
Medicinal Chemistry of Anti-HIV-1 Latency Chemotherapeutics: Biotargets, Binding Modes and Structure-Activity Relationship Investigation



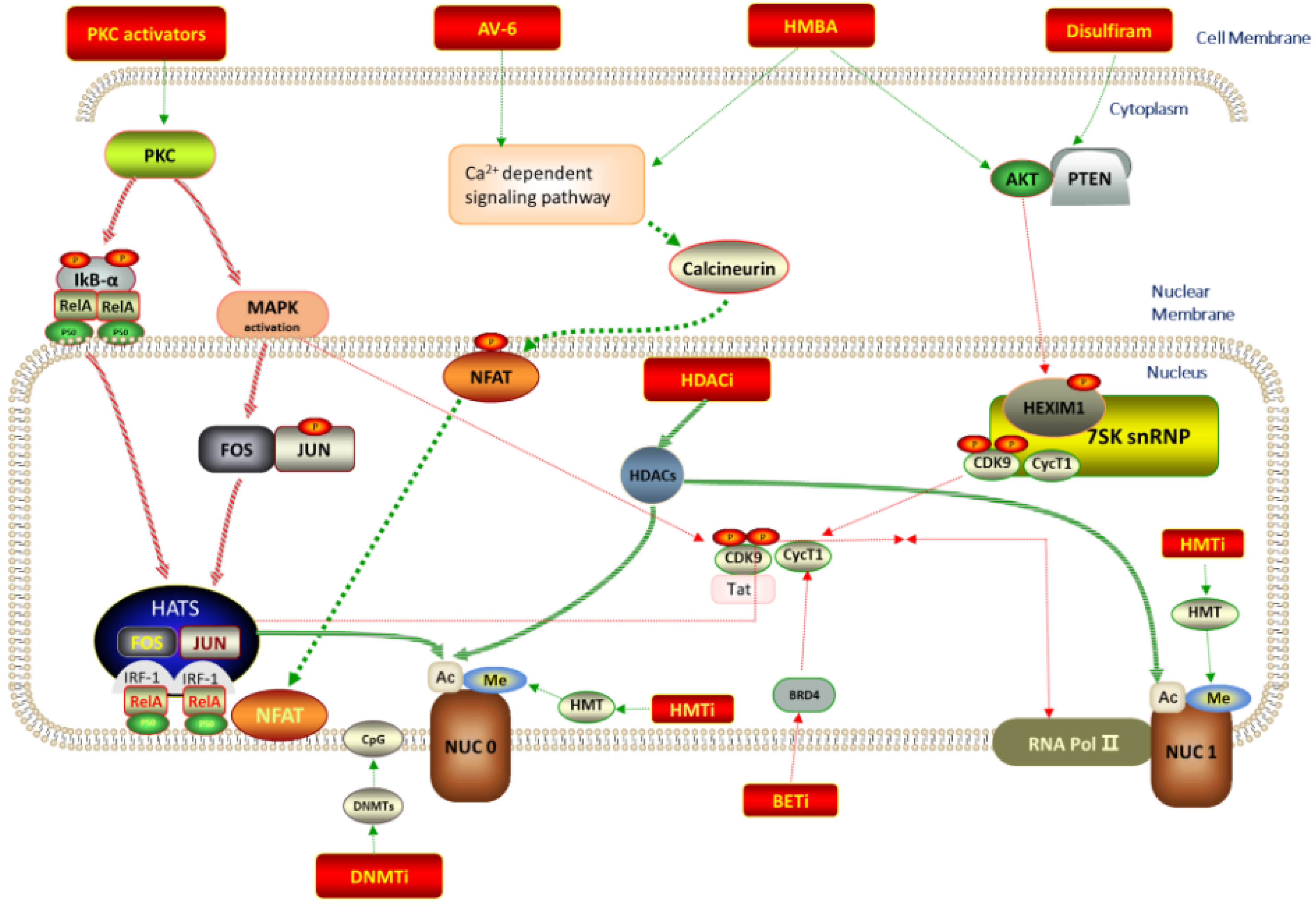{kind=link}
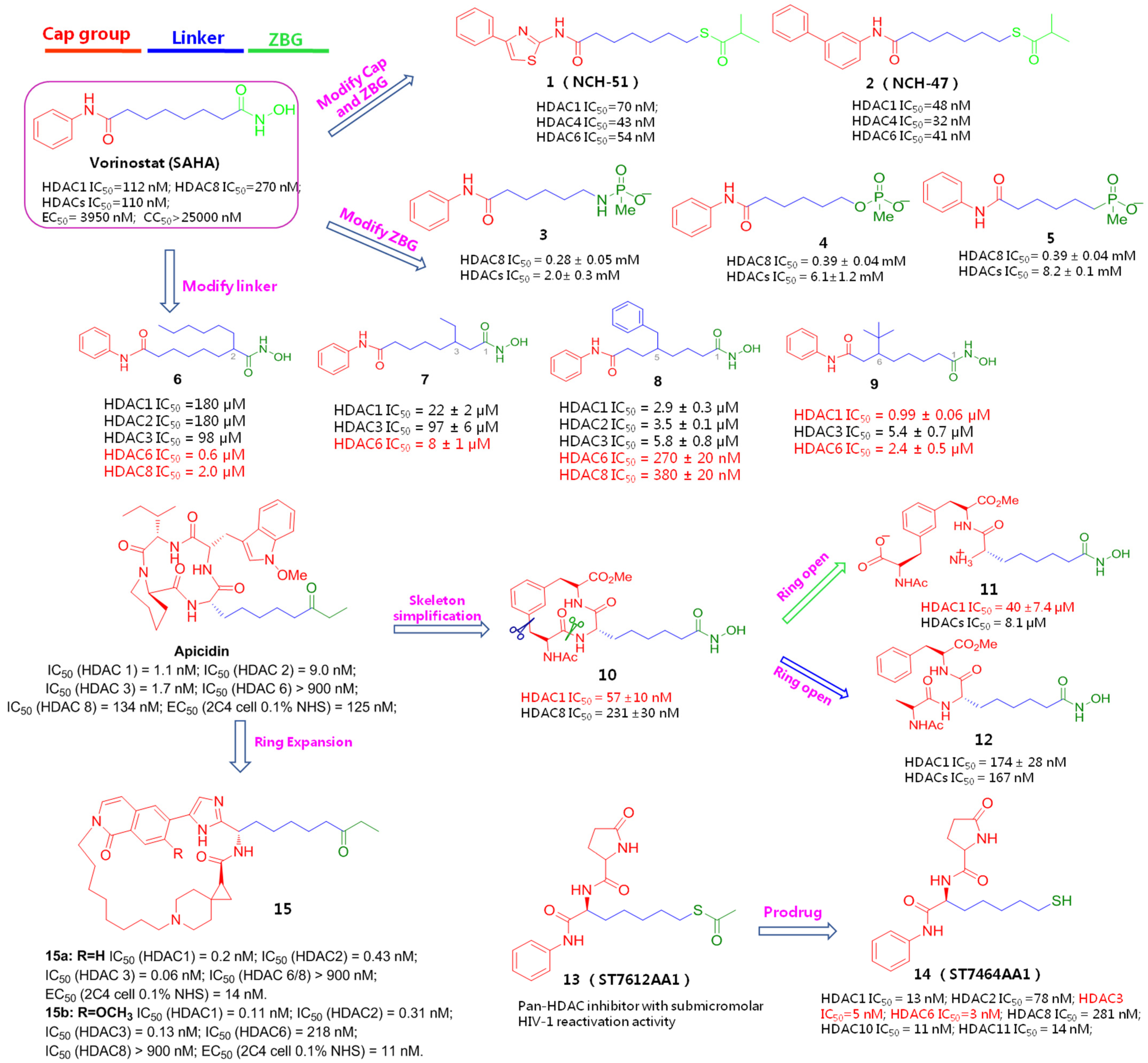{kind=link}
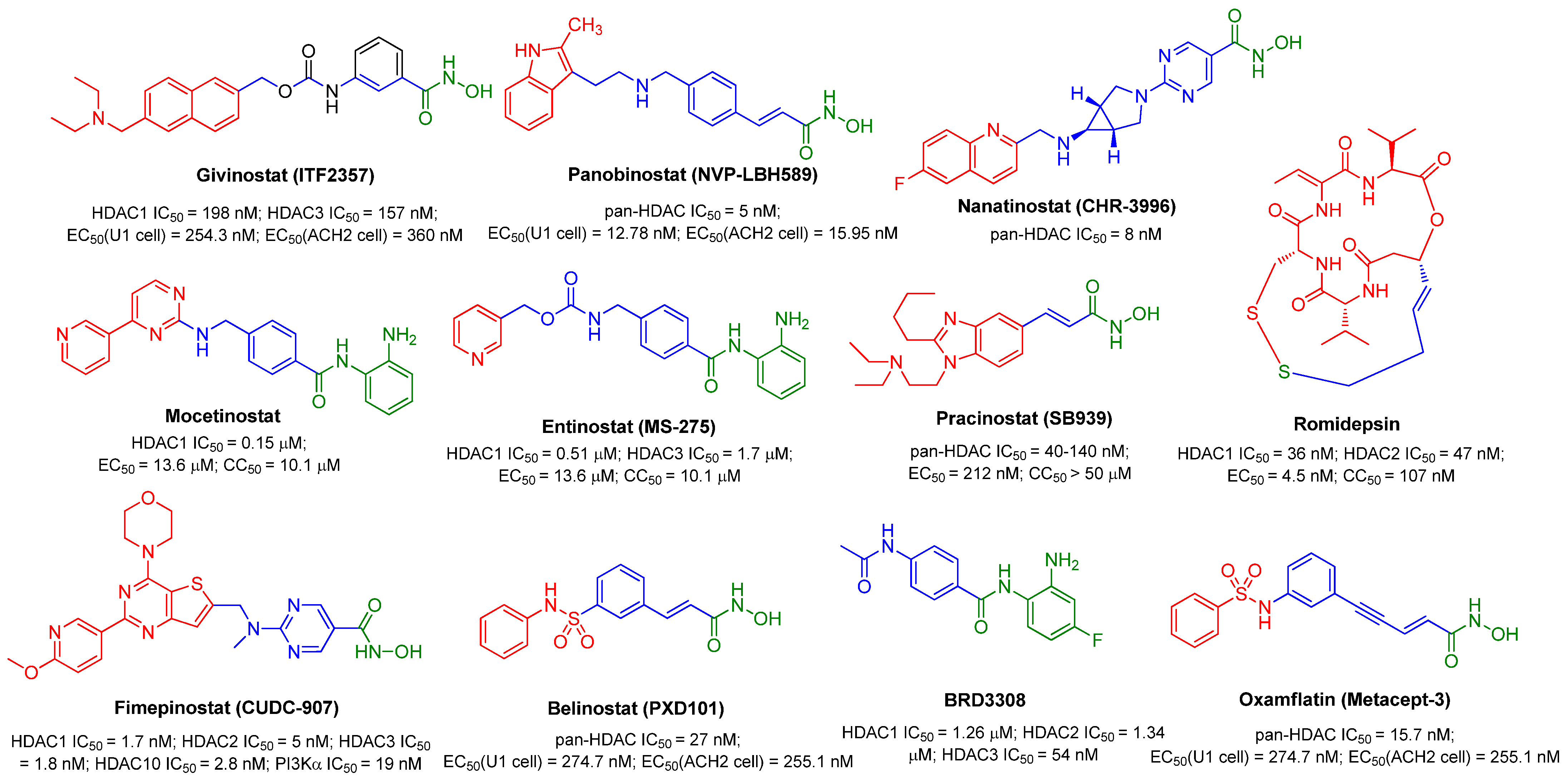{kind=link}
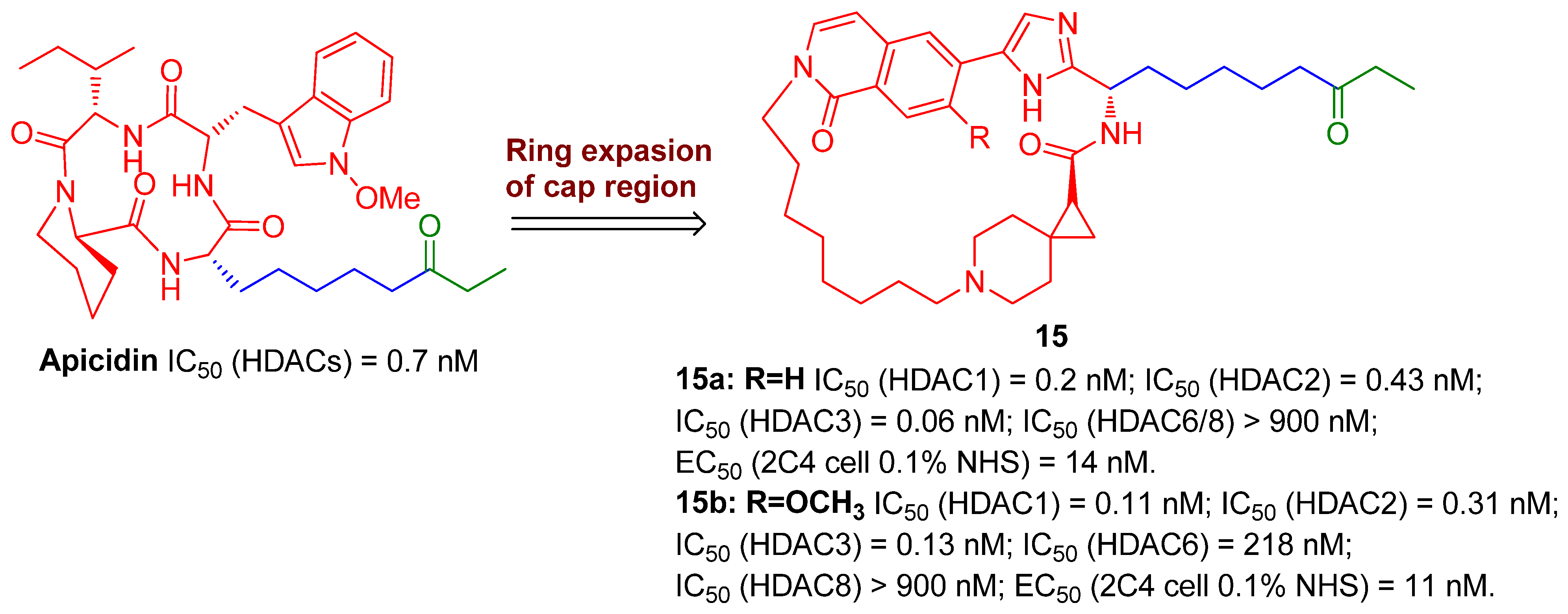{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Possible Biotargets and Related LRAs
2.1. Histone Deacetylase Inhibitors (HDACIs)
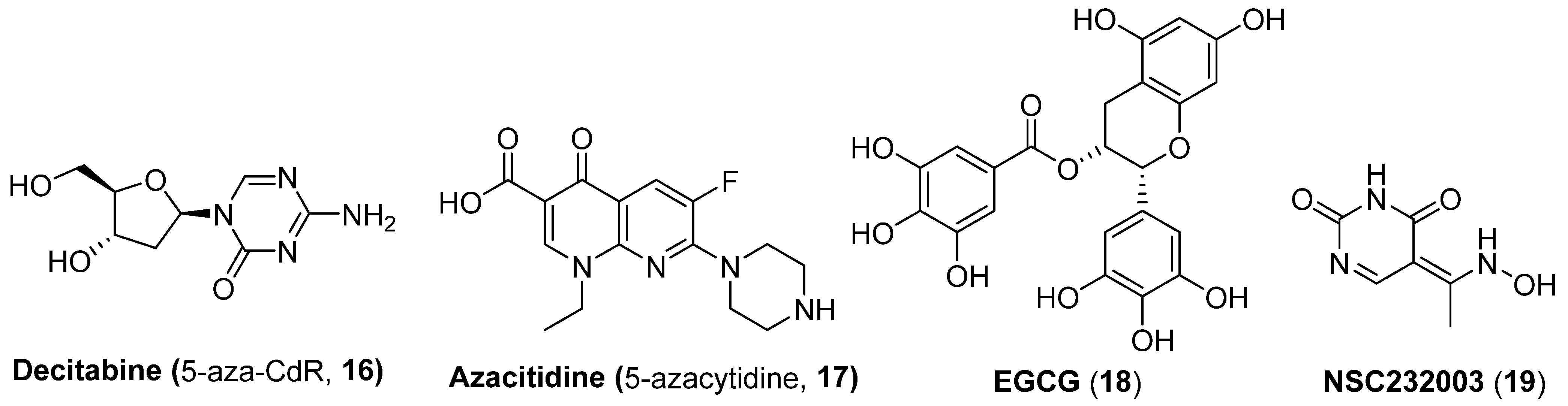
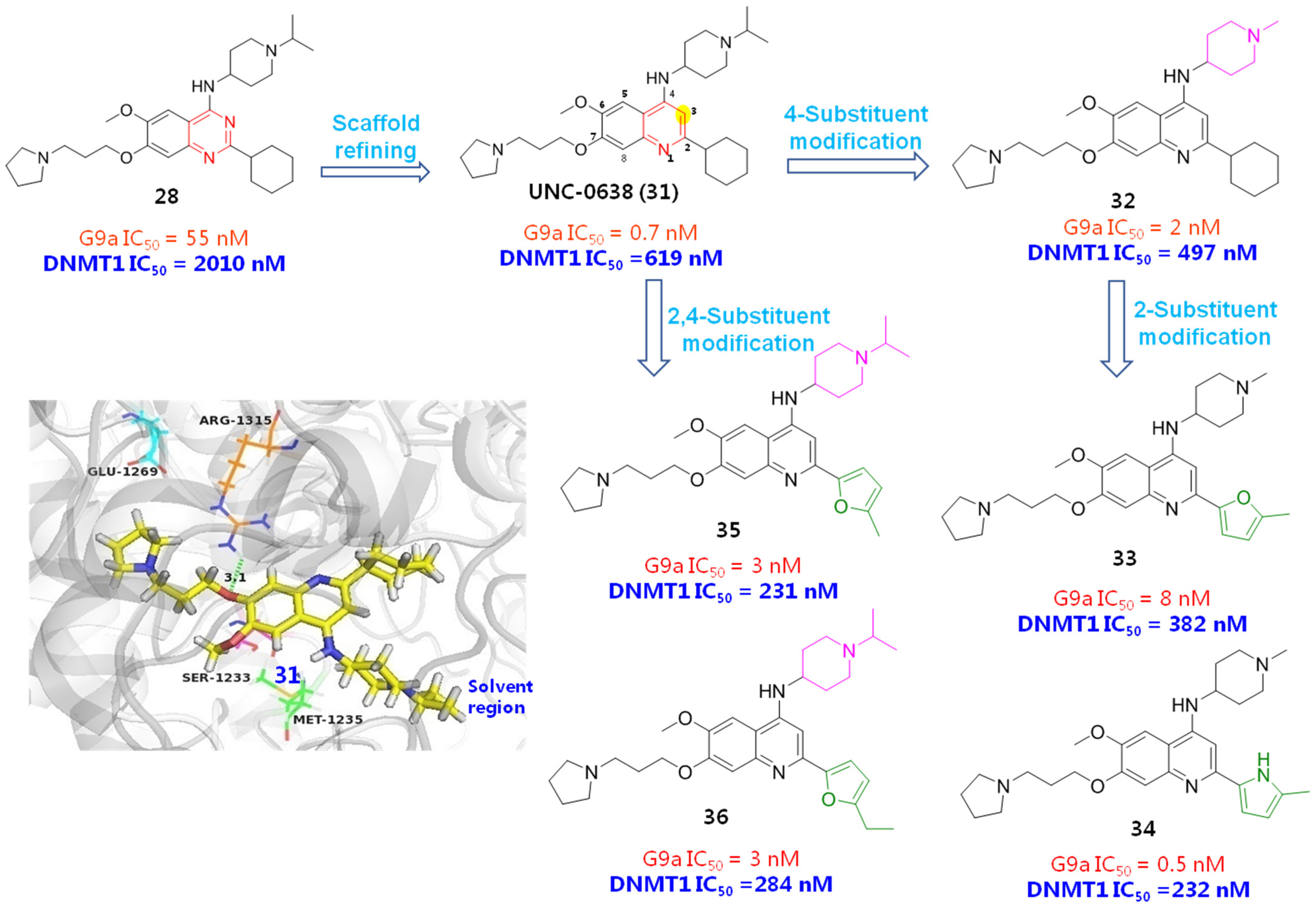
2.2. DNA Methyltransferase Inhibitors (DMTIs)
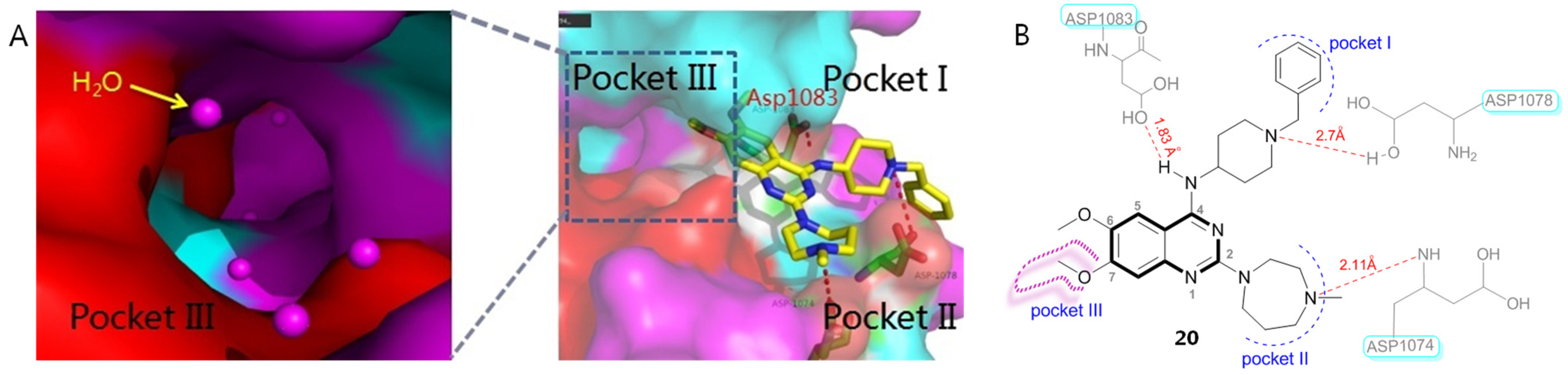
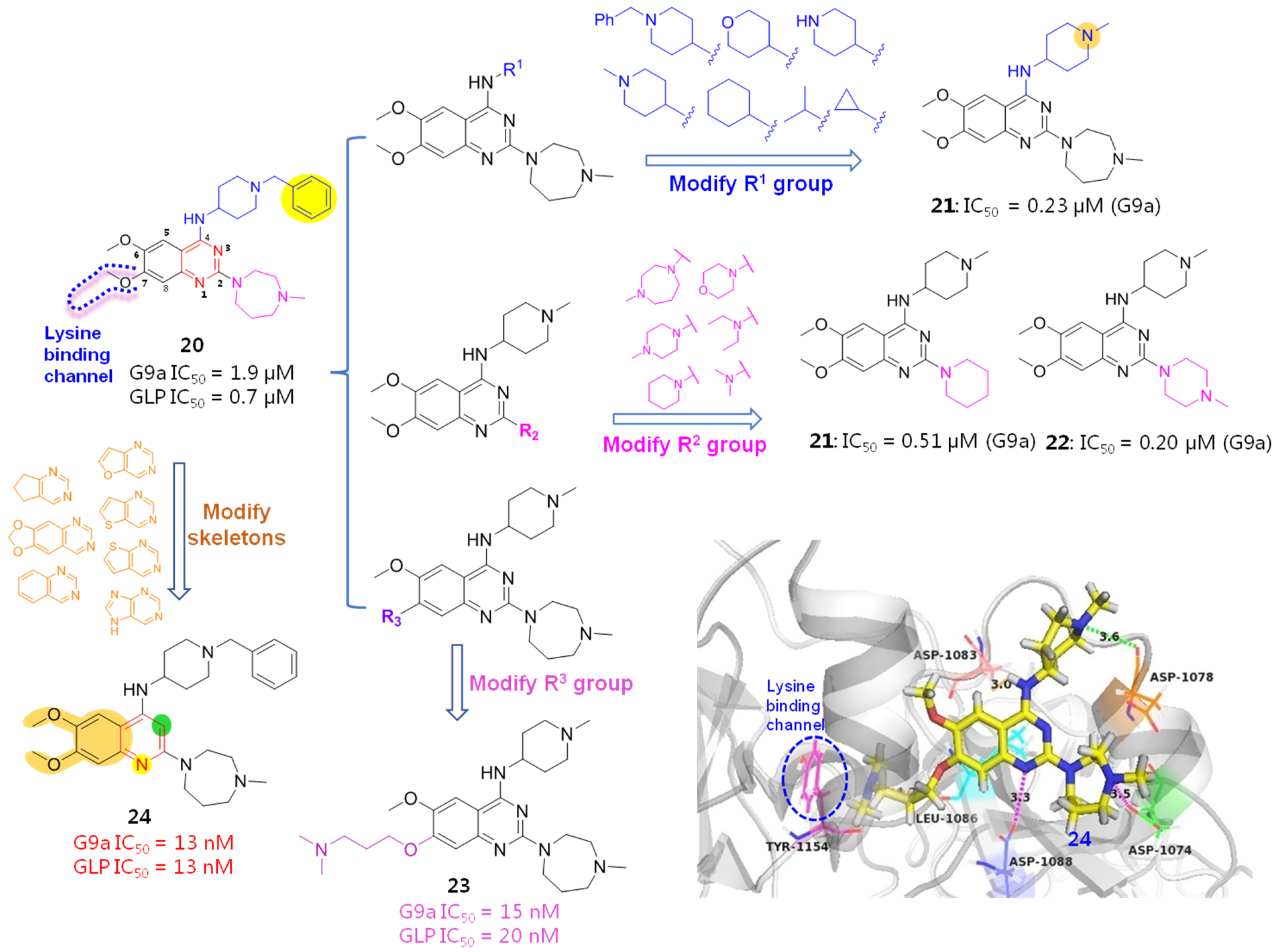
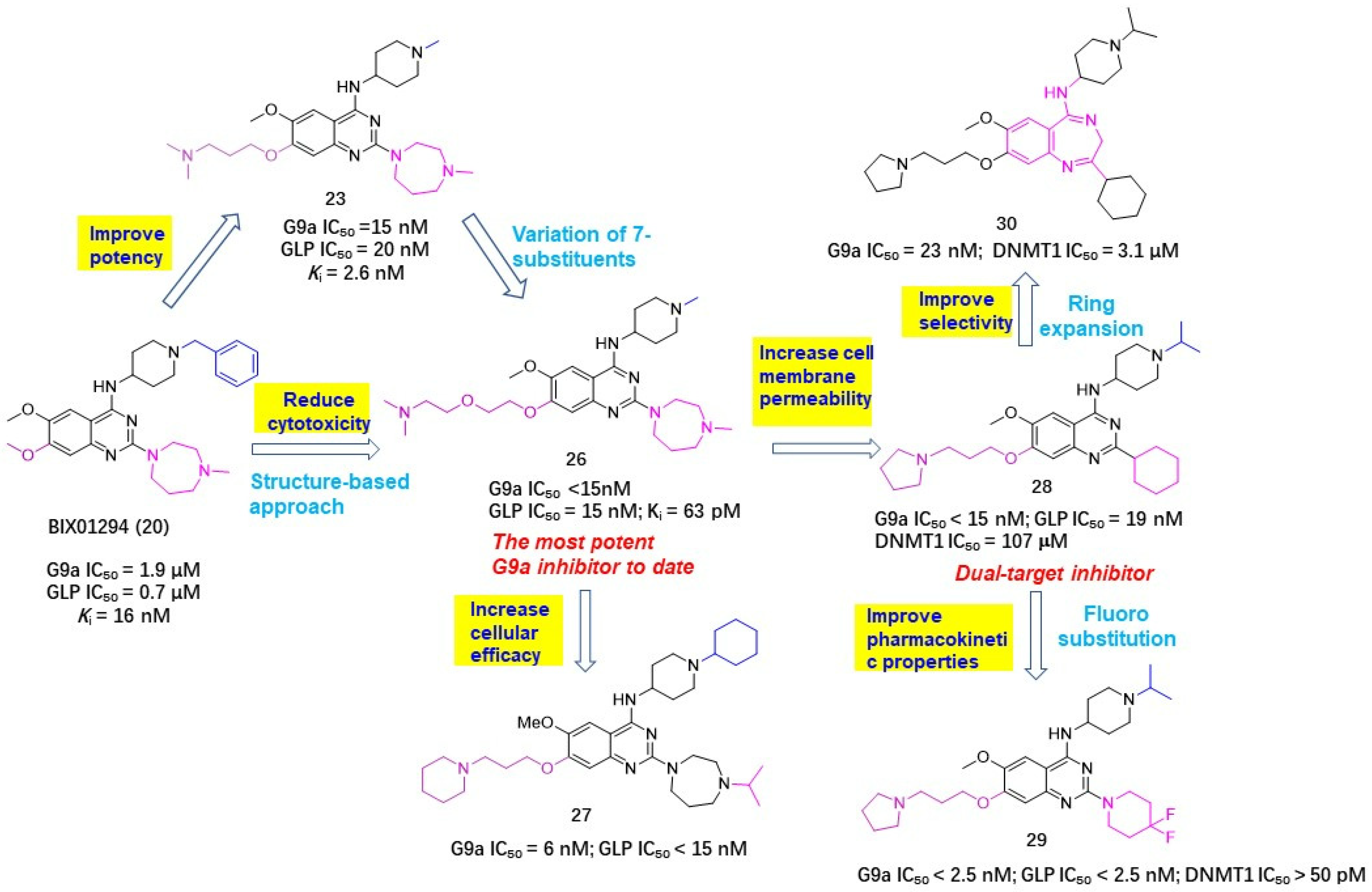
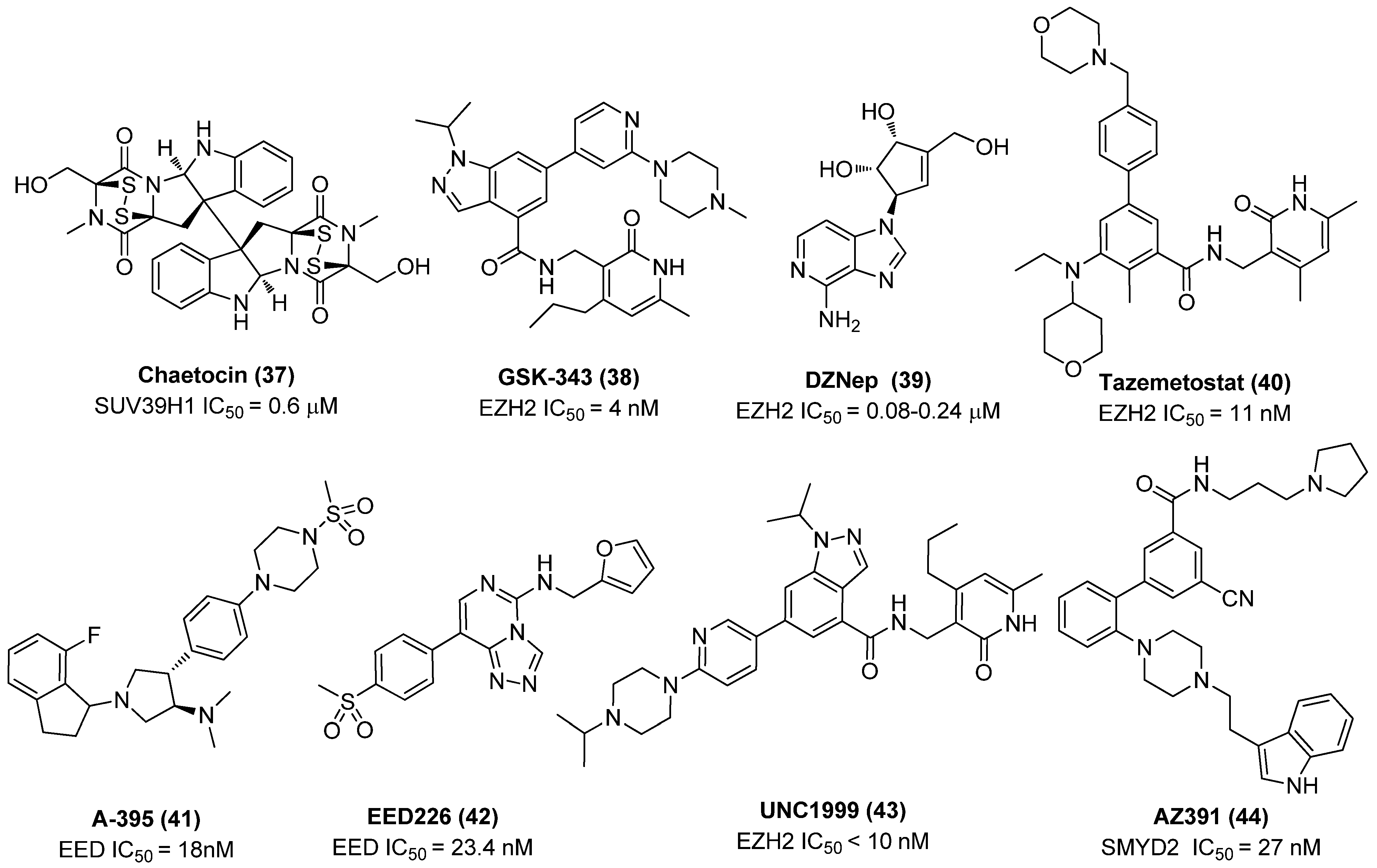
2.3. Histone Methyltransferase Inhibitors (HMTIs)
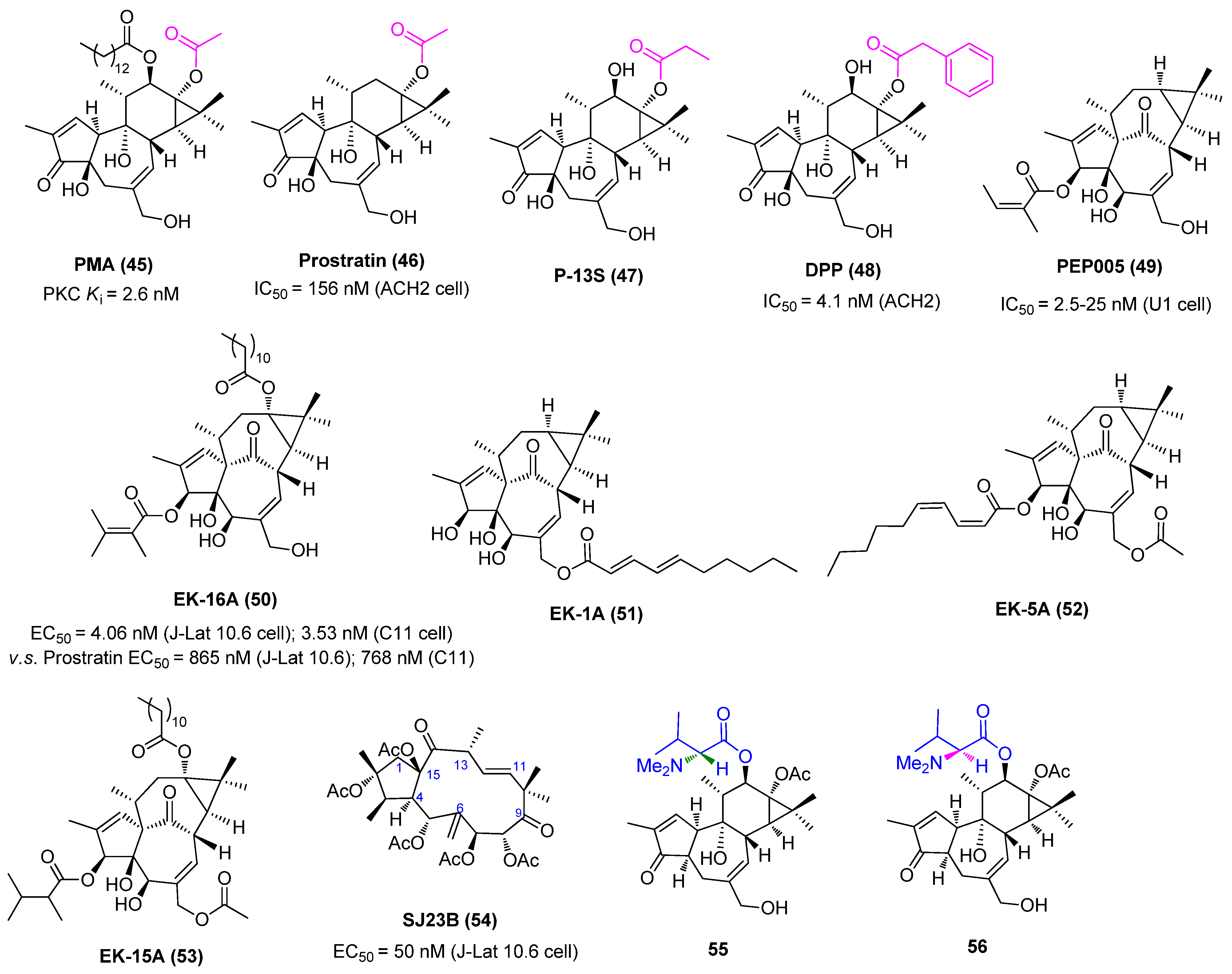
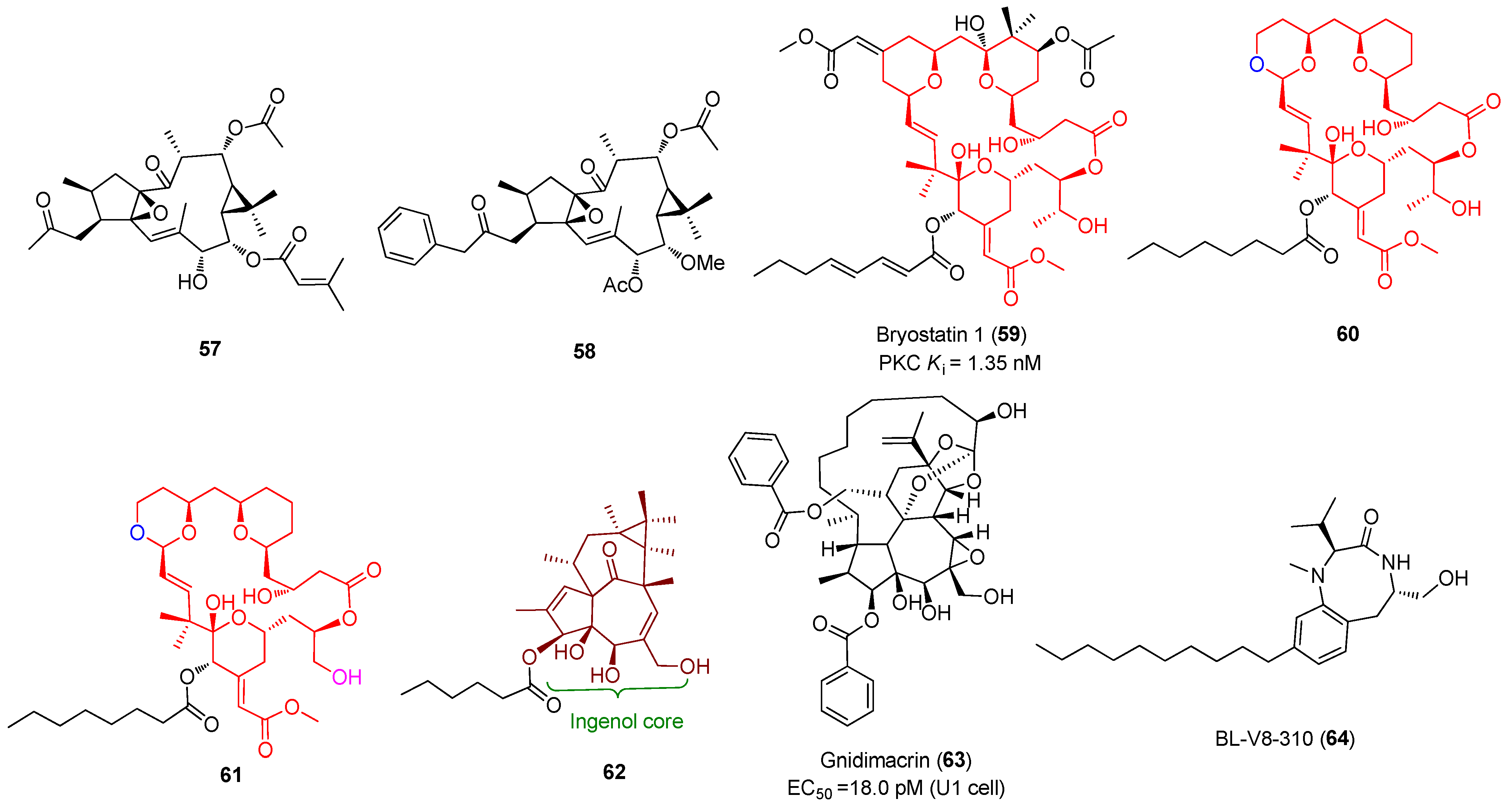
2.4. Protein Kinase C Activators
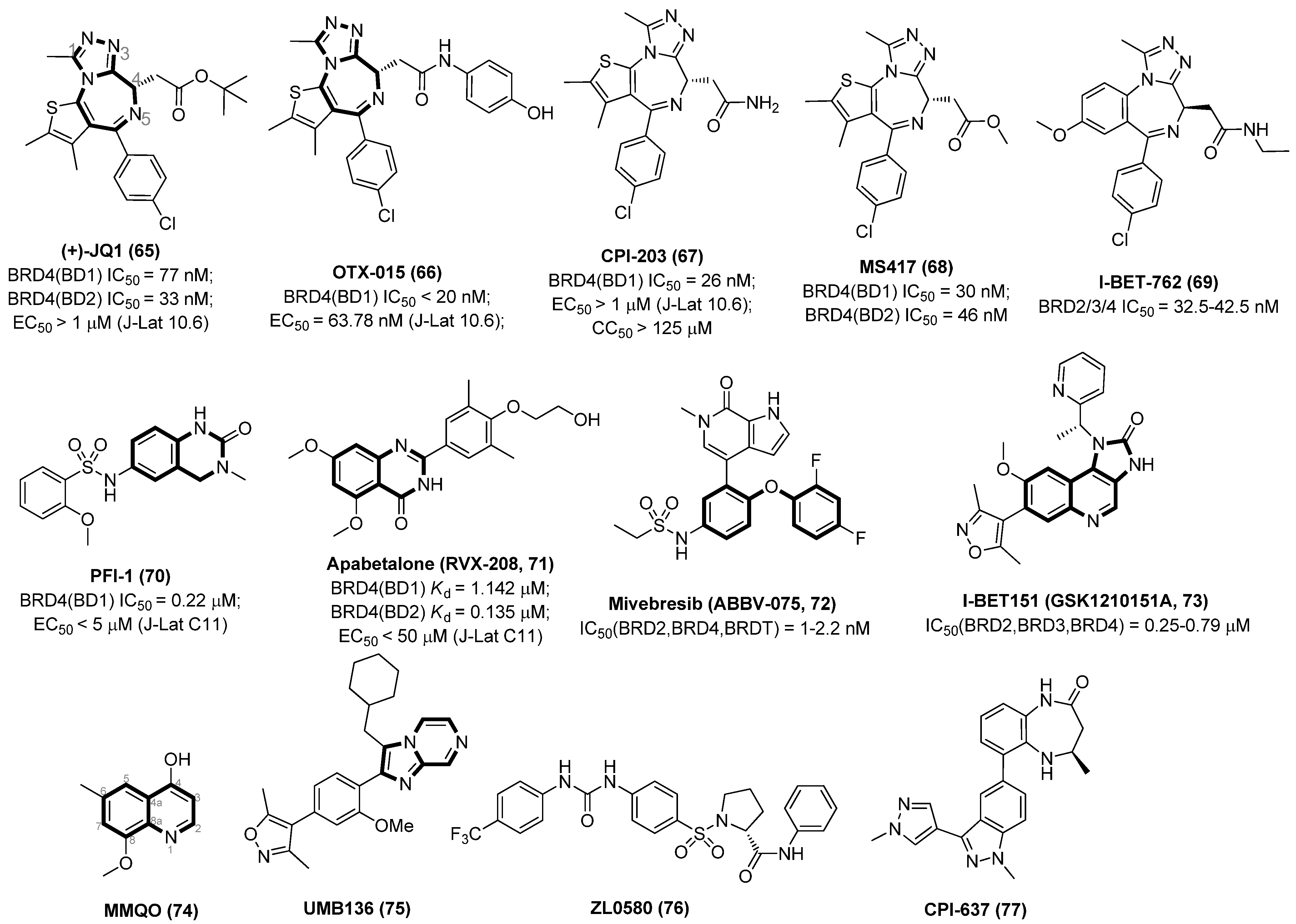
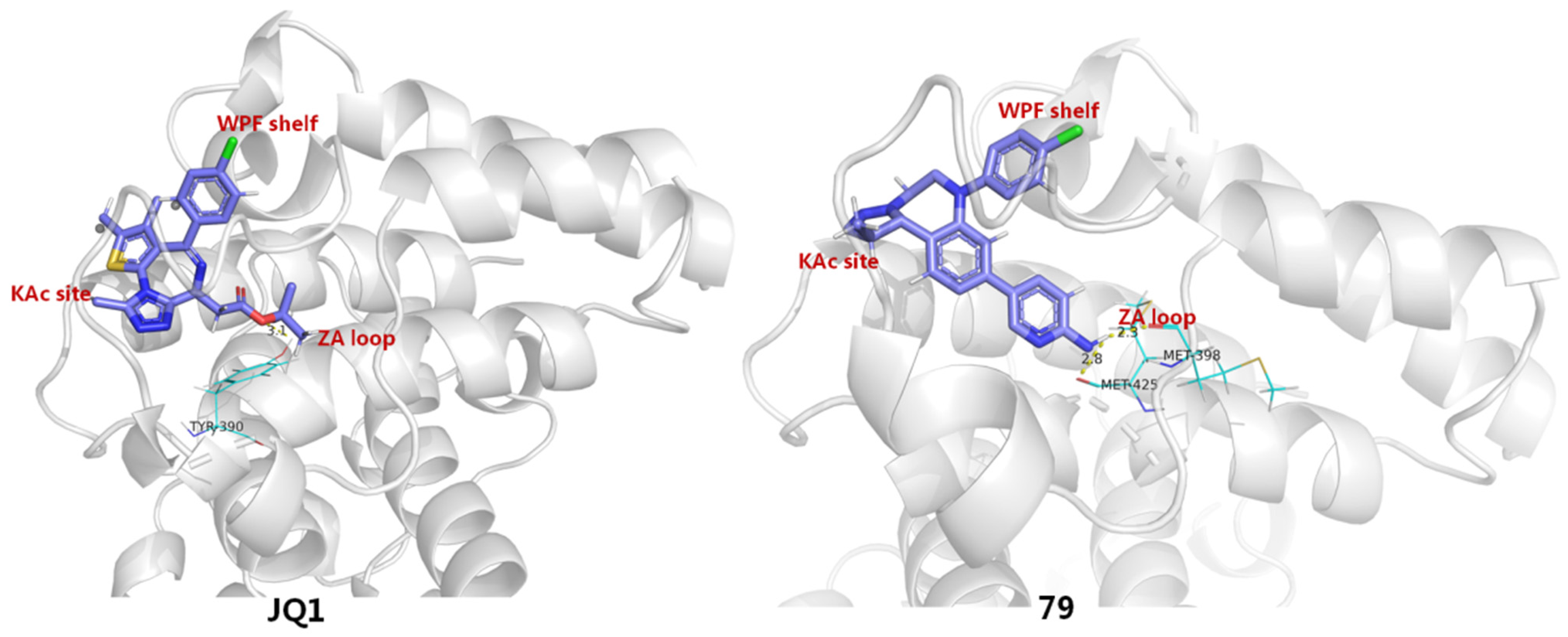
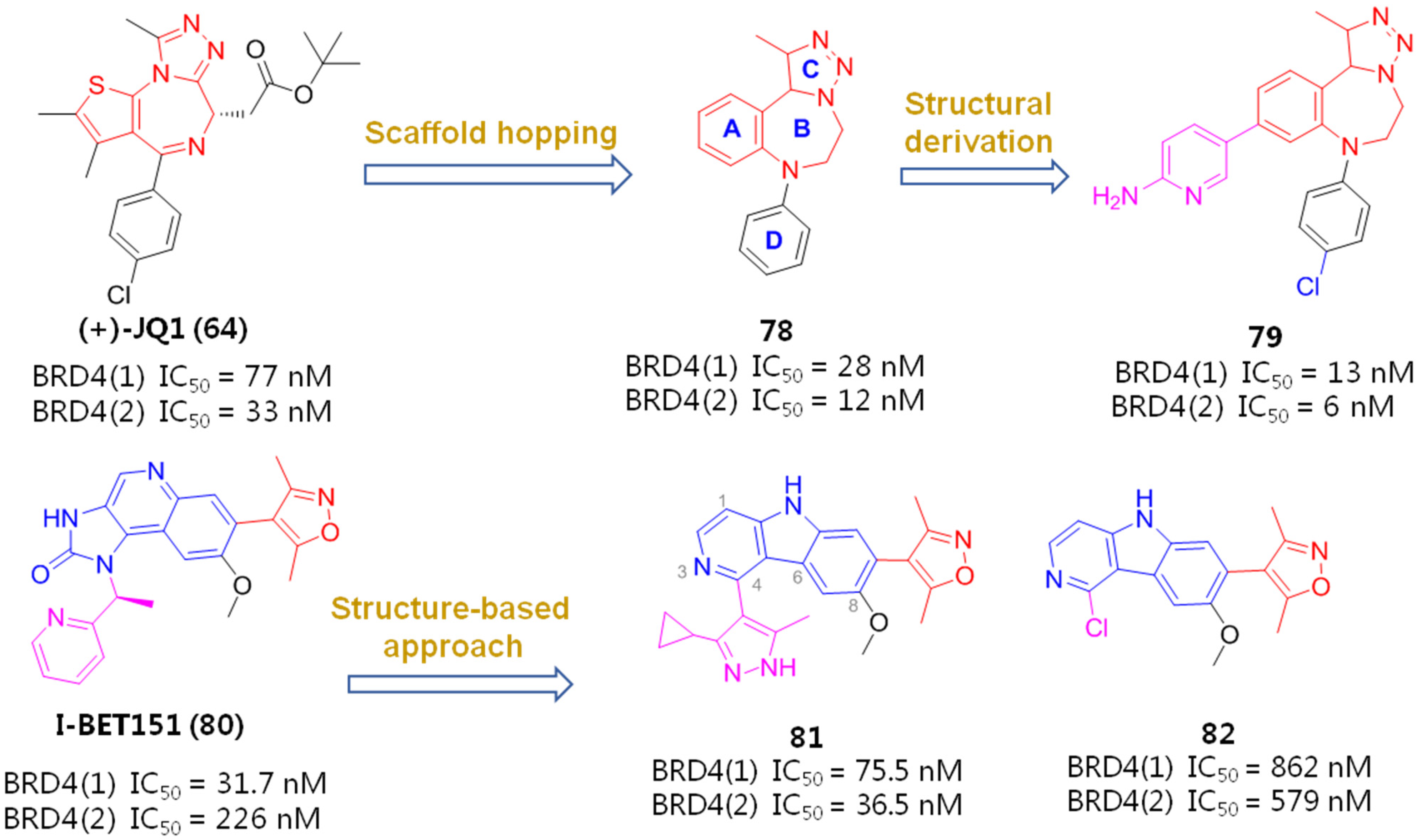
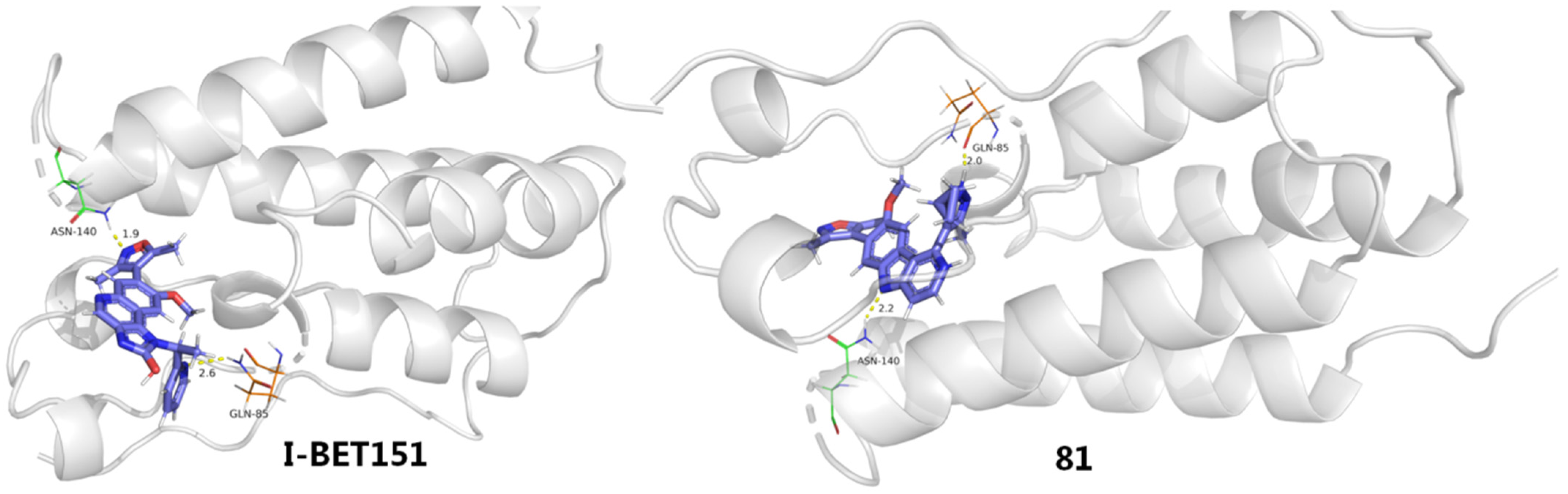
2.5. BET Inhibitors (BETIs)
2.6. P-TEFb Activators
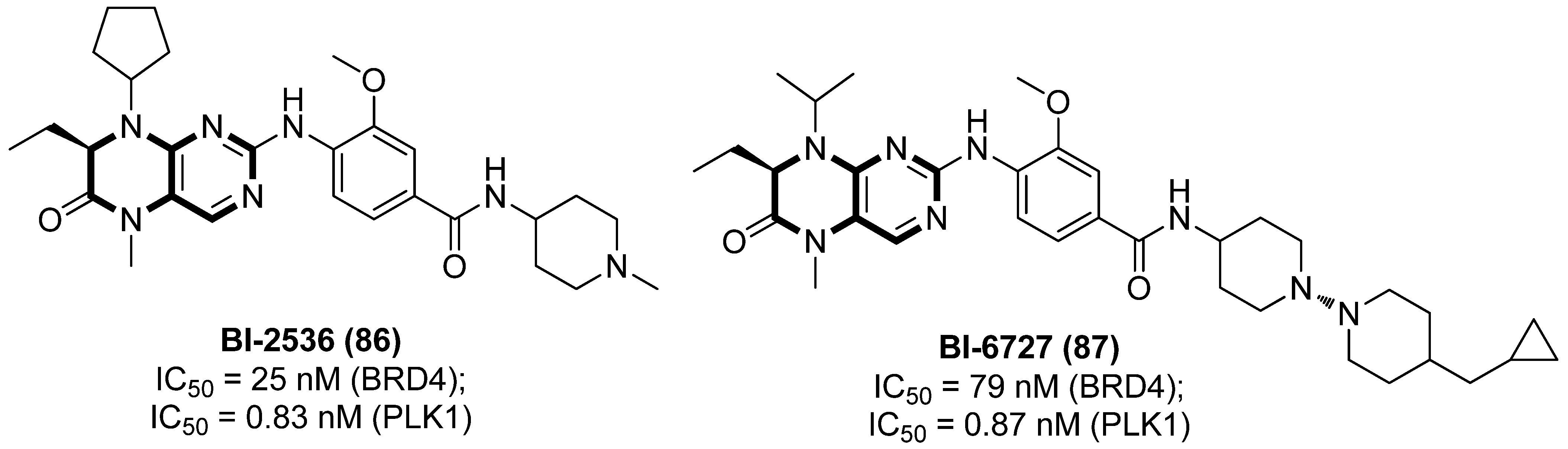
2.7. Polo-like Kinase 1 (PLK1) Inhibitors
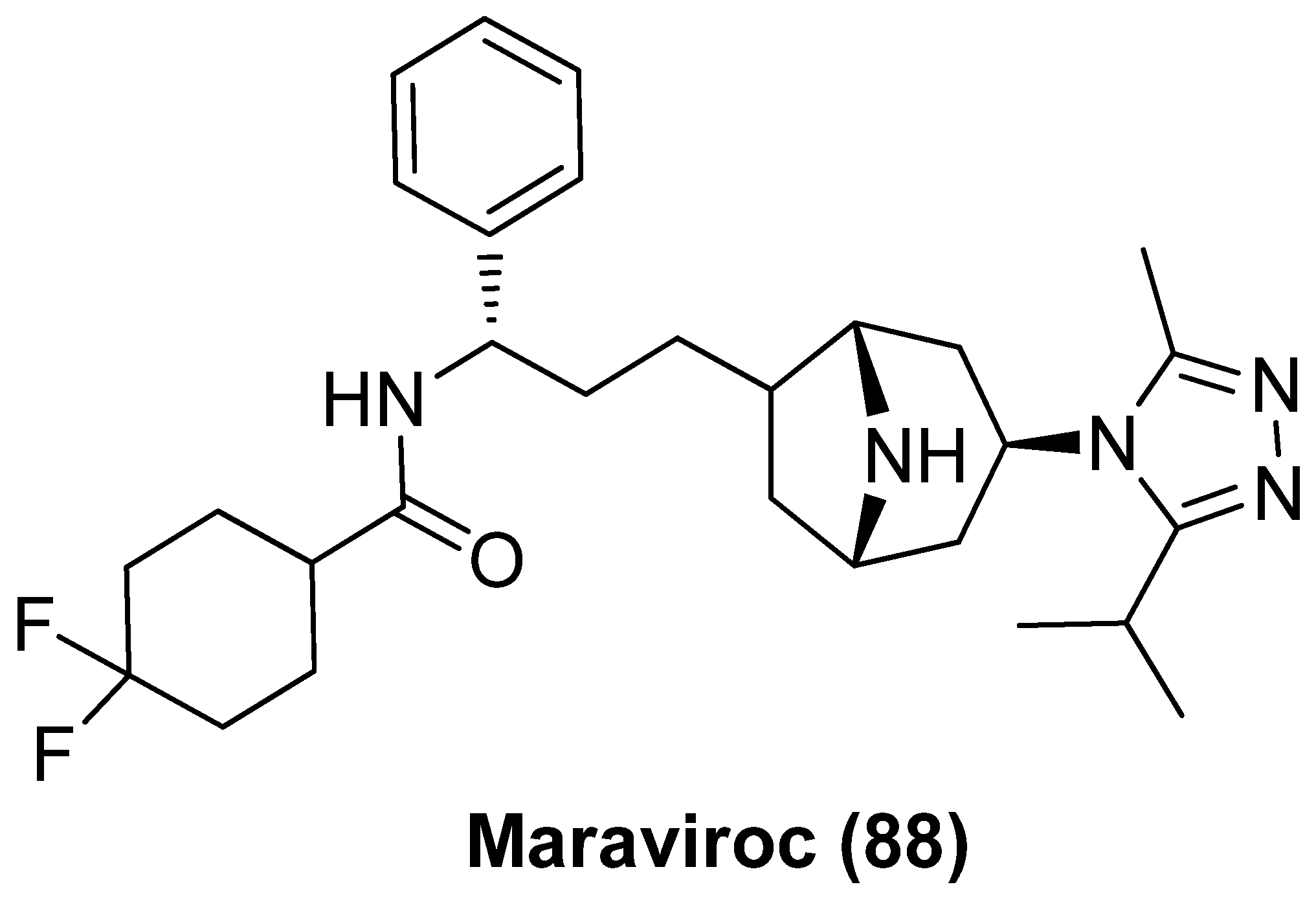
2.8. CCR5 Antagonist
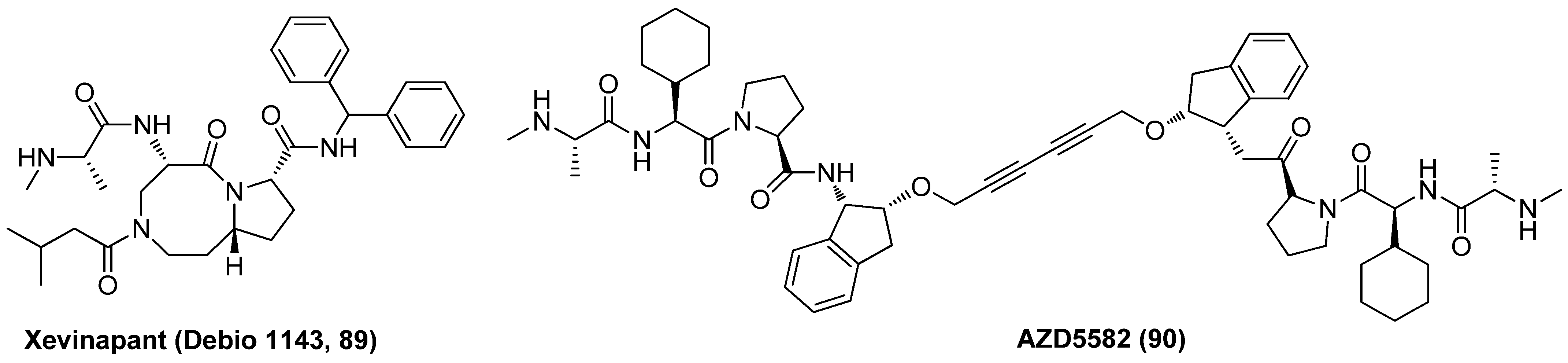
2.9. Inhibitor of Apoptosis Protein (IAP) Antagonist
2.10. Phosphatidylethanolamine-Binding Protein 1 (PEBP1) Inhibitor
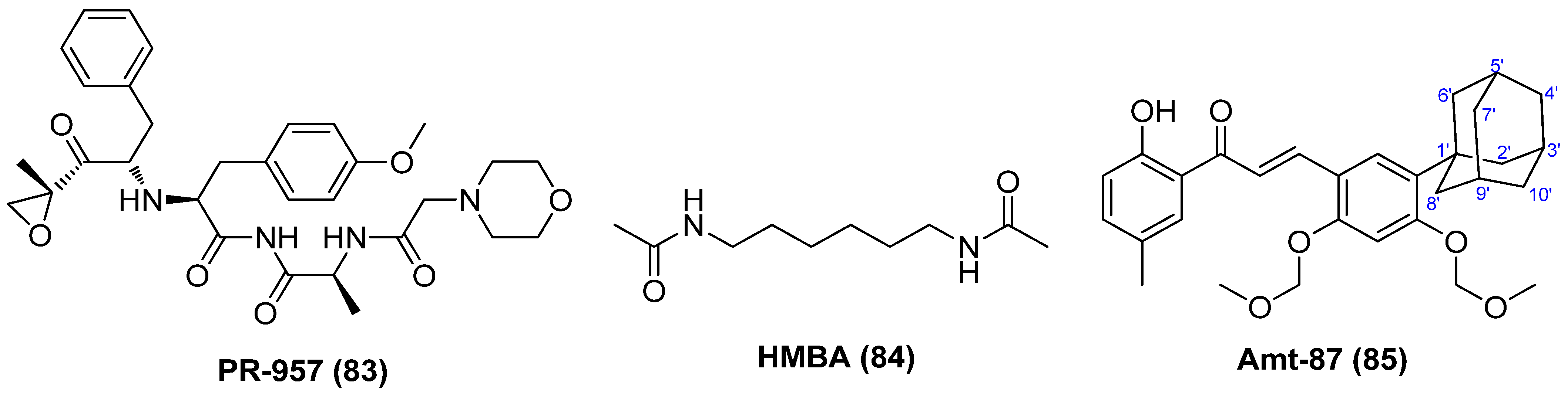
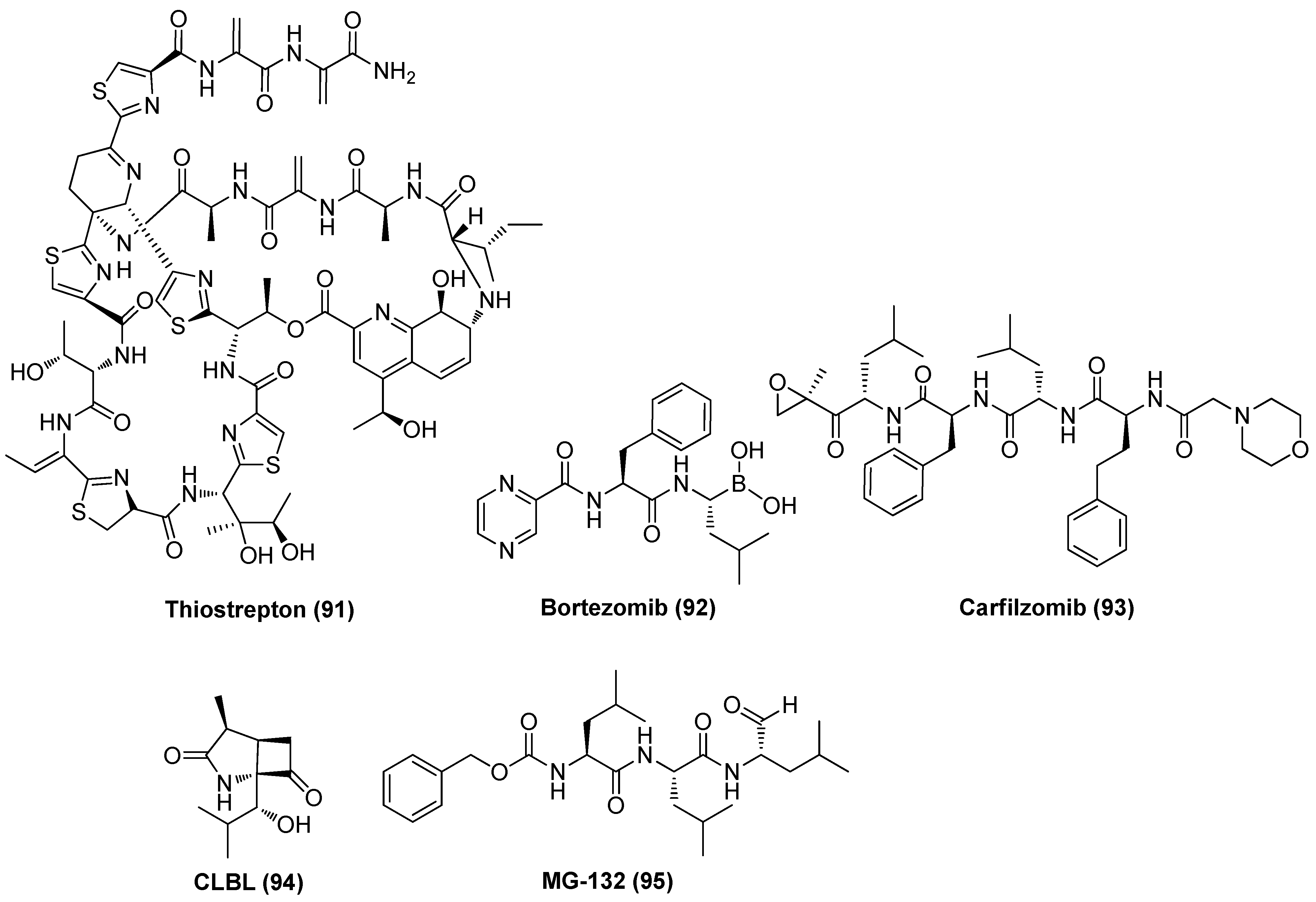
2.11. Proteasome Inhibitor (PI)
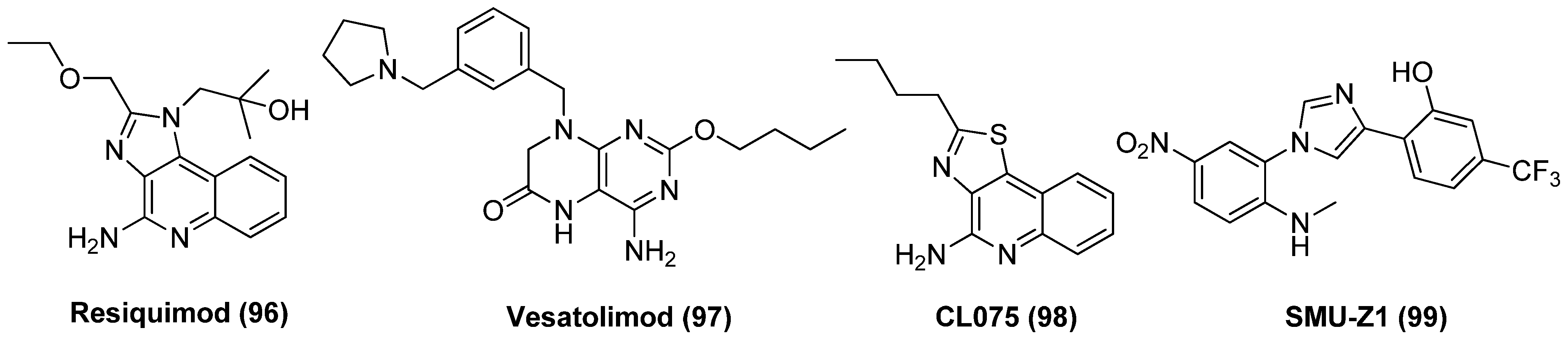
2.12. Toll-like Receptor (TLR) Agonist
2.13. Unclassified LRAs with Undefined Biotargets
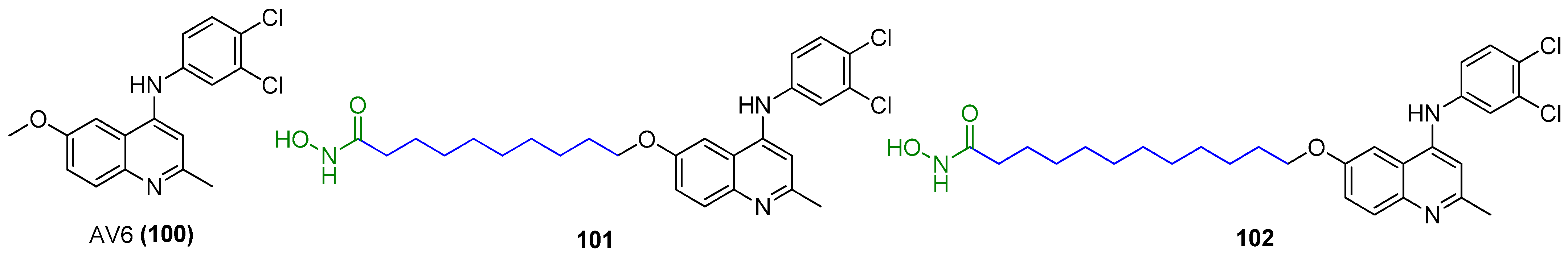
2.13.1. AV6 and Analogues
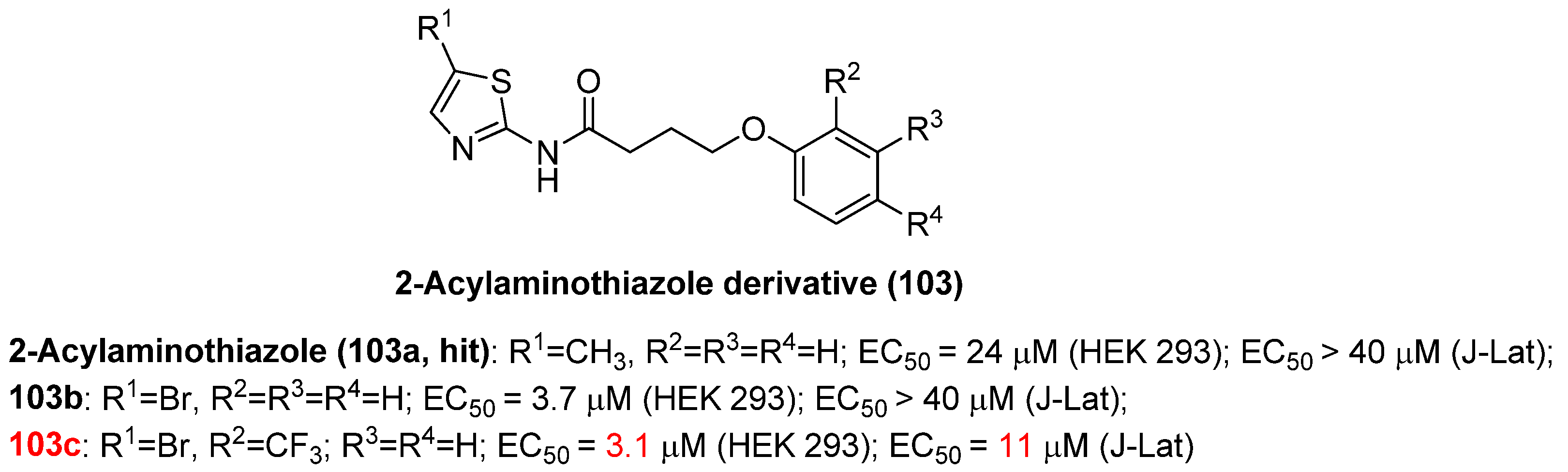
2.13.2. 2-Acylaminothiazole
2.13.3. Dihydropyranoindole Derivative
2.13.4. Carbazole Derivative
2.13.5. Benzazole Derivative
2.13.6. Pyridine Derivative
2.13.7. Polyphenols
3. LRA Combinations
4. “Block-and-Lock” Strategy
5. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chun, T.W.; Finzi, D.; Margolick, J.; Chadwick, K.; Schwartz, D.; Siliciano, R.F. In vivo fate of HIV-1-infected T cells: Quantitative analysis of the transition to stable latency. Nat. Med. 1995, 1, 1284−1290. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhou, T.; Zhang, Y.; Luo, S.; Chen, D.; Li, C.; Li, W. The reservoir of latent HIV. Front. Cell. Infect. Microbiol. 2022, 12, 945956. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Ramirez, N.P.; D’Orso, I. HIV-1 proviral transcription and latency in the new era. Viruses 2020, 12, 555. [Google Scholar] [CrossRef] [PubMed]
- Cary, D.C.; Fujinaga, K.; Peterlin, B.M. Molecular mechanisms of HIV latency. J. Clin. Invest. 2016, 126, 448−454. [Google Scholar] [CrossRef]
- Vanhamel, J.; Bruggemans, A.; Debyser, Z. Establishment of latent HIV-1 reservoirs: What do we really know? J. Virus Erad. 2019, 5, 3−9. [Google Scholar] [CrossRef]
- De-Scheerder, M.A.; Depelseneer, B.; Vandekerckhove, L.; Wim, T. Evolution of experimental design and research techniques in HIV reservoir studies: A systematic review. AIDS Rev. 2020, 22, 16−24. [Google Scholar] [CrossRef]
- Elsheikh, M.M.; Tang, Y.; Li, D.; Jiang, G. Deep latency: A new insight into a functional HIV cure. eBioMedicine 2019, 45, 624–629. [Google Scholar] [CrossRef]
- Ait-Ammar, A.; Kula, A.; Darcis, G.; Verdikt, R.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Rohr, O.; Lint, C.V. Current status of latency reversing agents facing the heterogeneity of HIV-1 cellular and tissue reservoirs. Front. Microbiol. 2019, 10, 3060. [Google Scholar] [CrossRef]
- McClure, J.J.; Li, X.; Chou, C.J. Advances and challenges of HDAC inhibitors in cancer therapeutics. Adv. Cancer Res. 2018, 138, 183−211. [Google Scholar]
- Margolis, D.M. Histone deacetylase inhibitors and HIV latency. Curr. Opin. HIV AIDS. 2011, 6, 25−29. [Google Scholar] [CrossRef]
- Melesina, J.; Simoben, C.V.; Praetorius, L.; Bülbül, E.F.; Robaa, D.; Sippl, W. Strategies to design selective histone deacetylase inhibitors. ChemMedChem 2021, 16, 1336−1359. [Google Scholar] [CrossRef] [PubMed]
- Boateng, A.T.; Abaidoo-Myles, A.; Bonney, E.Y.; Kyei, G.B. Isoform-selective versus nonselective histone deacetylase inhibitors in HIV latency reversal. AIDS Res. Hum. Retrovir. 2022, 38, 615−621. [Google Scholar] [CrossRef] [PubMed]
- Victoriano, A.F.B.; Imai, K.; Togami, H.; Ueno, T.; Asamitsu, K.; Suzuki, T.; Miyata, N.; Ochiai, K.; Okamoto, T. Novel histone deacetylase inhibitor NCH-51 activates latent HIV-1 gene expression. FEBS Lett. 2011, 585, 1103−1111. [Google Scholar] [CrossRef] [PubMed]
- Kapustin, G.V.; Fejé, G.; Gronlund, J.L.; McCafferty, D.G.; Seto, E.; Etzkorn, F.A. Phosphorus-based SAHA analogues as histone deacetylase inhibitors. Org. Lett. 2003, 5, 3053−3056. [Google Scholar] [CrossRef]
- Negmeldin, A.T.; Padige, G.; Bieliauskas, A.V.; Pflum, M.K. Structural requirements of HDAC inhibitors: SAHA analogues modified at the C2 position display HDAC6/8 selectivity. ACS Med. Chem. Lett. 2017, 8, 281−286. [Google Scholar] [CrossRef]
- Darkin-Rattray, S.J.; Gurnett, A.M.; Myers, R.W.; Dulski, P.M.; Crumley, T.M.; Allocco, J.J.; Cannova, C.; Meinke, P.T.; Colletti, S.L.; Bednarek, M.A.; et al. Apicidin: A novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc. Natl. Acad. Sci. USA 1996, 93, 13143–13147. [Google Scholar] [CrossRef]
- Tao, L.; Kapustin, G.; Etzkorn, F.A. Design and synthesis of a potent histone deacetylase inhibitor. J. Med. Chem. 2007, 50, 2003−2006. [Google Scholar]
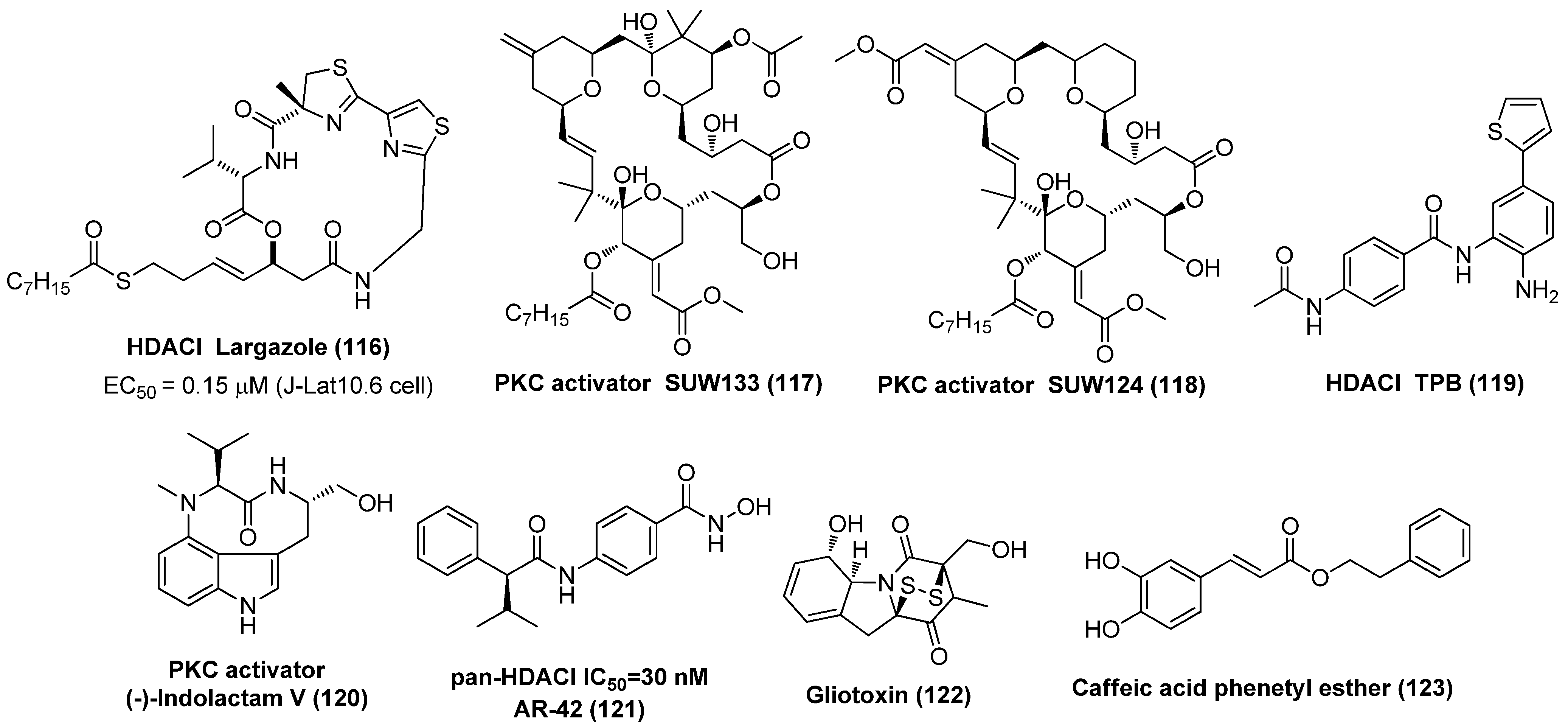
- Wei, D.G.; Chiang, V.; Fyne, E.; Balakrishnan, M.; Barnes, T.; Graupe, M.; Hesselgesser, J.; Irrinki, A.; Murry, J.P.; Stepan, G.; et al. Histone deacetylase inhibitor romidepsin induces HIV expression in CD4 T cells from patients on suppressive antiretroviral therapy at concentrations achieved by clinical dosing. PLoS Pathogens 2014, 10, e1004071. [Google Scholar] [CrossRef]
- Rasmussen, T.A.; Søgaard, O.S.; Brinkmann, C.; Wightman, F.; Lewin, S.R.; Melchjorsen, J.; Dinarello, C.; Østergaard, L.; Tolstrup, M. Comparison of HDAC inhibitors in clinical development: Effect on HIV production in latently infected cells and T-cell activation. Hum. Vaccin. Immunother. 2013, 9, 993−1001. [Google Scholar] [CrossRef]
- Matalon, S.; Rasmussen, T.A.; Dinarello, C.A. Histone deacetylase inhibitors for purging HIV-1 from the latent reservoir. Mol. Med. 2011, 17, 466–472. [Google Scholar] [CrossRef]
- Qi, J.; Ding, C.; Jiang, X.; Gao, Y. Advances in developing CAR T-cell therapy for HIV cure. Front. Immunol. 2020, 11, 361. [Google Scholar] [CrossRef] [PubMed]
- Gunst, J.D.; Kjær, K.; Olesen, R.; Rasmussen, T.A.; Østergaard, L.; Denton, P.W.; Søgaard, O.S.; Tolstrup, M. Fimepinostat, a novel dual inhibitor of HDAC and PI3K, effectively reverses HIV-1 latency ex vivo without T cell activation. J. Virus Erad. 2019, 5, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Zaikos, T.D.; Painter, M.M.; Sebastian-Kettinger, N.T.; Terry, V.H.; Collins, K.L. Class 1-selective histone deacetylase (HDAC) inhibitors enhance HIV latency reversal while preserving the activity of HDAC isoforms necessary for maximal HIV gene expression. J. Virol. 2018, 92, e02110-17. [Google Scholar] [CrossRef] [PubMed]
- Badia, R.; Grau, J.; Riveira-Muñoz, E.; Ballana, E.; Giannini, G.; Esté, J.A. The thioacetate-ω(γ-lactam carboxamide) HDAC inhibitor ST7612AA1 as HIV-1 latency reactivation agent. Antiviral Res. 2015, 123, 62–69. [Google Scholar] [CrossRef]
- Wightman, F.; Lu, H.K.; Solomon, A.E.; Saleh, S.; Harman, A.N.; Cunningham, A.L.; Gray, L.; Churchill, M.; Cameron, P.U.; Dear, A.E.; et al. Entinostat is a histone deacetylase inhibitor selective for class I histone deacety;ases and activates HIV production from latently infected primary T cells. AIDS 2013, 27, 2853–2862. [Google Scholar] [CrossRef]
- Barton, K.M.; Archin, N.M.; Keedy, K.S.; Espeseth, A.S.; Zhang, Y.L.; Gale, J.; Wagner, F.F.; Holson, E.B.; Margolis, D.M. Selective HDAC Inhibition for the Disruption of Latent HIV-1 Infection. PLoS ONE 2014, 9, e102684. [Google Scholar] [CrossRef]
- Yu, W.; Fells, J.; Clausen, D.; Liu, J.; Klein, D.J.; Chung, C.C.; Myers, R.W.; Wu, J.; Wu, G.; Howell, B.J.; et al. Discovery of macrocyclic HDACs 1, 2, and 3 selective inhibitors for HIV latency reactivation. Bioorg. Med. Chem. Lett. 2021, 47, 128168. [Google Scholar] [CrossRef]
- Mota, T.M.; McCann, C.D.; Danesh, A.; Huang, S.H.; Magat, D.B.; Ren, Y.; Leyre, L.; Bui, T.D.; Rohwetter, T.M.; Kovacs, C.M.; et al. Integrated assessment of viral transcription, antigen presentation, and CD8+ T cell function reveals multiple limitations of class I-selective histone deacetylase inhibitors during HIV-1 latency reversal. J. Virology 2020, 94, e01845-19. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramphal, U.; Adimulam, T.; Chinniah, R.; Ramsuran, V. Deciphering DNA methylation in HIV Infection. Front. Immunol. 2021, 12, 795121. [Google Scholar] [CrossRef]
- Fernandez, G.; Zeichner, S.L. Cell line-dependent variability in HIV activation employing DNMT inhibitors. Virol. J. 2010, 7, 266–276. [Google Scholar] [CrossRef]
- Bouchat, S.; Delacourt, N.; Kula, A.; Darcis, G.; Driessche, B.V.; Corazza, F.; Gatot, J.S.; Melard, A.; Vanhulle, C.; Kabeya, K.; et al. Sequential treatment with 5-aza-2’-deoxycytidine and deacetylase inhibitors reactivates HIV-1. EMBO Mol. Med. 2016, 8, 117−138. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P. Inhibitors of DNA methylation: Beyond myelodysplastic syndromes. Nat. Res. Clin. Oncol 2005, 2 (Suppl. 1), S36–S44. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Rahaman, S.; Islam, R.; Rahman, F.; Mithi, F.M.; Alqahtani, T.; Almikhlafi, M.A.; Alghamdi, S.Q.; Alruwaili, A.S.; Hossain, S.; et al. Role of phenolic compounds in human disease: Current knowledge and future prospects. Molecules 2022, 27, 233. [Google Scholar] [CrossRef] [PubMed]
- Verdikt, R.; Bendoumou, M.; Bouchat, S.; Nestola, L.; Pasternak, A.O.; Darcis, G.; Avettand-Fenoel, V.; Vanhulle, C.; AÏt-Ammar, A.; Santangelo, M.; et al. Novel role of UHRF1 in the epigenetic repression of the latent HIV-1. eBioMedicine 2022, 79, 103985. [Google Scholar] [CrossRef] [PubMed]
- Blazkova, J.; Murray, D.; Justement, J.S.; Funk, E.K.; Nelson, A.K.; Moir, S.; Chun, T.-W.; Fauci, A.S. Paucity of HIV DNA methylation in latently infected, resting CD4+ T cells from infected individuals receiving antiretroviral therapy. J. Virol. 2012, 86, 5390−5392. [Google Scholar] [CrossRef]
- Ding, D.; Qu, X.; Li, L.; Zhou, X.; Liu, S.; Lin, S.; Wang, P.; Liu, S.; Kong, C.; Wang, X.; et al. Involvement of histone methyltransferase GLP in HIV-1 latency through catalysis of H3K9 dimethylation. Virology 2013, 440, 182−189. [Google Scholar] [CrossRef]
- Nguyen, K.; Das, B.; Dobrowolski, C.; Karn, J. Multiple histone lysine methyltransferases are required for the establishment and maintenance of HIV-1 latency. mBio 2017, 8, e00133-17. [Google Scholar] [CrossRef]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef]
- Chang, Y.; Zhang, X.; Horton, J.R.; Upadhyay, A.K.; Spannhoff, A.; Liu, J.; Snyder, J.P.; Bedford, M.T.; Cheng, X. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat. Struct. Mol. Biol. 2009, 16, 312–317. [Google Scholar] [CrossRef]
- Liu, F.; Chen, X.; Allali-Hassani, A.; Quinn, A.M.; Wasney, G.A.; Dong, A.; Barsyte, D.; Kozieradzki, I.; Senisterra, G.; Chau, I.; et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a. J. Med. Chem. 2009, 52, 7950–7953. [Google Scholar] [CrossRef]
- Srimongkolpithak, N.; Sundriyal, S.; Li, F.; Vedadi, M.; Fuchter, M.J. Identification of 2,4-diamino-6,7-dimethoxyquinoline derivatives as G9a inhibitors. MedChemComm 2014, 5, 1821–1828. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Chen, X.; Allali-Hassani, A.; Quinn, A.M.; Wigle, T.J.; Wasney, G.A.; Dong, A.; Senisterra, G.; Chau, I.; Siarheyeva, A.; et al. Protein lysine methyltransferase G9a inhibitors: Design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines. J. Med. Chem. 2010, 53, 5844–5857. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Barsyte-Lovejoy, D.; Allali-Hassani, A.; He, Y.; Herold, J.M.; Chen, X.; Yates, C.M.; Frye, S.V.; Brown, P.J.; Huang, J.; et al. Optimization of cellular activity of G9a inhibitors 7-aminoalkoxy-quinazolines. J. Med. Chem. 2011, 54, 6139–6150. [Google Scholar] [CrossRef] [PubMed]
- Vedadi, M.; Barsyte-Lovejoy, D.; Liu, F.; Rival-Gervier, S.; Allali-Hassani, A.; Labrie, V.; Wigle, T.J.; Dimaggio, P.A.; Wasney, G.A.; Siarheyeva, A.; et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol. 2011, 7, 566–574. [Google Scholar] [CrossRef]
- Liu, F.; Barsyte-Lovejoy, D.; Li, F.; Xiong, Y.; Korboukh, V.; Huang, X.P.; Allali-Hassani, A.; Janzen, W.P.; Roth, B.L.; Frye, S.V.; et al. Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J. Med. Chem. 2013, 56, 8931–8942. [Google Scholar] [CrossRef]
- Milite, C.; Feoli, A.; Horton, J.R.; Rescigno, D.; Cipriano, A.; Pisapia, V.; Viviano, M.; Pepe, G.; Amendola, G.; Novellino, E.; et al. Discovery of a novel chemotype of histone lysine methyltransferase EHMT1/2 (GLP/G9a) inhibitors: Rational design, synthesis, biological evaluation, and co-crystal structure. J. Med. Chem. 2019, 62, 2666–2689. [Google Scholar] [CrossRef]
- Bernhard, W.; Barreto, K.; Saunders, A.; Dahabieh, M.S.; Johnson, P.; Sadowski, I. The Suv39H1 methyltransferase inhibitor chaetocin causes induction of integrated HIV-1 without producing a T cell response. FEBS Lett. 2011, 585, 3549–3554. [Google Scholar] [CrossRef]
- Bouchat, S.; Gatot, J.S.; Kabeya, K.; Cardona, C.; Colin, L.; Herbein, G.; Wit, S.D.; Clumeck, N.; Lambotte, O.; Rouzioux, C.; et al. Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4+ T cells from HIV-1-infected HAART-treated patients. Aids 2012, 26, 1473–1482. [Google Scholar] [CrossRef]
- Friedman, J.; Cho, W.K.; Chu, C.K.; Keedy, K.S.; Archin, N.M.; Margolis, D.M.; Karn, J. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J. Virol. 2011, 85, 9078–9089. [Google Scholar] [CrossRef]
- Turner, A.W.; Dronamraju, R.; Potjewyd, F.; James, K.S.; Winecoff, D.K.; Kirchherr, J.L.; Archin, N.M.; Browne, E.P.; Strahl, B.D.; Margolis, D.M.; et al. Evaluation of EED inhibitors as a class of PRC2-targeted small molecules for HIV latency reversal. ACS Infect. Dis. 2020, 6, 1719–1733. [Google Scholar] [CrossRef]
- Boehm, D.; Jeng, M.; Camus, G.; Gramatica, A.; Schwarzer, R.; Johnson, J.R.; Hull, P.A.; Montano, M.; Sakane, N.; Pagans, S.; et al. SMYD2-mediated histone methylation contributes to HIV-1 latency. Cell Host Microbe 2017, 21, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Nikolai, B.C.; Feng, Q. HIV latency gets a new histone mark. Cell Host Microbe 2017, 21, 549–550. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Dandekar, S. Targeting NF-κB signaling with protein kinase C agonists as an emerging strategy for combating HIV latency. AIDS Res. Hum. Retrovir. 2015, 31, 4−12. [Google Scholar] [CrossRef]
- Chang, S.N.; Dey, D.K.; Oh, S.T.; Kong, W.H.; Cho, K.H.; Al-Olayan, E.M.; Hwang, B.S.; Kang, S.C.; Park, J.G. Phorbol 12-myristate 13-acetate induced toxicity study and the role of tangeretin in abrogating HIF-1α-NF-κB crosstalk in vitro and in vivo. Int. J. Mol. Sci. 2020, 21, 9261. [Google Scholar] [CrossRef] [PubMed]
- Biancotto, A.; Grivel, J.C.; Gondois-Rey, F.; Bettendroffer, L.; Vigne, R.; Brown, S.; Margolis, L.B.; Hirsch, I. Dual role of prostratin in inhibition of infection and reactivation of human immunodeficiency virus from latency in primary blood lymphocytes and lymphoid tissue. J. Virol. 2004, 78, 10507–10515. [Google Scholar] [CrossRef] [PubMed]
- Ersvaer, E.; Kittang, A.O.; Hampson, P.; Sand, K.; Gjertsen, B.T.; Lord, J.M.; Bruserud, O. The protein kinase C agonist PEP005 (Ingenol 3-Angelate) in the treatment of human cancer: A balance between efficacy and toxicity. Toxins 2010, 2, 174–194. [Google Scholar] [CrossRef]
- Bocklandt, S.; Blumberg, P.M.; Hamer, D.H. Activation of latent HIV-1 expression by the potent anti-tumor promoter 12-deoxyphorbol 13phenylacetate. Antivir. Res. 2003, 59, 89–98. [Google Scholar] [CrossRef]
- Warrilow, D.; Gardner, J.; Darnell, G.A.; Suhrbier, A.; Harrich, D. HIV type 1 inhibition by protein kinase C modulatory compounds. AIDS Res. Hum. Retrovir. 2006, 22, 854–864. [Google Scholar] [CrossRef]
- Wang, P.; Lu, P.; Qu, X.; Shen, Y.; Zeng, H.; Zhu, X.; Zhu, Y.; Li, X.; Wu, H.; Xu, J.; et al. Reactivation of HIV-1 from latency by an ingenol derivative from Euphorbia Kansui. Sci. Rep. 2017, 7, 9451. [Google Scholar] [CrossRef]
- Yang, H.; Li, X.; Yang, X.; Lu, P.; Wang, Y.; Jiang, Z.; Pan, H.; Zhao, L.; Zhu, Y.; Khan, I.U.; et al. Dual effects of the novel ingenol derivatives on the acute and latent HIV-1 infections. Antiviral. Res. 2019, 169, 104555. [Google Scholar] [CrossRef]
- Bedoya, L.M.; Márquez, N.; Martínez, N.; Gutiérrez-Eisman, S.; Álvarez, A.; Calzado, M.A.; Rojas, J.M.; Appendino, G.; Muñoz, E.; Alcamí, J. SJ23B, a jatrophane diterpene activates classical PKCs and displays strong activity against HIV in vitro. Biochem. Pharmacol. 2009, 77, 965−978. [Google Scholar] [CrossRef] [PubMed]
- Pagani, A.; Navarrete, C.; Fiebich, B.L.; Munoz, E.; Appendino, G. Synthesis and biological evaluation of 12-aminoacylphorboids. J. Nat. Prod. 2010, 73, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Avila, L.; Perez, M.; Sanchez-Duffhues, G.; Hernández-Galán, R.; Muñoz, E.; Cabezas, F.; Quiñones, W.; Torres, F.; Echeverri, F. Effects of diterpenes from latex of Euphorbia lactea and Euphorbia laurifolia on human immunodeficiency virus type 1 reactivation. Phytochemistry 2010, 71, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Daoubi, M.; Marquez, N.; Mazoir, N.; Benharref, A.; Hernández-Galán, R.; Muñoz, E.; Collado, I.G. Isolation of new phenylacetylingol derivatives that reactivate HIV-1 latency and a novel spirotriterpenoid from Euphorbia officinarum latex. Bioorg. Med. Chem. 2007, 15, 4577–4584. [Google Scholar] [CrossRef]
- Pérez, M.; de Vinuesa, A.G.; Sanchez-Duffhues, G.; Marquez, N.; Bellido, M.L.; Muñoz-Fernandez, M.A.; Moreno, S.; Castor, T.P.; Calzado, M.A.; Muñoz, E. Bryostatin-1 synergizes with histone deacetylase inhibitors to reactivate HIV-1 from latency. Curr. HIV Res. 2010, 8, 418–429. [Google Scholar] [CrossRef]
- Hany, L.; Turmel, M.O.; Barat, C.; Ouellet, M.; Tremblay, M.J. Bryostatin-1 decreases HIV-1 infection and viral production in human primary macrophages. J. Virol. 2022, 96, e0195321. [Google Scholar] [CrossRef]
- Proust, A.; Barat, C.; Leboeuf, M.; Drouin, J.; Gagnon, M.T.; Vanasse, F.; Tremblay, M.J. HIV-1 infection and latency-reversing agents bryostatin-1 and JQ1 disrupt amyloid beta homeostasis in human astrocytes. Glia 2020, 68, 2212–2227. [Google Scholar] [CrossRef]
- Stone, J.C.; Stang, S.L.; Zheng, Y.; Dower, N.A.; Brenner, S.E.; Baryza, J.L.; Wender, P.A. Synthetic bryostatin analogues activate the RasGRP1 signaling pathway. J. Med. Chem. 2004, 47, 6638–6644. [Google Scholar] [CrossRef]
- Pandelo, J.D.; Bartholomeeusen, K.; da Cunha, R.D.; Abreu, C.M.; Glinski, J.; da Costa, T.B.; Bacchi Rabay, A.F.; Pianowski Filho, L.F.; Dudycz, L.W.; Ranga, U.; et al. Reactivation of latent HIV-1 by new semi-synthetic ingenol esters. Virology 2014, 462–463, 328–339. [Google Scholar] [CrossRef]
- Lai, W.; Huang, L.; Zhu, L.; Ferrari, G.; Chan, C.; Li, W.; Lee, K.H.; Chen, C.H. Gnidimacrin, a potent anti-HIV diterpene, can eliminate latent HIV-1 ex vivo by activation of protein kinase C beta. J. Med. Chem. 2015, 58, 8638–8646. [Google Scholar] [CrossRef]
- Matsuda, K.; Kobayakawa, T.; Tsuchiya, K.; Hattori, S.I.; Nomura, W.; Gatanaga, H.; Yoshimura, K.; Oka, S.; Endo, Y.; Tamamura, H.; et al. Benzolactam-related compounds promote apoptosis of HIV-infected human cells via protein kinase C-induced HIV latency reversal. J. Biol. Chem. 2019, 294, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Guest, E.E.; Pickett, S.D.; Hirst, J.D. Structural variation of protein-ligand complexes of the first bromodomain of BRD4. Org. Biomol. Chem. 2021, 19, 5632–5641. [Google Scholar] [CrossRef] [PubMed]
- Salahong, T.; Schwartz, C.; Sungthong, R. Are BET inhibitors yet promising latency-reversing agents for HIV-1 reactivation in AIDS therapy? Viruses 2021, 13, 1026. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Lu, P.; Qu, X.; Shen, Y.; Jiang, Z.; Wang, P.; Zeng, H.; Ji, H.; Deng, J.; Yang, X.; Li, X.; et al. The BET inhibitor OTX015 reactivates latent HIV-1 through P-TEFb. Sci. Rep. 2016, 6, 24100. [Google Scholar] [CrossRef]
- Liang, T.; Zhang, X.; Lai, F.; Lin, J.; Zhou, C.; Xu, X.; Tan, X.; Liu, S.; Li, L. A novel bromodomain inhibitor, CPI-203, serves as an HIV-1 latency-reversing agent by activating positive transcription elongation factor b. Biochem. Pharmacol. 2019, 164, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; Martins, L.; Aull, K.; Li, P.C.; Planelles, V.; Bradner, J.E.; et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle. 2013, 12, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Shen, Y.; Yang, H.; Wang, Y.; Jiang, Z.; Yang, X.; Zhong, Y.; Pan, H.; Xu, J.; Lu, H.; et al. BET inhibitors RVX-208 and PFI-1 reactivate HIV-1 from latency. Sci. Rep. 2017, 7, 16646. [Google Scholar] [CrossRef]
- Borthakur, G.; Odenike, O.; Aldoss, I.; Rizzieri, D.A.; Prebet, T.; Chen, C.; Popovic, R.; Modi, D.A.; Joshi, R.H.; Wolff, J.E.; et al. A phase 1 study of the pan-bromodomain and extraterminal inhibitor mivebresib (ABBV-075) alone or in combination with venetoclax in patients with relapsed/refractory acute myeloid leukemia. Cancer 2021, 127, 2943–2953. [Google Scholar] [CrossRef]
- Li, G.; Zhang, Z.; Reszka-Blanco, N.; Li, F.; Chi, L.; Ma, J.; Jeffrey, J.; Cheng, L.; Su, L. Specific activation in vivo of HIV-1 by a bromodomain inhibitor from monocytic cells in humanized mice under antiretroviral therapy. J. Virol. 2019, 93, e00233-19. [Google Scholar] [CrossRef]
- Abner, E.; Stoszko, M.; Zeng, L.; Chen, H.C.; Izquierdo-Bouldstridge, A.; Konuma, T.; Zorita, E.; Fanunza, E.; Zhang, Q.; Mahmoudi, T.; et al. A new quinoline BRD4 inhibitor targets a distinct latent HIV-1 reservoir for reactivation from other “shock” drugs. J. Virol. 2018, 92, e02056-17. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Liu, S.; Jean, M.; Simpson, S.; Huang, H.; Merkley, M.; Hayashi, T.; Kong, W.; Rodríguez-Sánchez, I.; Zhang, X.; et al. A novel bromodomain inhibitor reverses HIV-1 latency through specific binding with BRD4 to promote Tat and P-TEFb association. Front. Microbiol. 2017, 8, 1035. [Google Scholar] [CrossRef] [PubMed]
- Alamer, E.; Zhong, C.; Liu, Z.; Niu, Q.; Long, F.; Guo, L.; Gelman, B.B.; Soong, L.; Zhou, J.; Hu, H. Epigenetic suppression of HIV in myeloid cells by the BRD4-selective small molecule modulator ZL0580. J. Virol. 2020, 94, e01880-19. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Chen, P.; Huang, Y.; Qiu, J.; Zhou, C.; Wu, Z.; Li, L. CPI-637 as a potential bifunctional latency-reversing agent that targets both the BRD4 and TIP60 proteins. Front. Cell. Infect. Microbiol. 2021, 11, 686035. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.; Zhang, J.; Liu, J.; Chian, C.M.; Ouyang, L. Targeting bromodomain and extraterminal proteins for drug discovery: From current progress to technological development. J. Med. Chem. 2021, 64, 2419–2435. [Google Scholar] [CrossRef]
- Sharp, P.P.; Garnier, J.M.; Hatfaludi, T.; Xu, Z.; Segal, D.; Jarman, K.E.; Jousset, H.; Garnham, A.; Feutrill, J.T.; Cuzzupe, A.; et al. Design, synthesis, and biological activity of 1,2,3-triazolobenzodiazepine BET bromodomain inhibitors. ACS Med. Chem. Lett. 2017, 8, 1298–1303. [Google Scholar] [CrossRef]
- Ran, X.; Zhao, Y.; Liu, L.; Bai, L.; Yang, C.-Y.; Zhou, B.; Meagher, J.L.; Chinnaswamy, K.; Stuckey, J.A.; Wang, S. Structure-based design of γ-carboline analogues as potent and specific BET bromodomain inhibitors. J. Med. Chem. 2015, 58, 4927–4939. [Google Scholar] [CrossRef]
- Ma, X.; Yang, T.; Luo, Y.; Wu, L.; Jiang, Y.; Song, Z.; Pan, T.; Liu, B.; Liu, G.; Liu, J.; et al. TRIM28 promotes HIV-1 latency by SUMOylating CDK9 and inhibiting P-TEFb. eLife 2019, 8, e42426. [Google Scholar] [CrossRef]
- Das, B.; Dobrowolski, C.; Shahir, A.M.; Feng, Z.; Yu, X.; Sha, J.; Bissada, N.F.; Weinberg, A.; Karn, J.; Ye, F. Short chain fatty acids potently induce latent HIV-1 in T-cells by activating P-TEFb and multiple histone modifications. Virology 2015, 474, 65–81. [Google Scholar] [CrossRef][Green Version]
- Lin, J.; Zhang, X.; Lu, W.; Xu, X.; Pan, X.; Liang, T.; Duan, S.; Chen, Y.; Li, L.; Liu, S. PR-957, a selective immunoproteasome inhibitor, reactivates latent HIV-1 through P-TEFb activation mediated by HSF-1. Biochem. Pharmacol. 2018, 156, 511–523. [Google Scholar] [CrossRef]
- Chen, D.; Wang, H.; Aweya, J.J.; Chen, Y.; Chen, M.; Wu, X.; Chen, X.; Lu, J.; Chen, R.; Liu, M. HMBA enhances prostratin-induced activation of latent HIV-1 via suppressing the expression of negative feedback regulator A20/TNFAIP3 in NF-κB signaling. Biomed Res. Int. 2016, 2016, 5173205. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, E.; Desimio, M.G.; Doria, M. Hexamethylene bisacetamide impairs NK cell-mediated clearance of acute T lymphoblastic leukemia cells and HIV-1-infected T cells that exit viral latency. Sci. Rep. 2019, 9, 4373. [Google Scholar] [CrossRef]
- Wu, J.; Ao, M.; Shao, R.; Wang, H.; Yu, D.; Fang, M.; Gao, X.; Wu, Z.; Zhou, Q.; Xue, Y. A chalcone derivative reactivates latent HIV-1 transcription through activating P-TEFb and promoting Tat-SEC interaction on viral promoter. Sci. Rep. 2017, 7, 10657. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Hayashi, T.; Jean, M.; Kong, W.; Fiches, G.; Biswas, A.; Liu, S.; Yosief, H.O.; Zhang, X.; Zhu, J. Inhibition of Polo-like kinase 1 (PLK1) facilitates the elimination of HIV-1 viral reservoirs in CD4+ T cells ex vivo. Sci. Adv. 2020, 6, eaba1941. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Huang, Y.; Deng, M.; Liu, T.; Lai, W.; Ye, X. Polo-like kinase 1 inhibits the activity of positive transcription elongation factor of RNA pol II b (P-TEFb). PLoS ONE 2013, 8, e72289. [Google Scholar] [CrossRef] [PubMed]
- Gohda, J.; Suzuki, K.; Liu, K.; Xie, X.; Takeuchi, H.; Inoue, J.; Kawaguchi, Y.; Ishida, T. BI-2536 and BI-6727, dual Polo-like kinase/bromodomain inhibitors, effectively reactivate latent HIV-1. Sci. Rep. 2018, 8, 3521. [Google Scholar] [CrossRef]
- López-Huertas, M.R.; Jiménez-Tormo, L.; Madrid-Elena, N.; Gutiérrez, C.; Rodríguez-Mora, S.; Coiras, M.; Alcamí, J.; Moreno, S. The CCR5-antagonist Maraviroc reverses HIV-1 latency in vitro alone or in combination with the PKC-agonist Bryostatin-1. Sci. Rep. 2017, 7, 2385. [Google Scholar] [CrossRef]
- López-Huertas, M.R.; Jiménez-Tormo, L.; Madrid-Elena, N.; Gutiérrez, C.; Vivancos, M.J.; Luna, L.; Moreno, S. Maraviroc reactivates HIV with potency similar to that of other latency reversing drugs without inducing toxicity in CD8 T cells. Biochem. Pharmacol. 2020, 182, 114231. [Google Scholar] [CrossRef]
- López-Huertas, M.R.; Gutiérrez, C.; Madrid-Elena, N.; Hernández-Novoa, B.; Olalla-Sierra, J.; Plana, M.; Delgado, R.; Rubio, R.; Muñoz-Fernández, M.Á.; Moreno, S. Prolonged administration of maraviroc reactivates latent HIV in vivo but it does not prevent antiretroviral-free viral rebound. Sci. Rep. 2020, 10, 22286. [Google Scholar] [CrossRef]
- Madrid-Elena, N.; García-Bermejo, M.L.; Serrano-Villar, S.; Díaz-de Santiago, A.; Sastre, B.; Gutiérrez, C.; Dronda, F.; Díaz, M.C.; Domínguez, E.; López-Huertas, M.R.; et al. Maraviroc is associated with latent HIV-1 reactivation through NF-κB activation in resting CD4+ T cells from HIV-Infected individuals on suppressive antiretroviral therapy. J. Virol. 2018, 92, e01931-17. [Google Scholar] [CrossRef]
- Bobardt, M.; Kuo, J.; Chatterji, U.; Chanda, S.; Little, S.J.; Wiedemann, N.; Vuagniaux, G.; Gallay, P.A. The inhibitor apoptosis protein antagonist Debio 1143 is an attractive HIV-1 latency reversal candidate. PLoS ONE 2019, 14, e0211746. [Google Scholar] [CrossRef] [PubMed]
- Falcinelli, S.D.; Peterson, J.J.; Turner, A.M.; Irlbeck, D.; Read, J.; Raines, S.L.; James, K.S.; Sutton, C.; Sanchez, A.; Emery, A.; et al. Combined noncanonical NF-κB agonism and targeted BET bromodomain inhibition reverse HIV latency ex vivo. J. Clin. Invest. 2022, 132, e157281. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, Y.; Lu, P.; Shen, Y.; Zhao, X.; Zhu, Y.; Jiang, Z.; Yang, H.; Pan, H.; Zhao, L.; et al. PEBP1 suppresses HIV transcription and induces latency by inactivating MAPK/NF-κB signaling. EMBO Rep. 2020, 21, e49305. [Google Scholar] [CrossRef]
- Peng, W.; Hong, Z.; Chen, X.; Gao, H.; Dai, Z.; Zhao, J.; Liu, W.; Li, D.; Deng, K. Thiostrepton reactivates latent HIV-1 through the P-TEFb and NF-κB pathways mediated by heat shock response. Antimicrob. Agents Chemother. 2020, 64, e02328-19. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wu, J.; Chavez, L.; Hoh, R.; Deeks, S.G.; Pillai, S.K.; Zhou, Q. Reiterative enrichment and authentication of CRISPRi targets (REACT) identifies the proteasome as a key contributor to HIV-1 latency. PLoS Pathog. 2019, 15, e1007498. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.-Y.; Zhao, W.; Wang, C.-Y.; Lin, J.; Zeng, X.-Y.; Ren, R.-X.; Wang, K.; Xun, T.-R.; Shai, Y.; Liu, S.-W. Heat shock protein 90 facilitates latent hiv reactivation through maintaining the function of positive transcriptional elongation factor b (P-TEFb) under proteasome Inhibition. J. Biol. Chem. 2016, 291, 26177–26187. [Google Scholar] [CrossRef]
- Miller, L.K.; Kobayashi, Y.; Chen, C.-C.; Russnak, T.A.; Ron, Y.; Dougherty, J.P. Proteasome inhibitors act as bifunctional antagonists of human immunodeficiency virus type 1 latency and replication. Retrovirology 2013, 10, 120. [Google Scholar] [CrossRef]
- Tundo, G.R.; Sbardella, D.; Santoro, A.M.; Coletta, A.; Oddone, F.; Grasso, G.; Milardi, D.; Lacal, P.M.; Marini, S.; Purrello, R.; et al. The proteasome as a druggable target with multiple therapeutic potentialities: Cutting and non-cutting edges. Pharmacol. Ther. 2020, 213, 107579. [Google Scholar] [CrossRef]
- Timmons, A.; Fray, E.; Kumar, M.; Wu, F.; Dai, W.; Bullen, C.K.; Kim, P.; Hetzel, C.; Yang, C.; Beg, S.; et al. HSF1 inhibition attenuates HIV-1 latency reversal mediated by several candidate LRAs in vitro and ex vivo. Proc. Natl. Acad. Sci. USA 2020, 117, 15763–15771. [Google Scholar] [CrossRef]
- Martinsen, J.T.; Gunst, J.D.; Højen, J.F.; Tolstrup, M.; Søgaard, O.S. The use of toll-like receptor agonists in HIV-1 cure strategies. Front. Immunol. 2020, 11, 1112. [Google Scholar] [CrossRef]
- Macedo, A.B.; Novis, C.L.; Bosque, A. Targeting cellular and tissue HIV reservoirs with toll-like receptor agonists. Front. Immunol. 2019, 10, 2450. [Google Scholar] [CrossRef] [PubMed]
- Pache, L.; Marsden, M.D.; Teriete, P.; Portillo, A.J.; Heimann, D.; Kim, J.T.; Soliman, M.S.A.; Dimapasoc, M.; Carmona, C.; Celeridad, M.; et al. Pharmacological activation of non-canonical NF-κB signalling activates latent HIV-1 reservoirs in vivo. Cell Rep. Med. 2020, 1, 100037. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.M.; Jiang, G. NF-κB sub-pathways and HIV cure: A revisit. eBioMedicine 2021, 63, 103159. [Google Scholar] [CrossRef] [PubMed]
- Takahama, S.; Yamamoto, T. Pattern recognition receptor ligands as an emerging therapeutic agent for latent HIV-1 infection. Front. Cell. Infect. Microbiol. 2020, 10, 216. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, E.; Audige, A.; Joller, H.; Speck, R.F. TLR7/8 triggering exerts opposing effects in acute versus latent HIV infection. J. Immunol. 2006, 176, 2888–2895. [Google Scholar] [CrossRef]
- Hofmann, H.; Vanwalscappel, B.; Bloch, N.; Landau, N.R. TLR7/8 agonist induces a post-entry SAMHD1-independent block to HIV-1 infection of monocytes. Retrovirology 2016, 13, 83. [Google Scholar] [CrossRef]
- Tsai, A.; Irrinki, A.; Kaur, J.; Cihlar, T.; Kukolj, G.; Sloan, D.D.; Murry, J.P. Toll-like receptor 7 agonist GS-9620 induces HIV expression and HIV-specific immunity in cells from HIV-infected individuals on suppressive antiretroviral therapy. J. Virol. 2017, 91, e02166-16. [Google Scholar] [CrossRef]
- Rochat, M.A.; Schlaepfer, E.; Speck, R.F. Promising role of toll-like receptor 8 agonist in concert with prostratin for activation of silent HIV. J. Virol. 2017, 91, e02084-16. [Google Scholar] [CrossRef]
- Duan, S.; Xu, X.; Wang, J.; Huang, L.; Peng, J.; Yu, T.; Zhou, Y.; Cheng, K.; Liu, S. TLR1/2 agonist enhances reversal of HIV-1 latency and promotes NK cell-induced suppression of HIV-1-infected autologous CD4+ T cells. J. Virol. 2021, 95, e0081621. [Google Scholar] [CrossRef]
- Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Milne, S.; Das, B.; Dobrowolski, C.; Rojas, R.; Karn, J. Toll-like receptor 3 activation selectively reverses HIV latency in microglial cells. Retrovirology 2017, 14, 9. [Google Scholar] [CrossRef]
- Kula-Pacurar, A.; Rodari, A.; Darcis, G.; Lint, C.V. Shocking HIV-1 with immunomodulatory latency reversing agents. Semin. Immunol. 2021, 51, 101478. [Google Scholar] [CrossRef] [PubMed]
- Micheva-Viteva, S.; Kobayashi, Y.; Edelstein, L.C.; Pacchia, A.L.; Lee, H.L.; Graci, J.D.; Breslin, J.; Phelan, B.D.; Miller, L.K.; Colacino, J.M.; et al. High-throughput screening uncovers a compound that activates latent HIV-1 and acts cooperatively with a histone deacetylase (HDAC) inhibitor. J. Biol. Chem. 2011, 286, 21083–21091. [Google Scholar] [CrossRef] [PubMed]
- Ao, M.; Pan, Z.; Qian, Y.; Tang, B.; Feng, Z.; Fang, H.; Wu, Z.; Chen, J.; Xue, Y.; Fang, M. Design, synthesis, and biological evaluation of AV6 derivatives as novel dual reactivators of latent HIV-1. RSC Adv. 2018, 8, 17279. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, W.; Jacobson, J.; Jarman, K.E.; Sabroux, H.J.; Harty, L.; McMahon, J.; Lewin, S.R.; Purcell, D.F.; Sleebs, B.E. Identification of 5-substituted 2-acylaminothiazoles that activate Tat-mediated transcription in HIV-1 latency models. J. Med. Chem. 2019, 62, 5148–5175. [Google Scholar] [CrossRef] [PubMed]
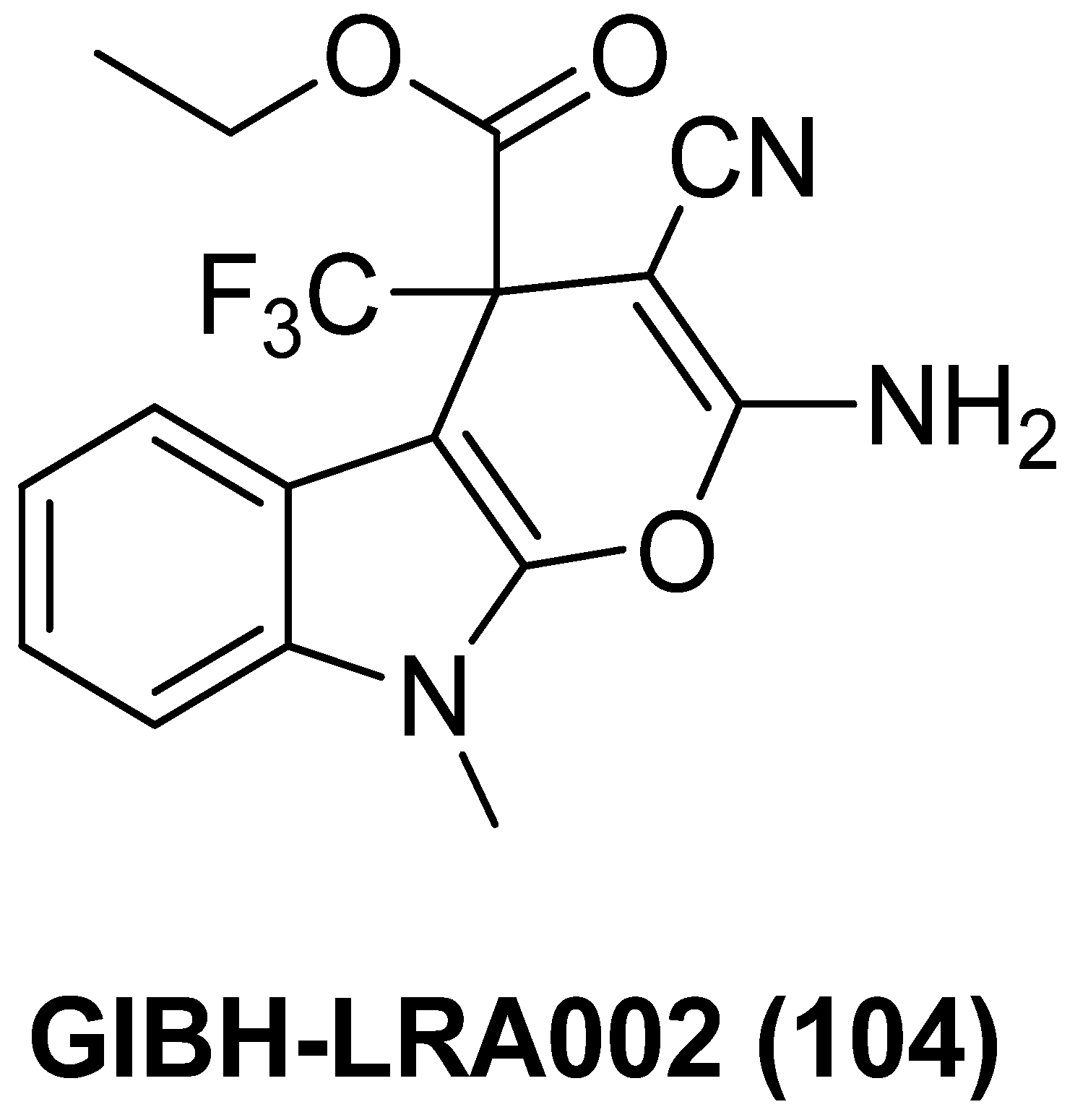
- Yang, Q.; Ding, Y.; Feng, F.; Pan, E.; Fan, X.; Ma, X.; Chen, L.; Zhao, J.; Sun, C. Structure-optimized dihydropyranoindole derivative GIBH-LRA002 potentially reactivated viral latency in primary CD4+ T lymphocytes of chronic HIV-1 patients. MedChemComm 2017, 8, 1806. [Google Scholar] [CrossRef] [PubMed]
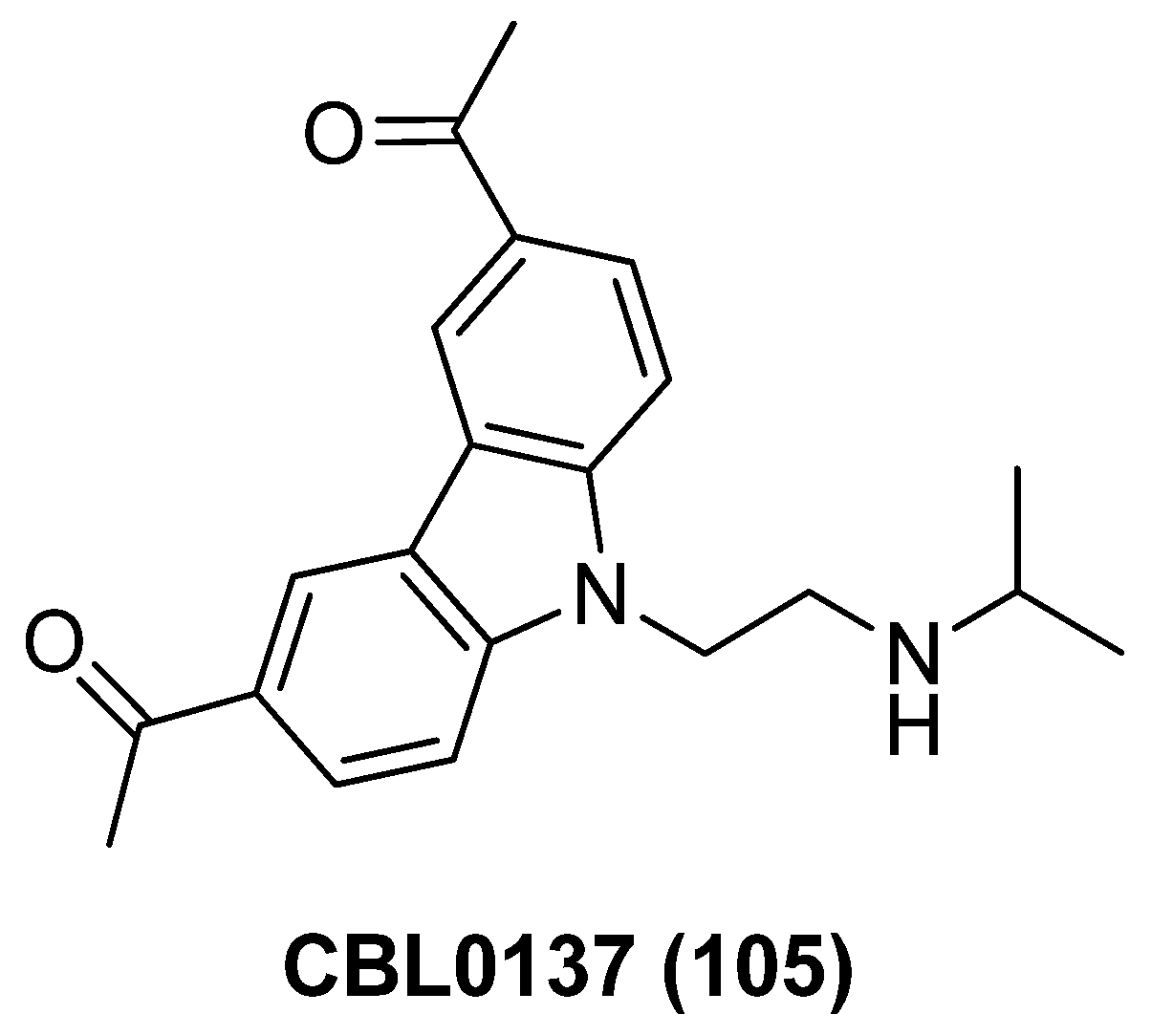
- Gasparian, A.V.; Burkhart, C.A.; Purmal, A.A.; Brodsky, L.; Pal, M.; Saranadasa, M.; Bosykh, D.A.; Commane, M.; Guryanova, O.A.; Pal, S.; et al. Curaxins: Anticancer compound that simultaneously suppress NF-κB and activate p53 by targeting FACT. Sci. Transl. Med. 2011, 3, 95ra74. [Google Scholar] [CrossRef]
- Jean, M.J.; Zhou, D.; Fiches, G.; Kong, W.; Huang, H.; Purmal, A.; Gurova, K.; Santoso, N.G.; Zhu, J. Curaxin CBL0137 has the potential to reverse HIV-1 latency. J. Med. Virol. 2019, 91, 1571–1576. [Google Scholar] [CrossRef]
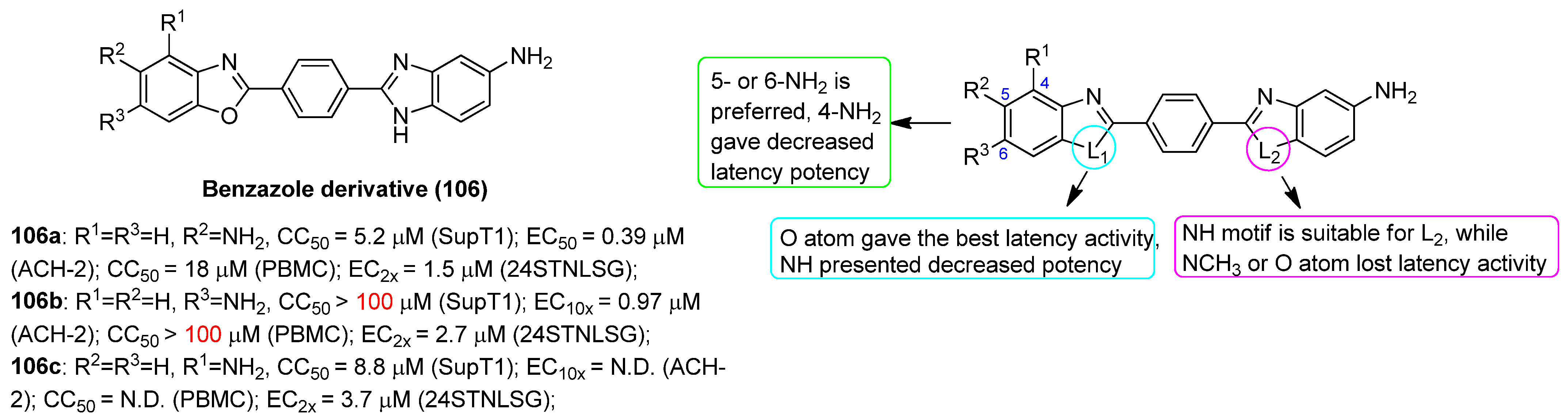
- Graci, J.D.; Michaels, D.; Chen, G.; Schiralli Lester, G.M.; Nodder, S.; Weetall, M.; Karp, G.M.; Gu, Z.; Colacino, J.M.; Henderson, A.J. Identification of benzazole compounds that induce HIV-1 transcription. PLoS ONE 2017, 12, e0179100. [Google Scholar] [CrossRef] [PubMed]
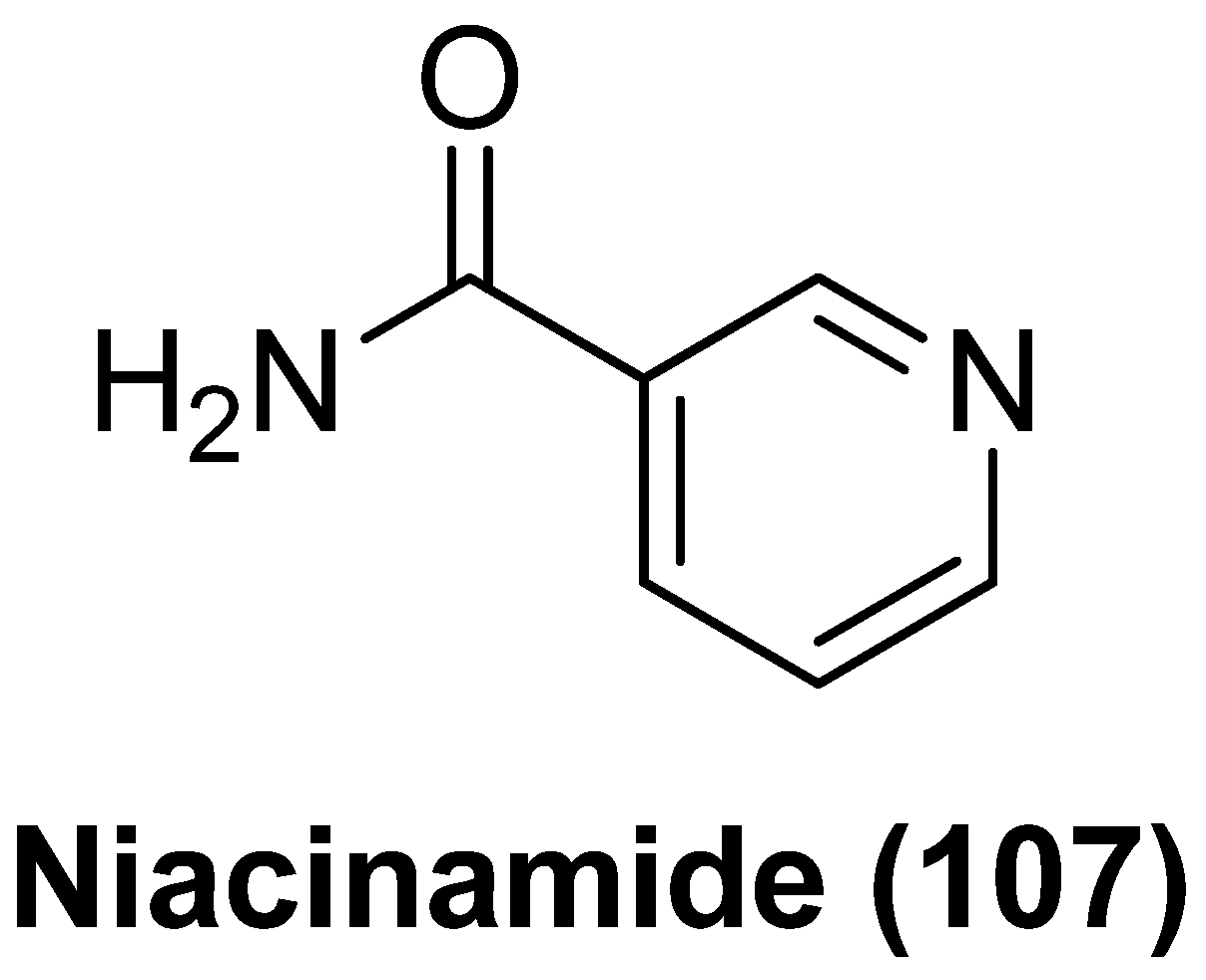
- Samer, S.; Arif, M.S.; Giron, L.B.; Zukurov, J.P.L.; Hunter, J.; Santillo, B.T.; Namiyama, G.; Galinskas, J.; Komninakis, S.V.; Oshiro, T.M.; et al. Nicotinamide activates latent HIV-1 ex vivo in ART suppressed individuals, revealing higher potency than the association of two methyltransferase inhibitors, chaetocin and BIX01294. Braz. J. Infect. Dis. 2020, 24, 150–159. [Google Scholar] [CrossRef]
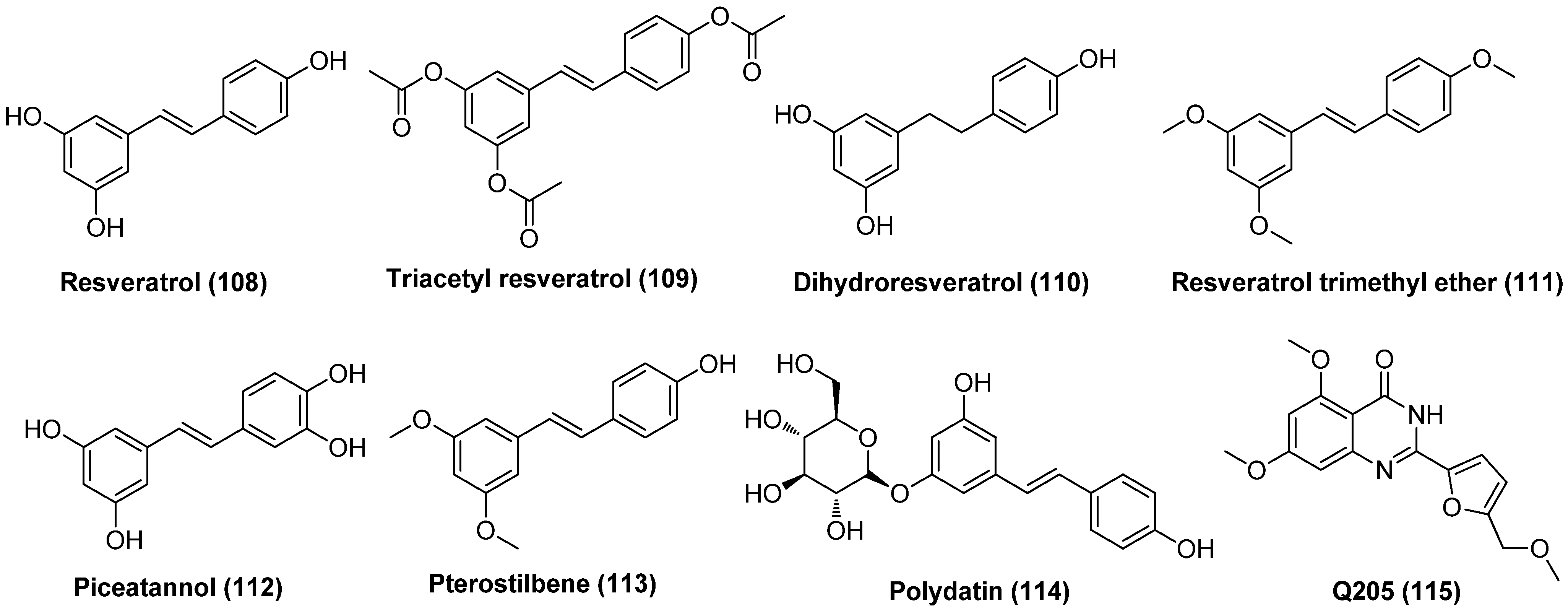
- Zeng, X.; Pan, X.; Xu, X.; Lin, J.; Que, F.; Tian, Y.; Li, L.; Liu, S. Resveratrol reactivates latent HIV through increasing histone acetylation and activating heat shock factor 1. J. Agric. Food Chem. 2017, 65, 4384–4394. [Google Scholar] [CrossRef]
- Liang, T.; Wu, Z.; Li, Y.; Li, C.; Zhao, K.; Qiao, X.; Duan, H.; Zhang, X.; Liu, S.; Xi, B.; et al. A synthetic resveratrol analog termed Q205 reactivates latent HIV-1 through activation of P-TEFb. Biochem. Pharmacol. 2022, 197, 114901. [Google Scholar] [CrossRef] [PubMed]
- Covino, D.A.; Desimio, M.G.; Doria, M. Combinations of histone deacetylase inhibitors with distinct latency reversing agents variably affect HIV reactivation and susceptibility to NK cell-mediated killing of T cells that exit viral latency. Int. J. Mol. Sci. 2021, 22, 6654. [Google Scholar] [CrossRef] [PubMed]
- Albert, B.J.; Niu, A.; Ramani, R.; Marshall, G.R.; Wender, P.A.; Williams, R.M.; Ratner, L.; Barnes, A.B.; Kyei, G.B. Combinations of isoform-targeted histone deacetylase inhibitors and bryostatin analogues display remarkable potency to activate latent HIV without global T-cell activation. Sci. Rep. 2017, 7, 7456. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Lai, W.-H.; Zhu, L.; Li, W.; Wei, L.; Lee, K.-H.; Xie, L.; Chen, C.-H. Elimination of HIV-1 latently infected cells by gnidimacrin and a selective HDAC inhibitor. ACS Med. Chem. Lett. 2018, 9, 268–273. [Google Scholar] [CrossRef]
- Curreli, F.; Ahmed, S.; Benedict Victor, S.M.; Debnath, A.K. Identification of combinations of protein kinase C activators and histone deacetylase inhibitors that potently reactivate latent HIV. Viruses 2020, 12, 609. [Google Scholar] [CrossRef]
- Stoszko, M.; Al-Hatmi, A.M.S.; Skriba, A.; Roling, M.; Ne, E.; Crespo, R.; Mueller, Y.M.; Najafzadeh, M.J.; Kang, J.; Ptackova, R.; et al. Gliotoxin, identified from a screen of fungal metabolites, disrupts 7SK snRNP, releases P-TEFb, and reverses HIV-1 latency. Sci. Adv. 2020, 6, eaba6617. [Google Scholar] [CrossRef]
- Moranguinho, I.; Valente, S.T. Block-and-lock: New horizons for a cure for HIV-1. Viruses 2020, 12, 1443. [Google Scholar] [CrossRef]
- Vansant, G.; Bruggemans, A.; Janssens, J.; Debyser, Z. block-and-lock strategies to cure HIV infection. Viruses 2020, 12, 84. [Google Scholar] [CrossRef]
- Ahlenstiel, C.; Mendez, C.; Lim, S.T.H.; Marks, K.; Turville, S.; Cooper, D.A.; Kelleher, A.D.; Suzuki, K. Novel RNA duplex locks HIV-1 in a latent state via chromatin-mediated transcriptional silencing. Mol. Ther. Nucl. Acids. 2015, 4, e261. [Google Scholar] [CrossRef]
- Umotoy, J.C.; de Taeye, S.W. Antibody conjugates for targeted therapy against HIV-1 as an emerging tool for HIV-1 cure. Front. Immunol. 2021, 12, 708806. [Google Scholar] [CrossRef]
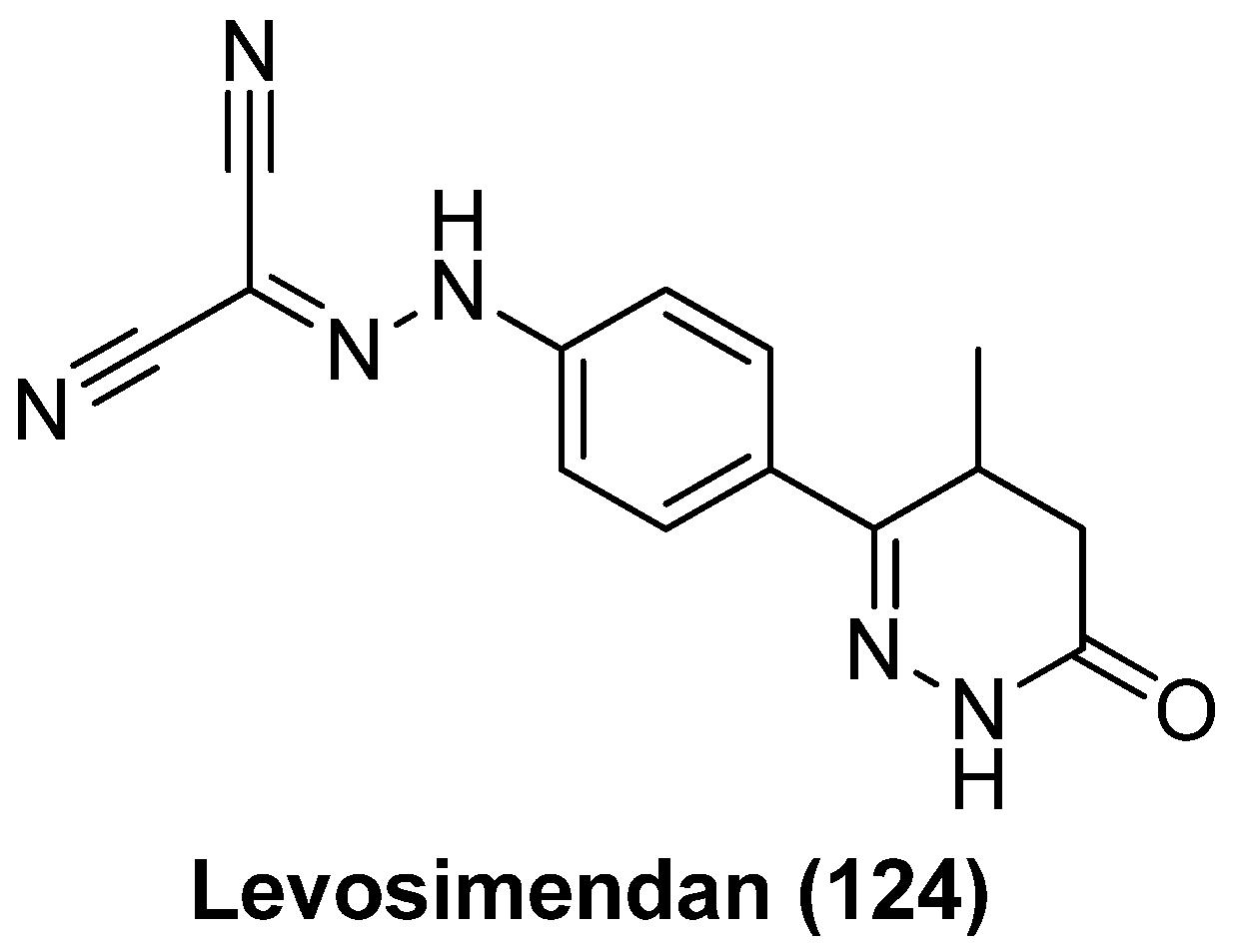
- Hayashi, T.; Jean, M.; Huang, H.; Simpson, S.; Santoso, N.G.; Zhu, J. Screening of an FDA-approved compound library identifies levosimendan as a novel anti-HIV-1 agent that inhibits viral transcription. Antiviral Res. 2017, 146, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Mori, L.; Valente, S.T. The block-and-lock strategy for human immunodeficiency virus cure: Lessons learned from didehydro–Cortistatin A. J. Infect. Dis. 2021, 223 (Suppl. 1), S46–S53. [Google Scholar] [CrossRef] [PubMed]































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.-K.; Wei, L.; Hu, W.; Yu, P.-X.; Li, Z.; Yu, H.-P.; Li, X. Medicinal Chemistry of Anti-HIV-1 Latency Chemotherapeutics: Biotargets, Binding Modes and Structure-Activity Relationship Investigation. Molecules 2023, 28, 3. https://doi.org/10.3390/molecules28010003
Wang Y-K, Wei L, Hu W, Yu P-X, Li Z, Yu H-P, Li X. Medicinal Chemistry of Anti-HIV-1 Latency Chemotherapeutics: Biotargets, Binding Modes and Structure-Activity Relationship Investigation. Molecules. 2023; 28(1):3. https://doi.org/10.3390/molecules28010003
Chicago/Turabian StyleWang, Yan-Kai, Long Wei, Wei Hu, Pei-Xia Yu, Zhong Li, Hai-Peng Yu, and Xun Li. 2023. "Medicinal Chemistry of Anti-HIV-1 Latency Chemotherapeutics: Biotargets, Binding Modes and Structure-Activity Relationship Investigation" Molecules 28, no. 1: 3. https://doi.org/10.3390/molecules28010003
APA StyleWang, Y.-K., Wei, L., Hu, W., Yu, P.-X., Li, Z., Yu, H.-P., & Li, X. (2023). Medicinal Chemistry of Anti-HIV-1 Latency Chemotherapeutics: Biotargets, Binding Modes and Structure-Activity Relationship Investigation. Molecules, 28(1), 3. https://doi.org/10.3390/molecules28010003