–50 μm). All reactions were carried out under Ar atmosphere unless otherwise noted. Commercially available organic and inorganic compounds were purchased from TCI, Kanto Chemical CO., INC. Wako Pure Chemical Industries, Ltd. or Nacalai Tesque, Inc., which had >95% purities, and used them without further purification. All dehydrated solvents were purchased from Wako Pure Chemical Industries, Ltd. or Nacalai Tesque, Inc., and were used without further purification. Calculation of binding constants were conducted by Bindfit program.
Imine substrates were synthesized according to the reported procedures [39,47,54,55,56,57]. Malonic esters and other active methylene species were purchased from the commercial source.
The raw data for the determination of binding constants (Table S1), 1H, 13C, and 19F NMR charts of the catalyst and their synthetic intermediate, and the products, HPLC charts of the products, DFT calculation results are available in the Supplementary Materials.
3.2. Synthesis of Chiral Bromonium Salts
3.2.1. Synthesis of 2
To a stirred solution of 1 (1.0 equiv) in CH2Cl2 (0.1 M) was added AcOH (10 equiv) and Ac2O (1.1 equiv) at 0 °C. The reaction was refluxed for 48 h before quenching with the addition of excess amount of NaOH aq. (2.0 M) at 0 °C. The reaction mixture was extracted with CH2Cl2, dried over Na2SO4 and filtered. After the removal of solvent by evaporation, the crude product was obtained, which was purified by column chromatography (Silica-gel, hexane/ethyl acetate) to give 2.
(R)-N-(2’-amino-3’-iodo-[1,1’-binaphthalen]-2-yl)acetamide (2)
White solid, 317.3 mg, 0.70 mmol, 90% yield.
m.p. = 188–190 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.58 (d, J = 9.0 Hz, 1H), 8.47 (s, 1H), 8.00 (d, J = 9.0 Hz, 1H), 7.90 (d, J = 8.1 Hz, 1H), 7.69–7.72 (m, 1H), 7.40–7.44 (m, 1H), 7.18–7.28 (m, 3H), 7.10 (d, J = 8.4 Hz, 1H), 6.93 (s, 1H), 6.86 (d, J = 8.2 Hz, 1H), 4.08 (s, 2H), 1.85 (s, 3H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 168.8, 142.1, 140.0, 134.9, 133.3, 131.9, 131.3, 129.6, 129.2, 128.3, 127.8, 127.15, 127.06, 125.3, 125.1, 123.7, 123.3, 121.1, 120.6, 110.6, 87.7, 24.7; HRMS (ESI+ in MeCN) calcd for C22H18ON2I [M + H] 453.0458 found 453.0453; IR (KBr) ν 2962, 1653, 1595, 1265, 819 cm−1; [α]20D = +43.9 (c = 0.2, CHCl3).
3.2.2. Synthesis of 4a–c
2 (1.0 equiv), 2-halophenyl boronic acid (1.1 equiv), K2CO3 (2.0 equiv), and Pd(PPh3)4 (5 mol%) were dissolved in degassed toluene/MeOH = 1/1 (0.1 M), which was refluxed for 2 h. The reaction was quenched by the addition of water, extracted with CH2Cl2, dried over Na2SO4 and filtered. After the removal of solvent by evaporation, the crude product was obtained, which was purified by column chromatography (Silica-gel, hexane/ethyl acetate) to give coupling product, which was used without further purification.
To a stirred solution of above obtained coupling product in THF (0.067 M) was added BH3 (1.0 M in THF, 5.0 equiv) at 0 °C, which was stirred at 60 °C for 2 h. The reaction was quenched by the addition of NaOH aq. (2.0 M) at 0 °C. The reaction mixture was evaporated and extracted with CH2Cl2, dried over Na2SO4 and filtered. After the removal of solvent by evaporation, the crude product was obtained, which was purified by column chromatography (Silica-gel, hexane/ethyl acetate) to give 3. 3 was obtained as a diastereomeric mixture, which was used in next step without isolation of both isomers.
To a stirred solution of 3 in CH2Cl2 (0.025 M) was added HBF4 (42% aq., 4.0 equiv) at 0 °C and stirred for 5 min at the same temperature. After that, tBuNO2 (8.0 equiv) was added at 0 °C, which was warmed to room temperature and stirred for 2 h. The reaction was quenched by the addition of water at 0 °C. The reaction mixture was extracted with CH2Cl2, dried over Na2SO4 and filtered. After the removal of solvent by evaporation at less than room temperature, the crude product was dissolved in CHCl3 (0.13 M) and stirred at 60 °C for 30 min. After the removal of the solvent by evaporation, crude product was obtained, which was purified by recrystallization (hexane/toluene) to give 4.
(R)-6-(2-(ethyl(nitroso)amino)naphthalen-1-yl)benzo[b]naphtho [2,3-d]bromol-5-ium Tetrafluoroborate (4a)
Pink solid, 9.9 mg, 0.017 mmol, 9% yield (from 2).
m.p. = 156–158 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.73 (s, 1H), 8.54 (d, J = 8.5 Hz, 1H), 8.37 (d, J = 8.5 Hz, 1H), 8.32 (d, J = 1.6 Hz, 1H), 8.22 (d, J = 8.1 Hz, 1H), 8.13 (d, J = 8.1 Hz, 1H), 7.85–7.89 (m, 1H), 7.70–7.79 (m, 3H), 7.64–7.68 (m, 1H), 7.55–7.59 (m, 1H), 7.38–7.45 (m, 2H), 7.04 (dd, J = 8.5, 0.9 Hz, 1H), 4.02 (qd, J = 14.0 Hz, 7.2 Hz, 1H), 3.79 (qd, J = 14.0 Hz, 7.2 Hz, 1H), 1.03 (t, J = 7.2 Hz, 3H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 138.6, 138.0, 137.5, 135.3, 134.0, 133.7, 133.5, 133.21, 133.15, 132.2, 131.8, 131.3, 130.8, 129.68, 129.61, 129.58, 129.37, 129.3, 128.1, 126.8, 126.4, 126.14, 125.96, 124.5, 121.6, 42.3, 12.3 (1 peak is overlapped with the other peak); 19F-NMR (377 MHz, CHLOROFORM-D) δ −149.9; HRMS (ESI+ in MeCN) calcd for C28H20ON2Br+ [M-BF4-] 479.0754 found 479.0754; IR (KBr) ν 2963, 1458, 1261, 1019, 800 cm−1; [α]20D = −52.2 (c = 0.2, CHCl3).
(R)-6-(2-(ethyl(nitroso)amino)naphthalen-1-yl)benzo[b]naphtho [2,3-d]iodol-5-ium Tetrafluoroborate (4b)
Pink solid, 10.6 mg, 0.017 mmol, 5% yield (from 2).
m.p. = 160–162 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.67 (d, J = 9.6 Hz, 1H), 8.61 (s, 1H), 8.34 (d, J = 8.8 Hz, 1H), 8.25 (dd, J = 7.9, 1.5 Hz, 1H), 8.14 (d, J = 8.4 Hz, 2H), 7.82 (td, J = 7.5, 0.6 Hz, 1H), 7.65–7.70 (m, 4H), 7.45–7.50 (m, 1H), 7.40–7.44 (m, 1H), 7.23–7.26 (m, 1H), 7.13 (dd, J = 8.6, 0.5 Hz, 1H), 3.76 (qd, J = 13.6 Hz, 7.2 Hz, 1H), 3.65 (qd, J = 13.6 Hz, 7.2 Hz, 1H), 0.91 (t, J = 7.2 Hz, 3H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 138.7, 138.0, 137.5, 135.3, 134.0, 133.7, 133.5, 133.22, 133.15, 132.16, 131.8, 131.3, 130.8, 129.7, 129.61, 129.59, 129.4, 129.3, 128.1, 126.8, 126.4, 126.14, 125.96, 124.5, 121.6, 42.3, 12.3 (1 peak is overlapped with the other peak); 19F-NMR (377 MHz, CHLOROFORM-D) δ −149.3; HRMS (ESI+ in MeCN) calcd for C28H20ON2I+ [M-BF4-] 527.0615 found 527.0603; IR (KBr) ν 2963, 1559, 1261, 1020, 800 cm−1; [α]20D = −20.3 (c = 0.2, CHCl3).
(R)-6-(2-(ethyl(nitroso)amino)naphthalen-1-yl)benzo[b]naphtho [2,3-d]chlorol-5-ium Tetrafluoroborate (4c)
Pink solid, 7.6 mg, 0.015 mmol, 9% yield (from 2).
m.p. = 152–154 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 8.83 (s, 1H), 8.53 (d, J = 8.8 Hz, 1H), 8.36–8.41 (m, 2H), 8.27 (d, J = 8.2 Hz, 1H), 8.12 (d, J = 8.2 Hz, 1H), 7.87–7.92 (m, 1H), 7.76–7.82 (m, 3H), 7.59–7.67 (m, 2H), 7.51 (dd, J = 8.2, 0.5 Hz, 1H), 7.40–7.45 (m, 1H), 7.02 (d, J = 8.5 Hz, 1H), 4.08 (qd, J = 14.0 Hz, 6.8 Hz, 1H), 3.85 (qd, J = 14.0 Hz, 6.8 Hz, 1H), 1.06 (t, J = 6.8 Hz, 3H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 140.6, 139.0, 138.6, 134.1, 133.8, 133.6, 133.06, 133.03, 132.4, 132.1, 131.5, 130.00, 129.99, 129.89, 129.7, 129.28, 129.25, 128.0, 127.4, 126.8, 125.44, 125.31, 124.6, 123.99, 123.86, 121.4, 42.4, 12.3; 19F-NMR (377 MHz, CHLOROFORM-D) δ −151.2; HRMS (ESI+ in MeCN) calcd for C28H20ON2Cl+ [M-BF4-] 435.1259 found 435.1259; IR (KBr) ν 3068, 1458, 1174, 1062, 831 cm−1; [α]20D = −17.4 (c = 0.2, CHCl3).
3.2.3. Synthesis of 4d
To a stirred solution of 4a (13.0 mg, 0.023 mmol, 1.0 equiv) in CHCl3 (3 mL) and water (0.2 mL) was added NaCl (1.3 mg, 0.023 mmol, 10 equiv) at room temperature and stirred for 35 min at the same temperature. The reaction was quenched by the addition of water. The reaction mixture was extracted with CH2Cl2, washed with brine, dried over Na2SO4 and filtered. After the removal of solvent by evaporation to give 4d.
(R)-6-(2-(ethyl(nitroso)amino)naphthalen-1-yl)benzo[b]naphtho [2,3-d]bromol-5-ium chloride (4d)
Yellow solid, 3.0 mg, 0.006 mmol, 25% yield.
m.p. = 137–139 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 9.63 (dd, J = 8.8, 0.7 Hz, 1H), 8.64 (s, 1H), 8.33 (d, J = 8.5 Hz, 1H), 8.27 (dd, J = 7.7, 1.7 Hz, 1H), 8.15 (d, J = 8.2 Hz, 1H), 8.11 (d, J = 8.2 Hz, 1H), 7.83–7.87 (m, 1H), 7.68–7.75 (m, 3H), 7.63–7.67 (m, 1H), 7.47–7.51 (m, 1H), 7.38–7.42 (m, 1H), 7.29 (d, J = 8.7 Hz, 1H), 7.09 (dd, J = 8.5, 0.8 Hz, 1H), 3.94 (qd, J = 14.0 Hz, 7.2 Hz, 1H), 3.70 (qd, J = 14.0 Hz, 7.2 Hz, 1H), 0.99 (t, J = 7.2 Hz, 3H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 139.8, 139.1, 138.7, 135.3, 133.7, 133.5, 133.2, 133.0, 132.2, 131.7, 131.5, 131.3, 130.9, 129.3, 129.16, 129.12, 129.09, 128.95, 128.4, 128.0, 127.4, 126.2, 125.1, 124.93, 124.83, 121.94, 42.3, 12.3; HRMS (ESI+ in MeCN) calcd for C28H20ON2Br+ [M-Cl] 479.0754 found 479.0748; IR (KBr) ν 2962, 1560, 1261, 1019, 801 cm−1; [α]20D = −22.1 (c = 0.2, CHCl3).
3.3. General Procedure for Mannich Reaction of Imines with Active Methylenes
Imine 5 (1.0 equiv), 4 (appropriate amount), and K2CO3 (2.0 equiv) were put into the test tube, which was cooled to –30 °C. After 5 min, the solvent (0.036 M) was slowly added and stirred for 5 min, which was added 6 (2.5 equiv) at the same temperature. The test tube was warmed to the appropriate temperature and stirred for the scheduled time, which was quenched by the adition of water. The reaction mixture was extracted with CH2Cl2, dried over Na2SO4 and filtered. After the removal of solvent by evaporation, the crude product was obtained, which was purified by column chromatography (Silica-gel, hexane/ethyl acetate) to give 7.
3.3.1. dimethyl (R)-2-(1-benzyl-3-((tert-butoxycarbonyl)amino)-2-oxoindolin-3-yl)malonate (7a)
White solid, 12.9 mg, 0.028 mmol, 55% yield, 62% ee.
m.p. = 114–116 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 7.41–7.43 (m, 2H), 7.31–7.38 (m, 3H), 7.24–7.28 (m, 1H), 7.19 (td, J = 7.8, 1.3 Hz, 1H), 6.99 (td, J = 7.8, 1.3 Hz, 1H), 6.71 (d, J = 7.8 Hz, 1H), 6.42 (s, 1H), 5.01 (d, J = 15.8 Hz, 1H), 4.87 (d, J = 15.8 Hz, 1H), 3.96 (s, 1H), 3.70 (s, 3H), 3.69 (s, 3H), 1.31 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 174.0, 166.3, 166.1, 153.9, 143.1, 135.6, 129.5, 128.7, 127.9, 127.6, 123.9, 122.7, 109.3, 80.5, 60.9, 55.3, 52.98, 52.95, 44.4, 28.1; HRMS (ESI+ in MeCN) calcd for C25H28O7N2Na [M + Na+] 491.1789 found 491.1783; IR (KBr) ν 2964, 1261, 1094, 800, 702 cm−1; [α]20D = +9.6 (c = 0.2, CHCl3); HPLC (CHIRALPAK IA column, hexane/2-propanol = 80/20, flow rate 1.0 mL/min, 25 °C, 254 nm) first peak: tR = 8.3 min (major), second peak: tR = 11.4 min (minor).
3.3.2. dimethyl (R)-2-(3-((tert-butoxycarbonyl)amino)-1-methyl-2-oxoindolin-3-yl)malonate (7b) [48]
White solid, 15.7 mg, 0.040 mmol, 80% yield, 31% ee.
1H-NMR (400 MHz, CHLOROFORM-D) δ 7.37 (d, J = 7.4 Hz, 1H), 7.32 (td, J = 7.7, 1.3 Hz, 1H), 7.03 (td, J = 7.7, 1.0 Hz, 1H), 6.83 (d, J = 7.4 Hz, 1H), 6.32 (s, 1H), 3.96 (s, 1H), 3.70 (s, 3H), 3.70 (s, 3H), 3.25 (s, 3H), 1.27 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 173.8, 166.4, 166.1, 153.8, 144.1, 129.7, 127.7, 123.9, 122.7, 108.3, 80.4, 60.8, 55.2, 53.0, 28.1, 26.6; HPLC (CHIRALCEL OD-H column, hexane/2-propanol = 80/20, flow rate 1.0 mL/min, 25 °C, 254 nm) first peak: tR = 6.8 min (major), second peak: tR = 10.7 min (minor).
3.3.3. dimethyl (R)-2-(3-((tert-butoxycarbonyl)amino)-2-oxo-1-phenylindolin-3-yl)malonate (7c)
White solid, 19.9 mg, 0.044 mmol, 88% yield, 55% ee.
m.p. = 129–131 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 7.48–7.53 (m, 4H), 7.38–7.45 (m, 2H), 7.23 (td, J = 7.8, 1.3 Hz, 1H), 7.06 (td, J = 7.5, 0.9 Hz, 1H), 6.80 (d, J = 8.0 Hz, 1H), 6.37 (s, 1H), 4.14 (s, 1H), 3.72 (s, 3H), 3.70 (s, 3H), 1.32 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 173.4, 166.5, 166.1, 153.9, 144.5, 134.4, 129.6, 128.1, 127.4, 126.6, 124.2, 123.1, 109.5, 80.6, 60.8, 55.6, 53.06, 53.00, 28.2; HRMS (ESI+ in MeCN) calcd for C24H26O7N2Na [M + Na+] 477.1632 found 477.1627; IR (KBr) ν 2963, 1261, 1018, 798, 702 cm−1; [α]20D = +17.1 (c = 0.2, CHCl3); HPLC (CHIRALPAK IA column, hexane/2-propanol = 80/20, flow rate 1.0 mL/min, 25 °C, 254 nm) first peak: tR = 7.2 min (major), second peak: tR = 8.2 min (minor).
3.3.4. dimethyl (R)-2-(1-benzyl-3-((tert-butoxycarbonyl)amino)-4-chloro-2-oxoindolin-3-yl)malonate (7d)
White solid, 14.3 mg, 0.028 mmol, 56% yield, racemic.
m.p. = 140–142 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 7.48–7.41 (m, 2H), 7.34–7.31 (m, 2H), 7.28–7.24 (m, 1H), 7.10 (t, J = 8.0 Hz, 1H), 6.90 (dd, J = 8.3, 0.8 Hz, 1H), 6.79 (s, 1H), 6.57 (d, J = 7.8 Hz, 1H), 5.10 (d, J = 16.1 Hz, 1H), 4.81 (d, J = 16.1 Hz, 1H), 4.44 (s, 1H), 3.86 (s, 3H), 3.49 (s, 3H), 1.29 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 173.4, 166.9, 165.4, 153.2, 146.1, 135.2, 130.8, 130.0, 128.7, 127.57, 127.52, 123.5, 107.8, 80.5, 61.2, 53.8, 53.6, 52.8, 44.7, 28.0; HRMS (ESI+ in MeCN) calcd for C25H27O7N2ClNa [M + Na+] 525.1399 found 525.1403; IR (KBr) ν 2980, 1729, 1498, 1257, 912 cm−1; HPLC (CHIRALPAK IA column, hexane/2-propanol = 80/20, flow rate 1.0 mL/min, 25 °C, 220 nm) first peak: tR = 7.2 min, second peak: tR = 11.0 min.
3.3.5. dimethyl (R)-2-(1-benzyl-3-((tert-butoxycarbonyl)amino)-5-methyl-2-oxoindolin-3-yl)malonate (7e)
White solid, 21.2 mg, 0.044 mmol, 88% yield, 60 % ee.
m.p. = 128–130 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 7.39–7.41 (m, 2H), 7.27–7.34 (m, 3H), 7.17 (d, J = 0.9 Hz, 1H), 6.97 (dd, J = 8.0, 0.9 Hz, 1H), 6.58 (d, J = 8.0 Hz, 1H), 6.43 (s, 1H), 4.97 (d, J = 16.5 Hz, 1H), 4.86 (d, J = 16.5 Hz, 1H), 3.93 (s, 1H), 3.70 (s, 3H), 3.69 (s, 3H), 2.26 (s, 3H), 1.32 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 174.0, 166.4, 166.2, 153.9, 140.7, 135.8, 132.2, 129.8, 128.7, 127.9, 127.5, 124.7, 109.1, 80.4, 61.0, 55.3, 52.96, 52.90, 44.4, 28.2, 21.1; HRMS (ESI+ in MeCN) calcd for C26H30O7N2K [M + K+] 521.1685 found 521.1676; IR (KBr) ν 2963, 1719, 1497, 1261, 803 cm−1; [α]20D = +19.8 (c = 0.2, CHCl3); HPLC (CHIRALPAK IA column, hexane/2-propanol = 80/20, flow rate 1.0 mL/min, 25 °C, 254 nm) first peak: tR = 7.8 min (major), second peak: tR = 11.0 min (minor).
3.3.6. dimethyl (R)-2-(1-benzyl-3-((tert-butoxycarbonyl)amino)-5-chloro-2-oxoindolin-3-yl)malonate (7f)
White solid, 22.9 mg, 0.046 mmol, 91% yield, 73% ee.
m.p. = 125–127 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 7.38–7.40 (m, 2H), 7.31–7.36 (m, 3H), 7.25–7.29 (m, 1H), 7.15 (dd, J = 8.3, 2.1 Hz, 1H), 6.60 (d, J = 8.3 Hz, 1H), 6.41 (s, 1H), 4.96 (d, J = 16.3 Hz, 1H), 4.89 (d, J = 16.3 Hz, 1H), 3.92 (s, 1H), 3.74 (s, 3H), 3.70 (s, 3H), 1.34 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 173.6, 166.2, 165.8, 153.8, 141.8, 135.2, 129.5, 128.8, 128.1, 127.7, 127.5, 124.5, 110.3, 80.8, 60.8, 55.2, 53.15, 53.08, 44.6, 28.1; HRMS (ESI+ in MeCN) calcd for C25H27O7N2ClNa [M + Na+] 525.1399 found 525.1396; IR (KBr) ν 2963, 1734, 1261, 1020, 801 cm−1; [α]20D = +15.7 (c = 0.2, CHCl3); HPLC (CHIRALPAK IA column, hexane/2-propanol = 80/20, flow rate 1.0 mL/min, 25 °C, 254 nm) first peak: tR = 8.4 min (major), second peak: tR = 10.8 min (minor).
3.3.7. dimethyl (R)-2-(1-benzyl-6-bromo-3-((tert-butoxycarbonyl)amino)-2-oxoindolin-3-yl)malonate (7g)
White solid, 26.9 mg, 0.049 mmol, 98% yield, 56% ee.
m.p. = 130–132 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 7.39–7.41 (m, 2H), 7.33–7.37 (m, 2H), 7.28–7.31 (m, 1H), 7.24 (d, J = 8.1 Hz, 1H), 7.13 (dd, J = 8.1, 1.8 Hz, 1H), 6.84 (d, J = 1.8 Hz, 1H), 6.39 (s, 1H), 4.96 (d, J = 16.3 Hz, 1H), 4.86 (d, J = 16.3 Hz, 1H), 3.93 (s, 1H), 3.72 (s, 3H), 3.68 (s, 3H), 1.33 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 173.9, 166.2, 165.9, 153.8, 144.5, 135.0, 128.8, 127.8, 127.4, 126.7, 125.6, 125.2, 123.3, 112.7, 80.7, 60.5, 55.0, 53.14, 53.08, 44.5, 28.1; HRMS (ESI+ in MeCN) calcd for C25H27O7N2BrNa [M + Na+] 569.0894 found 569.0892; IR (KBr) ν 2962, 1735, 1261, 1020, 800 cm−1; [α]20D = +14.5 (c = 0.2, CHCl3); HPLC (CHIRALPAK IA column, hexane/2-propanol = 80/20, flow rate 1.0 mL/min, 25 °C, 254 nm) first peak: tR = 7.5 min (major), second peak: tR = 10.0 min (minor).
3.3.8. dimethyl (R)-2-(1-benzyl-3-((tert-butoxycarbonyl)amino)-7-chloro-2-oxoindolin-3-yl)malonate (7h)
White solid, 20.9 mg, 0.042 mmol, 83% yield, 80% ee.
m.p. = 150–152 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 7.40 (d, J = 7.4 Hz, 2H), 7.29–7.33 (m, 2H), 7.22–7.27 (m, 2H), 7.18 (dd, J = 8.2, 1.1 Hz, 1H), 6.94 (t, J = 7.8 Hz, 1H), 6.54 (s, 1H), 5.38 (d, J = 16.3 Hz, 1H), 5.33 (d, J = 16.3 Hz, 1H), 3.86 (s, 1H), 3.72 (s, 3H), 3.71 (s, 3H), 1.33 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 174.8, 166.0, 165.9, 153.8, 139.3, 137.5, 132.2, 130.9, 128.4, 127.01, 126.90, 123.6, 122.2, 115.7, 80.8, 60.5, 55.3, 53.13, 53.08, 45.6, 28.1; HRMS (ESI+ in MeCN) calcd for C25H27O7N2ClNa [M + Na+] 525.1399 found 525.1393; IR (KBr) ν 2962, 1734, 1457, 1261, 802 cm−1; [α]20D = +6.7 (c = 0.2, CHCl3); HPLC (CHIRALPAK IA column, hexane/2-propanol = 80/20, flow rate 1.0 mL/min, 25 °C, 254 nm) first peak: tR = 7.8 min (major), second peak: tR = 11.3 min (minor).
3.3.9. dimethyl (R)-2-(1-benzyl-3-(((benzyloxy)carbonyl)amino)-2-oxoindolin-3-yl)malonate (7i)
White solid, 22.6 mg, 0.041 mmol, 90% yield, 47% ee
m.p. = 137–139 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 7.28–7.39 (m, 2H), 7.25–7.35 (m, 9H), 7.20 (td, J = 7.8, 1.2 Hz, 1H), 7.00 (td, J = 7.8, 1.2 Hz, 1H), 6.81 (s, 1H), 6.68 (d, J = 7.7 Hz, 1H), 4.95–5.03 (m, 4H), 3.93 (s, 1H), 3.73 (s, 3H), 3.67 (s, 3H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 173.6, 166.3, 166.0, 154.4, 143.2, 135.7, 135.5, 129.9, 128.7, 128.5, 128.20, 128.15, 127.6, 127.4, 127.2, 123.9, 122.9, 109.6, 67.2, 60.5, 55.4, 53.14, 53.03, 44.5; HRMS (ESI+ in MeCN) calcd for C28H26O7N2Na [M + Na+] 525.1632 found 525.1633; IR (KBr) ν 2962, 1733, 1260, 1021, 799 cm−1; [α]20D = +4.8 (c = 0.2, CHCl3); HPLC (CHIRALPAK IA column, hexane/2-propanol = 80/20, flow rate 1.0 mL/min, 25 °C, 254 nm) first peak: tR = 20.2 min (major), second peak: tR = 22.9 min (minor).
3.3.10. dibenzyl (R)-2-(1-benzyl-3-((tert-butoxycarbonyl)amino)-2-oxoindolin-3-yl)malonate (7j)
White solid, 27.4 mg, 0.044 mmol, 88% yield, 28% ee.
m.p. = 131–133 °C; 1H-NMR (400 MHz, CHLOROFORM-D) δ 7.20–7.35 (m, 16H), 7.14 (td, J = 7.8, 1.2 Hz, 1H), 6.88 (td, J = 7.8, 1.2 Hz, 1H), 6.61 (d, J = 7.8 Hz, 1H), 6.43 (s, 1H), 5.14 (d, J = 14.0 Hz, 1H), 5.11 (d, J = 14.0 Hz, 1H), 5.06 (d, J = 14.0 Hz, 1H), 5.04 (d, J = 14.0 Hz, 1H), 4.77–4.75 (m, 2H), 4.03 (s, 1H), 1.28 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 173.9, 165.6, 153.8, 143.0, 135.6, 134.61, 134.59, 129.5, 128.65, 128.62, 128.54, 128.52, 128.48, 127.8, 127.5, 124.0, 122.7, 109.3, 80.4, 67.98, 67.91, 61.0, 55.5, 44.2, 28.1; HRMS (ESI+ in MeCN) calcd for C37H36O7N2Na [M + Na+] 643.2415 found 643.2411; IR (KBr) ν 2926, 1734, 1507, 1168, 741 cm−1; [α]20D = −2.6 (c = 0.2, CHCl3); HPLC (CHIRALPAK IA column, hexane/2-propanol = 80/20, flow rate 1.0 mL/min, 25 °C, 254 nm) first peak: tR = 10.1 min (major), second peak: tR = 17.2 min (minor).
3.3.11. diethyl (R)-2-(1-benzyl-3-((tert-butoxycarbonyl)amino)-2-oxoindolin-3-yl)malonate (7k) [47]
White solid, 20.3 mg, 0.041 mmol, 82% yield, 3% ee.
1H-NMR (400 MHz, CHLOROFORM-D) δ 7.38–7.44 (m, 3H), 7.31–7.35 (m, 2H), 7.24–7.28 (m, 1H) 7.18 (td, J = 7.7, 1.3 Hz, 1H), 6.98 (td, J = 7.8, 1.3 Hz, 1H), 6.70 (d, J = 7.8 Hz, 1H), 6.46 (s, 1H), 5.00 (d, J = 15.9 Hz, 1H), 4.86 (d, J = 15.9 Hz, 1H), 4.20–4.12 (m, 4H), 3.91 (s, 1H), 1.31 (s, 9H), 1.187 (t, J = 7.2 Hz, 3H), 1.185 (t, J = 7.2 Hz, 3H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 174.1, 165.9, 165.7, 153.9, 143.2, 135.7, 129.5, 128.7, 128.0, 127.6, 124.1, 122.6, 109.2, 80.4, 62.15, 62.09, 60.9, 55.6, 44.4, 28.1, 13.85, 13.80; HPLC (CHIRALPAK IA column, hexane/2-propanol = 80/20, flow rate 1.0 mL/min, 25 °C, 254 nm) first peak: tR = 7.0 min (major), second peak: tR = 9.9 min (minor).
3.3.12. tert-butyl (S)-(1-benzyl-3-(2,4-dioxopentan-3-yl)-2-oxoindolin-3-yl)carbamate (7m) [48]
White solid, 11.2 mg, 0.026 mmol, 51% yield, 61% ee.
1H-NMR (400 MHz, CHLOROFORM-D) δ 7.42 (d, J = 8.8 Hz, 2H), 7.35 (t, J = 8.8 Hz, 2H), 7.26–7.28 (m, 2H), 7.18 (td, J = 7.8, 1.2 Hz, 1H), 6.98 (td, J = 7.8, 0.9 Hz, 1H), 6.72 (d, J = 7.8 Hz, 1H), 6.58 (s, 1H), 5.05 (d, J = 17.1 Hz, 1H), 4.82 (d, J = 17.1 Hz, 1H), 4.07 (s, 1H), 2.31 (s, 3H), 2.17 (s, 3H), 1.31 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 201.7, 201.3, 174.2, 153.9, 142.5, 135.6, 129.5, 128.8, 128.3, 127.7, 127.5, 123.6, 122.9, 109.5, 80.5, 68.6, 62.8, 44.4, 32.33, 32.26, 28.1; HRMS (ESI+ in MeCN) calcd for C25H28O7N2Na [M + Na+] 491.1789 found 491.1783; HPLC (CHIRALPAK IC column, hexane/2-propanol = 95/5, flow rate 1.0 mL/min, 25 °C, 210 nm) first peak: tR = 196.5 min (major), second peak: tR = 172.7 min (minor), or (CHIRALPAK IA column, hexane/2-propanol = 80/20, flow rate 1.0 mL/min, 25 °C, 254 nm) first peak: tR = 7.4 min (minor), second peak: tR = 15.8 min (major).
3.3.13. tert-butyl (S)-(1-benzyl-3-(1,3-dioxo-1,3-diphenylpropan-2-yl)-2-oxoindolin-3-yl)carbamate (7n) [58]
White solid, 20.3 mg, 0.021 mmol, 72% yield, 5% ee.
1H-NMR (400 MHz, CHLOROFORM-D) δ 7.79–7.81 (m, 2H), 7.65–7.66 (m, 2H), 7.47–7.54 (m, 2H), 7.30–7.39 (m, 10H), 7.16 (td, J = 7.7, 1.1 Hz, 1H), 7.07 (s, 1H), 6.90 (td, J = 7.7, 1.1 Hz, 1H), 6.75 (d, J = 7.7 Hz, 1H), 5.80 (s, 1H), 4.86 (d, J = 17.1 Hz, 1H), 4.69 (d, J = 17.1 Hz, 1H), 1.31 (s, 9H); 13C-NMR (101 MHz, CHLOROFORM-D) δ 192.1, 191.8, 174.4, 153.8, 142.4, 136.7, 136.5, 136.0, 133.8, 133.7, 129.2, 128.8, 128.7, 128.52, 128.46 127.61, 127.55, 125.1, 122.9, 109.1, 80.1, 64.1, 55.9, 44.2, 28.2; HPLC (CHIRALPAK IA column, hexane/2-propanol = 80/20, flow rate 1.0 mL/min, 25 °C, 254 nm) first peak: tR = 10.2 min (minor), second peak: tR = 14.5 min (major).
3.4. Synthesis of 8
Dibenzo[b,d]bromol-5-ium chloride [26] (1.0 equiv) and NaBF4 (10 equiv) were dissolved in CHCl3 (0.025 M) and H2O (0.05 M), which was stirred for 1.5 h at room temperature. The reaction was quenched by the addition of water. The reaction mixture was extracted with CH2Cl2, washed with brine, dried over Na2SO4 and filtered. After the removal of solvent by evaporation to give crude 8, which was washed by hexane to give 8.
dibenzo[b,d]bromol-5-ium Tetrafluoroborate (8) [28]
White solid, 11.1 mg, 0.035 mmol, 70% yield.
1H-NMR (400 MHz, DMSO-D6) δ 8.59 (d, J = 8.5 Hz, 2H), 8.49 (d, J = 8.5 Hz, 2H), 7.95 (t, J = 8.5 Hz, 2H), 7.83–7.87 (dd, J = 8.5 Hz, J = 1.6 Hz, 2H); 13C-NMR (101 MHz, DMSO-D6) δ 136.5, 135.3, 131.7, 131.1, 126.2, 125.5; 19F-NMR (377 MHz, DMSO-D6) δ −148.42.
3.5. Calculation Methods
All calculations were carried out with the Gaussian16 (Revision B.01). The M06-2X functional was employed, which was appropriate for the evaluation of weak non-covalent interactions. The 6–31G(d) was used as basis sets for all atoms. The calculations were carried out in gas phase at 25 °C. The nature of the calculated minimum was confirmed by the expected number of imaginary frequencies (Nimag = 0).
4. Conclusions
In conclusion, we have developed a chiral halonium salt with the N-nitrosamine moiety, which was applied to the Mannich reaction of malonic esters with ketimines to afford corresponding products with up to 80% ee. The catalyst screening, 1H NMR study, and DFT calculation of the catalyst suggested that the halogen bond from the halonium salt moiety and the N-nitrosamine part had a crucial effect on the product’s enantioselectivity. To the best of our knowledge, this is the first catalytic application of N-nitrosamines. The development of a novel reaction, which is to be achieved by the cooperative activation of both the halonium salt and the N-nitrosamine part is ongoing.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28010384/s1. The raw data for the determination of binding constants (Table S1), 1H, 13C, and 19F NMR charts of the catalyst and their synthetic intermediate, and the products, HPLC charts of the products, DFT calculation results.
Author Contributions
Conceptualization, Y.Y.; methodology, Y.Y., T.A., T.M. and M.S.; investigation, T.A.; data curation, Y.Y. and T.A.; writing—original draft preparation, Y.Y.; writing—review and editing, Y.Y. and T.A.; visualization, Y.Y. and T.A.; supervision, Y.Y.; project administration, Y.Y.; funding acquisition, Y.Y., T.M. and M.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a IAAR Research Support Program, Chiba University, Japan, a Grant-in-Aid for Early-Career Scientists (No. 22K14674) from the JSPS, and the Leading Research Promotion Program “Soft Molecular Activation” of Chiba University, Japan.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are included within the manuscript or the supplementary data file. All known compounds are prepared according to the reported procedures.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Lindblad, S.; Mehmeti, K.; Veiga, A.X.; Nekoueishahraki, B.; Gräfenstein, J.; Erdélyi, M. Halogen bond asymmetry in solution. J. Am. Chem. Soc. 2018, 140, 13503–13513. [Google Scholar] [CrossRef] [PubMed]
- Robidas, R.; Legault, C.Y.; Huber, S.M. A low cost, high accuracy method for halogen bonding complexes. Phys. Chem. Chem. Phys. 2021, 23, 3041–3049. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.J. The halogen bond: Nature and applications. Phys. Sci. Rev. 2017, 2, 20170136. [Google Scholar] [CrossRef]
- Nandy, A.; Kazi, I.; Guha, S.; Sekar, G. Visible-light-driven halogen-bond-assisted direct synthesis of heteroaryl thioethers using transition-metal-free one-pot C–I bond formation/C–S cross-coupling reaction. J. Org. Chem. 2021, 86, 2570–2581. [Google Scholar] [CrossRef]
- Decato, D.A.; Sun, J.; Bollera, M.R.; Berryman, O.B. Pushing the limits of the hydrogen bond enhanced halogen bond—The case of the C–H hydrogen bond. Chem. Sci. 2022, 13, 11156–11162. [Google Scholar] [CrossRef]
- Yang, H.; Wong, M.W. Application of halogen bonding to organocatalysis: A theoretical perspective. Molecules 2020, 25, 1045. [Google Scholar] [CrossRef]
- Jónsson, H.F.; Sethio, D.; Wolf, J.; Huber, S.M.; Fiksdahl, A.; Erdelyi, M. Halogen bond activation in gold catalysis. ACS Catal. 2022, 12, 7210–7220. [Google Scholar] [CrossRef]
- Minakata, S.; Miwa, H.; Yamamoto, K.; Hirayama, A.; Okumura, S. Diastereodivergent intermolecular 1,2-diamination of unactivated alkenes enabled by iodine catalysis. J. Am. Chem. Soc. 2021, 143, 4112–4118. [Google Scholar] [CrossRef]
- Riel, A.M.S.; Decato, D.A.; Sun, J.; Berryman, O.B. Halogen bonding organocatalysis enhanced through intramolecular hydrogen bonds. Chem. Commun. 2022, 58, 1378–1381. [Google Scholar] [CrossRef]
- Kniep, F.; Jungbauer, S.H.; Zhang, Q.; Walter, S.M.; Schindler, S.; Schnapperelle, I.; Herdtweck, E.; Huber, S.M. Organocatalysis by neutral multidentate halogen-bond donors. Angew. Chem. Int. Ed. 2013, 52, 7028–7032. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Nakatsuji, Y.; Li, S.; Tsuzuki, S.; Takemoto, Y. Direct N-glycofunctionalization of amides with glycosyl trichloroacetimidate by thiourea/halogen bond donor co-catalysis. Angew. Chem. Int. Ed. 2018, 57, 3646–3650. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Loh, C.C.J. A multistage halogen bond catalyzed strain-release glycosylation unravels new hedgehog signaling inhibitors. J. Am. Chem. Soc. 2019, 141, 5381–5391. [Google Scholar] [CrossRef] [PubMed]
- Kuwano, S.; Suzuki, T.; Yamanaka, M.; Tsutsumi, R.; Arai, T. Catalysis based on C−I⋅⋅⋅π halogen bonds: Electrophilic activation of 2-alkenylindoles by cationic halogen-bond donors for [4+2] cycloadditions. Angew. Chem. Int. Ed. 2019, 58, 10220–10224. [Google Scholar] [CrossRef]
- Sutar, R.L.; Huber, S.M. Catalysis of organic reactions through halogen bonding. ACS Catal. 2019, 9, 9622–9639. [Google Scholar] [CrossRef]
- Robidas, R.; Reinhard, D.L.; Huber, S.M.; Legault, C.Y. A quantum-chemical analysis on the lewis acidity of diarylhalonium ions. ChemPhysChem 2022. [Google Scholar] [CrossRef]
- Oishi, S.; Fujinami, T.; Masui, Y.; Suzuki, T.; Kato, M.; Ohtsuka, N.; Momiyama, N. Three-center-four-electron halogen bond enables non-metallic complex catalysis for Mukaiyama-Mannich-type reaction. iScience 2022, 25, 105220. [Google Scholar] [CrossRef]
- Zong, L.; Ban, X.; Kee, C.W.; Tan, C.-H. Catalytic Enantioselective alkylation of sulfenate anions to chiral heterocyclic sulfoxides using halogenated pentanidium salts. Angew. Chem. Int. Ed. 2014, 53, 11849–11853. [Google Scholar] [CrossRef]
- Lu, Y.H.; Nakatsuji, H.; Okumura, Y.; Yao, L.; Ishihara, K. Enantioselective halo-oxy- and halo-azacyclizations induced by chiral amidophosphate catalysts and halo-lewis acids. J. Am. Chem. Soc. 2018, 140, 6039–6043. [Google Scholar] [CrossRef]
- Kuwano, S.; Suzuki, T.; Hosaka, Y.; Arai, T. A chiral organic base catalyst with halogen-bonding-donor functionality: Asymmetric Mannich reactions of malononitrile with N-Boc aldimines and ketimines. Chem. Commun. 2018, 54, 3847–3850. [Google Scholar] [CrossRef]
- Zhang, X.; Ren, J.; Tan, S.M.; Tan, D.; Lee, R.; Tan, C.-H. An enantioconvergent halogenophilic nucleophilic substitution (SN2X) reaction. Science 2019, 363, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.-C.; Yeung, Y.-Y. Halogen-bond-catalyzed addition of carbon-based nucleophiles to N-acylimminium ions. Org. Lett. 2019, 21, 5665–5669. [Google Scholar] [CrossRef] [PubMed]
- Sutar, R.L.; Engelage, E.; Stoll, R.; Huber, S.M. Bidentate chiral Bis(imidazolium)-based halogen-bond donors: Synthesis and applications in enantioselective recognition and catalysis. Angew. Chem. Int. Ed. 2020, 59, 6806–6810. [Google Scholar] [CrossRef] [PubMed]
- Kuwano, S.; Nishida, Y.; Suzuki, T.; Arai, T. Catalytic asymmetric mannich-type reaction of malononitrile with N-Boc α-Ketiminoesters using chiral organic base catalyst with halogen bond donor functionality. Adv. Synth. Catal. 2020, 362, 1674–1678. [Google Scholar] [CrossRef]
- Ochiai, M.; Miyamoto, K.; Kaneaki, T.; Hayashi, S.; Nakanishi, W. Highly regioselective amination of unactivated alkanes by hypervalent sulfonylimino-λ3-bromane. Science 2011, 332, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Riedmüller, S.; Nachtsheim, B.J. Palladium-catalyzed synthesis of N-arylated carbazoles using anilines and cyclic diaryliodonium salts. Beilstein J. Org. Chem. 2013, 9, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Murray, J.S.; Politzer, P.; Pilati, T.; Ursini, M.; Resnati, G. Halogen bonding in hypervalent iodine and bromine derivatives: Halonium salts. IUCrJ 2017, 4, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Lanzi, M.; Dherbassy, Q.; Wencel-Delord, J. Cyclic diaryl λ3-bromanes as original aryne precursors. Angew. Chem. Int. Ed. 2021, 60, 14852–14857. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Saito, M.; Tsuji, S.; Takagi, T.; Shiro, M.; Uchiyama, M.; Ochiai, M. Benchtop-stable hypervalent Bromine(III) compounds: Versatile strategy and platform for air- and moisture-stable λ3-bromanes. J. Am. Chem. Soc. 2021, 143, 9327–9331. [Google Scholar] [CrossRef]
- Yoshimura, A.; Zhdankin, V.V. Advances in synthetic applications of hypervalent iodine compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef]
- Parra, A. Chiral hypervalent iodines: Active players in asymmetric synthesis. Chem. Rev. 2019, 119, 12033–12088. [Google Scholar] [CrossRef] [PubMed]
- Heinen, F.; Engelage, E.; Dreger, A.; Weiss, R.; Huber, S.M. Iodine(III) derivatives as halogen bonding organocatalysts. Angew. Chem. Int. Ed. 2018, 57, 3830–3833. [Google Scholar] [CrossRef] [PubMed]
- Heinen, F.; Engelage, E.; Cramer, C.J.; Huber, S.M. Hypervalent Iodine(III) compounds as biaxial halogen bond donors. J. Am. Chem. Soc. 2020, 142, 8633–8640. [Google Scholar] [CrossRef] [PubMed]
- Mayer, R.J.; Ofial, A.R.; Mayr, H.; Legault, C.Y. Lewis acidity scale of diaryliodonium ions toward oxygen, nitrogen, and halogen lewis bases. J. Am. Chem. Soc. 2020, 142, 5221–5233. [Google Scholar] [CrossRef]
- Heinen, F.; Reinhard, D.L.; Engelage, E.; Huber, S.M. A bidentate Iodine(III)-based halogen-bond donor as a powerful organocatalyst. Angew. Chem. Int. Ed. 2021, 60, 5069–5073. [Google Scholar] [CrossRef]
- Robidas, R.; Reinhard, D.L.; Legault, C.Y.; Huber, S.M. Iodine(III)-based halogen bond donors: Properties and applications. Chem. Rec. 2021, 21, 1912–1927. [Google Scholar] [CrossRef]
- Yoshida, Y.; Ishikawa, S.; Mino, T.; Sakamoto, M. Bromonium salts: Diaryl-λ3-bromanes as halogen-bonding organocatalysts. Chem. Commun. 2021, 57, 2519–2522. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, J.; Liu, Z.-J. Diaryliodonium salts as efficient Lewis acid catalysts for direct three component Mannich reactions. RSC Adv. 2015, 5, 25485–25488. [Google Scholar] [CrossRef]
- Yoshida, Y.; Mino, T.; Sakamoto, M. Chiral hypervalent Bromine(III) (bromonium salt): Hydrogen- and halogen-bonding bifunctional asymmetric catalysis by diaryl-λ3-bromanes. ACS Catal. 2021, 11, 13028–13033. [Google Scholar] [CrossRef]
- Yoshida, Y.; Fujimura, T.; Mino, T.; Sakamoto, M. Chiral binaphthyl-based iodonium salt (hypervalent Iodine(III)) as hydrogen- and halogen-bonding bifunctional catalyst: Insight into abnormal counteranion effect and asymmetric synthesis of N,S-acetals. Adv. Synth. Catal. 2022, 364, 1091–1098. [Google Scholar] [CrossRef]
- Beard, J.C.; Swager, T.M. An organic chemist’s guide to N-nitrosamines: Their structure, reactivity, and role as contaminants. J. Org. Chem. 2021, 86, 2037–2057. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Pi, C.; Cui, X.; Wu, Y. Rh(III)-catalyzed tandem acylmethylation/nitroso migration/cyclization of N-nitrosoanilines with sulfoxonium ylides in one pot: Approach to 3-nitrosoindoles. Org. Lett. 2020, 22, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Jiao, N. Cationic Cobalt(III) catalyzed indole synthesis: The regioselective intermolecular cyclization of N-nitrosoanilines and alkynes. Angew. Chem. Int. Ed. 2016, 55, 4035–4039. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fan, Y.; Gao, Y.; Sun, C.; Xu, C.; Zhu, J. Rhodium(III)-catalyzed N-nitroso-directed C-H olefination of arenes. High-yield, versatile coupling under mild conditions. J. Am. Chem. Soc. 2013, 135, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Chen, X.; Shao, Y.; Xie, H.; Deng, Y.; Ke, Z.; Jiang, H.; Zeng, W. Co(III)-catalyzed coupling-cyclization of aryl C–H Bonds with α-diazoketones involving wolff rearrangement. ACS Catal. 2018, 8, 1308–1312. [Google Scholar] [CrossRef]
- Yoshimura, M.; Muraoka, T.; Nakatsuka, H.; Huang, H.; Kitamura, M. Synthesis of 3,3′-diaryl-substituted 2,2′-diamino- 1,1′-binaphthyl and its derivatives. J. Org. Chem. 2010, 75, 4315–4318. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Wang, D.; Feng, J.; Li, P.; Zhao, D.; Wang, R. Synthesis of N-alkoxycarbonyl ketimines derived from isatins and their application in enantioselective synthesis of 3-aminooxindoles. Org. Lett. 2012, 14, 2512–2515. [Google Scholar] [CrossRef]
- Rao, K.S.; Ramesh, P.; Chowhan, L.R.; Trivedi, R. Asymmetric Mannich reaction: Highly enantioselective synthesis of 3-amino-oxindoles via chiral squaramide based H-bond donor catalysis. RSC Adv. 2016, 6, 84242–84247. [Google Scholar] [CrossRef]
- Il’in, M.V.; Sysoeva, A.A.; Novikov, A.S.; Bolotin, D.S. Diaryliodoniums as hybrid hydrogen- and halogen-bond-donating organocatalysts for the Groebke-Blackburn-Bienaymé reaction. J. Org. Chem. 2022, 87, 4569–4579. [Google Scholar] [CrossRef]
- Available online: http://supramolecular.org (accessed on 26 December 2022).
- Hibbert, D.B.; Thordarson, P. The death of the Job plot, transparency, open science and online tools, uncertainty estimation methods and other developments in supramolecular chemistry data analysis. Chem. Commun. 2016, 52, 12792–12805. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian, 16th ed.; revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, 16th ed.; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Hu, F.-L.; Wei, Y.; Shi, M.; Pindi, S.; Li, G. Asymmetric catalytic aza-Morita–Baylis–Hillman reaction for the synthesis of 3-substituted-3-aminooxindoles with chiral quaternary carbon centers. Org. Biomol. Chem. 2013, 11, 1921–1924. [Google Scholar] [CrossRef] [PubMed]
- Seayad, A.M.; Ramalingam, B.; Yoshinaga, K.; Nagata, T.; Chai, C.L.L. Highly enantioselective titanium-catalyzed cyanation of imines at room temperature. Org. Lett. 2010, 12, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Jing, C.; Zhai, C.; Hu, W. A novel method for synthesizing N-alkoxycarbonyl aryl α-imino esters and their applications in enantioselective transformations. Adv. Synth. Catal. 2012, 354, 301–307. [Google Scholar] [CrossRef]
- Da Silva, C.D.G.; Katla, R.; dos Santos, B.F.; Tavares, J.M.C., Jr.; Albuquerque, T.B.; Kupfer, V.L.; Rinaldi, A.W.; Domingues, N.L.C. Cobalt used as a novel and reusable catalyst: A new and one-pot synthesis of isatin-derived N,S-acetals using substituted isatins and thiols. Synthesis 2019, 51, 4014–4022. [Google Scholar] [CrossRef]
- Rodríguez-Ferrer, P.; Sanz-Novo, M.; Maestro, A.; Andrés, J.M.; Pedrosa, R. Synthesis of enantioenriched 3-amino-3-substituted oxindoles by stereoselective mannich reaction catalyzed by supported bifunctional thioureas. Adv. Synth. Catal. 2019, 361, 3645–3655. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).